Introduction
Chronic myelogenous leukemia (CML) is a chronic
myeloproliferative disorder, which causes uncontrolled growth of
immature myeloid cells (1). The BCR
RhoGEF and GTPase activating protein (BCR)-ABL proto-oncogene 1
non-receptor tyrosine kinase (ABL) gene rearrangement is the main
characteristic of CML, which expresses the oncogenic fusion protein
BCR-ABL (2). BCR-ABL is a
constitutively active tyrosine kinase, which activates multiple
signaling pathways and consequently promotes malignant
transformation, including uncontrolled cell proliferation (3), abnormal cell adhesion (4), and resistance to typical apoptotic
inducer anti-leukemic drugs (5,6). Thus,
formation of the BCR-ABL fusion gene serves an essential role in
the pathogenesis of CML (7).
Previously, imatinib, a specific ABL kinase inhibitor, was
established as the standard treatment for CML (8).
In addition to targeting BCR-ABL kinase, previous
studies have revealed that several pathways are important for CML
cell survival, which may be potential targets for developing novel
anti-leukemia drugs (9,10). Among these pathways, AMP-activated
protein kinase (AMPK) signaling has been reported to possess
anti-leukemia activity (11). AMPK
serves an important role in energy metabolism in response to
changes in cellular fuel levels (12). Furthermore, mounting evidences have
suggested that AMPK could be a target for tumor prevention and
treatment (7). Previous studies have
revealed that AMPK activators exhibit anti-leukemia effects in CML
cells by triggering apoptosis and autophagy (3,4). Thus, the
identification and characterization of AMPK activators is
important, and beneficial for the development of potential
anti-leukemia drugs.
The present study aimed to investigate whether
adenine, a purine compound that induces AMPK activation, exhibits
anti-leukemia effects on human CML cells. Furthermore, the
underlying mechanism, with emphasis on AMPK signaling was
evaluated. The results revealed that adenine suppressed cell
viability of K562 cells and induced accumulation of G0/G1 phase
cells. In parallel, it was observed that adenine triggered K562
cells autophagy. Finally, it was demonstrated that suppressed cell
viability, accumulation of G0/G1 phase cells
and induction of autophagy were associated with AMPK activation in
response to adenine.
Materials and methods
Reagents
All chemicals were obtained from Sigma-Aldrich
(Merck KGaA, Darmstadt, Germany) unless otherwise specified.
RPMI-1640 medium and fetal bovine serum (FBS) were purchased from
Invitrogen (Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Antibodies against β-actin, caspase-3, caspase-8, phosphorylated
(p)-AMPK (T172; cat. no. 2535), AMPK (cat. no. 2532),
poly-ADP-ribose polymerase (PARP) (cat. no. 9532), cell division
cycle (Cdc)25 (cat. no. 3652), cyclin-dependent kinase (CDK)2 (cat.
no. 2546), CDK4 (cat. no. 12790), CDK6 (cat. no. 13331), cyclin B
(cat. no. 4135), cyclin E (cat. no. 20808), Wee1-like protein
kinase (Wee1) (cat. no. 13084), autophagy protein (Atg)5 (cat. no.
12994), beclin-1 (cat. no. 3495) and microtubule associated protein
1 light chain 3 α (LC3) (cat. no. 3868) were purchased from Cell
Signaling Technologies, Inc. (Danvers, MA, USA). Anti-human
glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (cat. no. ab9485),
horseradish peroxidase-conjugated anti-mouse IgG (cat. no. ab6789)
and anti-rabbit IgG (cat. no. ab6721) antibodies were purchased
from Abcam (Cambridge, UK).
Cell culture and experimental
treatments
The human CML K562 cell line (CCL-243™) was obtained
from the American Type Culture Collection (Manassas, VA, USA) and
maintained in RPMI-1640 medium supplemented with 10% FBS at 37°C in
a humidified atmosphere with 5% CO2. Cells at the
density of 5×105 cells/ml were collected and were
passaged twice prior to being subcultured T-75 flasks (Corning
Incorporated, Corning, NY, USA) for subsequent treatments.
For treatments, cells were seeded in 6-well plates
at an initial density of 1×105 cells/ml and grown to
~80% confluence at 37°C. Treatments were performed by incubating
cells with various concentrations of adenine (0.5, 1.0, 2.0, 4.0 or
8.0 mM) in serum-free RPMI-1640 for 16 h. AMPK inhibition was
performed by pretreating cells with 5 µM dorsomorphin (cat. no.
P5499; Sigma-Aldrich; Merck KGaA) for 2 h, and then incubating the
pretreated cells with 4.0 mM adenine for 24 h or 48 h. Following
treatment, the cells were washed with PBS (25 mM sodium phosphate,
150 mM NaCl, pH 7.2) and collected for subsequent analyses.
Cell viability assay
Cell viability was assessed using an MTT assay
protocol as previously described (13) in the absence or presence of adenine.
After 24 or 48 h treatments, cells were harvested, and then
incubated with MTT (0.5 mg/ml) at 37°C for 4 h. The cell viability
was directly proportional to the production of formazan, which was
dissolved in isopropanol and determined by measuring the absorbance
at 570 nm using a microplate reader (SpectraMAX 360 pc; Molecular
Devices, LCC, Sunnyvale, CA, USA).
Cell cycle distribution analysis
Cells were starved for 16 h in serum-free medium,
and then cultured in fresh serum-containing medium at 37°C for 24 h
to allow cell-cycle progression. Following different treatments,
cells were fixed and analyzed by flow cytometry to determine cell
cycle distribution. At the end of each treatment, cells were
collected, fixed with 1 ml of ice-cold 70% ethanol on ice for 30
min, incubated at −20°C for 24 h and centrifuged at 380 × g at 4°C
for 5 min. Cell pellets were reacted with l ml cold staining
solution containing 20 µg/ml propidium iodide, 20 µg/ml RNase A and
1% Triton X-100, incubated at 4°C for 15 min in the dark, and then
analyzed using a FACS Calibur system with CellQuest™
software (version 2.0) (both BD Biosciences, Franklin Lakes, NJ,
USA). Representative results were acquired from three independent
experiments.
Western blot analysis
Cells were washed with PBS and then incubated with
lysis buffer (50 mM Tris-HCl, pH 7.5, 1% Nonidet P-40, 1 mM
phenylmethylsulfonyl fluoride and 1 mM NaF) containing complete
protease inhibitor cocktail (Roche Applied Science, Mannheim,
Germany) for 1 h at 4°C. The resulting supernatants were collected
for protein ßquantitation using a bicinchoninic acid kit (Pierce;
Thermo Fisher Scientific, Inc.). Crude proteins (20 µg) were
subjected to 12.5% SDS-PAGE, transferred to a nitrocellulose
membrane (EMD Millipore, Billerica, MA, USA) and then incubated
with 5% w/v skimmed milk/PBS at room temperature for 1 h to block
nonspecific binding. The blocked membrane was then incubated with
primary antibodies (1:1,000 dilution) at 25°C for 2 h, followed by
incubation with peroxidase-conjugated anti-IgG secondary antibodies
(1:2,000 dilution) at 25°C for 1 h. Detailed information for
antibodies were described in the reagents. The blocked membrane
without incubation with primary antibodies was also incubated with
secondary antibodies for specificity test, and a nonsignificant
signal was detected as the control. Signal development was
performed using an enhanced chemiluminescence reagent (EMD
Millipore). Luminescent signals were acquired and quantified using
an image analysis system (LAS-4000 with Image Reader LAS-4000
version 2.1, Fuji, Tokyo, Japan). GAPDH was used as an internal
control.
Quantification of acidic vesicular
organelles (AVOs) using acridine orange staining and flow
cytometric analysis
Cells (1×104) were incubated with
acridine orange at 25°C for 17 min, harvested with trypsin-EDTA,
and then collected in phenol red-free growth medium (Thermo Fisher
Scientific, Inc.). Green (510–530 nm) and red (650 nm) fluorescence
emission from cells illuminated with blue (488 nm) excitation light
was analyzed using FACS Calibur system using CellQuest software as
mentioned above. With acridine orange staining, bright green and
faint red fluorescence was observed in the cytoplasm and nucleolus,
respectively, and bright red fluorescence was observed in acidic
compartments (2,14). Since the intensity of the red
fluorescence is proportional to the degree of acidity, the volume
of the cellular acidic compartment could be quantified (15).
Statistical analysis
Data are expressed as the mean ± standard error of
the mean following three independent experiments. Statistical
analysis was performed by using SigmaStat (version 4.0, Systat
Software, Inc, San Jose, CA, USA). Statistical significance
analysis was determined using one-way analysis of variance followed
by Dunnett's test for multiple comparisons with the control.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Adenine suppresses the viability of
human CML K562 cells
To investigate the effect of adenine on human CML
K562 cells, cell viability was determined using MTT assay. As
presented in Fig. 1, for 24 h
treatments, the viability of K562 cells was significantly reduced
to 81.5±2.1, 75.4±2.2 and 69.3±2.5% of the control in response to
2, 4 and 8 mM adenine, respectively (all P<0.05). For 48 h
treatments, the cell viability was further reduced to 61.3±2.3 and
53.4±3.1% of the control in response to 4 and 8 mM adenine,
respectively (both P<0.01). These findings revealed that adenine
at concentrations of 2, 4 and 8 mM significantly inhibited the
viability of K562 cells.
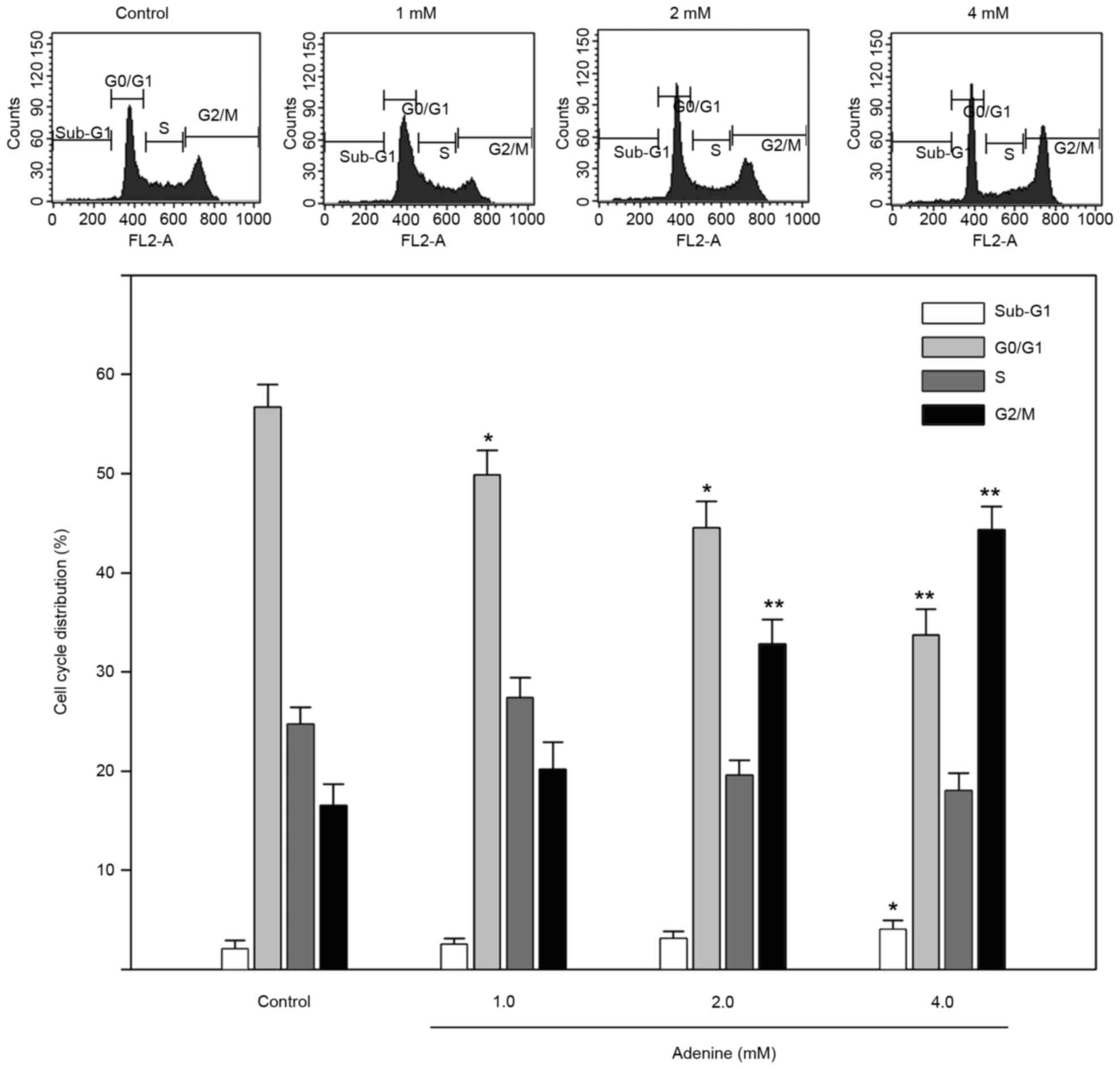
Adenine induces G2/M cell
cycle arrest and alters cell cycle regulators in human CML K562
cells
To explore the inhibited cell viability in response
to adenine, the cell cycle distribution of K562 cells treated with
adenine was determined using flow cytometric analysis. As presented
in Fig. 2, the sub-G1
phase ratio was increased by 4.1±0.7% in K562 cells treated with 4
mM adenine compared with the control group (P<0.05). In
addition, the G2/M phase ratio was significantly
increased in K562 cells treated with 2 mM (32.1±2.7%) and 4 mM
(43.8±2.1%) adenine compared with that in the untreated control
group (both P<0.01). The G0/G1 phase ratio
was significantly decreased in K562 cells treated with 2 mM
(44.6±2.7%) and 4 mM (33.8±2.4%) adenine compared with that in
untreated cells (both P<0.05).
Since cell viability and cell cycle distribution
were altered following adenine treatment, the effects of adenine on
caspase signaling cascades, and cell cycle regulators were then
investigated. As presented in Fig.
3A, cleavage of caspase-8, caspase-3 and PARP was
insignificantly affected by adenine treatments (1.0, 2.0 or 4.0
mM). Regarding cell cycle regulators, the protein expression levels
of Cyclin D1, CDK4, CDK6 and Cdc25A were markedly decreased in
response to adenine treatments (Fig.
3B). By contrast, the level of Wee1 was elevated upon adenine
stimulation. Notably, Cyclin E protein expression was
insignificantly affected by adenine. Taken together, these findings
suggest that adenine treatment induces G2/M cell cycle
arrest without involvement of the activation of caspase signaling
cascades and consequent apoptosis of K562 cells.
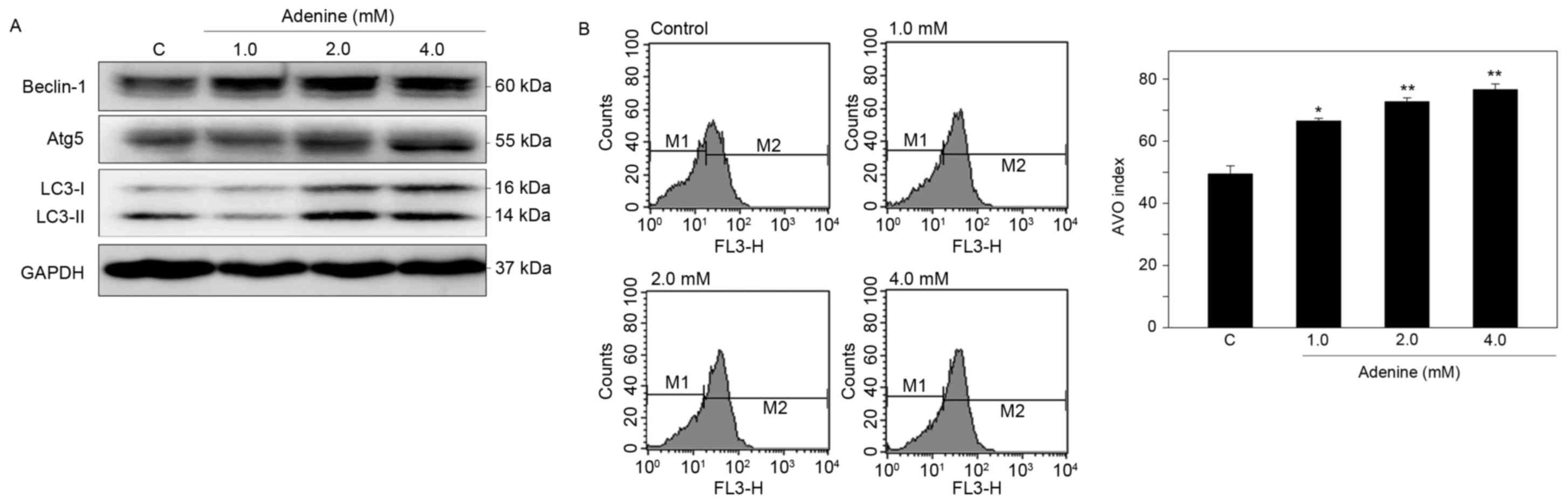
Adenine induces autophagy of human CML
K562 cells
To determine whether adenine suppresses cell
viability via autophagic cell death, induction of autophagy in K562
cells was then examined. Western blot analysis demonstrated that
adenine markedly upregulated the protein expression of Beclin-1 and
Atg5, and promoted the conversion of LC3-I to LC3-II in the cells
(Fig. 4A). In addition, using
acridine orange staining and flow cytometric quantification,
adenine was identified to significantly increase the number of AVOs
in K562 cells in a dose-dependent manner (Fig. 4B). These findings suggest that adenine
treatment promotes the formation of autophagosomes in K562
cells.
Adenine induces the phosphorylation of
AMPKα, contributing to inhibition of mTOR and autophagic signaling
cascades in K562 cells
Adenine has been reported to exert antitumoral
activity on human renal carcinoma 786-O cells via AMPK activation
(16). Thus, the roles of AMPK
signaling in autophagic cell death of K562 cells in response to
adenine were investigated. As presented in Fig. 5A, adenine markedly enhance the
phosphorylation of AMPKα at T172, reduced the phosphorylation of
mTOR at S2448 and elevated the protein expression level of
autophagic marker Atg5 compared with untreated cells. By contrast,
pretreatment with the AMPK inhibitor dorsomorphin (dm) reversed the
expression changes to p-AMPKa, p-mTOR and Atg5 in K562 cells
following exposure to adenine (Fig.
5B). In addition, pretreatment with dm significantly restored
the viability of K562 cells suppressed by 4.0 mM adenine (Fig. 5C). Collectively, these findings
suggest that the AMPK/mTOR signaling pathway is involved in
adenine-induced autophagy and consequent viability inhibition of
K562 cells.
Discussion
Clinically, the Philadelphia chromosome exists in
~95% of adults with CML and expresses the BCR-ABL fusion protein
with uncontrolled tyrosine kinase activity, resulting in abnormal
regulation of downstream signaling pathways (17). The aberrant activation of signaling
pathways, including the RAS/mitogen activated protein kinase
kinase/extracellular signal-related kinase and phosphoinositide
3-kinase/RAC-alpha serine/threonine-protein kinase pathways, may
promote cell proliferation, diminish cell apoptosis or contribute
to growth factor independence of cancer cells (18,19).
Therefore, tyrosine kinase inhibitors targeting the BCR-ABL fusion
protein, including imatinib, dasatinib and nilotinib, are used for
CML treatment, and have demonstrated improved outcomes in patients
with CML (20). However, high relapse
rates, drug resistance and high mortality rates associated with
transplantation remain challenges for CML treatment (21).
Previous studies have revealed that AMPK activators
can suppress several types of tumors, including pancreatic, bladder
and prostate cancer, through AMPK-dependent apoptosis (19–21). In
addition, AMPK has been reported as a potential target that
regulates various signaling pathways, subsequently exhibiting
antileukemia activity (22). Among
the signaling pathways regulated by AMPK, mTOR is known to perform
a central role in antitumor activity (23). Recently, several compounds, including
metformin and myrtucommulone A, have been reported to exert
antileukemia activity via activation of the AMPK/mTOR signaling
pathway (24,25). Similarly, the present results revealed
that adenine suppressed the viability of K562 cells through
activation of the AMPK/mTOR signaling pathway, resulting in
G2/M phase arrest and autophagic cell death.
Autophagy serves an important role in regulating
cellular physiology, removing senescent organelles and degrading
long-lived proteins (26,27). In starved cells, the fatty acids and
amino acids produced by hydrolysis of lipids, and proteins in
autophagolysosomes provide energy to maintain cell survival
(28). However, elongated autophagy
is proposed to trigger autophagic caspase-independent type II
programmed cell death (29). Thus,
the roles of autophagy in sustaining or killing cancer cells are
complicated. Although cytotoxicity of antitumor drugs is diminished
by autophagy to a certain extent (30), reduced expression of autophagic genes,
including Atg4, Atg5 and Atg7, has been reported to promote tumor
formation in genetically-engineered mice (31,32).
Collectively, the findings suggest that autophagy possesses tumor
suppressive activity. In the present study, it was demonstrated
that adenine can trigger autophagic cell death, but not apoptosis,
consequently suppressing the viability of K562 cells. Thus the
results of the current study indicate that adenine may serve as a
potential anti-leukemia agent.
In conclusion, the present study demonstrated that
adenine treatment significantly suppresses the viability of CML
K562 cells through activation of the AMPK/mTOR pathway and
synergistic induction of G2/M phase arrest, and
autophagy signaling. Thus, adenine represents a promising effective
antiproliferative agent against human leukemia cells.
References
|
1
|
Clarkson B, Strife A, Wisniewski D, Lambek
CL and Liu C: Chronic myelogenous leukemia as a paradigm of early
cancer and possible curative strategies. Leukemia. 17:1211–1262.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Traganos F and Darzynkiewicz Z: Lysosomal
proton pump activity: Supravital cell staining with acridine orange
differentiates leukocyte subpopulations. Methods Cell Biol.
41:185–194. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vakana E, Altman JK, Glaser H, Donato NJ
and Platanias LC: Antileukemic effects of AMPK activators on
BCR-ABL-expressing cells. Blood. 118:6399–6402. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sujobert P, Poulain L, Paubelle E,
Zylbersztejn F, Grenier A, Lambert M, Townsend EC, Brusq JM,
Nicodeme E, Decrooqc J, et al: Co-activation of AMPK and mTORC1
induces cytotoxicity in acute myeloid leukemia. Cell Rep.
11:1446–1457. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bedi A, Zehnbauer BA, Barber JP, Sharkis
SJ and Jones RJ: Inhibition of apoptosis by BCR-ABL in chronic
myeloid leukemia. Blood. 83:2038–2044. 1994.PubMed/NCBI
|
|
6
|
Klionsky DJ, Meijer AJ and Codogno P:
Autophagy and p70S6 kinase. Autophagy. 1:59–61. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim I and He YY: Targeting the
AMP-activated protein kinase for cancer prevention and therapy.
Front Oncol. 3:1752013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vakana E and Platanias LC: AMPK in BCR-ABL
expressing leukemias. Regulatory effects and therapeutic
implications. Oncotarget. 2:1322–1328. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Druker B, Okuda K, Matulonis U, Salgia R,
Roberts T and Griffin JD: Tyrosine phosphorylation of rasGAP and
associated proteins in chronic myelogenous leukemia cell lines.
Blood. 79:2215–2220. 1992.PubMed/NCBI
|
|
10
|
Gotoh A, Miyazawa K, Ohyashiki K, Tauchi
T, Boswell HS, Broxmeyer HE and Toyama K: Tyrosine phosphorylation
and activation of focal adhesion kinase (p125FAK) by BCR-ABL
oncoprotein. Exp Hematol. 23:1153–1159. 1995.PubMed/NCBI
|
|
11
|
Fernandes A, Azevedo MM, Pereira O,
Sampaio-Marques B, Paiva A, Correia-Neves M, Castro I and Ludovico
P: Proteolytic systems and AMP-activated protein kinase are
critical targets of acute myeloid leukemia therapeutic approaches.
Oncotarget. 6:31428–31440. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Long YC and Zierath JR: AMP-activated
protein kinase signaling in metabolic regulation. J Clin Invest.
116:1776–1783. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Denizot F and Lang R: Rapid colorimetric
assay for cell growth and survival. Modifications to the
tetrazolium dye procedure giving improved sensitivity and
reliability. J Immunol Methods. 89:271–277. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stankiewicz M, Jonas W, Hadas E, Cabaj W
and Douch PG: Supravital staining of eosinophils. Int J Parasitol.
26:445–446. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Paglin S, Hollister T, Delohery T, Hackett
N, McMahill M, Sphicas E, Domingo D and Yahalom J: A novel response
of cancer cells to radiation involves autophagy and formation of
acidic vesicles. Cancer Res. 61:439–444. 2001.PubMed/NCBI
|
|
16
|
Hsu CY, Lin CH, Lin JT, Cheng YF, Chen HM
and Kao SH: Purine analogue ENERGI-F706 induces apoptosis of 786-O
renal carcinoma cells via 5′-adenosine monophosphate-activated
protein kinase activation. Mol Med Rep. 12:4566–4571. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sattler M and Griffin JD: Molecular
mechanisms of transformation by the BCR-ABL oncogene. Semin
Hematol. 40 (2 Suppl 2):S4–S10. 2003. View Article : Google Scholar
|
|
18
|
Skorski T, Kanakaraj P,
Nieborowska-Skorska M, Ratajczak MZ, Wen SC, Zon G, Gewirtz AM,
Perussia B and Calabretta B: Phosphatidylinositol-3 kinase activity
is regulated by BCR/ABL and is required for the growth of
Philadelphia chromosome-positive cells. Blood. 86:726–736.
1995.PubMed/NCBI
|
|
19
|
Cortez D, Reuther G and Pendergast AM: The
Bcr-Abl tyrosine kinase activates mitogenic signaling pathways and
stimulates G1-to-S phase transition in hematopoietic cells.
Oncogene. 15:2333–2342. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mace ML, Dahl J and Jabbour EJ: Which
tyrosine-kinase inhibitor to use first in chronic phase chronic
myelogenous leukemia? Expert Opin Pharmacother. 16:999–1007. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Adekola K, Popat U and Ciurea SO: An
update on allogeneic hematopoietic progenitor cell transplantation
for myeloproliferative neoplasms in the era of tyrosine kinase
inhibitors. Bone Marrow Transplant. 49:1352–1359. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Karnevi E, Said K, Andersson R and
Rosendahl AH: Metformin-mediated growth inhibition involves
suppression of the IGF-I receptor signalling pathway in human
pancreatic cancer cells. BMC Cancer. 13:2352013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zheng QY, Jin FS, Yao C, Zhang T, Zhang GH
and Ai X: Ursolic acid-induced AMP-activated protein kinase (AMPK)
activation contributes to growth inhibition and apoptosis in human
bladder cancer T24 cells. Biochem Biophys Res Commun. 419:741–747.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sauer H, Engel S, Milosevic N, Sharifpanah
F and Wartenberg M: Activation of AMP-kinase by AICAR induces
apoptosis of DU-145 prostate cancer cells through generation of
reactive oxygen species and activation of c-Jun N-terminal kinase.
Int J Oncol. 40:501–508. 2012.PubMed/NCBI
|
|
25
|
Borthakur G, Duvvuri S, Ruvolo V, Tripathi
DN, Piya S, Burks J, Jacamo R, Kojima K, Ruvolo P, Fueyo-Margareto
J, et al: MDM2 inhibitor, nutlin 3a, induces p53 dependent
autophagy in acute leukemia by AMP kinase activation. PLoS One.
10:e01392542015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Levine B and Kroemer G: Autophagy in
aging, disease and death: The true identity of a cell death
impostor. Cell Death Differ. 16:1–2. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Settembre C, Fraldi A, Medina DL and
Ballabio A: Signals from the lysosome: A control centre for
cellular clearance and energy metabolism. Nat Rev Mol Cell Biol.
14:283–296. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gozuacik D and Kimchi A: Autophagy as a
cell death and tumor suppressor mechanism. Oncogene. 23:2891–2906.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Aredia F and Scovassi AI: Manipulation of
autophagy in cancer cells: An innovative strategy to fight drug
resistance. Future Med Chem. 5:1009–1021. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yue Z, Jin S, Yang C, Levine AJ and Heintz
N: Beclin 1, an autophagy gene essential for early embryonic
development, is a haploinsufficient tumor suppressor. Proc Natl
Acad Sci USA. 100:15077–15082. 2003. View Article : Google Scholar : PubMed/NCBI
|