Introduction
Death associated protein kinase 1 (DAPK1) is a
notable multifunctional protein kinase involved in the regulation
of apoptosis, autophagy, membrane blebbing and inflammation
(1–4).
Conventionally, DAPK1 is considered to be a tumor suppressor, and
its expression is often downregulated in various types of cancer,
including leukemia, lung cancer and ovarian cancer due to DNA
methylation (5). In 2015, Zhao et
al (6) reported that endogenous
DAPK1 functioned as a survival factor in p53-mutant cancer types,
particularly triple negative breast cancers (TNBC). This oncogenic
function of DAPK1 was mediated by its stimulatory effect towards
mammalian target of rapamycin complex 1 (mTORC1) and subsequent
proliferation induction (6), which
was consistent with a previous study demonstrating that DAPK1 may
activate mTORC1 via tuberous sclerosis 2 (TSC2) phosphorylation
(7). However, the effect of DAPK1 on
colony formation, migration or invasion of breast cancer cells were
not evaluated (6). DAPK1 has been
demonstrated to inhibit cell mobility by inhibiting the
integrin-mediated polarity pathway (8). Therefore, it is important to examine the
functions of DAPK1 aside from proliferation induction in p53
wild-type and mutant breast cancer cells.
In the present study, the prognostic value of DAPK1
was evaluated using a Chinese breast cancer patient cohort. The
regulatory function of endogenous DAPK1 for breast cancer cell
viability, colony formation and migration was assessed using a
stable inducible knockdown system.
Materials and methods
Cell culture
Human breast cancer epithelial cell lines MCF7
[estrogen receptor (ER)-positive] and MDA-MB-231 derived from a
metastatic site pleural effusion were purchased from the Institute
of Biochemistry and Cell Biology at the Chinese Academy of Sciences
(Shanghai, China). Both of MCF7 and MDA-MB-231 cell lines were
verified using short tandem repeat genotyping and tested negative
for mycoplasma. The MCF7 cells were maintained in RPMI-1640,
whereas MDA-MB-231 cells were maintained in Dulbecco's modified
Eagle's medium (DMEM; both from Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), containing 10% fetal bovine
serum (FBS; Equitech-Bio, Inc., Kerrville, TX, USA), 100 U/ml
penicillin G, and 100 µg/ml streptomycin (Invitrogen; Thermo Fisher
Scientific, Inc.). All cell lines were incubated at 37°C in a
humidified incubator with 5% CO2.
Clinical samples
A total of 66 tissue samples (with a median age of
49 and an age range of 26 to 77 years old) from patients with
breast cancer were obtained from patients with invasive ductal
carcinoma at The First Affiliated Hospital of Fujian Medical
University (Fuzhou, China). All patients were admitted between
January 2010 and December 2010. The pathological and clinical data
including histological grade, invasion, and metastasis were
collected for all samples. Evaluation of the statuses of ER,
progesterone receptor and human epidermal growth factor receptor-2
was performed during patient recruitment by the Pathology
Department of The First Affiliated Hospital of Fujian Medical
University. The present study was approved by the Ethics Committee
of the First Affiliated Hospital of Fujian Medical University and
written informed consent was obtained from all patients
involved.
Immunohistochemistry (IHC)
analysis
Sample preparation and IHC staining was performed as
previously described (9). Briefly,
IHC staining for death-associated protein kinase (DAPK) was
performed on formalin-fixed, paraffin-embedded in tissue sections
(4-µm-thick) The sections were deparaffinized with 100%
dimethylbenzene and rehydrated through 100, 100, 95, 85, and 75%
ethanol series. Endogenous peroxidase was blocked by incubation in
3% H2O2 for 10 mins at room temperature. The
sections were then washed in PBS and blocked with 10% goat serum
(Beijing Zhongshan Jinqiao Biotechnology Co., Ltd., Beijing, China)
for 30 mins and incubated with anti-DAPK primary antibody (cat. no.
3008; 1:150 dilution; Cell Signaling Technology, Inc., Danvers, MA,
USA) in a humidified chamber at 4°C overnight. Following 3
additional washes in PBS, the sections were incubated with
HRP-conjugated secondary antibody (cat. no. sc-2031; 1:2,000
dilution; Santa Cruz Biotechnology, Inc. TX, USA) for 30 mins at
room temperature. The visualization signal was developed with
diaminobenzidine (DAB) solution and all slides were counterstained
with 20% hematoxylin for 30s at room temperature. Finally, all
slides were dehydrated using a series of ethanol (85% ethanol for 5
min, 95% ethanol for 5 min and 100% ethanol for 5 mins) and mounted
on cover slips. The IHC-stained tissue sections were reviewed with
a light microscope (BX51; Olympus Corporation, Tokyo, Japan) at
magnifications, ×200 and ×400. The staining intensity was
classified as negative, and positive. Briefly, the protein
expression was scored independently according to the intensity of
cellular staining and the proportion of stained tumor cells. The
staining intensity was scored as 0, no staining; 1, weak staining,
light yellow; 2, moderate staining, yellow brown; and 3, strong
staining, brown. The proportions of stained tumor cells were
classified as 0, ≤5% positive cells; 1, 6–25% positive cells; 2,
26–50% positive cells; and 3, ≥51% positive cells. The total scores
for intensity and proportion were used to signify the level of
protein expression. A score of 3 or less was considered negative
DAPK expression, and a score of 4 or more was considered positive
DAPK expression.
Construction of stable inducible cell
lines
Target cells (MCF7 and MDA-MB-231) were seeded at a
density of 1×105 cells/ml into 24-well plates.
Subsequent to growing to 50–70% confluence, cells were infected
with 250 µl short hairpin RNA (shRNA) viruses mixed with serum-free
culture medium (RPMI-1640 for MCF 7, DMEM for MDA-MB-231). A total
of 2 h later, 250 µl 10% FBS normal culture media was added. After
48 h, cells were cultured for 2 days with screening culture medium
containing 0.4 µg/ml puromycin, which was used to select for
infected cells to further culture. The stable inducible cell lines
were successfully established.
Western blot analysis
Cells were homogenized in 100–200 µl
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology, Shanghai, China) containing protease inhibitors
(Roche Diagnostics, Basel, Switzerland) and incubated at 4°C for 30
min. Protein concentrations were measured using the Micro BCA
protein assay kit (Pierce; Thermo Fisher Scientific, Inc.). Cell
lysate (60 µg per lane) was separated on a 10% SDS-PAGE gel and
subsequently transferred to a Hybond C nitrocellulose membrane
(Thermo Fisher Scientific, Inc.). The nitrocellulose membrane was
blocked with 5% fat-free milk in TBS-T buffer (20 mM Tris-HCl pH
8.0, 150 mM NaCl, 0.1% Tween-20) for 1 h at room temperature and
subsequently incubated overnight at 4°C with the primary antibodies
anti-human DAPK (1:1,000 dilution; cat. no. 3008; Cell Signaling
Technology, Inc.) and anti-human GAPDH (1:5,000 dilution; cat. no.
2118; Cell Signaling Technology, Inc.), then washing (3× 10 min) in
TBS-T buffer, followed by incubating for 1 h at room temperature
with IRDye 800CW-conjugated goat-anti-Rabbit secondary antibodies
(cat. no. C60607-15; LI-COR Biosciences, Lincoln, NE, USA). The
membrane was finally washed (3× 10 min) in TBS-T buffer again. The
signals were detected and measured using LICOR Odyssey system
(LI-COR, Nebraska, USA).
Sulforhodamine B (SRB) assay
Cells were first seeded in 6-cm dishes with or
without 400 ng/ml doxycycline (Dox) (cat. no. 324385; EMB
Millipore, Billerica, MA, USA) and cultured for 5 days. Then, for
the SRB assay, MCF7 and MDA-MB-231 cells were seeded at a density
of 7×103 and 1×104 cells/well into 96-well
plates, respectively. Next, cells continued to grow with or without
Dox for another 96 h. Subsequent to culturing, total cell amounts
were detected using an Sulforhodamine B (cat. no. S1402;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) assay as previously
described (10).
Colony formation
Cells were first seeded in 6 cm dishes with or
without Dox and allowed to grow for 5 days. Then for the colony
formation assay, MCF7 and MDA-MB-231 cells were seeded at a density
of 500 cells/well into six-well plates. Next, cells continued to
grow with or without Dox for 7 days. Following culturing, the
number of clones formed was detected using a colony formation assay
as previously described (10).
Cell migration assays
Cells were first seeded in 6 cm dishes with or
without Dox for 5 days. Cells were then seeded in six-well plates
with or without Dox and allowed to grow to confluence. The cell
migration was then measured using a scratch wound healing assay as
previously described (9). Briefly,
the monolayer cells were scratched with a 10 µl sterile pipette tip
to create a ~10 µm wound, washed twice with serum-free media to
remove floating cells, and left to grow in serum-free media. The
cells migrating from the leading edge were photographed at 0 and 24
h. Multiple views of each well were documented with an inverted
light microscope (CKX-41; Olympus Corporation, Tokyo, Japan) at
magnification, ×200 and three independent experiments were
performed. The percentage of reduced cover area was calculated as
(g0-gt)/g0 ×100%. The g0 and gt were the wound width at 0 and 24 h,
respectively. For the cell migration assay, Transwell chambers
(polycarbonate filters of 8 µm porosity; BD Biosciences, Franklin
Lakes, NJ, USA) were used. The bottom chamber was filled with a
culture medium (RPMI-1640 for MCF 7 cells and DMEM for MDA-MB-231
cells) containing 10% FBS and the upper chamber was filled with
serum-free medium. A total of 2×105 stable cells were
suspended in serum-free medium and plated in the upper chamber.
Following incubation for 24 h, the cells were removed from the
upper chamber using a cotton swab. Cells that had penetrated and
attached to the bottom of the filter were fixed for 10 min at room
temperature with 4% formaldehyde in PBS, followed by 20 min
staining with 0.5% crystal violet at room temperature and then
subjected to imaging under a ×20 objective lens and photographed.
Statistical results of cell numbers per each image field were
obtained from three independent experiments averaging from five
image fields.
Statistical analysis
Analyses of the association between the protein
expression of DAPK1 and clinicopathological variables were
performed using Fisher's exact test. Progression-free survival was
defined as the interval from the first day of surgery until tumor
progression, mortality or the end of follow-up. Time-to-progress
comparisons were performed using a Log-rank test. Univariate
Cox-regression and multivariate analysis were additionally
performed for available clinicopathological parameters.
Physiological functional data of DAPK1 in breast cancer were
analyzed using GraphPad Prism version 7.0 software (Graphpad
Software, Inc., La Jolla, CA, USA). All data are presented as the
mean ± standard deviation of three independent experiments. The
significance of difference in colony formation and cell migration
of MCF7 and MDA-MB-231 cells with or without Dox induction were
examined using a two-tailed Student's t-test. Multiple comparisons
were performed using one-way analysis of variance followed by
Dunnett's post-hoc test. For all analyses, P<0.05 was considered
to indicate a statistically significant difference.
Results
Association between DAPK1 expression
and clinicopathological characteristics of patients with breast
cancer
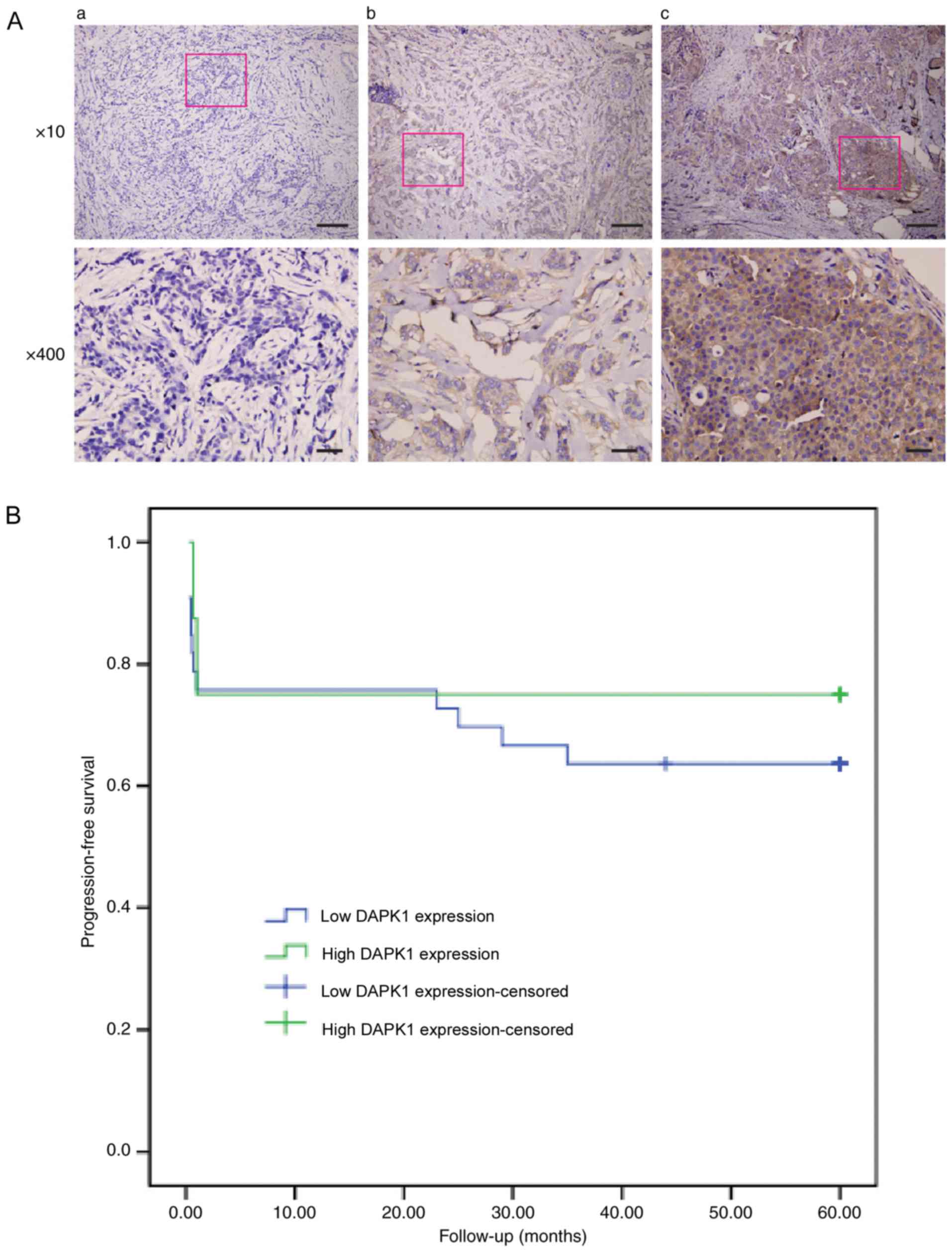
First, the prognostic function of DAPK1 in breast
cancer was evaluated using the IHC staining of 66 samples from
patients with breast cancer (Fig.
1A). The general profile of this patient cohort was listed on
Table I. Of all the
clinicopathological parameters available, DAPK1 expression was only
significantly associated with histological grade (P=0.036; Table II). Additionally, when patients were
grouped as ER-positive and ER-negative, DAPK1 expression was only
significantly associated with histological grade in ER-negative
patients (P=0.042; Table III).
Furthermore, DAPK1 expression was not significantly associated with
the 5-year progression-free survival of patients with breast cancer
(Fig. 1B). These data demonstrated
that despite the previous study that observed that DAPK1 may
function as a survival factor in breast cancer to promote cell
growth (6), DAPK1 did not
significantly affect the overall survival rate in the present
cohort. Unfortunately, the p53 mutation was not routinely examined
at Fujian Medical Hospital. Therefore, it was difficult to
distinguish if p53 status affected the prognostic value of DAPK1 in
the present cohort.
 | Table I.General profile of 66 patients with
invasive breast ductal carcinoma. |
Table I.
General profile of 66 patients with
invasive breast ductal carcinoma.
| Characteristics | Number of
patients/total (%) |
|---|
| Age (years) |
|
|
<55 | 41/66 (62.1) |
| ≥55 | 25/66 (37.9) |
| Histological
grade |
|
| I | 3/66 (4.5) |
| II | 53/66 (80.3) |
| III | 10/66 (15.2) |
| Axillary nodal
status |
|
|
Negative | 25/66 (37.9) |
|
Positive | 33/66 (50.0) |
| NA | 8/66 (12.1) |
| ER |
|
|
Negative | 29/66 (43.9) |
|
Positive | 31/66 (46.9) |
| NA | 6/66 (9.2) |
| PR |
|
|
Negative | 38/66 (57.5) |
|
Positive | 22/66 (33.3) |
| NA | 6/66 (9.2) |
| Her2 |
|
|
Negative | 30/66 (45.4) |
|
Positive | 30/66 (45.4) |
| NA | 6/66 (9.2) |
| DAPK |
|
|
Positive | 53/66 (80.3) |
|
Negative | 13/66 (19.7) |
| Table II.Association between DAPK and other
clinicopathological characteristics. |
Table II.
Association between DAPK and other
clinicopathological characteristics.
|
| DAPK |
|
|---|
|
|
|
|
|---|
| Characteristics | Negative | Positive | P-value |
|---|
| Age (years) |
|
| 0.185 |
|
<55 | 6 | 35 |
|
| ≥55 | 7 | 18 |
|
| Histological
grade |
|
| 0.036a |
| I | 2 | 1 |
|
| II | 11 | 42 |
|
|
III | 0 | 10 |
|
| Axillary nodal
status |
|
| 0.910 |
|
Negative | 5 | 20 |
|
|
Positive | 7 | 26 |
|
| ER |
|
| 0.438 |
|
Negative | 7 | 22 |
|
|
Positive | 5 | 26 |
|
| PR |
|
| 0.688 |
|
Negative | 7 | 31 |
|
|
Positive | 5 | 17 |
|
| Her2 |
|
| 0.519 |
|
Negative | 5 | 25 |
|
|
Positive | 7 | 23 |
|
| Table III.Association between DAPK and other
clinicopathological characteristics in ER positive and negative
patient samples. |
Table III.
Association between DAPK and other
clinicopathological characteristics in ER positive and negative
patient samples.
|
| ER-negative |
| ER-positive |
|
|---|
|
|
|
|
|
|
|---|
|
Characteristics | DAPK-negative | DAPK-positive | P-value | DAPK-negative | DAPK-positive | P-value |
|---|
| Age (years) |
|
| 0.403 |
|
| 0.998 |
|
<55 | 3 | 14 |
| 3 | 17 |
|
|
≥55 | 4 | 8 |
| 2 | 9 |
|
| Histological
grade |
|
| 0.066 |
|
| 0.042a |
| I | 0 | 0 |
| 2 | 1 |
|
| II | 7 | 13 |
| 3 | 24 |
|
|
III | 0 | 9 |
| 0 | 1 |
|
| Axillary nodal
status |
|
| 0.997 |
|
| 0.998 |
|
Negative | 2 | 7 |
| 2 | 12 |
|
|
Positive | 4 | 11 |
| 3 | 11 |
|
| PR |
|
| 0.998 |
|
| 0.368 |
|
Negative | 6 | 19 |
| 1 | 12 |
|
|
Positive | 1 | 3 |
| 4 | 14 |
|
| Her2 |
|
| 0.382 |
|
| 0.997 |
|
Negative | 1 | 8 |
| 4 | 17 |
|
|
Positive | 6 | 14 |
| 1 | 9 |
|
Expression of DAPK1 in breast cancer
cell lines
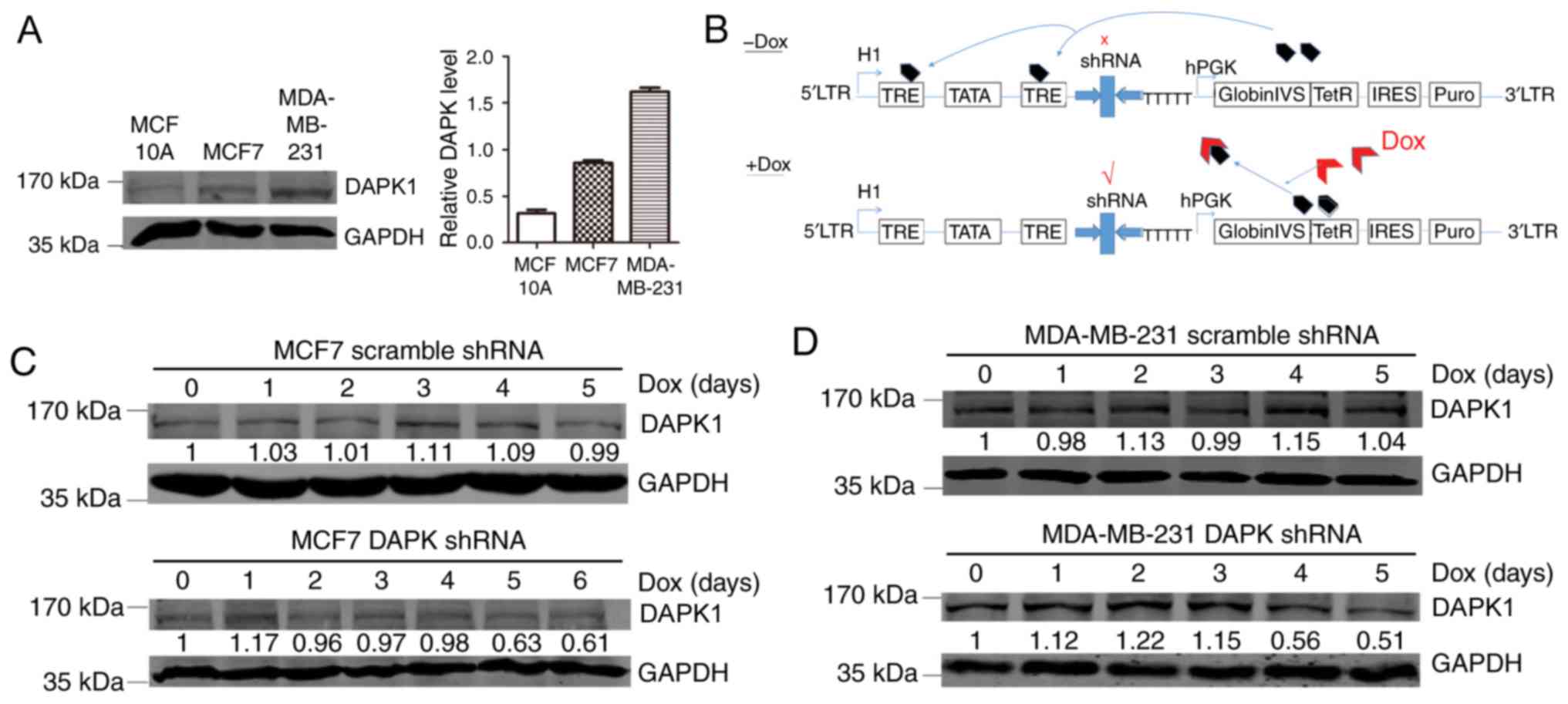
Next, the functions of DAPK1 in breast cancer cell
lines were investigated. The p53 wild-type MCF7 and p53 mutant TNBC
MDA-MB-231 cell lines were employed. Compared with the normal
breast epithelial cell line MCF10A, MCF7 and MDA-MB-231 cell lines
exhibited enhanced DAPK1 expression, suggesting that DAPK1 may be
overexpressed in breast cancer cell lines (Fig. 2A).
Effect of DAPK1 knockdown in MCF7 and
MDA-MB-231 cells
In order to evaluate the physiological function of
DAPK1 in these breast cancer cell lines, a Dox inducible shRNA
plasmid was used to create stable inducible DAPK1 knockdown cell
lines (Fig. 2B). When exposed to Dox,
stable MCF7 and MDA-MB-231 cells revealed substantial DAPK1
downregulation, indicating successful establishment of the stable
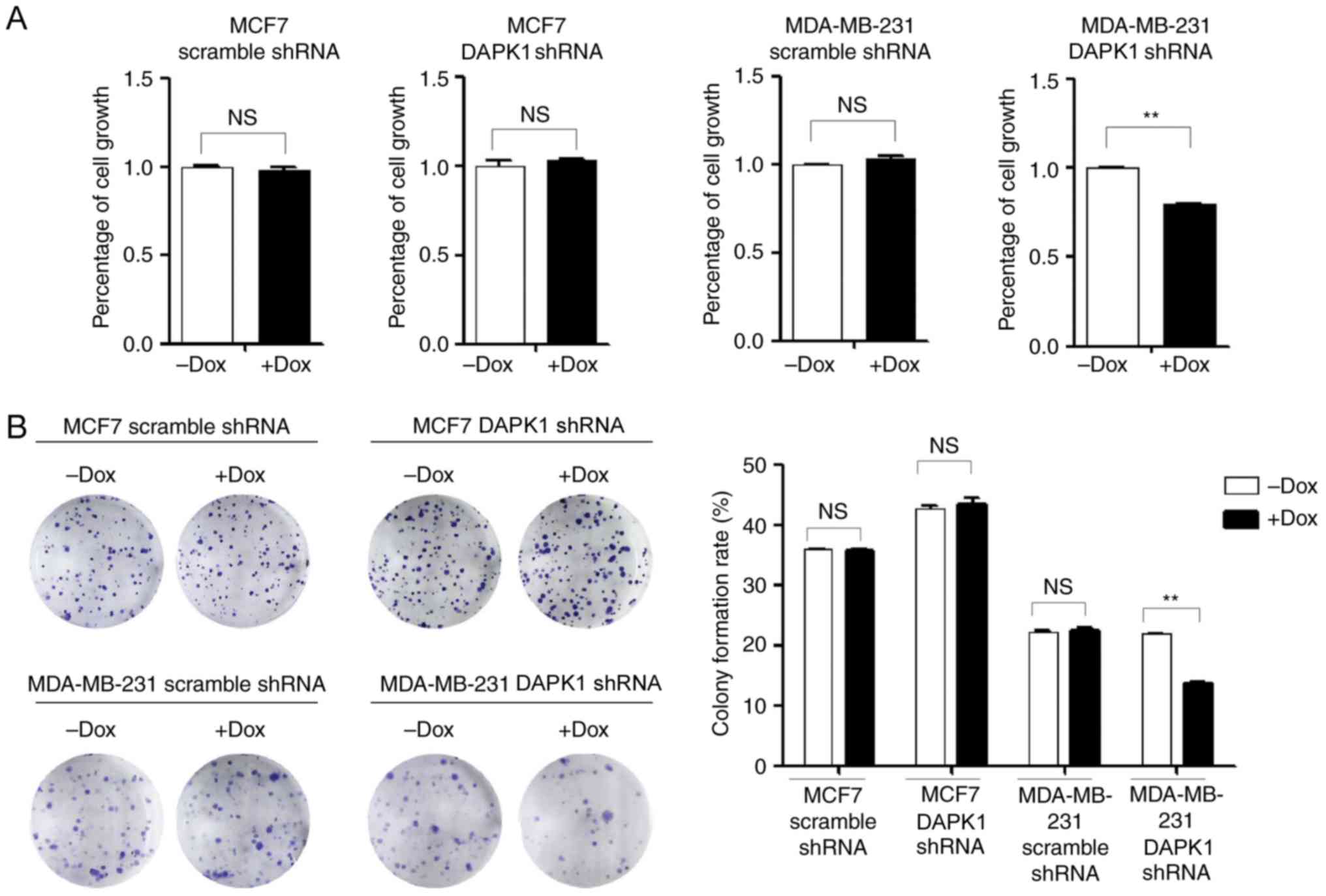
cells (Fig. 2C and D). Next, SRB and
colony formation assays were performed to investigate the function
of endogenous DAPK1 on cell viability, and colonization ability.
The knockdown of DAPK1 only significantly reduced the viability and
colony forming ability of MDA-MB-231 cells treated with Dox
(P<0.01), but not MCF7 cells (Fig.
3), consistent with the previous report that DAPK1 only affects
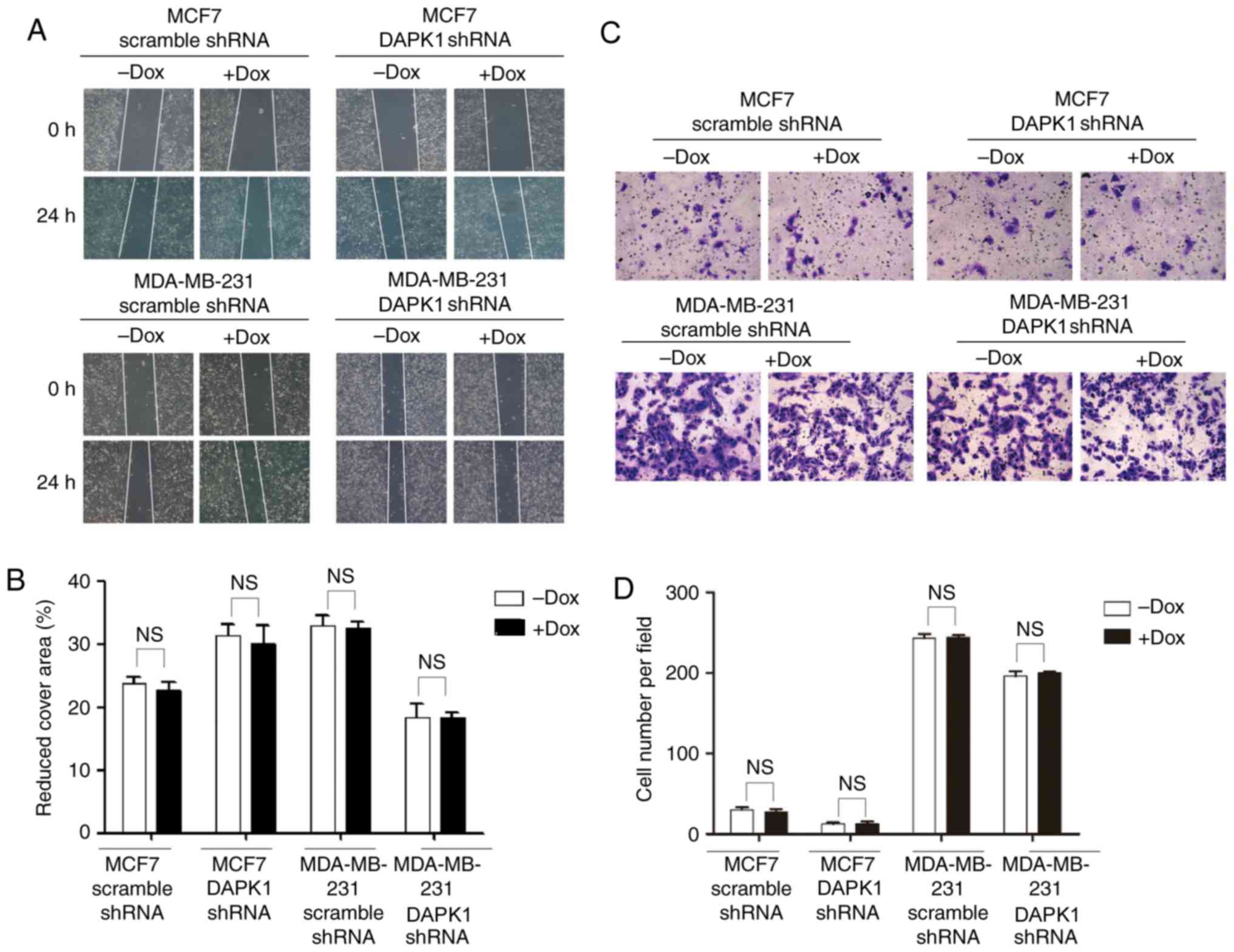
breast cancer viability in p53 mutant cells (6). Then, the effect of DAPK1 on cell
migration was investigated. Following the knockdown of DAPK1, no
significant difference was observed in MCF7 or MDA-MB-231 cells
(Fig. 4), suggesting endogenous DAPK1
does not regulate cell motility in these cells.
Discussion
In the present study, expression of DAPK1 protein
was identified in 53/66 samples from patients with breast cancer.
The observation that the two breast cancer cell lines exhibited
enhanced DAPK1 protein expression in comparison with the normal
breast epithelial cell line MCF10A, as demonstrated by western
blotting, was concordant with the IHC results. DAPK1 DNA
methylation and the loss of its mRNA expression had been previously
reported in breast cancer (11). The
results of the present study indicated that post-transcriptional
regulatory pathways notably affect the protein expression of DAPK1.
Although the DNA methylation of DAPK1 was widely reported in
multiple types of cancer, only a few of them examined mRNA
expression or the protein expression of DAPK1 (5,11–13). Therefore, extra caution is required
when interpreting the prognostic value of DAPK1 DNA methylation, as
it may not be associated with DAPK1 protein expression and may
actually reflect the activity of other proteins involved in DNA
methylation.
It took ~5 days to effectively knockdown DAPK1
protein expression in MCF7 and MDA-MB-231 cell lines. Whereas in
previous studies, exogenous DAPK1 protein had a relatively short
half-life and its expression dropped to 10–20% once DAPK
translation was stopped using cycloheximide within 48 h (14). Furthermore, the knockdown of DAPK1 in
colon cancer cell lines using small interfering RNA significantly
decreased DAPK1 expression levels 48 h post transfection (15). These data indicated that the stability
of exogenous and endogenous DAPK1 proteins in different tissues is
different, which may be caused by the diverse range of binding
partners, and cellular environment. At present, DAPK1 is known to
be degraded via proteasomal and lysosomal signaling pathways
(14,16). The ubiquitin E3s for DAPK1 include
mind bomb 1 (16), C-terminus of
Hsc70-interacting protein (17) and
kelch like family member 20-cullin3-ring-box 1 complex (18). TSC2 and a splice variant of DAPK
mediate the lysosomal degradation of DAPK1 (3,14).
Furthermore, a lysosomal protease, cathepsin B, is able to cleave
DAPK1 in response to tumor necrosis factor receptor 1
overexpression (19). The tissue
specific DAPK1 stability control may be due to the differential
expression of these proteins. The inducible knockdown of DAPK1
resulted in the reduced viability of MDA-MB-231, but not MCF7
cells, supporting the model that DAPK1 positively regulates the
viability in p53 mutant breast cancer cells (6). In addition to p53, a number of
components in the ER signaling pathway, including extracellular
signal-regulated kinase were reported to interact and regulate
DAPK1 activity (20,21). It is unclear whether the status of
these upstream signaling molecules of DAPK1 is different in MCF7
and MDA-MB-231 cells, and contributes to the differential viability
responses. Of note, DAPK1 did not function as a prognostic factor
for patient survival in the present dataset regardless of the ER
status. One potential explanation may be that the patient cohort of
the present study was limited in size. There were only patients
with TNBC in our dataset and the p53 mutation status was not clear.
Furthermore, the scoring system used in the present study is
different from that used in a previous publication (6) as patients were classified into negative
and positive, which were considered to be least likely to create
any confusion. Further studies are required in order to elucidate
the clinical function of DAPK1 in breast cancer.
However, the inducible knockdown of DAPK1 did not
affect migration in either of the breast cancer cell lines.
However, the ability of DAPK1 to suppress the migration of breast
cancer cell lines, including MDA-MB-231 has been previously
demonstrated (8). This discrepancy
may be due to the difference technique used to regulate DAPK1
expression. Previous studies have typically reported a clear
migration or invasion suppressive effect of overexpressed DAPK1
(15,22). Although DAPK1 is capable of
suppressing cell motility, this function may not be triggered
during the physiological stage and requires extra signal
stimulation (22). Furthermore,
previously, Ivanovska et al (15) reported that DAPK1 did not affect
proliferation in colon cancer, but was strongly associated with the
migration and invasion of colon cancer cells, suggesting that the
physiological function of DAPK1 may be tissue specific.
In conclusion, the present study investigated the
ability of DAPK1 to regulate cell viability, colony formation and
migration in breast cancer cell lines. Endogenous DAPK1 was only
able to regulate the viability and colony formation of MDA-MB-231
cells. Further studies are required to investigate the
physiological functions of DAPK1 and the key factors determining
the tissue specific functions of DAPK1.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Scientific
Research Innovation Team Construction Program of Fujian Normal
University (grant no. IRTL1702), the Natural Science Foundation of
Fujian Province (grant nos. 2016Y0029, 2016J01146 and 2016J01538)
and the Key Clinical Specialty Discipline Construction Program of
Fujian, P.R. China [grant no. (2013) 544].
Availability of data and materials
All data and materials generated or analyzed in the
present study are included in this manuscript.
Authors' contributions
YH, YZ and YL were responsible for study conception
and design. YH, ML, XC, CH, XZ, LC, KW and YC performed data
collection and assembly. YH, ML, XC, YZ and YL performed data
analysis and interpretation. All authors approved the final version
of this manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the First Affiliated Hosptal of Fujian Medical
University, and informed consent to participate in the study was
obtained from all patients involved.
Consent for publication
No identifying patient information is included in
the published manuscript.
Competing interests
All the authors declare that they have no competing
interests.
References
|
1
|
Shiloh R, Bialik S and Kimchi A: The DAPK
family: A structure-function analysis. Apoptosis. 19:286–297. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lin Y, Hupp TR and Stevens C:
Death-associated protein kinase (DAPK) and signal transduction:
Additional roles beyond cell death. FEBS J. 277:48–57. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lin Y, Stevens C, Harrison B, Pathuri S,
Amin E and Hupp TR: The alternative splice variant of DAPK-1,
s-DAPK-1, induces proteasome-independent DAPK-1 destabilization.
Mol Cell Biochem. 328:101–107. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lin Y, Stevens C, Hrstka R, Harrison B,
Fourtouna A, Pathuri S, Vojtesek B and Hupp T: An alternative
transcript from the death-associated protein kinase 1 locus
encoding a small protein selectively mediates membrane blebbing.
FEBS J. 275:2574–2584. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huang Y, Chen L, Guo L, Hupp TR and Lin Y:
Evaluating DAPK as a therapeutic target. Apoptosis. 19:371–386.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhao J, Zhao D, Poage GM, Mazumdar A,
Zhang Y, Hill JL, Hartman ZC, Savage MI, Mills GB and Brown PH:
Death-associated protein kinase 1 promotes growth of p53-mutant
cancers. J Clin Invest. 125:2707–2720. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stevens C, Lin Y, Harrison B, Burch L,
Ridgway RA, Sansom O and Hupp T: Peptide combinatorial libraries
identify TSC2 as a death-associated protein kinase (DAPK) death
domain-binding protein and reveal a stimulatory role for DAPK in
mTORC1 signaling. J Biol Chem. 284:334–344. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kuo JC, Wang WJ, Yao CC, Wu PR and Chen
RH: The tumor suppressor DAPK inhibits cell motility by blocking
the integrin-mediated polarity pathway. J Cell Biol. 172:619–631.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xie JW, Chen PC, Zheng CH, Li P, Wang JB,
Lin JX, Lu J, Chen QY, Cao LL, Lin M, et al: Evaluation of the
prognostic value and functional roles of CD44v6 in gastric cancer.
J Cancer Res Clin Oncol. 141:1809–1817. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lin Y, Richards FM, Krippendorff BF,
Bramhall JL, Harrington JA, Bapiro TE, Robertson A, Zheleva D and
Jodrell DI: Paclitaxel and CYC3, an aurora kinase A inhibitor,
synergise in pancreatic cancer cells but not bone marrow precursor
cells. Br J Cancer. 107:1692–1701. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lehmann U, Celikkaya G, Hasemeier B,
Langer F and Kreipe H: Promoter hypermethylation of the
death-associated protein kinase gene in breast cancer is associated
with the invasive lobular subtype. Cancer Res. 62:6634–6638.
2002.PubMed/NCBI
|
|
12
|
Christoph F, Kempkensteffen C, Weikert S,
Köllermann J, Krause H, Miller K, Schostak M and Schrader M:
Methylation of tumour suppressor genes APAF-1 and DAPK-1 and in
vitro effects of demethylating agents in bladder and kidney cancer.
Br J Cancer. 95:1701–1707. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Satoh A, Toyota M, Itoh F, Kikuchi T,
Obata T, Sasaki Y, Suzuki H, Yawata A, Kusano M, Fujita M, et al:
DNA methylation and histone deacetylation associated with silencing
DAP kinase gene expression in colorectal and gastric cancers. Br J
Cancer. 86:1817–1823. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin Y, Henderson P, Pettersson S, Satsangi
J, Hupp T and Stevens C: Tuberous sclerosis-2 (TSC2) regulates the
stability of death-associated protein kinase-1 (DAPK) through a
lysosome-dependent degradation pathway. FEBS J. 278:354–370. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ivanovska J, Zlobec I, Forster S,
Karamitopoulou E, Dawson H, Koelzer VH, Agaimy A, Garreis F, Söder
S, Laqua W, et al: DAPK loss in colon cancer tumor buds:
implications for migration capacity of disseminating tumor cells.
Oncotarget. 6:36774–36788. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jin Y, Blue EK and Gallagher PJ: Control
of death-associated protein kinase (DAPK) activity by
phosphorylation and proteasomal degradation. J Biol Chem.
281:39033–39040. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang L, Nephew KP and Gallagher PJ:
Regulation of death-associated protein kinase. Stabilization by
HSP90 heterocomplexes. J Biol Chem. 282:11795–11804. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee YR, Yuan WC, Ho HC, Chen CH, Shih HM
and Chen RH: The Cullin 3 substrate adaptor KLHL20 mediates DAPK
ubiquitination to control interferon responses. EMBO J.
29:1748–1761. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lin Y, Stevens C and Hupp T:
Identification of a dominant negative functional domain on DAPK-1
that degrades DAPK-1 protein and stimulates TNFR-1-mediated
apoptosis. J Biol Chem. 282:16792–16802. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Stevens C, Lin Y, Sanchez M, Amin E,
Copson E, White H, Durston V, Eccles DM and Hupp T: A germ line
mutation in the death domain of DAPK-1 inactivates ERK-induced
apoptosis. J Biol Chem. 282:13791–13803. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen CH, Wang WJ, Kuo JC, Tsai HC, Lin JR,
Chang ZF and Chen RH: Bidirectional signals transduced by DAPK-ERK
interaction promote the apoptotic effect of DAPK. EMBO J.
24:294–304. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kuo JC, Wang WJ, Yao CC, Wu PP and Chen
RH: The tumor suppressor DAPK inhibits cell motility by blocking
the integrin-mediated polarity pathway. J cell Biol. 172:619–631.
2006. View Article : Google Scholar : PubMed/NCBI
|