Introduction
Colorectal cancer (CRC) is the third most deadly
cancer worldwide accounting for more than 600,000 deaths annually
(1). At the diagnosis, a quarter of
the patients with primary CRC have synchronous hepatic metastasis,
and more than 50% of the patients with CRC will develop liver
metastases in the course. Almost half of the patients undergoing
resection for primary CRC eventually develop metachronous liver
metastasis. Survival in metastatic cases is rarely longer than
three years (2). Interestingly,
although caudal type homeobox 2 (CDX2) is widely used in the daily
routine diagnostic, there are less than sixty publications in the
last sixty years performed on human tissue investigating the role
of CDX2 (3).
The Cdx family of transcription factors contributes
also to the CRC phenotype, but a mechanism by which CDX2 expression
is lost or downregulated in colorectal tumors is currently not
clear. The CDX2 is necessary for the proper development of the
intestinal tract and is crucial for development and homeostasis of
the intestinal epithelium throughout life (1). The role of Cdx2 in colorectal
carcinogenesis is multi-sided. The CDX2 expression is reduced in
CRC and its expression is inversely correlated to tumor grade,
tumor stage and lymph node metastasis (4). Loss of CDX2 expression can strongly
predict high level CpG island methylation phenotype (CIMP-H)
independently from microsatellite status of CRCs. Thus Cdx2 was
proposed as a surrogate marker for CIMP-H (5). In addition, CDX2 was attributed to play
a regulatory role in apoptosis and DNA repair. Colon epithelium
with decreased CDX2 expression lead to impaired apoptosis potential
after γ-irradiation, thus resulting in higher resistance to
genotoxic stress. Besides, the effect of CDX2 in DNA repair
activity can contribute to its attributed tumor suppressor function
(6).
DNA methylation of tumor suppressor genes resulting
in its transcriptional inactivation and has been identified as an
important mechanism. CIMP characterized by the extensive
hypermethylation of multiple CpG islands, and belongs to one of the
major mechanisms in the colorectal carcinogenesis (7). O6-methylguanine DNA methyltransferase
(MGMT), a surrogate marker for CIMP, gene promoter methylation
plays an important role in colorectal carcinogenesis. Loss of MGMT
expression, which is secondary to gene promoter methylation, occurs
in approximately 30–40% of metastatic CRC. In addition, loss of
MGMT expression results in high response to alkylating agents
(i.e., dacarbazine or temozolomide) (8). Thus, MGMT is believed to have predictive
potential for therapy.
A further level of DNA damage defence mechanism is
represented by the mismatch repair (MMR) system, which take part
not only in the DNA repair processes, but also in the regulation of
cell cycle check-points and apoptosis (9). Deficiency of MMR proteins (i.e., MLH1
and MSH2) is responsible for resistance to various chemotherapeutic
drugs and subsequently for resistance to apoptosis (9). Interestingly, loss of MGMT expression is
more frequent in CRC with microsatellite instability, suggesting
that methylated MGMT selects cellular clones with MMR deficient
status (8). Moreover, MMR deficiency
is also correlated with loss of CDX2 (10).
Excision repair cross-complementing 1 (ERCC1) is a
structure specific DNA repair endonuclease responsible for 5′
incision (5′-endonuclease), a key enzyme in nucleotide excision
repair (NER) pathway and is essential for repair of platinum-DNA
adducts, thus associated with therapy resistance to
platinum-containing compounds (i.e., cisplatin) (11,12).
Aberrant β-catenin expression and disturbed Wnt
signaling is recognized as an important event in the genesis of
several malignancies, especially in CRC. β-catenin mutations or
loss-of-function mutations of the adenomatous polyposis coli (APC)
tumor suppressor gene appear to be crucial steps in the progression
of this disease (13). APC and
β-catenin were found to traffic independently from each other into
and out of the nucleus in response to internal and external
signals. This fact has prompted debate about the previously
proposed role of APC as a β-catenin chaperone (14). Germline mutations in the APC gene
cause familial adenomatous polyposis (FAP), and over 80% of CRCs
(both inherited and sporadic) carry truncating mutations that
inactivate the APC protein. Most of these mutations occur in the
so-called ‘mutation cluster region’ of the APC gene, accounting for
a truncated protein incapable of binding regulatory proteins (i.e.,
Axin) or associating with microtubules. The relevance of truncating
mutations for β-catenin is enormous: Mutated APC cannot stimulate
its degradation (because of its failure to bind Axin), although APC
still can bind to β-catenin (albeit less efficiently) (14,15).
β-catenin has been observed to accumulate in the nuclei of colon
cancer cells, which results from the inability of APC to promote
β-catenin degradation, rather than a lack of export function,
leading to nuclear accumulation of β-catenin in APC-mutant tumor
cells (14). There are only few
studies that focused on interactions between CDX2 and Wnt
signalling in colon cancer. It has been demonstrated that CDX2 can
inhibit the transcriptional activity of β-catenin/TCF lines in a
non-transcriptional way (4).
Expression of CDX2 in association with DNA repair
proteins and members of Wnt signaling pathway has not been studied
previously in liver metastasis of CRC. In this study, we analysed
the expression distribution of CDX2 in matters of expression status
of DNA repair proteins (MMR proteins, MGMT and ERCC1), APC, and
β-catenin. Furthermore, we correlated CDX2 protein expression with
clinical data.
Materials and methods
Tissue samples
Formalin-fixed paraffin-embedded surgical specimens
of liver metastasis of CRC were selected from the archives of the
Institute of Pathology at the University Hospital of Heidelberg.
Hundred and one patients without neo-adjuvant chemotherapy (64
male, 37 female; mean age 62 years) were included. Tumor size was
between 5 mm and 16 cm in diameter. 12 cases showed mucinous
adenocarcinoma histology and 89 cases showed histology of
adenocarcinoma NOS. We had only two cases with grade 1
adenocarcinoma, 83 cases had grade 2 and 12 cases grade 3
histology. Serial paraffin sections were cut at 4 µm for
immunohistochemistry. Important clinical data, such as: Age,
gender, size and number of metastases were collected from
histological reports. Tissue samples were provided by the tissue
bank of the National Centre for Tumor Diseases (NCT, Heidelberg,
Germany) in accordance with the regulations of the tissue bank and
the approval of the ethics committee of Heidelberg University
according to ethical standards formulated in the Declaration of
Helsinki 1975 (revised in 1983).
Tissue microarray (TMA)
TMA blocks were punched from paraffin-embedded human
liver specimens with a tissue microarrayer (Beecher Instruments,
Sun Prairie, WI, USA). From each case, two cores of tumor tissue
were punched with a diameter size of 1.6 mm and two muscle cores
were used for orientation of the TMA slides. Therefore serial
sections were cut from the TMA block. So far, there is no
standardised operating protocol or universal agreement for sampling
and staining of TMA blocks and slides. The general consensus is
that at least two 0.6 mm cores adequately represent for
immunohistochemical changes (16,17).
Immunohistochemistry
4 µm thick slides were obtained from TMA blocks.
Slides were then deparaffinised according to standard protocol by
xylene, and dehydrated with 95–96% ethanol, 70% ethanol and
distilled water. All slides were stained simultaneously using a
computer-controlled autostainer (Dako TechMate 500 cytomation) and
Dako EnVision-Sytem (Dako; Agilent Technologies, Inc., Santa Clara,
CA, USA) and pretreated with 3% Hydrogen Peroxide prior to antibody
incubation. MLH1 [M1, ready-to-use (RTU), Ventana Medical Systems,
Inc.; Roche Diagnostics, Basel, Switzerland], MSH6 (44, RTU;
Ventana Medical Systems, Inc.; Roche Diagnostics), PMS2 (EPR3947,
RTU; Ventana Medical Systems, Inc.; Roche Diagnostics), MSH2
(G219-1129, RTU; Ventana Medical Systems, Inc.; Roche Diagnostics),
MGMT (MT-23.2; Thermo Fisher Scientific, Inc., Waltham, MA, USA;
1:20) and CDX2 (EPR2764Y; Thermo Fisher Scientific, Inc.; 1:200)
antibodies were used. Secondary antibody binding (all Dako, 1:200)
was visualised using a streptavidin ABC-kit (Dako), followed by
3,3′-diaminobenzidine (Vector, Peterborough, UK). For ERCC1 (8F1,
Neomarkers; dilution: 1:100) and β-catenin (14, RTU; Ventana
Medical Systems, Inc.; Roche Diagnostics) slides were stained by a
computer-controlled autostainer (Ventana BenchMark Ultra; Ventana
Medical Systems, Inc.; Roche Diagnostics). Polyclonal rabbit
anti-APC antibody (DP2.5 1:200 Fa; Acris Antibodies; OriGene
Technologies, Inc., Rockville, MD, USA) were used for APC staining.
Staining was performed using ChemMate Detection kit (Dako)
according to recommendations of the manufacturer. The antibodies
were incubated overnight at 4°C followed by avidin-biotin complex
peroxidease technique using aminoethylcarbazole for visualization
and hematoxylin for nuclear counterstaining. All slides were
covered with Aquatex (Merck KGaA, Darmstadt, Germany).
Evaluation of
immunohistochemistry
For the semi-quantitative assessment of staining
intensity, we adjusted a previously published scoring system for
each protein and fitted to TMA dots. For MSI proteins and for MGMT
the staining was evaluated according to Bethesda guidelines
(18): score 1, more than 10% of
tumor cell nuclei are positive; score 0, less than 10% of tumor
cell nuclei positive (but: positive internal control, i.e., stromal
cells and lymphocytes). The immunostained TMA sections were
evaluated and scored under a light microscope independently by two
pathologists in a blinded fashion. Discordant cases were reviewed
and re-evaluated based on a consensus opinion.
Immunostaining for CDX2, ERCC1 and nuclear β-catenin
was scored in a three-graded scale: score 0, weak staining in less
than 10% of the tumor cells; score 1, moderate staining in up to
75% of the tumor cells; and score 2, strong nuclear staining in
more than 75% of the tumor cells. For cytoplasmic β-catenin
staining a two-graded scale was used: score 0, no or weak staining
in less than 10% of tumor cells and weaker staining compared to
normal colonic mucosa; score 1, nuclear staining in more than 10%
of the tumor cells.
Cytoplasmic and nuclear APC staining was separately
scored. For nuclear APC staining a two-graded scale was used: score
0, No or weaker staining in less than 10% of tumor cells and weaker
staining compared to normal colonic mucosa; score 1, nuclear
staining in more than 10% of the tumor cells. For cytoplasmic APC
staining a three-graded scale was used: score 0, no cytoplasmic
staining or weak staining in less than 10% of tumor cells; score 1,
10–75% of the tumor cells with moderate intensity; and score 2,
more than 75% of the tumor cells are positive with high staining
intensity. Normal colorectal mucosa was set as baseline expression
level for APC (score 2).
Statistical analysis
The statistical analyses were performed with SAS
software (SAS Institute, Inc., Cary, NC, USA). Spearman-Rho test
was used to evaluate the relationship between clinical data, CDX2,
MLH1, MSH2, MSH6, PMS2, MGMT, ERCC1, APC and β-catenin.
Results
CDX2 expression and its correlation
with clinical data
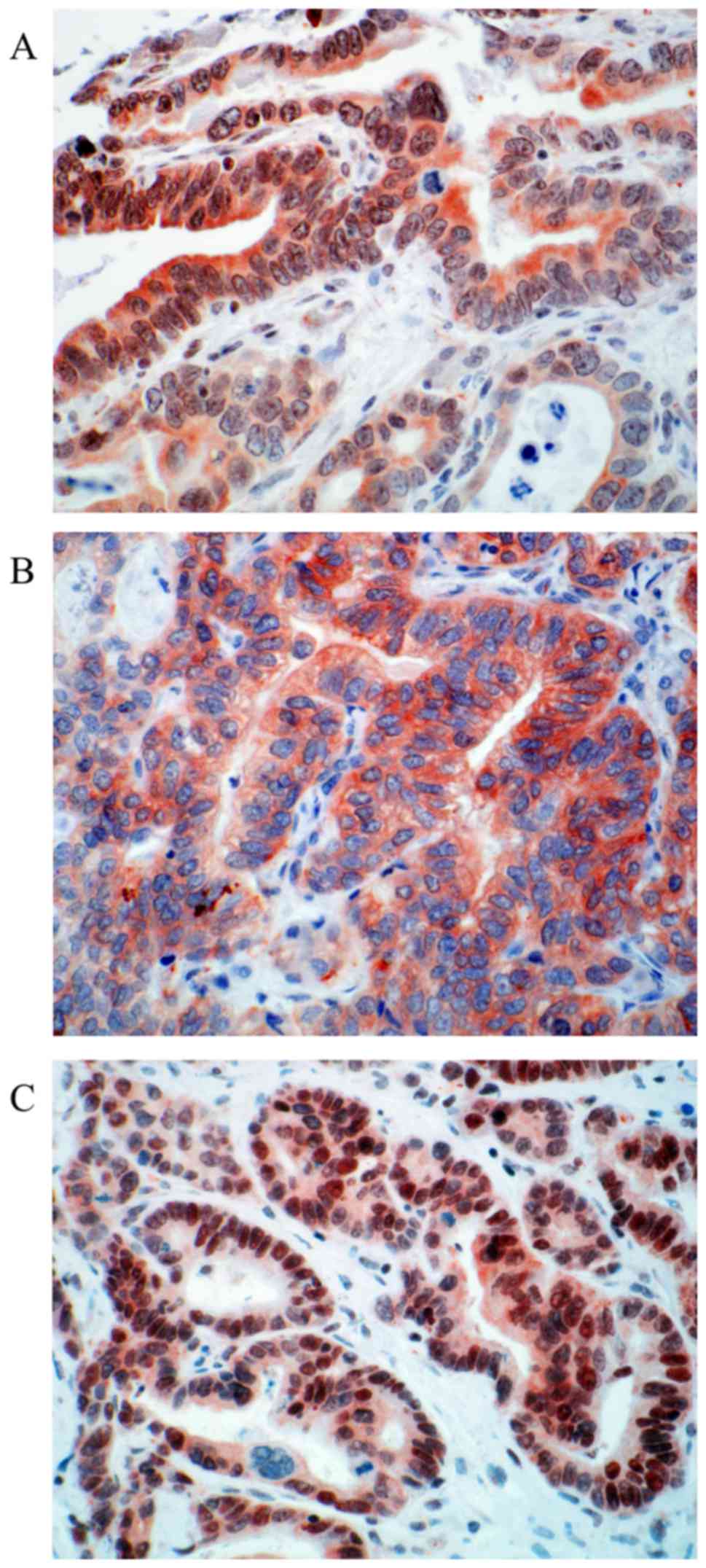
We could reach valid expression data for CDX2
(Table I) in 83 of 101 cases. 32
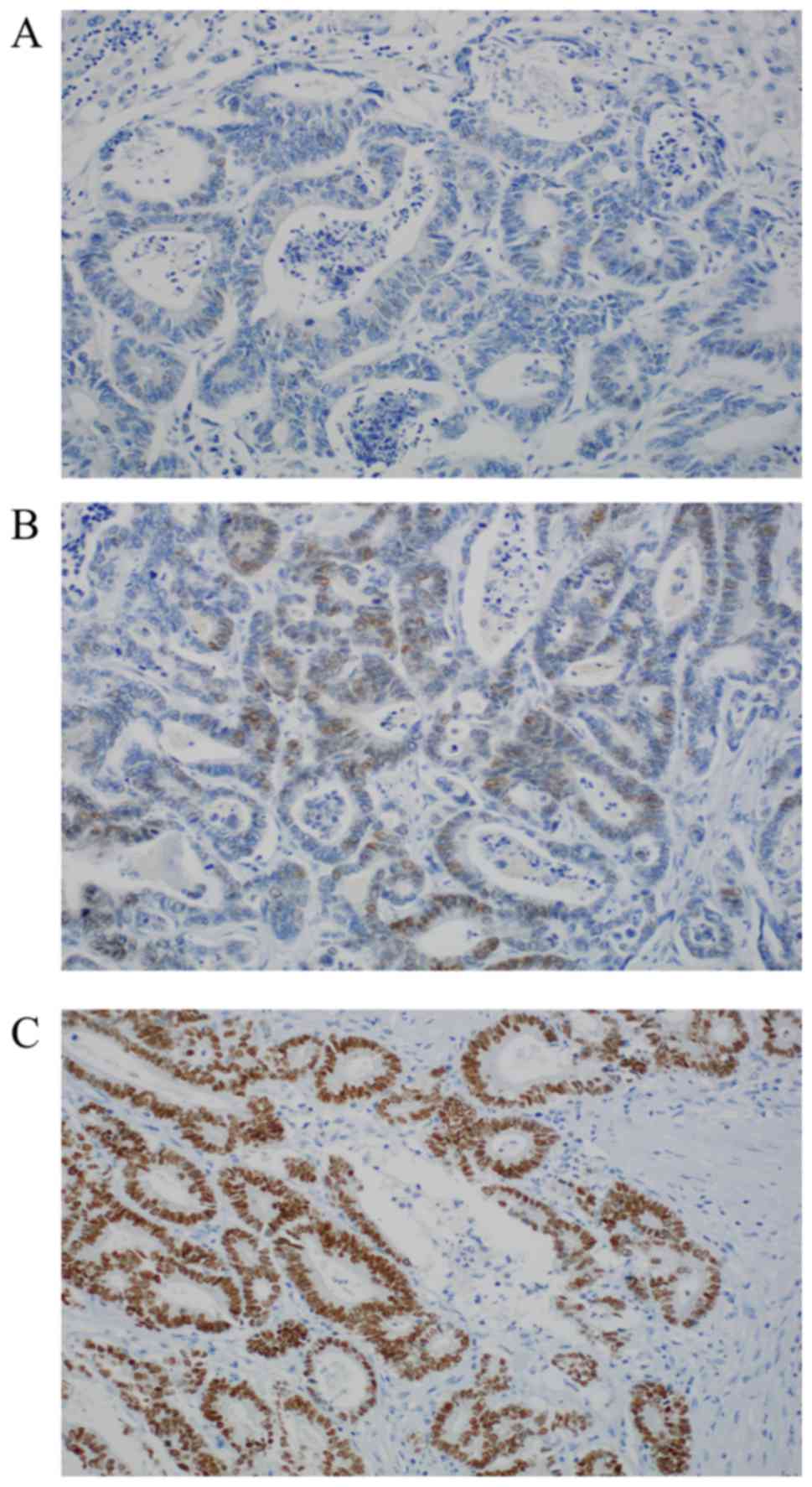
cases (38.55%) show no nuclear expression. Positive stainings
(61.45%, n=51/83) can be subdivided into two groups: Moderate
nuclear expression with score 1 (16.87% n=14); and strong
positivity with score 2 (44.58% %, n=37). Representative
photomicrographs of CDX2 immunohistochemistry are depicted in
Fig. 1.
 | Table I.Distribution of immunostaining
results of CDX2, APC and β-catenin. |
Table I.
Distribution of immunostaining
results of CDX2, APC and β-catenin.
| Protein | Score 0 (%) | Score 1 (%) | Score 2 (%) | No. of valid cases
(%) |
|---|
| CDX2 | 32 (38.55) | 14 (16.87) | 37 (44.58) | 83
(100) |
| Nuclear APC | 62 (61.38) | 39 (38.62) | – | 101 (100) |
| Cytoplasmic
APC | 13 (12.87) | 75 (74.26) | 13 (12.87) | 101 (100) |
| Cytoplasmic
β-catenin | 37 (38.14) | 60 (61.86) | – | 97
(100) |
| Nuclear
β-catenin | 60 (61.86) | 21 (21.65) | 16 (16.49) | 97
(100) |
Concerning clinical parameters like: Age, gender of
the patients, grading of the tumor and the number of metastases,
there was no significant correlation to CDX2 expression. Regarding
the size of the metastasis a strong negative correlation could be
detected (P=0.038). In addition to CDX2, ERCC1 expression was also
strongly correlated with the size of the metastases (P=0.027).
Bigger metastasis size diameter was seen in cases with CDX2 and
ERCC1 loss.
Expression distribution of DNA repair
proteins and proteins involved in Wnt-signaling
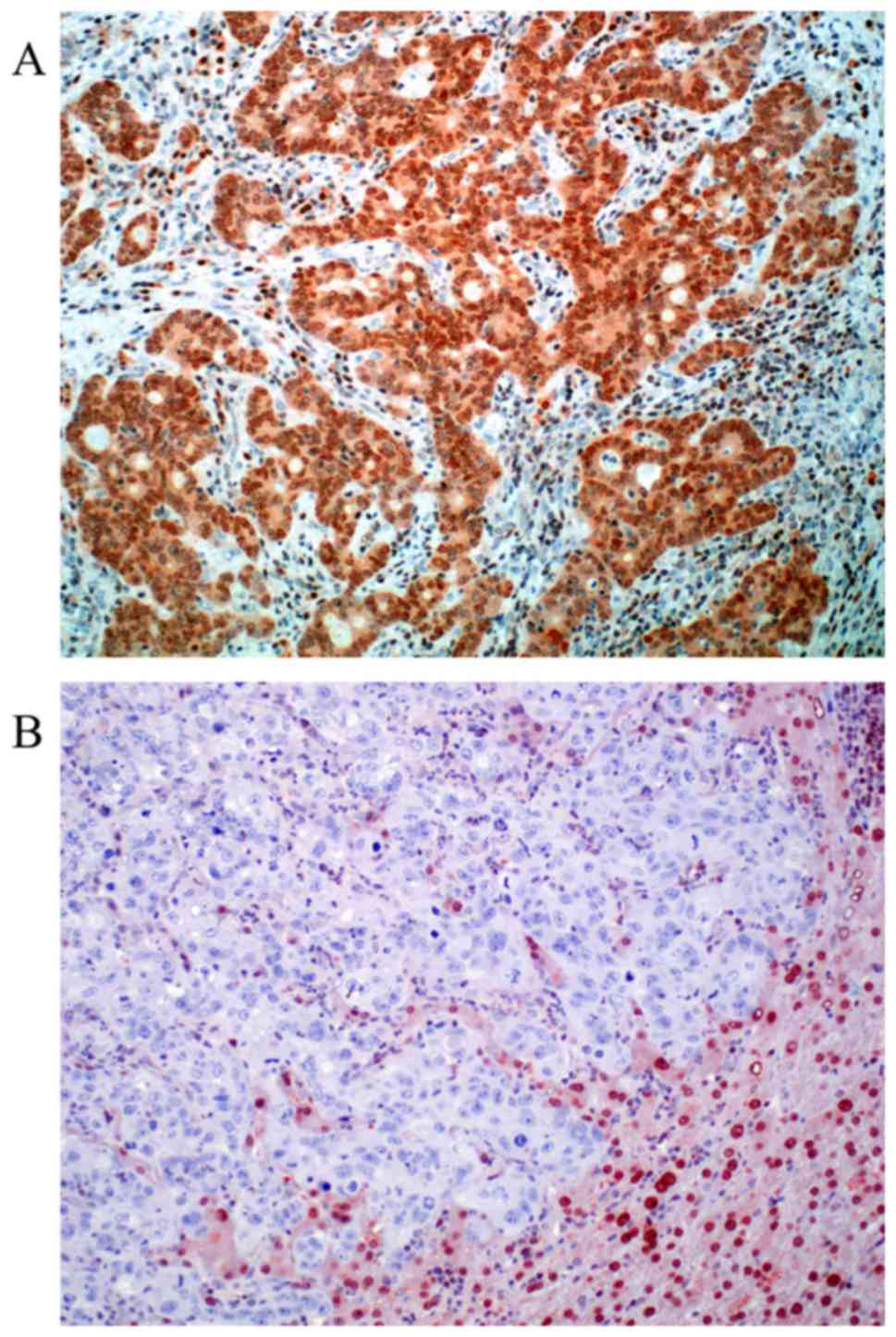
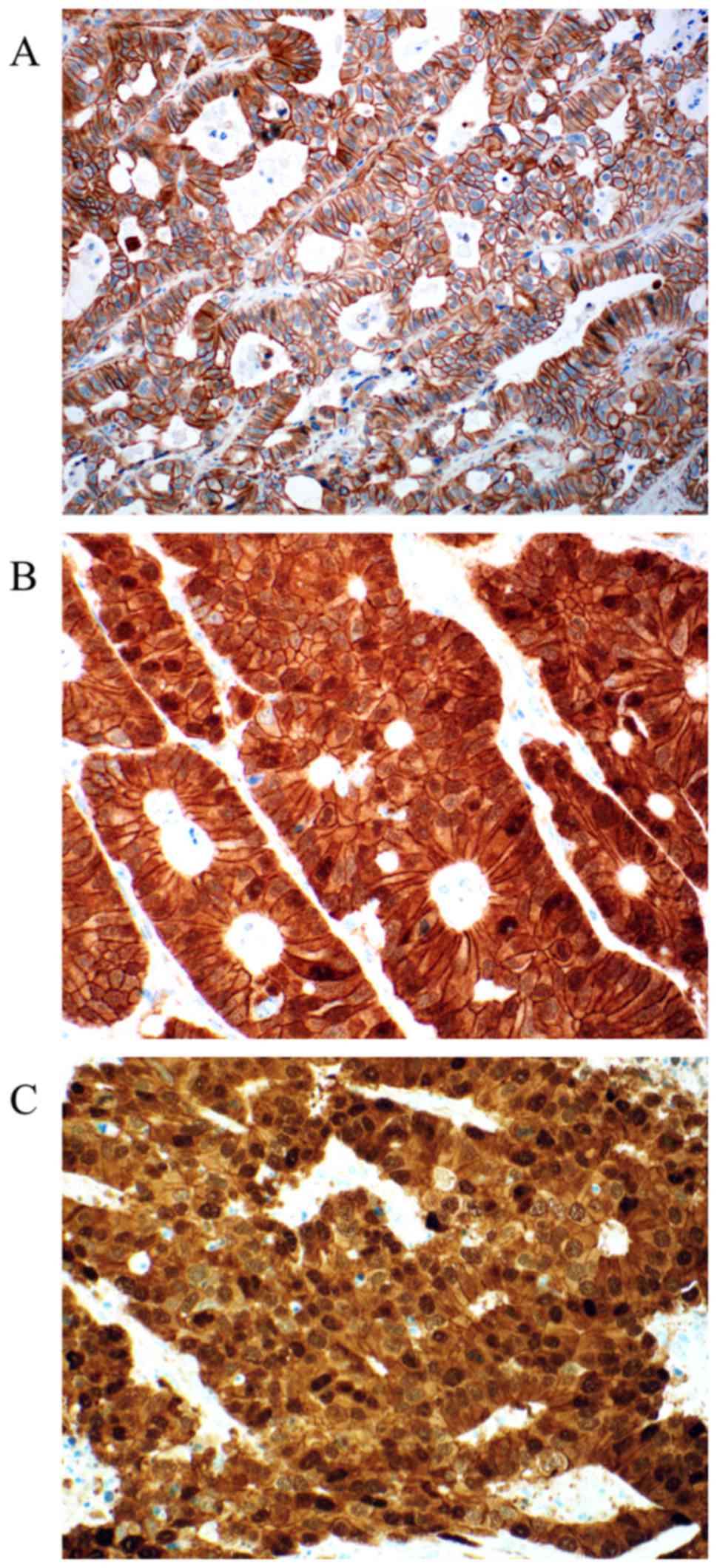
For MGMT 97 valid cases were obtained. Loss of MGMT
expression was found in 24 cases (24.75%). Representative
photomicrographs of MGMT immunohistochemistry are depicted in
Fig. 2. Nuclear positivity was
sustained in 73 cases (75.25%). Out of 94 valid cases for ERCC1 we
found 29.8% of the cases negative (score 0). Positive ERCC1
staining could be in 70.2% of the cases detected (30.8% score 1 and
39.4% score 2). Representative photomicrographs of ERCC1
immunohistochemistry are depicted in Fig.
3. Both MGMT and ERCC1 loss is strongly associated with female
gender (P=0.011, and P=0.047, respectively).
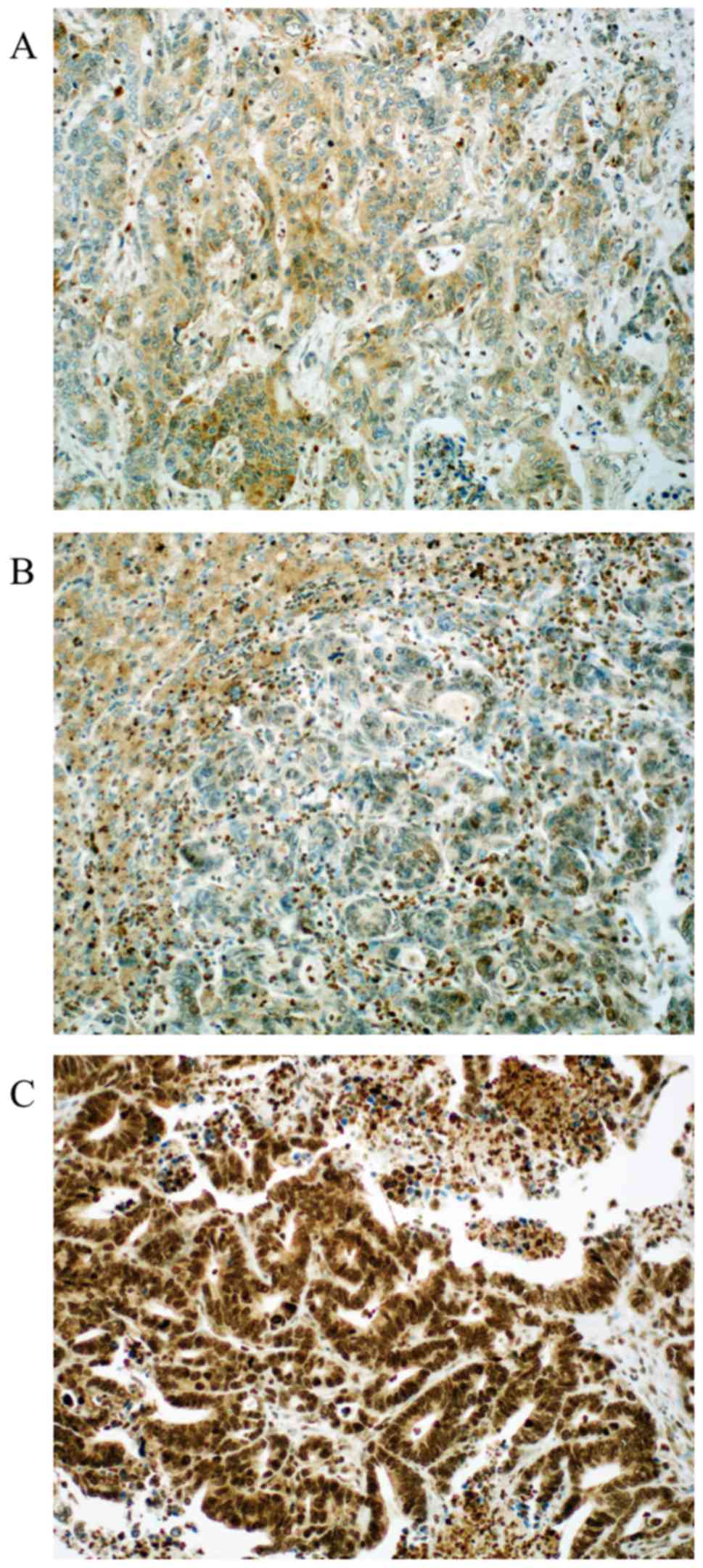
Regarding MMR proteins, the following distribution
was seen: Loss of expression was detected in 4.2 to 26% of the
cases (MLH1 4.2%, MSH2 26%, MSH6 24% and PMS2 9.5%, respectively)
as published before (19). Loss of
PMS2 is associated with loss of MGMT (P=0.014) and loss of MLH1 and
MSH2 were also associated with loss of ERCC1 (P<0.01, and
P<0.01, respectively). Representative photomicrographs of MMR
protein immunohistochemistry are depicted in Fig. 4. Expression distribution of β-catenin,
and APC proteins are depicted in Table
I.
Statistical correlations between CDX2
and DNA repair proteins
We found statistically strong positive correlation
between CDX2 and all of analysed DNA repair proteins (Table II). These results mean that loss of
CDX2 expression is strongly associated with loss of expression of
DNA repair proteins (MMR proteins, MGMT and ERCC1).
| Table II.Results of statistical analysis
between CDX2, DNA repair proteins and tumor size. |
Table II.
Results of statistical analysis
between CDX2, DNA repair proteins and tumor size.
|
|
|
| DNA repair
proteins |
|---|
|
|
|
|
|
|---|
| Gene | Analysis | Tumor size
(mm) | MLH1 | MSH2 | MSH6 | PMS2 | MGMT | ERCC1 |
|---|
| CDX2 | Correlation
coefficient | −0.247a | 0.388b | 0.334b | 0.317b | 0.228a | 0.236a | 0.574b |
|
| Significance
(2-sided) | 0.038 | <0.001 | 0.002 | 0.004 | 0.040 | 0.039 | <0.001 |
|
| Number of valid
cases | 71 | 77 | 82 | 82 | 82 | 77 | 74 |
Statistical correlations between CDX2,
APC and β-catenin
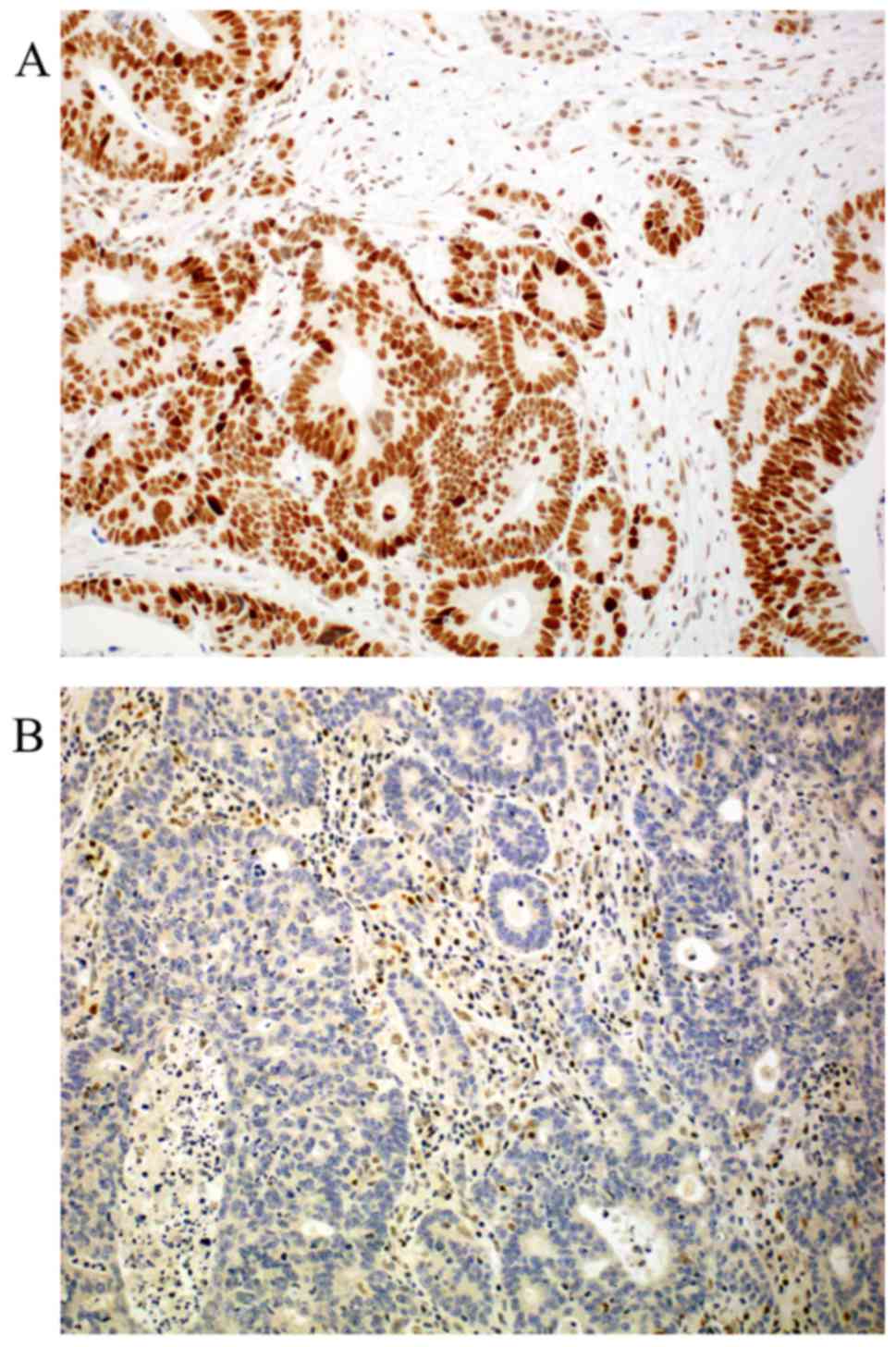
We analysed the possible statistical correlation
between CDX2 and β-catenin, and APC (Table III). Cytoplasmic, but not nuclear
β-catenin expression is associated with sustained nuclear CDX2
expression (P=0.042). In addition, CDX2 is positively correlated
with nuclear APC expression (P<0.01). Cytoplasmic and nuclear
β-catenin is associated also positive with each other (P<0.01).
Representative photomicrographs of APC and β-catenin
immunohistochemistry are depicted in Figs. 5 and 6,
respectively.
| Table III.Results of statistical analysis
between CDX2, APC and β-catenin. |
Table III.
Results of statistical analysis
between CDX2, APC and β-catenin.
| Gene | Analysis |
Membranous/cytoplasmic β-catenin | Nuclear
β-catenin | Cytoplasmic
APC | Nuclear APC |
|---|
| CDX2 | Correlation
coefficient |
0.231a | 0.152 | 0.065 | 0.415b |
|
| Significance
(2-sided) | 0.042 | 0.183 | 0.567 | <0.001 |
|
| Number of valid
cases | 78 | 78 | 79 | 79 |
Discussion
In this study, we have demonstrated significant
correlations between CDX2, DNA repair proteins and crucial members
of Wnt signaling. To our knowledge, this is the first report
performed on human tissue of CRC liver metastasis presenting
statistically significant correlations between expression of CDX2
referring to expression of MMR proteins and key proteins of base
and nuclear excision repair. Furthermore, we show, for the first
time, significant correlation between CDX2, APC and β-catenin in
liver metastasis of CRC.
Loss of CDX2 expression is seen in approximately 30%
of human CRC and is associated with higher tumor grade (1). We found loss of CDX2 expression in
38.55% of the cases. Loss of CDX2 expression was negatively
correlated with tumor size, but no correlation with age, gender of
the patients, grade of the tumor and the number of metastases.
Interestingly, ERCC1 expression loss was also correlated with tumor
size. Furthermore, loss of CDX2 is strongly correlated with loss of
ERCC1. Thus, we can conclude, that loss of CDX2 or ERCC1 expression
is strongly associated with bigger metastatic tumor size. Similar
results for ERCC1 were found recently in breast cancer (20), but the exact mechanisms are still
unclear.
We can demonstrate statistically significant
correlations between CDX2 and DNA repair proteins: Loss of CDX2
expression is associated with loss of MMR proteins, MGMT, and
ERCC1. These results are consistent with literature data from
primary CRC: MMR-deficient or MSI high CRCs have significant losses
of CDX2 expression. In addition, loss of CDX2 is associated with
CIMP-high, more aggressive histomorphological features, and
unfavourable survival (21). In a
study on primary CRC and its lymph node metastasis reduced
expression of CDX2 were found to be as predictor of MMR-deficiency
in CRC. Moreover, loss of CDX2 is a poor prognostic factor, even
among patients with MMR-proficient cancers (22).
Mutations in DNA repair genes are rare in sporadic
cancers with DNA repair deficiency. However, DNA repair deficiency
occurs in a majority of sporadic cancers caused by epigenetic
alterations that reduce or silence DNA repair gene expression. For
example, a majority of primary CRCs have reduced MGMT expression
due to i.e., methylation of the MGMT promoter region (an epigenetic
alteration) (23). MGMT can be
epigenetically depressed in many ways. Beside hypermethylation,
MGMT can be depressed by di-methylation of lysine 9 of histone 3
(24) or by over-expression of a
number of microRNAs including miR-181d, miR-767-3p and miR-603
(25).
Methylation of MGMT promoter region plays a
significant role not only in carcinogenesis but also predictive for
therapy response. In glioblastoma multiforme, the methylation state
of the MGMT gene determined whether patients would be responsive to
temozolomide therapy (26). On a
clinical level, this translates into a prolonged survival of
glioblastoma patients with a methylated MGMT promoter. In addition,
MGMT methylation can be used to predict patient survival in
clinical prediction models (27).
Loss of MGMT and ERCC1 expression was associated
with female sex in our study. Similar data were demonstrated in
primary CRC for MGMT (28) and for
ERCC1 in lung cancer (29), thus we
can conclude that this phenomenon stay maintained in liver
metastasis. For ERCC1 our study is the first demonstrating
statistically significant correlation with female gender in CRC.
ERCC1 is essential for a functional NER system and ERCC1 expression
loss may contribute to impaired DNA repair capacity thus increasing
cancer risk. Reduced expression or loss of ERCC1 and MGMT were
reported in vast majority of CRCs (30,31), and
ERCC1 promoter hypermethylation in 38% of gliomas, resulting in
reduced mRNA and protein expression (32). Disturbed ERCC1 protein expression
appears to be an early event in colorectal carcinogenesis: reduced
or loss of ERCC1 expression was detected in 40% of the colonic
crypts within early field defects in colorectal mucosa (30). Similarly to MGMT, ERCC1 silencing can
be resulted not only from promoter methylation, but can also be
evocated by miRNAs repressing its expression (33). Whether epigenetic mechanisms reduce
ERCC1 and MGMT protein expression in liver metastasis of CRC has to
be determined in methylation studies. In general, the exact role of
ERCC1 should be further elucidated because of its predictive role
in chemotherapy. Pre-clinical studies have demonstrated its
important role in determining cisplatin resistance (34).
In summary, loss of CDX2 is associated with each DNA
repair protein, which we analysed and our results in liver
metastasis are in accordance with the literature data originated
from primary CRC (21,22). Loss of CDX2 has also been found to be
an independent predictor of the CIMP-high phenotype (22). We used MGMT as surrogate marker for
CIMP phenotype, but it has been noted that studies about MGMT
methylation and CIMP had inconsistent findings, thus tumors with
loss of MGMT cannot be clearly classified as CIMP phenotype
(35). CIMP-high CRCs have been
reported to have a different clinicopathological features than
CIMP-low ones. CIMP-high phenotype is associated with older age,
cigarette smoking, proximal tumor location, female gender, poorly
differentiated or mucinous adenocarcinoma, MSI, and BRAF mutation.
In addition, CIMP-high cancers regardless of microsatellite status
show a poorer outcome (36). We
suggest that MGMT is an adequate marker to detect CIMP
phenotype.
The Wnt-β-catenin pathway is a crucial signalling
pathway in control of embryonic development and tissue homeostasis.
Its deregulation is observed in many cancers (i.e., CRC,
non-hepatitis-related hepatocellular cancers, cholangiocarcinoma,
desmoid tumor, breast cancer, osteosarcoma etc) (37). The pathway is over-activated in almost
all colon cancer because of mutations of APC tumor suppressor gene,
which actually represent the initiating event in colorectal
carcinogenesis (38). Nevertheless,
the actual mechanisms, which regulate β-catenin still remain highly
controversial. Furthermore, the exact role of APC in particular is
unclear, and the consequences of the mutations found in cancer
cells are still poorly defined (38).
Subcellular localisation of APC protein is differentially regulated
in normal tissues and cell lines: in normal human colorectal
epithelium, APC is located in the nuclei at basal segment of the
crypts; in HT29 colon cancer cells, truncated APC translocated to
the nucleus during early apoptosis (39), and cellular APC accumulates in the
nucleus of sub-confluent cells but is partly excluded in
super-confluent cells (14). Although
there is consensus in many areas in the field of nuclear APC
localization and function, there have also been some conflicting
results with no apparent resolution. Moreover, the specificity of
several APC antibodies has been investigated, with no clear
consensus about the ‘best’ antibody to detect APC protein (40). The nuclear transport of APC in tumor
cells occurs independently of β-catenin translocation to the
nucleus or plasma membrane (41).
Nuclear accumulation of β-catenin is also observed
in cancers resulting from mutations in the β-catenin, APC or Axin
genes (15,42). The APC tumor suppressor binds to
β-catenin and the scaffold protein Axin to form a complex promoting
GSK-3β phosphorylation of β-catenin. However, overexpression of APC
(1–1309), the most frequently
occurring APC cancer mutant, translocates β-catenin from the
nucleus to the cytoplasm. This mutant therefore has the ability to
bind and regulate localization but lacks the Axin binding sites
required for β-catenin degradation. Therefore, it seems more likely
that it is the inability of APC to promote β-catenin degradation,
rather than a lack of export function, that causes the nuclear
accumulation of β-catenin in APC-mutant tumor cells (14).
Little is known about the connections between CDX2
and Wnt signaling pathway. In a study on Caco-2 cells lower CDX2
expression is associated with endogenous downregulation of APC
expression, but did not affect GSK3β expression (4). Our analysis led to similar results:
Reduced expression or loss of CDX2 is associated with reduced
nuclear APC expression (P<0.01). In our study, the cytoplasmic
APC expression was not associated with CDX2 expression. We assume
that although CDX2 induce APC expression, which is already proven
(4), the truncated APC protein cannot
be shifted to cytoplasm, but we could detect this truncated protein
with our antibody. In conclusion, truncated APC can be detected
with immunohistochemistry and has certainly not lost its full
function and can still participate in β-catenin regulation. Thus,
APC can still fulfil an unexpectedly large spectrum of APC function
(38). Furthermore, we found
statistically significant correlation between CDX2 and cytoplasmic
β-catenin. We think this correlation can be explained through the
Mucdhl, a common interaction partner for β-catenin and CDX2. It has
been shown that β-catenin interacts with a protocadherin Mucdhl,
which is regulated by CDX2 in mice. Membrane-bound β-catenin is a
consequence of interactions to membranous-expressed Mucdhl. Thus,
Mucdhl can inhibit β-catenin translocation to the nucleus (4).
CDX2 is indeed expressed in all stages of CRC,
little is known about its expression manner in association with
other established prognostic or predictive proteins. In this
report, we have directly demonstrated that CDX2 gene expression is
strongly associated with DNA repair proteins and crucial members of
Wnt signaling. Our results further strengthen the role of CDX2 in
DNA repair and in regulation of APC and β-catenin expression. In
fact, our analysis is restricted only for metastasis, our results
strongly suggest potential (functional) interactions between the
investigated proteins. To our knowledge, this is the first study to
investigate CDX2 in this context on human liver metastasis of CRC.
Although, CDX2 is a useful marker in routine diagnostics for CRC,
its exact role in liver metastasis remains to be further
elucidated. In further studies should be investigated if primary
CRC differs from liver metastasis regarding CDX2 expression.
Acknowledgements
This study was supported by GINOP
2.3.2-15-2016-00020 project, which is co-financed by the European
Regional Developmental Fund of the European Union.
References
|
1
|
Hryniuk A, Grainger S, Savory JG and
Lohnes D: Cdx1 and Cdx2 function as tumor suppressors. J Biol Chem.
289:33343–33354. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Misiakos EP, Karidis NP and Kouraklis G:
Current treatment for colorectal liver metastases. World J
Gastroenterol. 17:4067–4075. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Olsen J, Espersen ML, Jess P, Kirkeby LT
and Troelsen JT: The clinical perspectives of CDX2 expression in
colorectal cancer: A qualitative systematic review. Surg Oncol.
23:167–176. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Olsen AK, Coskun M, Bzorek M, Kristensen
MH, Danielsen ET, Jørgensen S, Olsen J, Engel U, Holck S and
Troelsen JT: Regulation of APC and AXIN2 expression by intestinal
tumor suppressor CDX2 in colon cancer cells. Carcinogenesis.
34:1361–1369. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zlobec I, Bihl M, Foerster A, Rufle A and
Lugli A: Comprehensive analysis of CpG island methylator phenotype
(CIMP)-high, -low and -negative colorectal cancers based on protein
marker expression and molecular features. J Pathol. 225:336–343.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Renouf B, Soret C, Saandi T, Delalande F,
Martin E, Vanier M, Duluc I, Gross I, Freund JN and Domon-Dell C:
Cdx2 homeoprotein inhibits non-homologous end joining in colon
cancer but not in leukemia cells. Nucleic Acids Res. 40:3456–3469.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li X, Hu F, Wang Y, Yao X, Zhang Z, Wang
F, Sun G, Cui BB, Dong X and Zhao Y: CpG island methylator
phenotype and prognosis of colorectal cancer in Northeast China.
Biomed Res Int. 2014:2363612014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Inno A, Fanetti G, Di Bartolomeo M, Gori
S, Maggi C, Cirillo M, Iacovelli R, Nichetti F, Martinetti A and de
Braud F: Role of MGMT as biomarker in colorectal cancer. World J
Clin Cases. 2:835–839. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hassen S, Ali N and Chowdhury P: Molecular
signaling mechanisms of apoptosis in hereditary non-polyposis
colorectal cancer. World J Gastrointest Pathophysiol. 3:71–79.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sayar I, Akbas EM, Isik A, Gokce A, Peker
K, Demirtas L and Gürbüzel M: Relationship among mismatch repair
deficiency, CDX2 loss, p53 and E-cadherin in colon carcinoma and
suitability of using a double panel of mismatch repair proteins by
immunohistochemistry. Pol J Pathol. 66:246–253. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li Z, Qing Y, Guan W, Li M, Peng Y, Zhang
S, Xiong Y and Wang D: Predictive value of APE1, BRCA1, ERCC1 and
TUBB3 expression in patients with advanced non-small cell lung
cancer (NSCLC) receiving first-line platinum-paclitaxel
chemotherapy. Cancer Chemother Pharmacol. 74:777–786. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ruzzo A, Graziano F, Loupakis F, Rulli E,
Canestrari E, Santini D, Catalano V, Ficarelli R, Maltese P,
Bisonni R, et al: Pharmacogenetic profiling in patients with
advanced colorectal cancer treated with first-line FOLFOX-4
chemotherapy. J Clin Oncol. 25:1247–1254. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Persad S, Troussard AA, McPhee TR,
Mulholland DJ and Dedhar S: Tumor suppressor PTEN inhibits nuclear
accumulation of beta-catenin and T cell/lymphoid enhancer factor
1-mediated transcriptional activation. J Cell Biol. 153:1161–1174.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Henderson BR and Fagotto F: The ins and
outs of APC and beta-catenin nuclear transport. EMBO Rep.
3:834–839. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Polakis P: Wnt signaling and cancer. Genes
Dev. 14:1837–1851. 2000.PubMed/NCBI
|
|
16
|
Camp RL, Charette LA and Rimm DL:
Validation of tissue microarray technology in breast carcinoma. Lab
Invest. 80:1943–1949. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Torhorst J, Bucher C, Kononen J, Haas P,
Zuber M, Köchli OR, Mross F, Dieterich H, Moch H, Mihatsch M, et
al: Tissue microarrays for rapid linking of molecular changes to
clinical endpoints. Am J Pathol. 159:2249–2256. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Umar A, Boland CR, Terdiman JP, Syngal S,
de la Chapelle A, Rüschoff J, Fishel R, Lindor NM, Burgart LJ,
Hamelin R, et al: Revised bethesda guidelines for hereditary
nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite
instability. J Natl Cancer Inst. 96:261–268. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Toth C, Meinrath J, Herpel E, Derix J,
Fries J, Buettner R, Schirmacher P and Heikaus S: Expression of the
apoptosis repressor with caspase recruitment domain (ARC) in liver
metastasis of colorectal cancer and its correlation with DNA
mismatch repair proteins and p53. J Cancer Res Clin Oncol.
142:927–935. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gerhard R, Carvalho A, Carneiro V, Bento
RS, Uemura G, Gomes M, Albergaria A and Schmitt F:
Clinicopathological significance of ERCC1 expression in breast
cancer. Pathol Res Pract. 209:331–336. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dawson H, Galvan JA, Helbling M, Muller
DE, Karamitopoulou E, Koelzer VH, Economou M, Hammer C, Lugli A and
Zlobec I: Possible role of Cdx2 in the serrated pathway of
colorectal cancer characterized by BRAF mutation, high-level CpG
Island methylator phenotype and mismatch repair-deficiency. Int J
Cancer. 134:2342–2351. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dawson H, Koelzer VH, Lukesch AC, Mallaev
M, Inderbitzin D, Lugli A and Zlobec I: Loss of Cdx2 expression in
primary tumors and lymph node metastases is specific for mismatch
repair-deficiency in colorectal cancer. Front Oncol. 3:2652013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Halford S, Rowan A, Sawyer E, Talbot I and
Tomlinson I: O(6)-methylguanine methyltransferase in colorectal
cancers: Detection of mutations, loss of expression and weak
association with G:C>A:T transitions. Gut. 54:797–802. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nakagawachi T, Soejima H, Urano T, Zhao W,
Higashimoto K, Satoh Y, Matsukura S, Kudo S, Kitajima Y, Harada H,
et al: Silencing effect of CpG island hypermethylation and histone
modifications on O6-methylguanine-DNA methyltransferase (MGMT) gene
expression in human cancer. Oncogene. 22:8835–8844. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cabrini G, Fabbri E, Lo Nigro C, Dechecchi
MC and Gambari R: Regulation of expression of O6-methylguanine-DNA
methyltransferase and the treatment of glioblastoma (Review). Int J
Oncol. 47:417–428. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hegi ME, Diserens AC, Gorlia T, Hamou MF,
de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani
L, et al: MGMT gene silencing and benefit from temozolomide in
glioblastoma. N Engl J Med. 352:997–1003. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Molenaar RJ, Verbaan D, Lamba S, Zanon C,
Jeuken JW, Boots-Sprenger SH, Wesseling P, Hulsebos TJ, Troost D,
van Tilborg AA, et al: The combination of IDH1 mutations and MGMT
methylation status predicts survival in glioblastoma better than
either IDH1 or MGMT alone. Neuro Oncol. 16:1263–1273. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shen L, Kondo Y, Rosner GL, Xiao L,
Hernandez NS, Vilaythong J, Houlihan PS, Krouse RS, Prasad AR,
Einspahr JG, et al: MGMT promoter methylation and field defect in
sporadic colorectal cancer. J Natl Cancer Inst. 97:1330–1338. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kalogeraki A, Karvela-Kalogeraki I,
Tamiolakis D, Petraki P, Saridaki Z and Tzardi M: ERCC1 expression
correlated with EGFR and clinicopathological variables in patients
with non-small cell lung cancer. An immunocytochemical study on
fine-needle aspiration biopsies samples. Rev Port Pneumol.
20:200–207. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Facista A, Nguyen H, Lewis C, Prasad AR,
Ramsey L, Zaitlin B, Nfonsam V, Krouse RS, Bernstein H, Payne CM,
et al: Deficient expression of DNA repair enzymes in early
progression to sporadic colon cancer. Genome Integr. 3:32012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Smith DH, Fiehn AM, Fogh L, Christensen
IJ, Hansen TP, Stenvang J, Nielsen HJ, Nielsen KV, Hasselby JP,
Brünner N and Jensen SS: Measuring ERCC1 protein expression in
cancer specimens: Validation of a novel antibody. Sci Rep.
4:43132014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen HY, Shao CJ, Chen FR, Kwan AL and
Chen ZP: Role of ERCC1 promoter hypermethylation in drug resistance
to cisplatin in human gliomas. Int J Cancer. 126:1944–1954.
2010.PubMed/NCBI
|
|
33
|
Borrmann L, Schwanbeck R, Heyduk T,
Seebeck B, Rogalla P, Bullerdiek J and Wisniewski JR: High mobility
group A2 protein and its derivatives bind a specific region of the
promoter of DNA repair gene ERCC1 and modulate its activity.
Nucleic Acids Res. 31:6841–6851. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Garofalo M and Croce CM: MicroRNAs as
therapeutic targets in chemoresistance. Drug Resist Updat.
16:47–59. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hawkins NJ, Lee JH, Wong JJ, Kwok CT, Ward
RL and Hitchins MP: MGMT methylation is associated primarily with
the germline C>T SNP (rs16906252) in colorectal cancer and
normal colonic mucosa. Mod Pathol. 22:1588–1599. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kang KJ, Min BH, Ryu KJ, Kim KM, Chang DK,
Kim JJ, Rhee JC and Kim YH: The role of the CpG island methylator
phenotype on survival outcome in colon cancer. Gut Liver.
9:202–207. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pai SG, Carneiro BA, Mota JM, Costa R,
Leite CA, Barroso-Sousa R, Kaplan JB, Chae YK and Giles FJ:
Wnt/beta-catenin pathway: Modulating anticancer immune response. J
Hematol Oncol. 10:1012017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang L, Liu X, Gusev E, Wang C and Fagotto
F: Regulation of the phosphorylation and nuclear import and export
of β-catenin by APC and its cancer-related truncated form. J Cell
Sci. 127:1647–1659. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Efstathiou JA, Noda M, Rowan A, Dixon C,
Chinery R, Jawhari A, Hattori T, Wright NA, Bodmer WF and
Pignatelli M: Intestinal trefoil factor controls the expression of
the adenomatous polyposis coli-catenin and the E-cadherin-catenin
complexes in human colon carcinoma cells. Proc Natl Acad Sci USA.
95:pp. 3122–3127. 1998; View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Neufeld KL: Nuclear APC. Adv Exp Med Biol.
656:13–29. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fagman H, Larsson F, Arvidsson Y, Meuller
J, Nordling M, Martinsson T, Helmbrecht K, Brabant G and Nilsson M:
Nuclear accumulation of full-length and truncated adenomatous
polyposis coli protein in tumor cells depends on proliferation.
Oncogene. 22:6013–6022. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fodde R, Smits R and Clevers H: APC,
signal transduction and genetic instability in colorectal cancer.
Nat Rev Cancer. 1:55–67. 2001. View Article : Google Scholar : PubMed/NCBI
|