Introduction
Acute promyelocytic leukemia (APL), a unique subtype
of acute myeloid leukemia, is characterized by a translocation
between chromosomes 15 and 17 that encodes the oncogenic fusion
protein promyelocytic leukemia/retinoic acid receptor-α (PML/RARα)
(1). PML-RARα has an essential role
in the development of APL by interfering with target genes that
control differentiation, proliferation and apoptosis of APL cells
(2). Considerable success in treating
APL has been achieved using all-trans retinoic acid
(3) and arsenic trioxide (4) in clinical settings. However, the
toxicity of these molecules and the prevalence of drug-resistant
forms of APL limit the clinical application of these drugs
(5). Therefore, novel therapeutics to
treat APL are urgently required.
Src homology 1 domain-containing protein tyrosine
phosphatase (SHP-1), also known as PTPN6 (6), consists of 17 exons and 16 introns and
spans ~17 kb (7). SHP-1 controls the
changes in the levels of intracellular phosphorylation, including
JAK/STAT (8). SHP-1 exerts multiple
biological functions through the alteration of several signaling
pathways (9,10). A number of agonists and inhibitors of
SHP-1 have been applied in clinical cancer therapies. For example,
γ-tocotrienol (11) and regorafenib
(12) have been used to treat breast
tumors and colorectal cancer, respectively. Studies have reported
that SHP-1 is highly expressed in normal hematopoietic cells
(13) but weakly expressed in
hematological malignancies, including Burkitt's lymphoma (14), APL (15)
and chronic myeloid leukemia (16).
Therefore, the present authors hypothesize that increases in SHP-1
expression may have notable roles in APL treatment.
Epigallocatechin-3-gallate (EGCG), a major
constituent of green tea (17)
induces cell death in AML (18) via
cellular mechanisms that currently remain unclear. A previous
report demonstrated that EGCG induced apoptosis in chronic myeloid
leukemia cells by increasing SHP-1 expression and dephosphorylating
the fusion protein breakpoint cluster region
protein-tyrosine-protein kinase ABL1 (19). An interesting question is whether
SHP-1 can be increased by EGCG in NB4 cells. Previous studies
suggested that EGCG mediates increased SHP-1 expression, which
subsequently activates the p38α mitogen-activated protein kinase
(MAPK)-Bcl-2-like protein 4 (Bax) cascade via phosphorylation
(20). The p38 MAPK signaling pathway
has a notable role in differentiation, proliferation, apoptosis and
invasion (21–23), and is also known to affect the
development of APL (24).
Therefore, the present study hypothesize that EGCG
induces apoptosis in NB4 cells by increasing SHP-1 expression and
activating the p38α MAPK-Bax cascade.
Materials and methods
Materials
EGCG was purchased from MedChem Express (Monmouth
Junction, NJ, USA). The p38MAPK inhibitor PD 169316 was purchased
from MedChem Express. The SHP-1 inhibitor NSC87877 was purchased
from Tocris Bioscience (Bristol, UK).
Cell culture
NB4 cells (Shanghai Institutes for Biological
Sciences of the Chinese Academy of Sciences, Shanghai, China) were
cultured in RPMI 1640 medium (Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) containing 10% fetal bovine serum (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 100 mg/ml
penicillin and streptomycin (Beyotime Institute of Biotechnology,
Haimen, China), in an incubator with 5% CO2 at 37°C.
Western blot analysis
The cells were collected and washed with chilled PBS
for three times. Then, the cells were lysed using
radioimmunoprecipitation assay buffer with protease and phosphatase
inhibitors (Beyotime Institute of Biotechnology). Discarding the
supernatant following centrifugation at 13,000 × g at 4°C for 30
min. Protein lysate concentrations were quantified using a
bicinchoninic acid assay kit (Beyotime Institute of Biotechnology).
Equal quantities (50 µg) of the protein were isolated by 10%
SDS-PAGE and transferred to polyvinylidene fluoride membranes. The
membranes were blocked with 5% skimmed milk suspended by
Tris-Buffered Saline with 0.05% Tween-20 (TBST) for 2 h and then
probed with anti-SHP-1 (1:1,000; cat. no. ab32559; Abcam,
Cambridge, UK), anti-p38α MAPK (1:1,000; cat. no. 9218; Cell
Signaling Technology, Inc., Danvers, MA, USA), anti-p-p38α MAPK
(1:1,000; cat. no. 09-272; Merck KGaA, Darmstadt, Germany), Bax
(1:1,000; cat. no. wl01637; Wanleibio Co., Ltd., Shanghai, China)
and β-actin (1:500; cat. no. BM0627; Boster Biological Technology,
Pleasanton, CA, USA) at 4°C overnight. Following washing with TBST,
the membranes were incubated with horseradish peroxidase-conjugated
secondary antibodies, goat anti-rabbit (cat. no. ZA-0448) and
HRP-conjugated goat anti-mouse (cat. no. ZM-0491) IgG (1:1,000;
Beijing Zhongshan Jinqiao Biotechnology Co., Ltd., Beijing, China)
for 1 h. The membranes were then washed with TBS and TBST and the
protein levels were detected by an enhanced chemiluminescence kit
(EMD Millipore, Billerica, MA, USA). β-actin (1:500; cat. no.
BM0627; Boster Biological Technology) protein levels were used as a
control to verify equal protein loading. Protein bands were
visualized using the Quantity One Software version 4.5.2 (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Cell viability assay
NB4 cells (1×104 cells/well) were seeded
into wells of a 96-well plate. NB4 cells were pretreated with
different concentrations of EGCG (0, 10, 20 or 30 µM), 10 µM
PD169316 (MedChem Express, Monmouth Junction, NJ, USA) or 10 µM
NSC87877 (Tocris Bioscience, Bristol, UK) and equal volumes of a
solvent control (PBS) for 24 h. Cell viability was quantified using
a Cell Counting kit-8 (CCK-8; 7Sea Biotech Co., Ltd., Shanghai,
China). The cell number index was calculated at 450 nm using a
spectrophotometer (Bio-Rad Laboratories, Inc.) as follows: Cell
number index=(ABS of treated/ABS of blank)/(ABS of control/ABS of
blank) ×100.
Flow cytometric analysis of
apoptosis
NB4 cells (1×104 cells/well) were seeded
into wells of a 6-well plate. NB4 cells were pretreated at 37°C
with different concentrations of EGCG (0, 10, 20 or 30 µM), 10 µM
PD169316 (MedChem Express) or 10 µM NSC87877 (Tocris Bioscience)
and equal volumes of PBS for 24 h. Cells were then collected and
washed using chilled PBS. Apoptosis was detected using propidium
iodide and annexin V double labeling assay kit (Sigma-Aldrich;
Merck KGaA) by a FACsorter (BD Biosciences, San Jose, CA, USA).
Statistical analysis
All results are expressed as the mean ± standard
error. Statistical analysis was performed using SPSS (version 23.0;
IBM Corp., Armonk, NY, USA). The data were analyzed using one-way
analysis of variance. P<0.05 was considered to indicate a
statistically significant difference.
Results
EGCG induces apoptosis in NB4
cells
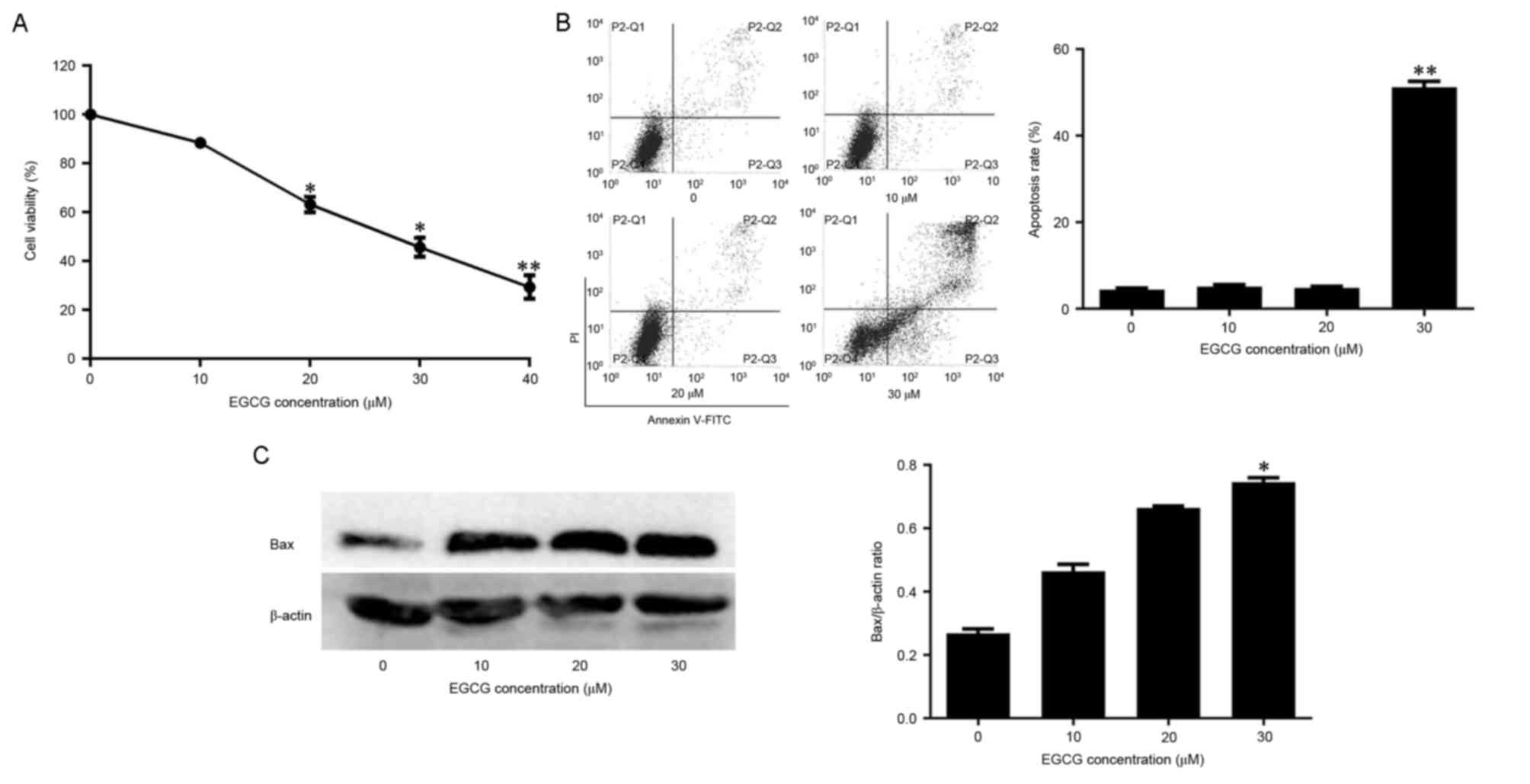
CCK-8 assay was used to detect the effect of EGCG on
the viability of NB4 cells. Cell viability was negatively
associated with EGCG concentration (Fig.
1A). Whether EGCG-induced cell death was associated with
apoptosis was also assessed. In normal cells, the
phosphatidylserine (PS) groups in the plasma membrane are directed
towards the inside of the cell. However, the PS groups are exposed
to the environment upon apoptosis (25). The propidium iodide and annexin-V
double-labeling assay indicated that this observed cell death was
due to apoptosis. The apoptotic rates of cells exposed to 10 or 20
µM EGCG did not differ significantly from the apoptotic rate of the
control group. However, the apoptotic rate of cells exposed to 30
µM EGCG was significantly higher compared with the control group
(Fig. 1B). The levels of the
apoptosis-associated protein Bax were also quantified in
EGCG-treated cells. Bax expression increased with EGCG in a
dose-dependent manner (Fig. 1C).
Therefore, these data suggest that EGCG was able to induce
apoptosis in NB4 cells.
EGCG increases SHP-1 expression and
levels of phosphorylated (p)-p38α MAPK
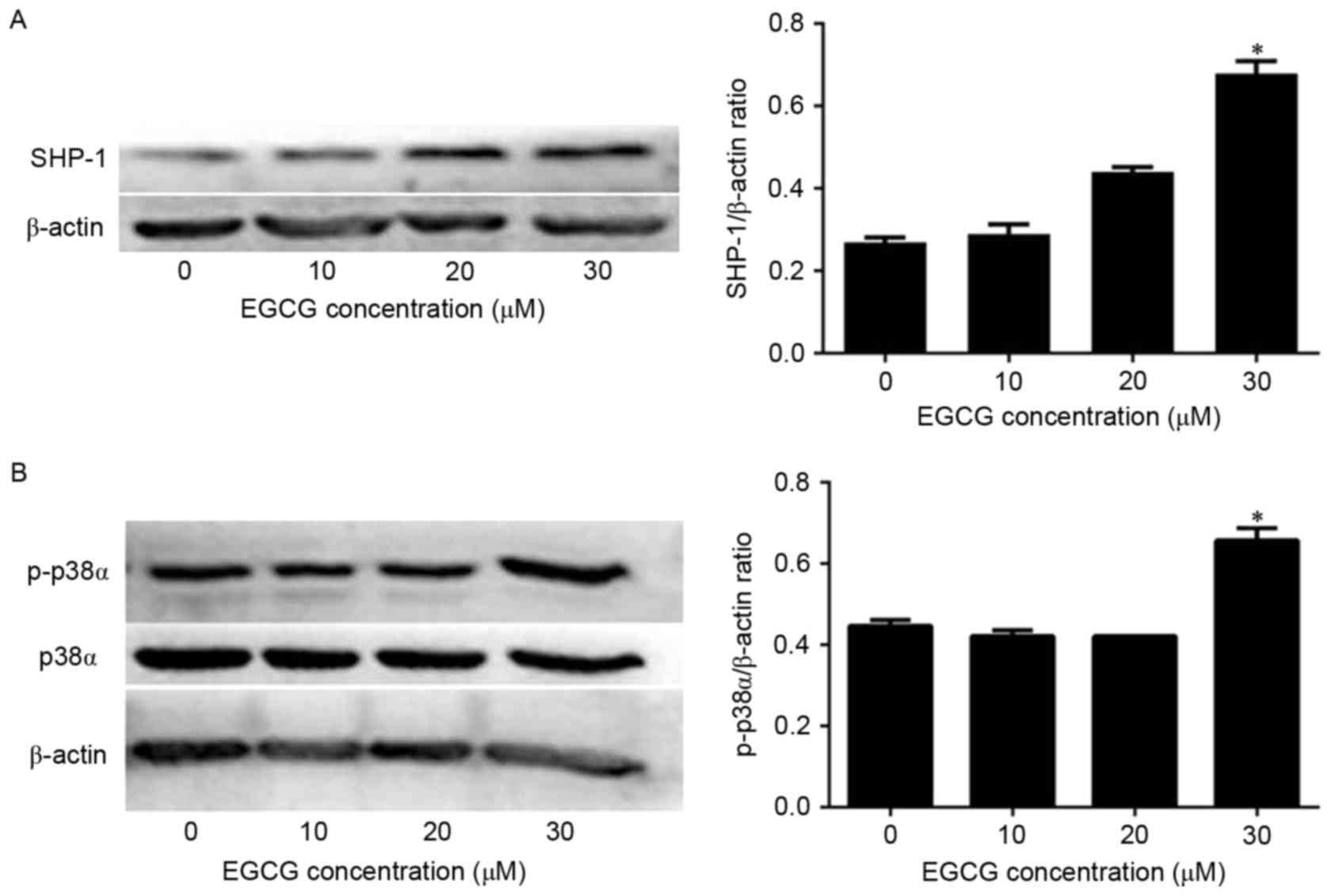
Next, the mechanism of EGCG-induced apoptosis in NB4
cells was investigated by quantifying SHP-1 expression via western
blot analysis. SHP-1 protein levels were not affected when the
cells were treated with 10 or 20 µM EGCG compared with the
expression in untreated cells. However, SHP-1 expression was
significantly increased when cells were treated with 30 µM EGCG
compared with the expression in untreated cells (Fig. 2A). Since p38α MAPK acts downstream of
SHP-1, the present authors hypothesized that p38α MAPK levels would
also be affected by EGCG treatment. In fact, p38α MAPK expression
was not affected by EGCG treatment (Fig.
2B). However, p-p38α MAPK levels were significantly increased
when the cells were treated with 30 µM EGCG compared with the
expression in untreated cells (Fig.
2B). Therefore, these data indicate that EGCG was able to
increase SHP-1 expression and trigger the phosphorylation of p38α
MAPK to p-p38α MAPK in NB4 cells.
Inhibition of p38α MAPK partially
blocks EGCG-induced NB4 cell apoptosis
As p-p38α MAPK levels were increased by EGCG
treatment, the role of p38α MAPK in EGCG-induced NB4 cell apoptosis
was investigated further. NB4 cells were pretreated with PD169316,
an inhibitor of p38 MAPK, and subsequently treated with EGCG.
Protein levels of p38α MAPK were not markedly affected by the
combined treatment compared with the levels in cells treated with
EGCG alone, but p-p38α MAPK levels were reduced compared with the
levels in cells treated with EGCG alone (Fig. 3A). However, the viability of NB4 cells
when treated with PD169316 and EGCG increased compared with the
viability of cells treated with EGCG alone (Fig. 3B). Since PD169316 treatment increased
EGCG-induced viability of NB4 cells, whether PD169316 affected
EGCG-induced apoptosis was assessed. The apoptotic rate of PD169316
inhibitor and EGCG-pretreated cells was reduced compared with the
rate of the cells treated with EGCG alone (Fig. 3C). Protein levels of the
apoptosis-associated protein Bax were also reduced in the PD169316
inhibitor and EGCG-pretreated cells in comparison with the cells
treated with EGCG alone (Fig. 3D).
Therefore, these data suggest that EGCG may induce apoptosis via
p38α MAPK in NB4 cells.
 | Figure 3.Effects of p38α inhibition on
EGCG-mediated apoptosis and protein expression levels of Bax and
p-p38α in NB4 cells. NB4 cells were pretreated with 10 µM PD169316
for 0.5 h and then treated with 30 µM EGCG for 24 h. (A and D)
Western blot analysis was used to detect the protein expression
level of p-p38α and Bax. (B) Cell-Counting Kit-8 assay was used to
measure the cell viability of NB4 cells. (C) Flow cytometric
analysis was used to determine apoptotic rate in NB4 cells. a,
EGCG− PD169316−; b, EGCG+
PD169316−; c, EGCG+ PD169316+; d,
EGCG− PD169316+. All experiments were
performed in triplicate. *P<0.05 vs. control group. EGCG,
epigallocatechin-3-gallate; Bax, Bcl-2-like protein 4; E, 30 µM
EGCG. P, 10 µM PD169316; PI, propidium iodide; FITC, fluorescein
isothiocyanate. |
Inhibition of SHP-1 partially blocks
EGCG-induced apoptosis and decreases levels of p-p38α MAPK and
Bax
As the levels of SHP-1 were increased by EGCG
treatment, the effect of SHP-1 inhibition on EGCG-induced NB4
apoptosis was subsequently investigated. When NB4 cells were
pretreated with NSC87877 and subsequently treated with EGCG, SHP-1
protein levels were reduced compared with the levels in the cells
treated with EGCG alone (Fig. 4A).
Similarly, the viability of NSC87877 inhibitor and EGCG-pretreated
NB4 cells increased (Fig. 4B).
Notably, pretreatment with NSC87877 reduced the EGCG-induced
apoptotic rate of NB4 cells, compared with the apoptotic rate of
cells treated with EGCG alone (Fig.
4C). To investigate the potential mechanisms underlying changes
in viability and apoptotic rates upon SHP-1 inhibition, the changes
in expression of proteins associated with the apoptotic cascades
were quantified.
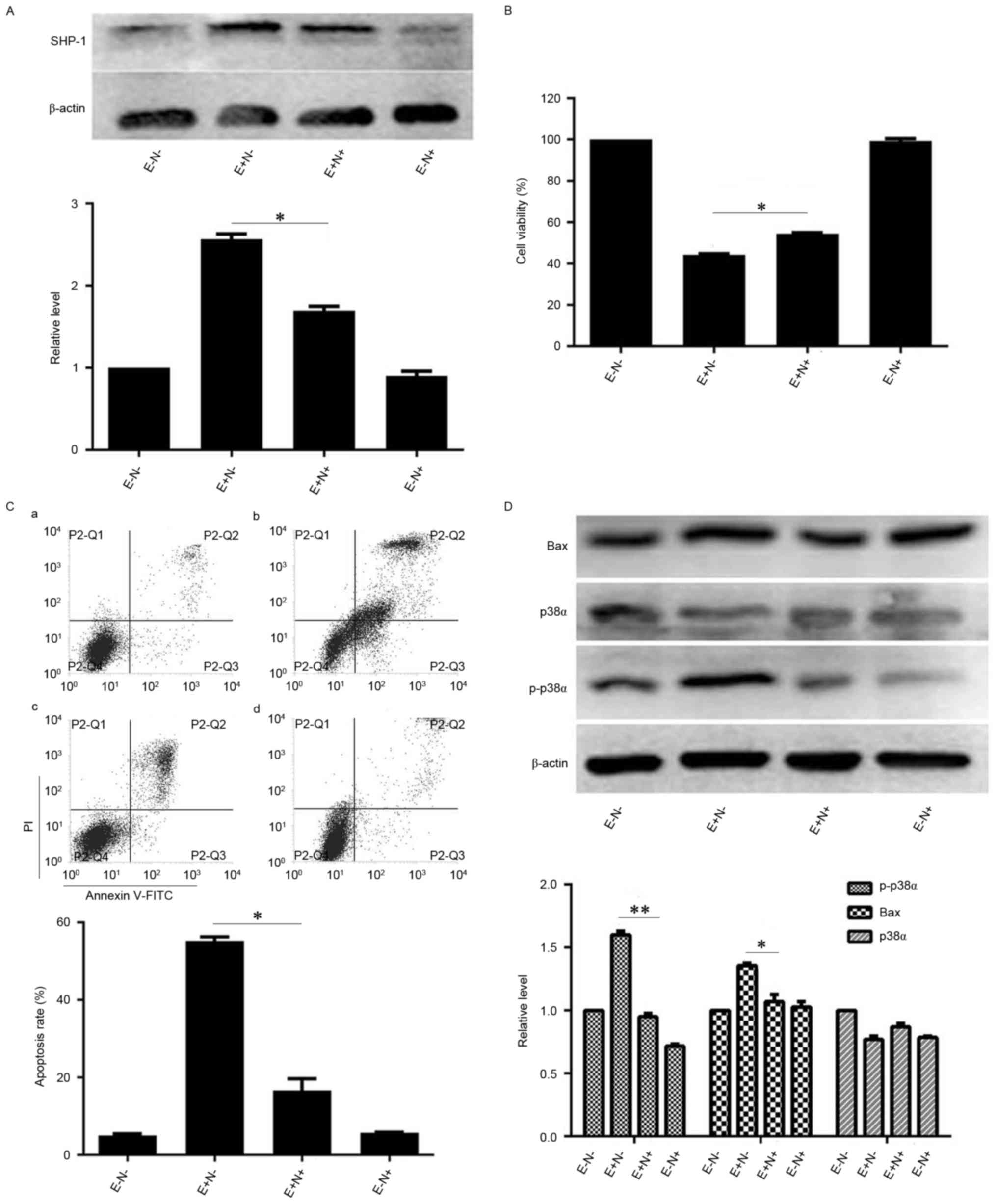 | Figure 4.Effects of SHP-1 inhibition on
EGCG-mediated apoptosis and expression levels of associated
proteins in NB4 cells. NB4 cells were pretreated with 10 µM
NSC87877 for 0.5 h and then treated with 30 µM EGCG for 24 h. (A)
Western blot analysis was used to detect the expression level of
SHP-1. (B) Cell Counting Kit-8 assay was used to measure the
viability of NB4 cells. (C) Flow cytometric analysis was used to
determine the apoptotic rate of NB4 cells. a, EGCG−
NSC87877−; b, EGCG+ NSC87877−; c,
EGCG+ NSC87877+; d, EGCG−
NSC87877+. (D) p38α, p-p38α and Bax protein levels were
assessed by western blot analysis. All experiments were performed
in triplicate. *P<0.05, **P<0.01 vs. control group. EGCG,
epigallocatechin-3-gallate; SHP-1, Src homology region 2
domain-containing phosphatase-1; p38α, p38α mitogen activated
protein kinase; E, 30 µM EGCG. N, 10 µM NSC87877; p-,
phosphorylated. |
When the cells were pretreated with SHP-1 inhibitor
and EGCG, the levels of p-p38α MAPK and Bax proteins were lower
compared with the expression in the cells that were treated with
EGCG alone. However, the expression level of p38α MAPK was
unaffected by pretreatment with SHP-1 inhibitor (Fig. 4D). Therefore, these data suggest that
SHP-1 may have an important role in EGCG-induced NB4 cell
apoptosis.
Discussion
The purpose of the present study was to investigate
whether EGCG induces apoptosis of NB4 cells through a SHP-1-p38α
MAPK-Bax cascade. EGCG treatment increased the levels of p-p38α
MAPK and Bax expression compared with control group, although p38α
MAPK expression was unaffected. The observed increase in p-p38α
MAPK and Bax expression was associated with the expression level of
SHP-1. It was observed that the inhibition of p38α MAPK was able to
reduce EGCG-induced apoptosis of NB4 cells. Additionally,
inhibition of SHP-1 reduced EGCG-induced apoptosis of NB4 cells and
EGCG-mediated increase in p-p38α MAPK and Bax expression.
EGCG, a catechin, has been demonstrated to exhibit
antitumor activities in multiple studies on solid tumor (26,27) and
leukemia (28,29) cells, particularly in APL cells
(30). EGCG mediates its
anti-leukemic activity primarily through the induction of
apoptosis, which has been indicated by increased levels of Bax in
this study (30). However, the exact
mechanisms underlying antitumor activities in APL were unclear.
The present study reports, to the best of our
knowledge for the first time, that SHP-1 expression is increased in
NB4 cells treated with EGCG, suggesting that SHP-1 has a pivotal
role in mediating the antitumor activity of EGCG. Inhibition of
SHP-1 partially blocked EGCG-induced apoptosis and triggered a
reduction in the levels of p-p38αMAPK and Bax. SHP-1 is known to be
a key modulator of protein phosphorylation levels in cells, and
protein phosphorylation has an important role in numerous
biological functions, including the differentiation, apoptosis and
invasion of cells (9,10). Therefore, on the basis of the findings
detailed in the present study, the present authors hypothesize that
SHP-1 contributes to EGCG-induced apoptosis by modifying the
phosphorylation patterns of key apoptosis regulators, including
p38α MAPK. Although a number of agonists and inhibitors of SHP-1
have been employed to treat solid tumors, these treatments are
rarely used for leukemia (11,12). The
finding that SHP-1 affects apoptosis in NB4 cells suggests that
agonists and inhibitors of SHP-1, either alone or combination with
EGCG, may also be used to treat leukemia in the future.
As SHP-1 has an important role in EGCG-induced
apoptosis of NB4 cells, the downstream mechanism of SHP-1 was
investigated. A number of studies indicated that p38α MAPK is a
downstream target of SHP-1 (31,32).
However, whether the p38α MAPK signaling pathway contributes to
EGCG-induced NB4 cell apoptosis has not been demonstrated. In the
present study, p38α MAPK was activated upon treatment with EGCG. In
addition, pretreatment with PD169316 (p38α MAPK inhibitor)
partially blocked EGCG-induced apoptosis of NB4 cells and decreased
Bax expression. Therefore, the findings of the present study
indicate that p38α MAPK activation was associated with apoptosis in
EGCG-treated NB4 cells. However, whether p38α MAPK is activated
directly by SHP-1 remains unclear. Further investigation is
therefore required to understand the association between SHP-1 and
p38α MAPK better.
In conclusion, the present study revealed the
molecular mechanism underlying EGCG-induced apoptosis in NB4 cells.
Although the effect of EGCG on APL cells had been studied
previously, the findings of the present study indicate that
EGCG-mediated apoptosis in NB4 cells is dependent on the SHP-1-p38α
MAPK-Bax cascade.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81171658)
and the Natural Science Foundation of Major Project of Chongqing of
China (grant no. 2011BA5037).
References
|
1
|
de Thé H, Le Bras M and
Lallemand-Breitenbach V: The cell biology of disease: Acute
promyelocytic leukemia, arrsenic, and PML bodies. J Cell Biol.
198:11–21. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Laurenzana A, Pettersson F and Miller WH:
Role of PML/RARAα in the pathogenesis of APL. Drug Discov Today Dis
Mech. 3:499–505. 2006. View Article : Google Scholar
|
|
3
|
Congleton J, MacDonald R and Yen A: Src
inhibitors, PP2 and dasatinib, increase retinoic acid-induced
association of Lyn and c-Raf (S259) and enhance MAPK-dependent
differentiation of myeloid leukemia cells. Leukemia. 26:1180–1188.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liang H, Li X, Wang L, Yu S, Xu Z, Gu Y,
Pan Z, Li T, Hu M, Cui H, et al: MicroRNAs contribute to
promyelocyte apoptosis in As2O3-treated APL cells. Cell Physiol
Biochem. 32:1818–1829. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Petrie K, Zelent A and Waxman S:
Differentiation therapy of acute myeloid leukemia: Past, present
and future. Curr Opin Hematol. 16:84–91. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Neel BG, Gu H and Pao L: The “Shp”ing
news: SH2 domain-containing tyrosine phosphatases in cell
signaling. Trends Biochem Sci. 28:284–293. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stocco DR and Shen SH: Human protein
tyrosine phosphatase 1C (PTPN6) gene structure: Alternate promoter
usage and exon skipping generate multiple transcripts. Genomics.
27:165–173. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tseng CYLM, Su JC, Chang KC, Chu PY, Tai
WT, Shiau CW and Chen KF: Novel sorafenib analogues induces
apoptosis through SHP-1 dependent STAT3 inactivation in human
breast cancer cells. Breast Cancer Res. 15:R632013.PubMed/NCBI
|
|
9
|
Dong G, You M, Ding L, Fan H, Liu F, Ren D
and Hou Y: STING negatively regulates Double-Stranded DNA-activated
JAK1-STAT1 signaling via SHP-1/2 in B cells. Mol Cells. 38:441–451.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Al-Jamal HA, Jusoh Mat SA, Hassan R and
Johan MF: Enhancing SHP-1 expression with 5-azacytidine may inhibit
STAT3 activation and confer sensitivity in lestaurtinib
(CEP-701)-resistant FLT3-ITD positive acute myeloid leukemia. BMC
Canner. 15:8692015. View Article : Google Scholar
|
|
11
|
Xiong A, Yu W, Liu Y, Sanders BG and Kline
K: Elimination of ALDH+ breast tumor initiating cells by
docosahexanoic acid and/or gamma tocotrienol through SHP-1
inhibition of Stat3 signaling. Mol Carcinog. 55:420–430. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fan LC, Teng HW, Shiau CW, Lin H, Hung MH,
Chen YL, Huang JW, Tai WT, Yu HC and Chen KF: SHP-1 is a target of
regorafenib in colorectal cancer. Oncatarget. 5:6243–6251. 2014.
View Article : Google Scholar
|
|
13
|
Nakase K, Cheng J, Zhu Q and Marasco WA:
Mechanisms of SHP-1 P2 promoter regulation in hematopoietic cells
and its silencing in HTLV-1-transformed T cells. J Leukoc Biol.
85:165–174. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Delibrias CC, Floettmann JE, Rowe M and
Fearon DT: Downregulated expression of SHP-1 in Burkitt lymphomas
and germinal center B lymphocytes. J Exp Med. 186:1575–1583. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Uesugi Y, Fuse I, Toba K, Kishi K,
Furukawa T, Koike T and Aizawa Y: Involvement of SHP-1, a
phosphotyrosine phosphatase, during myeloid cell differentiation in
acute promyelocytic leukemia cell lines. Eur J Haematol.
62:239–245. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Amin HM, Hoshino K, Yang H, Lin Q, Lai R
and Garcia-Manero G: Decreased expression level of SH2
domain-containing protein tyrosine phosphatase-1 (Shp1) is
associated with progression of chronic myeloid leukaemia. J Pathol.
212:402–410. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nakazato T, Ito K, Miyakawa Y, Kinjo K,
Yamada T, Hozumi N, Ikeda Y and Kizaki M: Catechin, a green tea
component, rapidly induces apoptosis of myeloid leukemia cells via
modulation of reactive oxygen species production in vitro
andinhibits tumor growth in vivo. Haematologica. 90:317–325.
2005.PubMed/NCBI
|
|
18
|
Britschgi A, Simon HU, Tobler A, Fey MF
and Tschan MP: Epigallocatechin-3-gallate induces cell death in
acute myeloid leukaemia cells and supports all-trans retinoic
acid-induced neutrophil differentiation via death-associated
protein kinase 2. Br J Haematol. 149:55–64. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jung JH, Yun M, Choo EJ, Kim SH, Jeong MS,
Jung DB, Lee H, Kim EO, Kato N, Kim B, et al: A derivative of
epigallocatechin-3-gallate induces apoptosis via SHP-1-mediated
suppression of BCR-ABL and STAT3 signalling in chronic myelogenous
leukaemia. Br J Pharmacol. 172:3565–3578. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Geraldes P, Hiraoka-Yamamoto J, Matsumoto
M, Clermont A, Leitges M, Marette A, Aiello LP, Kern TS and King
GL: Activation of PKC-delta and SHP-1 by hyperglycemia causes
vascular cell apoptosis and diabetic retinopathy. Nat Med.
15:1298–1306. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Harsha Raj M, Yashaswini B, Rössler J and
Salimath BP: Combinatorial treatment with anacardic acid followed
by TRAIL augments induction of apoptosis in TRAIL resistant cancer
cells by the regulation of p53, MAPK and NFκβ pathways. Apoptosis.
21:578–593. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang JG, Yang S, Qiao J, Li T, Yang S and
Hong Y: Macrophage migration inhibitory factor regulating the
expression of VEGF-C through MAPK signal pathways in breast cancer
MCF-7 cell. World J Surg Oncol. 14:512016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rodríguez-Berriguete G, Torrealba N,
Fraile B, Paniagua R and Royuela M: Epidermal growth factor induces
p38 MAPK-dependent G0/G1-to-S transition in prostate cancer cells
upon androgen deprivation conditions. Growth Factor. 34:5–10. 2016.
View Article : Google Scholar
|
|
24
|
Mandegary A, Hosseini R, Ghaffari SH,
Alimoghaddam K, Rostami S, Ghavamzadeh A and Ghahremani MH: The
expression of p38, ERK1 and Bax proteins has increased during the
treatment of newly diagnosed acute promyelocytic leukemia with
arsenic trioxide. Ann Oncol. 21:1884–1890. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tyurin VA, Balasubramanian K, Winnica D,
Tyurina YY, Vikulina AS, He RR, Kapralov AA, Macphee CH and Kagan
VE: Oxidatively modified phosphatidylserines om the surface of
apoptotic cells are essential phagocytic ‘eat-me’ signals: Cleavage
and inhibition of phagocytosis by Lp-PLA2. Cell Death Differ.
21:825–835. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Toden S, Tran HM, Tovar-Camargo OA,
Okugawa Y and Goel A: Epigallocatechin-3-gallate targets cancer
stem-like cells and enhances 5-fluorouracil chemosensitivity in
colorectal cancer. Oncotarget. 7:16158–16171. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li M, Li JJ, Gu QH, An J, Cao LM, Yang HP
and Hu CP: EGCG induces lunch canner A549 cells apoptosis by
regulating Ku70 acetylation. Oncol Rep. 35:2339–2347. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tofolean IT, Ganea C, Ionescu D, Filippi
A, Garaiman A, Goicea A, Gaman MA, Dimancea A and Baran I: Cellular
determinants involving mitochondrial dysfunction, oxidative stress
and apoptosis correlate with the synergic cytotoxicity of
epigallocatechin-3-gallate and menadione in human leukemia Jurkat T
cells. Pharmacol Res. 103:300–317. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vézina A, Chokor R and Annabi B: EGCG
targeting efficacy of NF-κB downstream gene products is dictated by
the monocytic/macrophagic differentiation status of promyelocytic
leukemia cells. Cancer Immunol Immunother. 16:2321–2331. 2012.
View Article : Google Scholar
|
|
30
|
Elbling L, Herbacek I, Weiss RM,
Jantschitsch C, Micksche M, Gerner C, Pangratz H, Grusch M,
Knasmüller S and Berger W: Hydrogen peroxide mediates EGCG-induced
antioxidant protection in human keratinocytes. Free Radic Biol Med.
49:1444–1452. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Khan TH, Srivastava N, Srivastava A,
Sareen A, Mathur RK, Chande AG, Musti KV, Roy S, Mukhopadhyaya R
and Saha B: SHP-1 plays a crucial role in CD40 signaling
reciprocity. J Immunol. 193:3644–3653. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen YY, Hsieh CY, Jayakumar T, Lin KH,
Chou DS, Lu WJ, Hsu MJ and Sheu JR: Andrographolide induces
vascular smooth muscle cell apoptosis through a
SHP-1-PP2A-p38MAPK-p53 cascade. Sci Rep. 4:56512014. View Article : Google Scholar : PubMed/NCBI
|