Introduction
Non-small cell lung cancer (NSCLC) accounts for ~80%
of lung cancer cases and is the leading cause of cancer-associated
mortalities worldwide in 2008 (1). In
clinical practice, chemotherapy remains the principal treatment due
to the poor prognosis of this type of malignancy. Cisplatin is the
most commonly used in chemotherapy for patients with NSCLC
(2). Although cisplatin treatment
leads to initially successful responses, it typically results in
the development of chemoresistance and results in therapeutic
failure (3). Therefore, elucidating
the molecular mechanisms underlying cisplatin resistance in NSCLC
may contribute to identify novel therapeutic targets for
attenuating cisplatin resistance, which has been a focus in
research in recent years (4).
Previous studies have demonstrated that inactivation of cell
apoptosis signaling pathways, activation of cell survival signaling
pathways, abnormal expression of tumor associated genes and
non-coding RNAs contribute to the cisplatin resistance of NSCLC
(5–7).
MicroRNAs (miRNA/miR), small endogenous non-coding RNAs between 21
and 25 nucleotides, may regulate post-transcriptional gene
expression by targeting the 3′-untranslated regions (3′-UTR) of
mRNAs (8). Previous studies have
revealed that miRNAs are involved in human cancer as diagnostic and
prognostic cancer biomarkers, and exhibit therapeutic tools
(9–11). Furthermore, it has been demonstrated
that dysregulation of miRNAs may contribute to the chemoresistance
in human tumor cells, including cisplatin resistance (12–14). c-Jun
N-terminal kinases (JNKs) function as a signaling hub in
mitogen-activated protein kinase (MAPK) signaling pathways and are
involved in inflammation, tumorigenesis, and cell proliferation,
differentiation, migration and apoptosis (15,16). JNK1,
JNK2 and JNK3 are major isoforms of JNK proteins and are encoded by
MAPK8, MAPK9 and MAPK10, respectively (17). JNK1 and JNK2 are widely expressed in
numerous tissue types, whereas JNK3 is primarily located in the
nervous system (17,18). The JNK-mediated signaling pathway is
involved in a variety of processes in human cancers (19). A previous study has identified that
chemotherapy resistance is not only the result of deregulation of
miRNAs targeting drug efflux transporter, but is a multifactorial
process (20). A number of miRNAs are
associated with cell apoptotic and survival signaling pathways, and
cells exhibiting altered expression profiles of pro- and
anti-apoptotic miRNAs are typically involved in resistance to
cytotoxic agents (21,22). miR-200c has been identified as a
regulator of tumorigenesis and tumor metastasis, and demonstrated
to attenuate P-glycoprotein 1-mediated multi-drug resistance and
metastasis by targeting JNK2/c-Jun signaling pathway in colorectal
cancer (23). Upregulation of miR-21
serves key roles in cisplatin-resistant ovarian cancer via
JNK-1/c-Jun pathway (24). In recent
years, miR-146a has been identified to exhibit functions in a
number of types of disease; in particular, in the proliferation,
metastasis and apoptosis of distinct types of cancer cells
(25). For chemotherapy resistance,
only Tomokuni et al (26) has
revealed an increased expression of miR-146a in hepatocellular
carcinoma cell lines resistant to interferon-α. The association
between miR-146a and cisplatin resistance, and the underlying
molecular mechanism, remain unknown.
To the best of our knowledge, the results of the
present study identified that miR-146a was significantly
downregulated in NSCLC cisplatin-resistant A549 cells and that
miR-146a targeted the 3′UTR of the JNK2 gene directly, which
affected the phosphorylated JNK-mediated signaling pathway
(27). Furthermore, overexpression of
miR-146a was demonstrated to downregulate the levels of cisplatin
resistance via the JNK signaling pathway, resulting in increased
sensitivity to cisplatin and induced apoptosis in vitro.
Therefore, the present study may identify a novel mechanism for
NSCLC cell cisplatin resistance and provide a new way of treating
NSCLC cisplatin resistance, by targeting miR-146a.
Materials and methods
Cell culture
A549 cells and A549/cisplatin (A549/DDP) cells
(Guangzhou Zixiutang Biotechnology Co., Ltd., Guangzhou, China)
were cultured in RPMI-1640 medium (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), supplemented with 10% fetal
bovine serum, 100 U/ml penicillin and 100 µg/ml streptomycin. All
cell lines were maintained at 37°C in a humidified atmosphere
containing 5% CO2.
Target prediction
For miR-146a target prediction, PicTar (http://pictar.mdc-berlin.de/), miRBase (http://www.mirbase.org/), miRanda (http://www.microrna.org/microrna/home.do), Bibiserv
(https://bibiserv.cebitec.uni-bielefeld.de/agenda)
and TargetScan (http://www.targetscan.org/vert_71/) were used to
identify and analyze potential targets of JNK2.
miRNA microarray analysis
Total RNA was extracted from culture cells with
guanidine thiocyanate, 2-mercaptoethanol and chloroformin GenElute
Mammalian Total RNA Purification kit (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) for 3 min on ice and the RNA concentration was
read using the NanoDrop 2000 spectrophotometer. RNA quality was
validated using a Bioanalyzer (Agilent Technologies, Inc., Santa
Clara, CA, USA). Total RNA (100 ng) was used for cDNA synthesis.
Subsequently, cDNA was labeled with fluorescence, fragmented and
hybridized to the Affymetrix GeneChip Gene 1.0 ST Human Array
(Thermo Fisher Scientific, Inc.). Following incubation at 45°C for
16 h, the arrays were washed with 1X saline sodium citrate (SSC),
0.1% SDS, 0.1X SSC, 0.1% SDS and 0.1X SSC and water for 2 min of
each at room temperature, and then scanned (Affymetrix Model 3000
scanner; Thermo Fisher Scientific, Inc.). The data adjustments
included data filtering, log2 transformation, gene
centering. In addition, the signal was normalized using a
Locally-weighted Regression filter.
RNA and miRNA extraction for reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated with buffer RLT and purified
from A549 and A549/DDP cells using the RNeasy Mini kit (Qiagen
Inc., Valencia, CA, USA) for 5 min on ice. miRNA was prepared using
the miRcute miRNA isolation kit (Tiangen Biotech Co., Ltd.,
Beijing, China) and cDNA was synthesized from the RNA by reverse
transcription employing the Qiagen OneStep RT-PCR kit (Qiagen GmbH,
Hilden, Germany). Sequences of all the primers are presented in
Table I. qPCR amplification was
performed to enable the fluorescence-based quantitation of gene
expression using the SYBR Green Quantitative RT-PCR kit
(Sigma-Aldrich; Merck KGaA). U6 was regarded as the reference gene.
The PCR volume and conditions were set up as previously described
(28) The primers for miR-182-5p
(catalog no., MIRAP00211), miR-106b (catalog no., MIRAP00129) and
miR-146a (catalog no., MIRAP00183) were from the
MystiCq® microRNA qPCR Assay Primer purchased for
Sigma-Aldrich (Merck KGaA). In additon, 2−∆∆Cq (29) was applied for gene quantification. The
primer sequences of U6 were: Forward, 5′-AAAGCAAATCATCGGACGACC and
reverse, 3′-GTACAACACATTGTTTCCTCGGA.
 | Table I.Primer sequences used in the present
study. |
Table I.
Primer sequences used in the present
study.
| Primer | Sequence |
|---|
| miRNA-146a
mimics |
|
|
Forward |
5′-UGAGAACUGAAUUCCAUGGGUU-3′ |
|
Reverse |
5′-CCCAUGGAAUUCAGUUCUCAUU-3′ |
| miRNA-146a
inhibitor |
5′-AACCCAUGGAAUUCAGUUCUCA-3′ |
| JNK2 primers |
|
|
Forward |
5′-CTGCGTCACCCATACATC-3′ |
|
Reverse |
5′-TGGCGTTGCTACTTACTGC-3′ |
| p53 primers |
|
|
Forward |
5′-TGCGTGTGGAGTATTTGGATG-3′ |
|
Reverse |
5′-TGGTACAGTCAGAGCCAACCAG-3′ |
| Bcl-2 primers |
|
|
Forward |
5′-CTGGTGGACAACATCGCTCTG-3′ |
|
Reverse |
5′-GGTCTGCTGACCTCACTTGTG-3′ |
Transfection of plasmids, miRNA mimics
and miRNA inhibitor
miR-146a mimic and inhibitor targeting JNK2 (1 ng)
were synthesized by GenePharma, Inc. (Sunnyvale, CA, USA). While,
the A549/DDP cells infected with empty plasmid were used as
controls (MOCK group). The sequences are presented in Table I. The transfection in A549/DDP cells
was performed using Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol
(23).
Cell proliferation assay
A549/cisplatin (A549/DDP) cells (1×104
cells/well) were seeded in 96-well plates in RPMI-1640 medium. An
MTT assay was performed using a Cell proliferation kit I (GE
Healthcare Life Sciences, Little Chalfont, UK) according to the
manufacturer's protocol. Subsequently, absorbance was determined at
570 nm using VersaMax (Molecular Devices; Thermo Fisher Scientific,
Inc.) to estimate MTT-formazan production dissolved with dimethyl
sulfoxide after 24, 48 and 72 h incubation. The OD490 was detected
using a spectrodensitometer under 490 nm wavelength.
Apoptosis rate and cell cycle
determination in vitro
The FITC Annexin V Apoptosis Detection kit with PI
(BioLegend, Inc., San Diego, CA, USA) was used for cell apoptosis
assay. After 72 h incubation, the transfected A549/cisplatin
(A549/DDP) cells (1×105) were fixed with 2.5%
glutaraldehyde for 30 min at room temperature. Annexin
V-fluorescein isothiocyanate (FITC) and propidium iodide (PI)
staining flow cytometry was used to determine apoptotic rates, by
identifying the relative amount of Annexin V-FITC positive and PI
negative cells. Unstained cells and cells stained with Annexin
V-FITC or PI alone were used as controls. PI staining flow
cytometry was used to determine the cell cycle. CYTOSPEC™ version
7.0 from Purdue University Cytometry Laboratories was applied for
data analysis.
Cell invasion assay
The cell invasion assay was performed using 24-well
Transwell plates, as previously described (30). A total of 8×104 cells were
seeded in serum-free RPMI-1640 medium in the upper and lower
Transwell chambers and incubated for 24 h. Matrigel (BD
Biosciences) was pre-coated on the upper side of the membrane and
incubated at 37°C for 1 h for gel formation. Following the removal
of the Transwell inserts, the cells that had not migrated through
the filter and remained inside each Transwell were removed by
wiping with a cotton swab. Cells that had migrated through the
filter and adhered to the other side of the filter were stained
with crystal violet at room temperature for 5 min for image
capture, and counted using a light microscope with ×200
magnification (30).
JNK2-3′-UTR luciferase reporter
assay
Human JNK2 cDNA, containing putative and mutant
target sites for miR-146a, was chemically synthesized and inserted
into a pMIR-REPORT™ vector (Ambion; Thermo Fisher Scientific,
Inc.). For the luciferase reporter assay, miR-146a mimic, miRNA
mimic control, miR-146a inhibitor and miRNA inhibitor control were
transfected into wild-(pMIR-jnk2-wt) or mutant-type (pMIR-jnk2-mut)
reporter vectors using Lipofectamine 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.). Luciferase activity was determined 48 h
after transfection using a dual-luciferase assay kit (Promega
Corporation, Madison, WI, USA) (31).
Additionally, Renilla luciferase activity was applied for
luciferase intensity normalization.
Protein extraction and western blot
analysis
The total protein was extracted from A549/DDP cells
and separated using SDS-PAGE (10% gel), as previously described
(32). Subsequently, the gel was
transferred to a polyvinylidene difluoride membrane (Solvay
Chemicals, Brussels, Belgium) and blocked with 5% skim milk at room
temperature for 1 h. The rabbit anti-JNK2 (catalog no., PA528664),
-p53 (catalog no., PA527822) and -B cell lymphoma (Bcl-)2 primary
antibodies (catalog no., PA520069) purchased from Wuhan Khayal
Bio-Technology Co., Ltd. (Wuhan, China) were used at a dilution of
1:1,000. The incubation with primary antibodies was at room
temperature for 1 h. Subsequently, the goat anti-rabbit
immunoglobulin G secondary antibody conjugated with horseradish
peroxidase (cat. no. Ab97051, Abcam, Cambridge, MA, USA) was used
at a dilution of 1:10,000. In addition, the incubation with
secondary antibodies was at room temperature for 45 min. The signal
was visualized using the enhanced chemiluminescence kit (GE
Healthcare Life Sciences). Image J 1.41 software (NIH, Bethesda,
MD, USA) was used to compare the gray values between the proteins
of interest and the internal control protein (β-actin), and between
the phosphorylated protein and the total protein.
RT-qPCR analysis
The expression levels of JNK2, p53 and Bcl-2 mRNA in
A549/DDP cells were assessed using RT-qPCR. Total RNA was extracted
from the cells using the TRIzol method (Thermo Fisher Scientific,
Inc.) at 4°C for 15 min. Subsequently, cDNA was synthesized from
the RNA by reverse transcription using SYBR Green Quantitative
RT-PCR kit (Sigma-Aldrich; Merck KGaA). PCR amplification was
performed to enable fluorescence-based quantitation of gene
expression. PCR reaction volumes were 10 µl and composed of cDNA (1
µl), primers (0.2 µl each), 2X Premix Ex Taq (5 µl) and
H2O (3.6 µl). The primer sequences used are presented in
Table I. For cDNA synthesis, samples
were incubated at 40°C for 30 min, 98°C for 5 min and 5°C for 5
min. The PCR conditions were as follows: Pre-denaturation at 96°C
for 5 min, followed by initiation at 94°C for 30 sec, annealing at
60°C for 30 sec and elongation at 78°C for 1.5 min for 35 cycles,
after which samples were stored at 4°C. Additionally,
2−∆∆Cq (29) was applied
for gene quantification. β-actin was selected as the internal
reference.
Statistical analysis
All data are presented as the mean ± standard
deviation. Statistical analysis was performed using GraphPad Prism
version 5.01 (GraphPad Software, Inc., La Jolla, CA, USA) via
Student's t-test. For all comparisons, P<0.05 was considered to
indicate a statistically significant difference.
Results
miR-146a is downregulated in A549/CDDP
cells compared with A549 cells
miRNA microarray analyzed 30 miRNAs and PicTar,
miRBase, miRanda, Bibiserv and TargetScan were used to identify and
analyze potential targets of JNK2. A total of 3 miRNAs, including
miR-146a, miR-182-5p and miR-106b, were identified to be potential
targets of JNK2 in >2 databases. In addition, the expression
levels of these 3 miRNAs in A549 and A549/DDP cells were
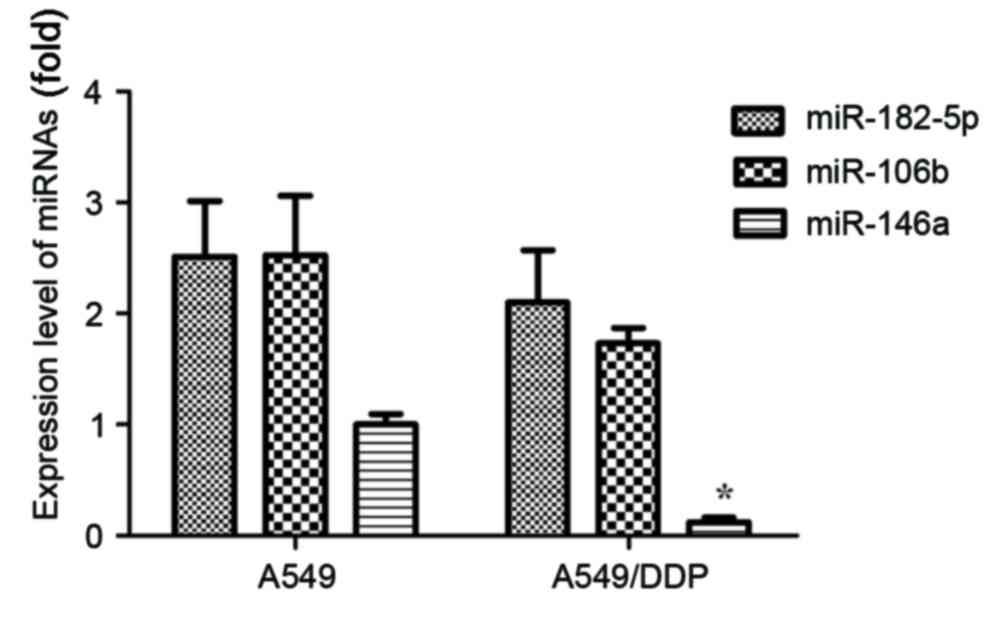
determined. As presented in Fig. 1,
the expression levels of miR-182-5p, miR-106b and miR-146a were
demonstrated to be downregulated in NSCLC A549/DDP cells, compared
with A549 cells. Only the expression of miR-146a revealed
statistical significance (P<0.05).
JNK2 is a direct target of
miR-146a
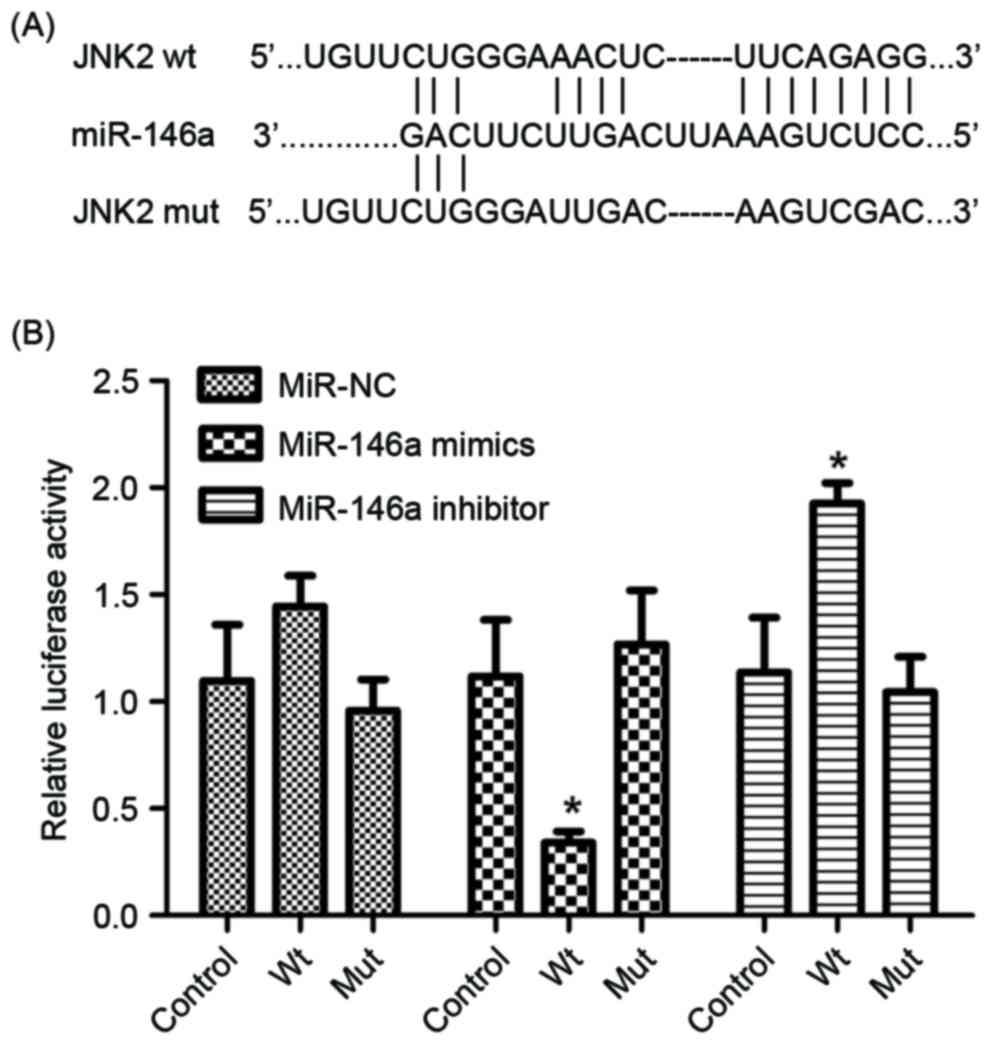
The luciferase reporter assay was performed to
determine whether miR-146a directly targets JNK2 by binding to the
3′-UTR of the JNK2 gene. Wild-type and mutant JNK2 3′UTR sequences,
which contained the miR-146a binding site, were constructed and
inserted into vectors (Fig. 2A). The
reporter construct and miR-control, miR-146a mimics, miR-146a
inhibitor and relative controls were transfected into the A549/DDP
cell line. As presented in Fig. 2B,
the luciferase reporter activity represented a significant decrease
in JNK2 3′-UTR in the presence of miR-146a mimics in A549/DDP
cells, compared with the miR-control. Furthermore, a significant
increase was observed in the presence of miR-146a inhibitor
(Fig. 2B). Therefore, JNK2 was
identified as a direct target of miR-146a.
miR-146a inhibits proliferation and
invasion, and induces apoptosis of A549/DDP cells
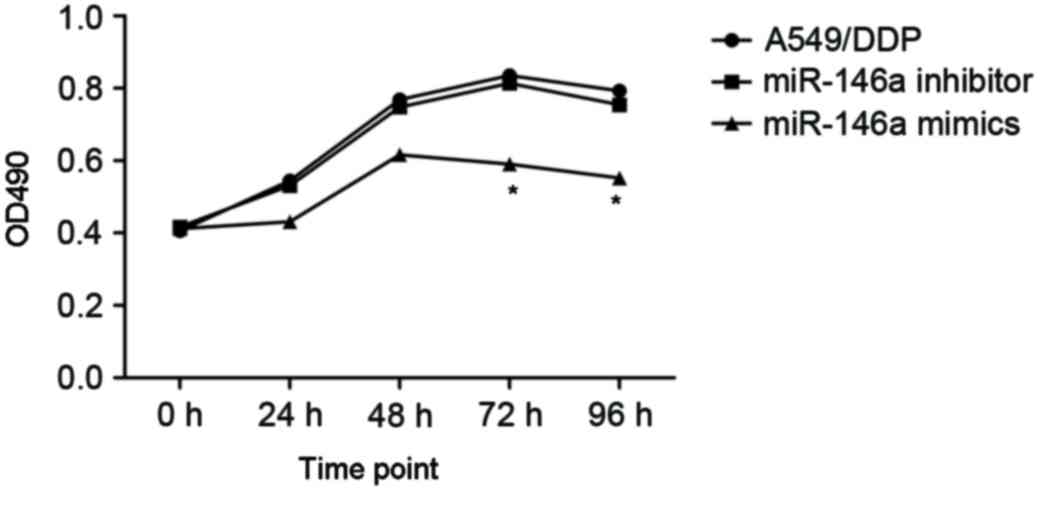
To investigate the effects of miR-146a on the
cisplatin sensitivity, the A549/DDP cells were transfected with
miR-146a mimics and inhibitors. The A549/DDP cells infected with
empty plasmid were used as controls (MOCK group). The proliferation
of cells was determined after 24, 48 and 72 h incubation, and the
results are presented in Fig. 3.
After 24 h incubation, upregulated miR-146a A549/DDP cells revealed
a significantly decreased proliferation rate, compared with the
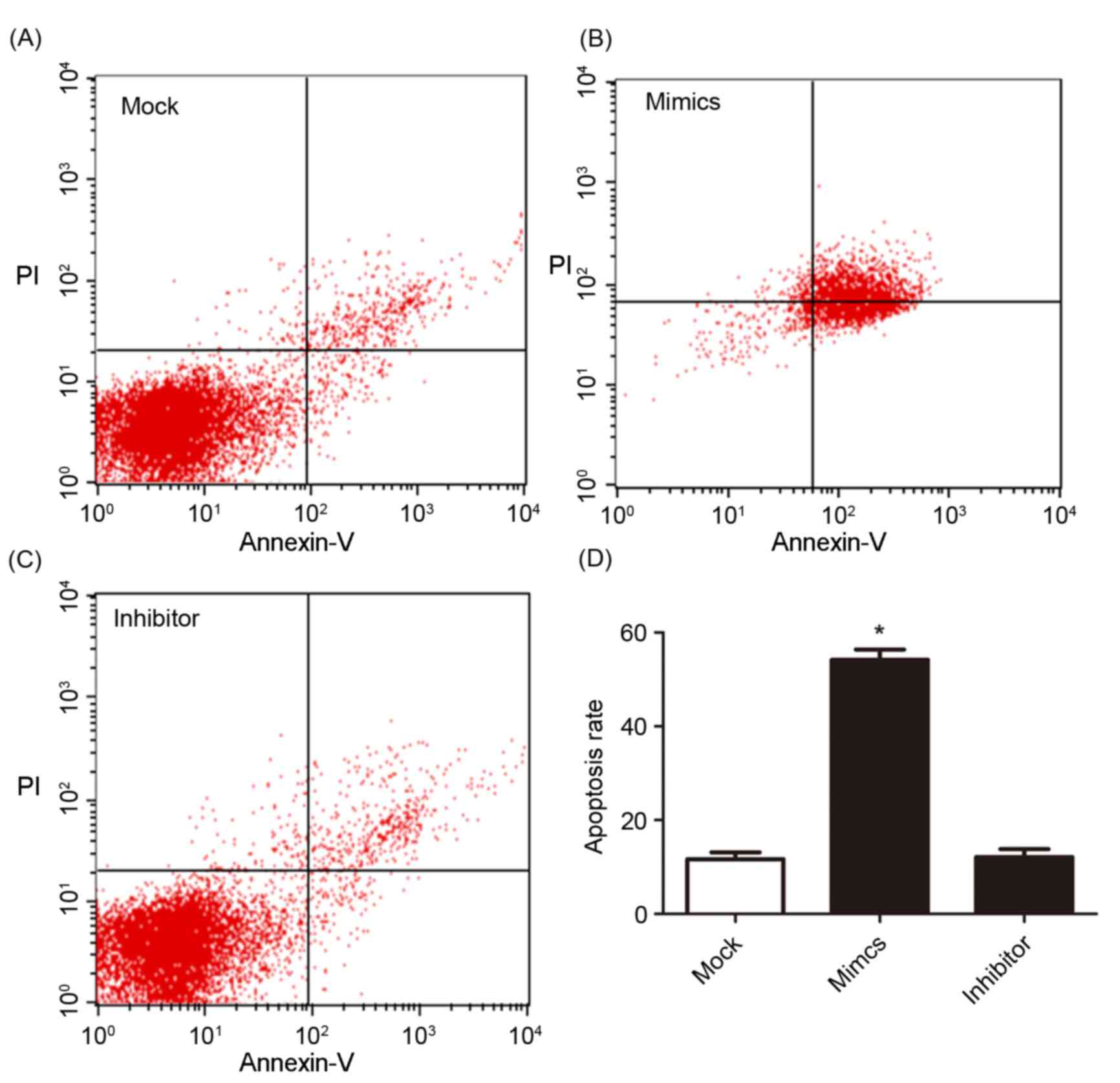
other two groups. The cell apoptosis and cell cycle were determined
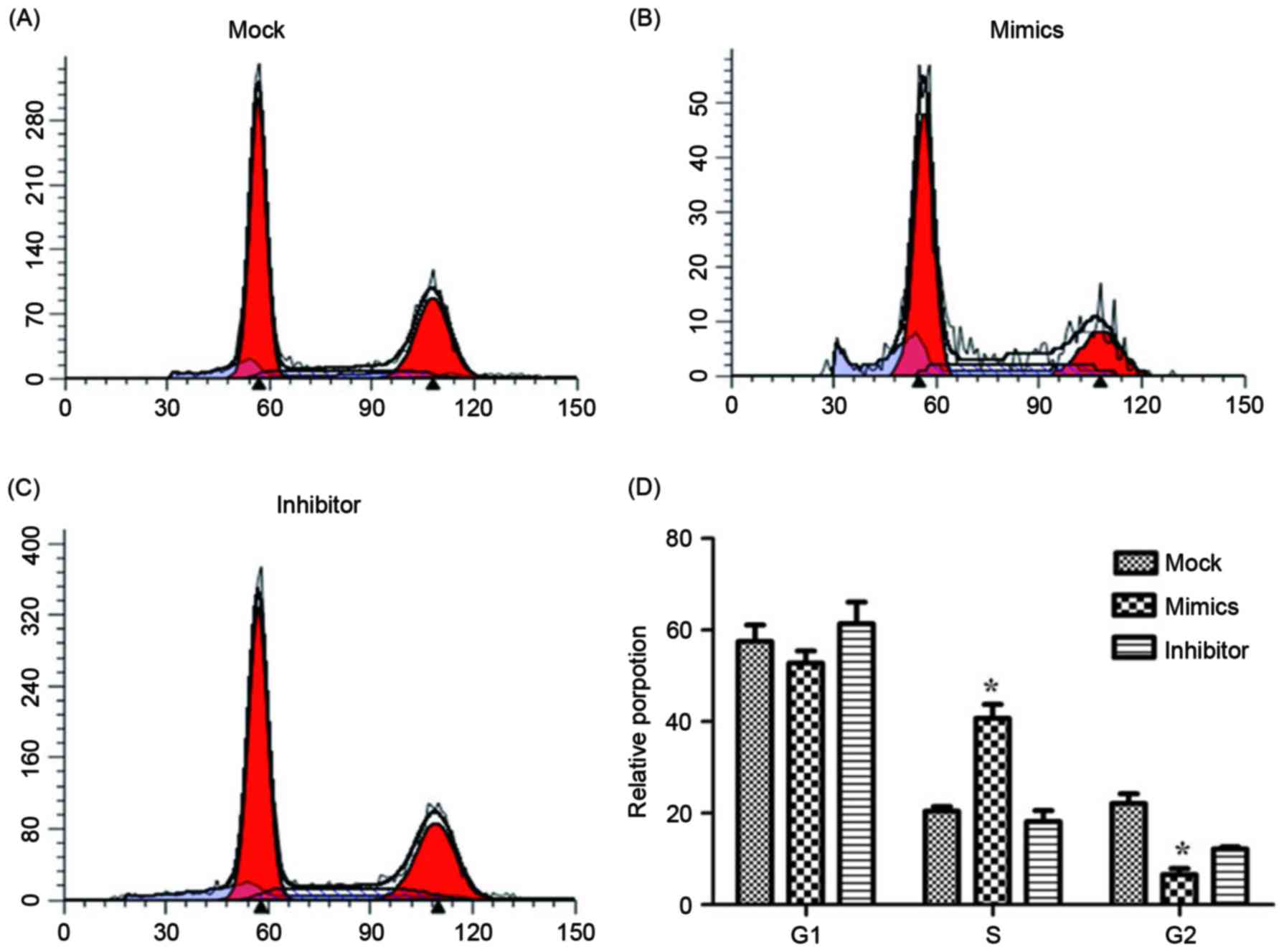
using flow cytometry and are presented in Figs. 4 and 5.
Fig. 4A, B and C present cell
apoptosis in the mock, mimic and inhibitor groups, respectively,
and Fig. 4D demonstrates the
apoptosis rate of A549/DDP cells. The results of the present study
revealed that, in the miR-146a overexpression group, the apoptosis
rate was significantly increased compared with the other two
groups. The relative proportion of A549/DDP cells in G1,
S and G2 period were calculated and are presented in
Fig. 5. Cells in the miR-146a mimic
group were arrested in the S period, which resulted in cells
failing to enter the G2 period and complete mitosis. For
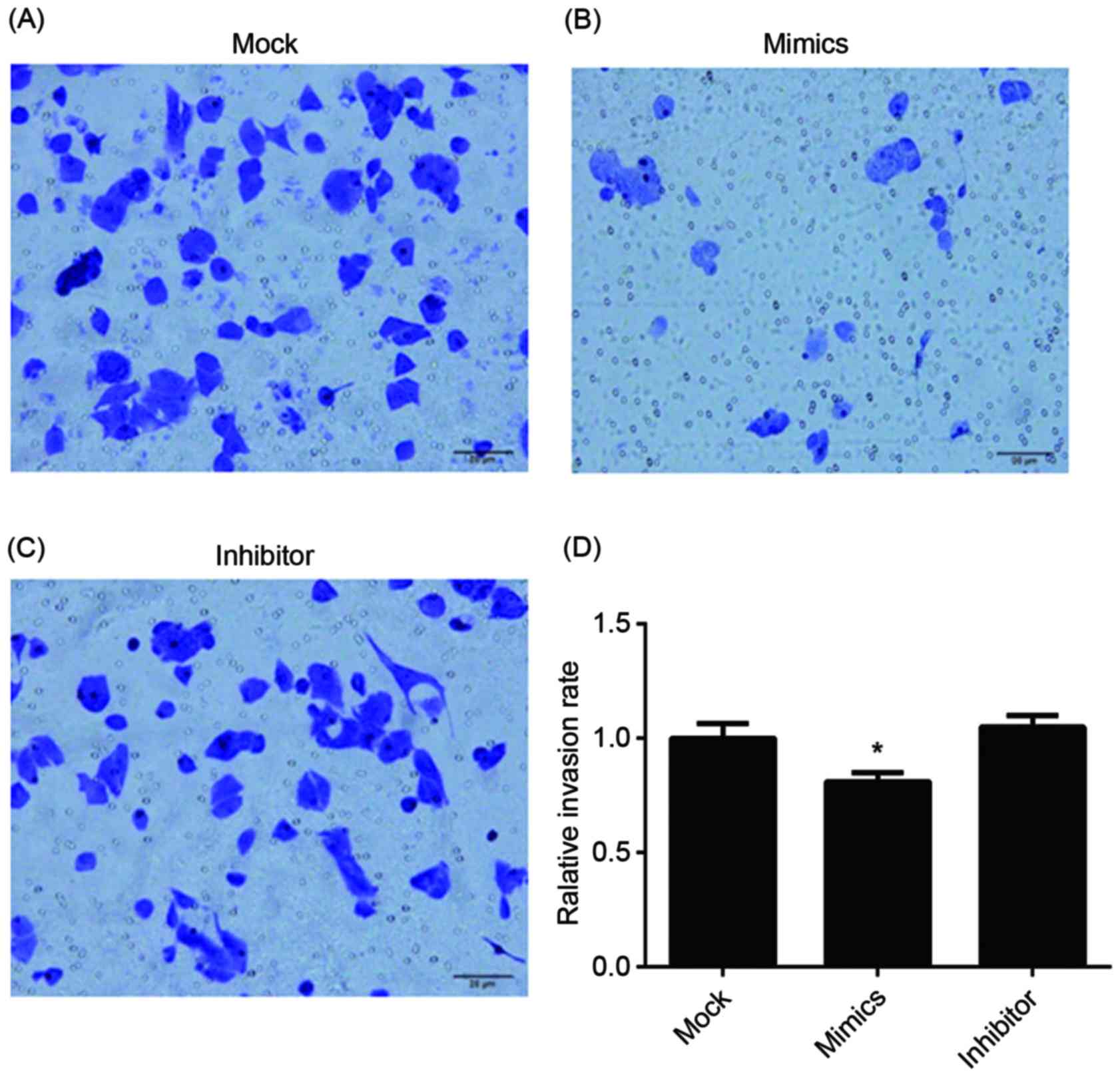
invasion, the results of the Transwell assay of infected A549/DDP
cells are presented in Fig. 6. The
relative invasion rate of A549/DDP cells in the miR-146a mimic
group was significantly decreased, compared with the other two
groups.
Expression level of tumor-associated
factors
Expression levels of JNK2, P53 and Bcl-2 mRNA and
proteins in infected A549/DDP cells were determined using RT-qPCR
and western blot analysis (Fig. 7).
JNK2, as the direct target of miR-146a, was identified to be
significantly upregulated by miR-146a overexpression. Additionally,
p53 was demonstrated to be significantly upregulated by miR-146a
overexpression, whereas Bcl-2 was revealed to be significantly
downregulated by miR-146a.
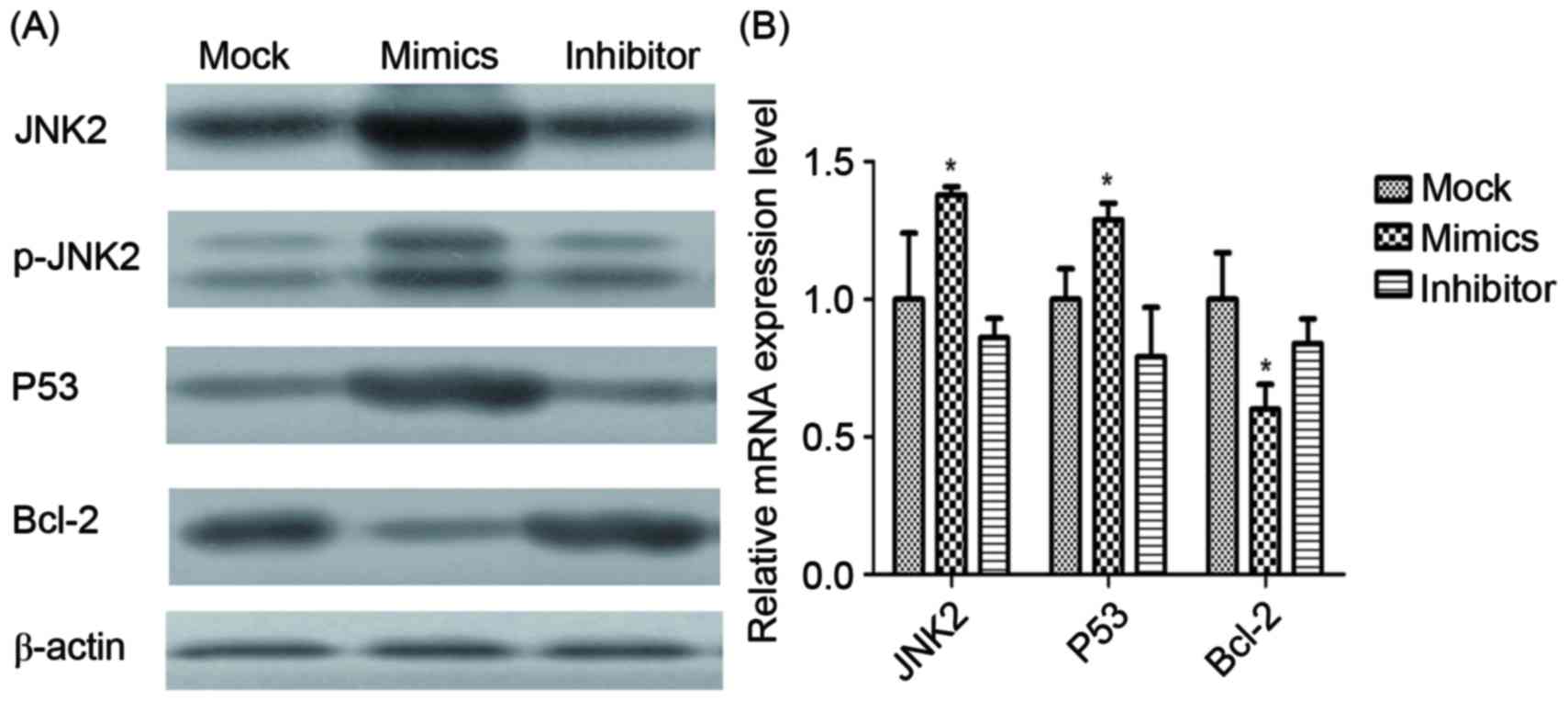 | Figure 7.mRNA and protein expression of
different factors. (A) The protein expression of JNK2, p-JNK2, p53,
Bcl-2 and β-actin in the mock, miR-146a mimic and miR-146a
inhibitor groups, determined using western blot analysis. β-actin
was used as the loading control. (B) The mRNA expression of JNK2,
p53 and Bcl-2, determined using the reverse
transcription-quantitative polymerase chain reaction. *P<0.05
vs. mock group. JNK2, Jun kinase 2; p-, phosphorylated; Bcl-2, B
cell lymphoma 2. |
Discussion
The results of the present study demonstrated that
miR-146a was downregulated in NSCLC A549/DDP cells and may affect
the phosphorylated JNK-mediated signaling pathway by targeting JNK2
gene 3′-UTR directly. Additionally, the overexpression of miR-146a
was revealed to induce apoptosis, and inhibit the proliferative and
invasive capabilities of A549/DDP cells, which resulted in an
increased sensitivity of NSCLC cells to cisplatin. miR-146a, one of
the most common downregulated miRNAs in tumor cells, may inhibit
cell proliferation, invasion and migration, and induce apoptosis of
cancer cells, thereby functioning as a tumor suppressor miRNA
(33–35). To the best of our knowledge, only
Tomokuni et al (26) has
demonstrated that miR-146a suppresses the sensitivity to
interferon-α in hepatocellular carcinoma cells. Furthermore, to the
best of our knowledge, the present study was the first to
investigate the association between miR-146a and cisplatin
resistance, and the mechanism of its function.
Numerous miRNAs have been identified as
differentially expressed in cisplatin-sensitive and
cisplatin-resistant tumor cells; for example, miR-214
overexpression was associated with cisplatin resistance of ovarian
cancer cells (36). The
overexpression of miR-141 and downregulation of its target gene,
the Kelch-Like ECH-Associated Protein 1, have been associated with
cisplatin resistance in esophageal squamous cell carcinoma
(37). In addition, overexpression of
miR-21 in cisplatin resistant ovarian cancer cells has been
identified to be a secondary event associated with the activation
of the JNK-1/c-Jun pathway in these cells (24). miR-146a has been revealed to be
upregulated in a number of types of cancer including papillary
thyroid carcinoma, anaplastic thyroid cancer and cervical cancer
(38,39), which suggests that miR-146a may
function as an oncogenic miRNA. However, decreased expression of
miR-146a was identified in prostate, pancreatic and gastric cancers
(32,40–42).
Therefore, miR-146a may have opposing functions in different types
of cancer. In NSCLC, miR-146a was downregulated, inhibited cell
growth and migration, and induced apoptosis (34). In the present study, miR-146a was
downregulated in A549/DDP NSCLC cells compared with A549 NSCLC
cells. Furthermore, the overexpression of miR-146a in the present
study resulted in an increased apoptosis rate, and decreased
proliferation and migration of A549/DDP cells, compared with
control A549/DDP cells. Therefore, it may be concluded that
miR-146a is associated with cisplatin resistance of A549 cells and
the overexpression of miR-146a may attenuate cisplatin resistance
of A549/DDP cells.
To study the underlying molecular mechanism of
miR-146a in A549/DDP cells, the target of miR-146a was determined
using a luciferase reporter assay. The results of the present study
identified that JNK2 was a direct target of miR-146a. Cisplatin, a
DNA-damaging drug, induces cell apoptosis by binding to DNA. JNK is
involved in the regulation of apoptosis and responds to stress
signals by phosphorylating transcription factors, including c-Jun
and ATF-2 (43), or by activating
other target proteins involved in the initiation or execution of
apoptosis, including p53 (44), c-Myc
(45) and proteins of the Bcl-2
family (46). The p53 gene, encoding
a transcriptional factor, may induce apoptosis of tumor cells and
serve as a tumor suppressor gene. The Bcl-2 protein, a member of
the Bcl-2 family, may inhibit the cell apoptosis in a number of
types of cancer (47). In the present
study, expression of JNK2 and the phosphorylation protein p-JNK2
were upregulated by the overexpression of miR-146a, which indicated
that JNK2 may be activated by miR-146a. Furthermore, JNK2 activated
the expression of p53 and inhibited Bcl-2. The results of the
present study demonstrated that miR-146a increased cisplatin
sensitivity of NSCLC A549/DDP cells by targeting JNK2 and inducing
cell apoptosis. The present study may provide a novel method for
treating NSCLC cisplatin resistance, by targeting miR-146a.
Acknowledgements
The present study was supported by the Program for
Social Development Research of Yinzhou District of Ningbo (grant
no. 2013-107).
References
|
1
|
Eaton KD and Martins RG: Maintenance
chemotherapy in non-small cell lung cancer. J Natl Compr Canc Netw.
8:815–821. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Socinski MA, Crowell R, Hensing TE, Langer
CJ, Lilenbaum R, Sandler AB and Morris D: American College of Chest
Physicians: Treatment of Non-small Cell Lung Cancer, Stage IV: ACCP
Evidence-based Clinical Practice Guidelines (2nd edition). Chest.
132 3 Suppl:277S–289S. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Korita PV, Wakai T, Shirai Y, Matsuda Y,
Sakata J, Takamura M, Yano M, Sanpei A, Aoyagi Y, Hatakeyama K and
Ajioka Y: Multidrug resistance-associated protein 2 determines the
efficacy of cisplatin in patients with hepatocellular carcinoma.
Oncol Rep. 23:965–972. 2010.PubMed/NCBI
|
|
4
|
Rose MC, Kostyanovskaya E and Huang RS:
Pharmacogenomics of cisplatin sensitivity in non-small cell lung
cancer. Genomics Proteomics Bioinformatics. 12:198–209. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Galluzzi L, Senovilla L, Vitale I, Michels
J, Martins I, Kepp O, Castedo M and Kroemer G: Molecular mechanisms
of cisplatin resistance. Oncogene. 31:1869–1883. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang Y, Li H, Hou S, Hu B, Liu J and Wang
J: The noncoding RNA expression profile and the effect of lncRNA
AK126698 on cisplatin resistance in non-small-cell lung cancer
cell. PLoS One. 8:e653092013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lin Y, Wang Z, Liu L and Chen L: Akt is
the downstream target of GRP78 in mediating cisplatin resistance in
ER stress-tolerant human lung cancer cells. Lung Cancer.
71:291–297. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Edwards JK, Pasqualini R, Arap W and Calin
GA: MicroRNAs and ultraconserved genes as diagnostic markers and
therapeutic targets in cancer and cardiovascular diseases. J
Cardiovasc Transl Res. 3:271–279. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fabbri M: miRNAs as molecular biomarkers
of cancer. Expert Rev Mol Diagn. 10:435–444. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jackson A and Linsley PS: The therapeutic
potential of microRNA modulation. Discov Med. 9:311–318.
2010.PubMed/NCBI
|
|
12
|
Ma J, Dong C and Ji C: MicroRNA and drug
resistance. Cancer Gene Ther. 17:523–531. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu ZW, Zhong LP, Ji T, Zhang P, Chen WT
and Zhang CP: MicroRNAs contribute to the chemoresistance of
cisplatin in tongue squamous cell carcinoma lines. Oral Oncol.
46:317–322. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sorrentino A, Liu CG, Addario A, Peschle
C, Scambia G and Ferlini C: Role of microRNAs in drug-resistant
ovarian cancer cells. Gynecol Oncol. 111:478–486. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Johnson GL and Nakamura K: The c-jun
kinase/stress-activated pathway: Regulation, function and role in
human disease. Biochim Biophys Acta. 1773:1341–1348. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dhanasekaran DN and Johnson GL: MAPKs:
Function, regulation, role in cancer and therapeutic targeting.
Oncogene. 26:3097–3099. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cuevas BD, Abell AN and Johnson GL: Role
of mitogen-activated protein kinase kinase kinases in signal
integration. Oncogene. 26:3159–3171. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bode AM and Dong Z: The functional
contrariety of JNK. Mol Carcinog. 46:591–598. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bubici C and Papa S: JNK signalling in
cancer: In need of new, smarter therapeutic targets. Br J
Pharmacol. 171:24–37. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Panczyk M: Pharmacogenetics research on
chemotherapy resistance in colorectal cancer over the last 20
years. World J Gastroenterol. 20:9775–9827. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Subramanian S and Steer CJ: MicroRNAs as
gatekeepers of apoptosis. J Cell Physiol. 223:289–298.
2010.PubMed/NCBI
|
|
22
|
Haenisch S and Cascorbi I: miRNAs as
mediators of drug resistance. Epigenomics. 4:369–381. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sui H, Cai GX, Pan SF, Deng WL, Wang YW,
Chen ZS, Cai SJ, Zhu HR and Li Q: miR200c attenuates P-gp-mediated
MDR and metastasis by targeting JNK2/c-Jun signaling pathway in
colorectal cancer. Mol Cancer Ther. 13:3137–3151. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Echevarría-Vargas IM, Valiyeva F and
Vivas-Mejía PE: Upregulation of miR-21 in cisplatin resistant
ovarian cancer via JNK-1/c-Jun pathway. PLoS One. 9:e970942014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen G, Umelo IA, Lv S, Teugels E, Fostier
K, Kronenberger P, Dewaele A, Sadones J, Geers C and De Grève J:
miR-146a inhibits cell growth, cell migration and induces apoptosis
in non-small cell lung cancer cells. PLoS One. 8:e603172013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tomokuni A, Eguchi H, Tomimaru Y, Wada H,
Kawamoto K, Kobayashi S, Marubashi S, Tanemura M, Nagano H, Mori M
and Doki Y: miR-146a suppresses the sensitivity to interferon-α in
hepatocellular carcinoma cells. Biochem Biophys Res Commun.
414:675–680. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hansen PR, Holm AM, Svendsen UG, Olsen PS
and Andersen CB: Apoptosis and formation of peroxynitrite in the
lungs of patients with obliterative bronchiolitis. J Heart Lung
Transplant. 19:160–166. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sui H, Zhou S, Wang Y, Liu X, Zhou L, Yin
P, Fan Z and Li Q: COX-2 contributes to P-glycoprotein-mediated
multidrug resistance via phosphorylation of c-Jun at Ser63/73 in
colorectal cancer. Carcinogenesis. 32:667–675. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Park JE, Tan HS, Datta A, Lai RC, Zhang H,
Meng W, Lim SK and Sze SK: Hypoxic tumor cell modulates its
microenvironment to enhance angiogenic and metastatic potential by
secretion of proteins and exosomes. Mol Cell Proteomics.
9:1085–1099. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sharma A, Kumar M, Aich J, Hariharan M,
Brahmachari SK, Agrawal A and Ghosh B: Posttranscriptional
regulation of interleukin-10 expression by hsa-miR-106a. Proc Natl
Acad Sci USA. 106:pp. 5761–5766. 2009, View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kamiya T, Kobayashi Y, Kanaoka K,
Nakashima T, Kato Y, Mizuno A and Sakai H: Fluorescence microscopic
demonstration of cathepsin K activity as the major lysosomal
cysteine proteinase in osteoclasts. J Biochem. 123:752–759. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li Y, Vandenboom TG II, Wang Z, Kong D,
Ali S, Philip PA and Sarkar FH: miR-146a suppresses invasion of
pancreatic cancer cells. Cancer Res. 70:1486–1495. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen G, Umelo IA, Lv S, Teugels E, Fostier
K, Kronenberger P, Dewaele A, Sadones J, Geers C and De Grève J:
miR-146a inhibits cell growth, cell migration and induces apoptosis
in non-small cell lung cancer cells. PLoS One. 8:e603172013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Stevenson M, Joyner J, Dildar K, et al:
The role of miR-146a and novel Rhenium compounds on prostate cancer
cell lines derived from African Americans and European American
patients. Cancer Res. 75:4840. 2015. View Article : Google Scholar
|
|
36
|
Yang H, Kong W, He L, Zhao JJ, O'Donnell
JD, Wang J, Wenham RM, Coppola D, Kruk PA, Nicosia SV and Cheng JQ:
MicroRNA expression profiling in human ovarian cancer: miR-214
induces cell survival and cisplatin resistance by targeting PTEN.
Cancer Res. 68:425–433. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Imanaka Y, Tsuchiya S, Sato F, Shimada Y,
Shimizu K and Tsujimoto G: MicroRNA-141 confers resistance to
cisplatin-induced apoptosis by targeting YAP1 in human esophageal
squamous cell carcinoma. J Hum Genet. 56:270–276. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pacifico F, Crescenzi E, Mellone S,
Iannetti A, Porrino N, Liguoro D, Moscato F, Grieco M, Formisano S
and Leonardi A: Nuclear factor-{kappa}B contributes to anaplastic
thyroid carcinomas through up-regulation of miR-146a. J Clin
Endocrinol Metab. 95:1421–1430. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang X, Tang S, Le SY, Lu R, Rader JS,
Meyers C and Zheng ZM: Aberrant expression of oncogenic and
tumor-suppressive microRNAs in cervical cancer is required for
cancer cell growth. PLoS One. 3:e25572008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lin SL, Chiang A, Chang D and Ying SY:
Loss of mir-146a function in hormone-refractory prostate cancer.
RNA. 14:417–424. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kogo R, Mimori K, Tanaka F, Komune S and
Mori M: Clinical significance of miR-146a in gastric cancer cases.
Clin Cancer Res. 17:4277–4284. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hou Z, Xie L, Yu L, Qian X and Liu B:
MicroRNA-146a is down-regulated in gastric cancer and regulates
cell proliferation and apoptosis. Med Oncol. 29:886–892. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hayakawa J, Depatie C, Ohmichi M and
Mercola D: The activation of c-Jun NH2-terminal kinase (JNK) by
DNA-damaging agents serves to promote drug resistance via
activating transcription factor 2 (ATF2)-dependent enhanced DNA
repair. J Biol Chem. 278:20582–20592. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Buschmann T, Potapova O, Bar-Shira A,
Ivanov VN, Fuchs SY, Henderson S, Fried VA, Minamoto T,
Alarcon-Vargas D, Pincus MR, et al: Jun NH2-terminal kinase
phosphorylation of p53 on Thr-81 is important for p53 stabilization
and transcriptional activities in response to stress. Mol Cell
Biol. 21:2743–2754. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Noguchi K, Kitanaka C, Yamana H, Kokubu A,
Mochizuki T and Kuchino Y: Regulation of c-Myc through
phosphorylation at Ser-62 and Ser-71 by c-Jun N-terminal kinase. J
Biol Chem. 274:32580–32587. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Park J, Kim I, Oh YJ, Lee K, Han PL and
Choi EJ: Activation of c-Jun N-terminal kinase antagonizes an
anti-apoptotic action of Bcl-2. J Biol Chem. 272:16725–16728. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lee DH, Ha JH, Kim Y, Jang M, Park SJ,
Yoon HS, Kim EH, Bae KH, Park BC, Park SG, et al: A conserved
mechanism for binding of p53 DNA-binding domain and anti-apoptotic
Bcl-2 family proteins. Mol Cells. 37:264–269. 2014. View Article : Google Scholar : PubMed/NCBI
|