Introduction
Cervical cancer is the fourth most common cancer in
women worldwide, with an estimated 527,600 new cases and 265,600
deaths in 2012 (1). Although the
association between persistent high-risk human papillomavirus
(HR-HPV) infection and the development of cervical cancer has been
demonstrated by molecular and functional studies, the specific
molecular network mechanisms from HPV infection to tumorigenesis
have not been fully elucidated. Therefore, investigating the
potential mechanism underlying tumorigenesis may be crucial for
prolonging patient survival.
Tumorigenesis is a complex pathological process
involving a variety of genetic alterations, including the
overexpression of oncogenes and/or the inactivation of tumor
suppressor genes (2). The development
of cervical cancer is a stepwise process from a low-grade cervical
intraepithelial neoplasia (CIN1) to high-grade CIN (CIN2 and 3)
that ultimately develops into carcinoma (3), involving multiple genetic and epigenetic
events. The identification of dysregulated genes in
cancer-associated pathways may shed light on the molecular
mechanisms underlying tumorigenesis, thus helping to develop new
strategies for tumor therapy.
Recently, gene analysis using the high-throughput
platforms has been developed as a promising tool with various
clinical applications, such as the molecular diagnosis and
classification of cancers, and the prediction of tumor response and
patient prognosis (4). Several gene
expression profiles related to cervical carcinogenesis have been
studied with microarray technology, revealing hundreds of
differentially expressed genes (DEGs) that are involved in the
process of tumorigenesis, serving a potential role in the
identification of novel therapeutic targets (5). The present study applied bioinformatics
analysis to identify DEGs involved in the progression from normal
cervical epithelium tissue to high-grade CIN and cervical cancer,
and explored the significant GO terms, KEGG pathways and
protein-protein interaction (PPI) networks, with a particular focus
on possible hub genes that are likely to play key roles in the
progression of cervical cancer.
Materials and methods
Microarray datasets
The cervical cancer microarray datasets GSE7803 and
GSE63514 were downloaded from the NCBI GEO database (http://www.ncbi.nlm.nih.gov/geo). The dataset
GSE7803 was based on the GPL96 platform (Affymetrix Human Genome
U133A Array; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
including 10 normal squamous cervical epithelium (NE), 7 high-grade
squamous intraepithelial cervical lesion (HSIL), and 21 invasive
squamous cell carcinoma (SCC) of the cervix samples. The dataset
GSE63514, produced using the GPL570 Affymetrix Human Genome U133
Plus 2.0 Array, included 24 NE samples, 2 CIN2 lesion samples, 40
CIN3 lesion samples, and 28 cancer specimens. CIN2 and CIN3 were
considered to be HSIL in our study.
Identification of DEGs
GEO2R, an interactive web tool for comparing two or
more groups of samples, and identifying genes that are
differentially expressed across experimental conditions, was used
to identify DEGs in the GSE7803 and GSE63514 datasets with the
limma package, which had been processed, normalized and
transformed. An adjusted P-value was obtained by applying the
Benjamini-Hochberg false discovery rate (FDR) correction on the
original P-value, and a fold change threshold was selected based on
our aim to focus on statistically significant DEGs (6). Only genes with a fold change >2 and
adjusted P-value <0.05 were considered as statistically
significant DEGs. In addition, the selected DEGs were divided into
two groups: DEGs between the NE and HSIL samples were considered as
pre-invasive DEGs, whereas DEGs between the HSIL and invasive SCC
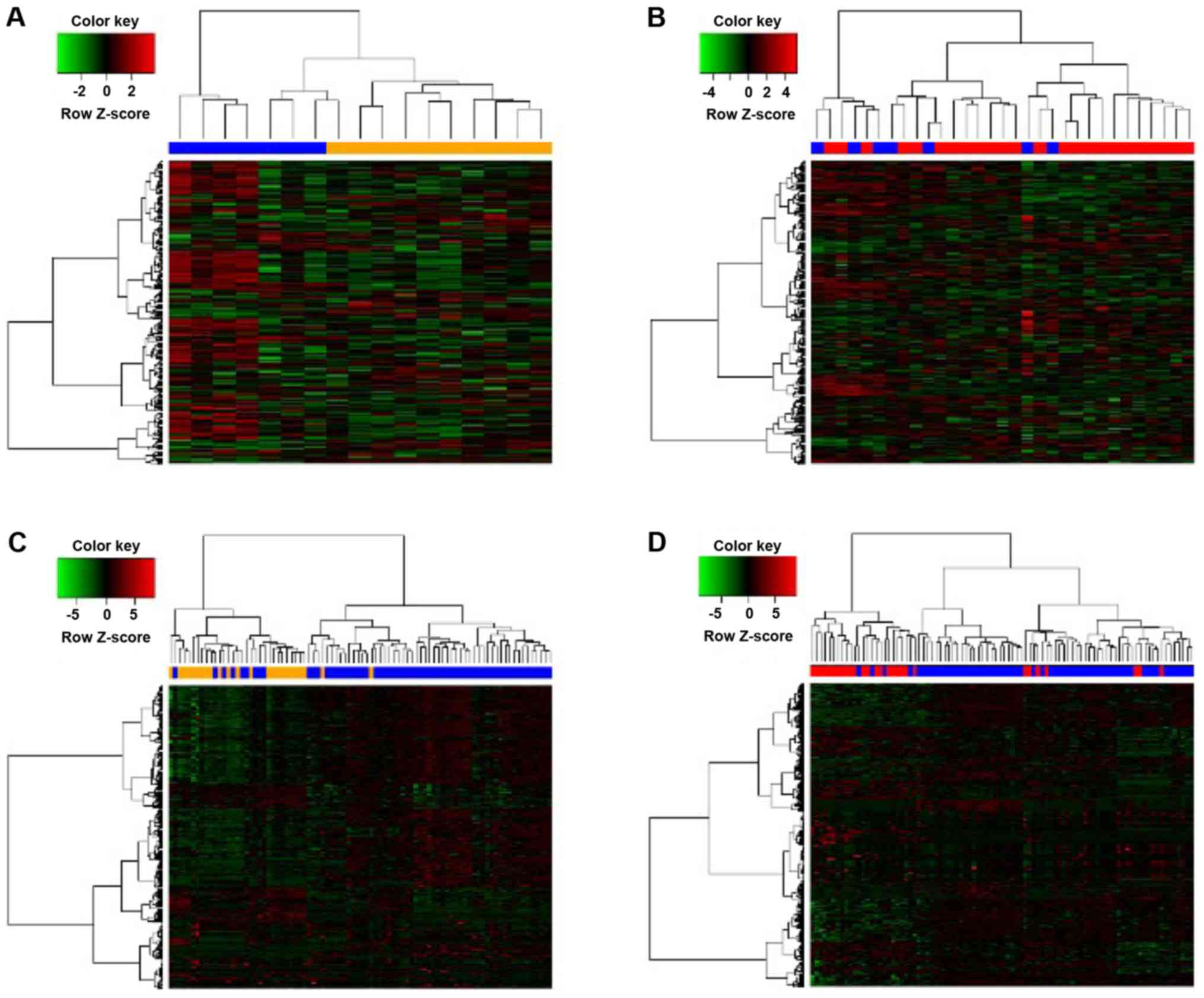
samples were considered as invasive DEGs. A heat map of the
identified DEGs was also constructed, using an R package.
Gene ontology and pathway enrichment
analysis of DEGs
In the present study, the significant enrichment
analysis of the two groups of DEGs was assessed based on the Gene
Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG)
using the Database for Annotation, Visualization and Integrated
Discovery (DAVID), an online tool for functional annotation
analysis (7). GO analysis is a
common, useful method for annotating genes and gene products, and
for identifying characteristic biological attributes of
high-throughput genome or transcriptome data (8), including 3 categories: Biological
process (BP), cellular component (CC) and molecular function (MF).
KEGG (http://www.genome.jp/) is a knowledge
database for the assignment of specific pathways to sets of DEGs,
thus linking-omics data with higher-order functional information
(9). Comprehensively mapping genes to
relevant biological annotations in databases such as DAVID is
critical for the success of any high-throughput gene functional
analysis. An FDR of <0.05 was set as the cut-off.
Construction of biological
network
To evaluate the interactions among the two groups of
identified DEGs, we mapped them to the STRING database, a database
of known and predicted protein-protein interactions (PPIs), and
constructed two PPI networks; only experimentally validated
interactions with a combined score >0.7 were considered
significant. Subsequently, the PPI networks were imported into
Cytoscape, an open-source software platform for visualizing
molecular interaction networks and integrating data, for further
analysis (10). A plugin of
Cytoscape, CytoHubba was used to predict and explore the important
nodes and subnetworks in the network with 12 topological
algorithms, including degree, edge percolated component (EPC),
maximum neighborhood component (MNC) and density of maximum
neighborhood component (DMNC), among others (11). CytoHubba was used to rank nodes in a
network by their network features, select the top 10 genes from
each method, and eliminate the duplicate genes. Finally, all the
identified hub genes and imported into STRING to construct a
complete PPI network.
Results
Identification of DEGs
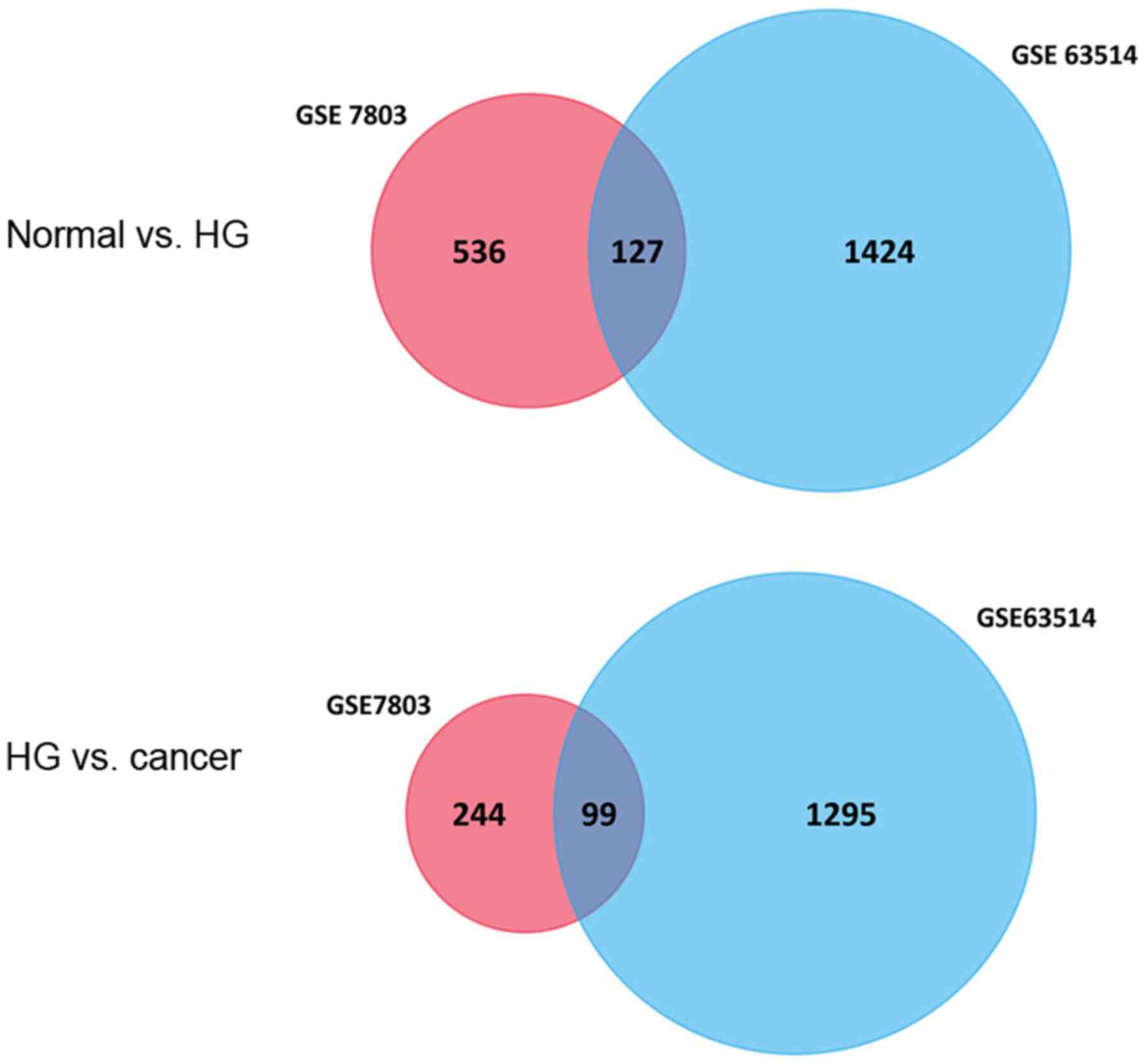
In the present study, a total of 663 and 1,551 genes
were identified as the DEGs between NE and HSIL, among which 127
DEGs were co-expressed, of which 52 genes were upregulated and 75
were downregulated. Furthermore, 343 and 1,394 genes were
identified as the DEGs between HSIL and SCC, with 99 DEGs
overlapping, of which 32 were upregulated and 67 were downregulated
(Fig. 1). A corresponding heat map is
shown in Fig. 2.
Function and pathway enrichment
analysis
To uncover the biological significance of the
screened DEGs in the progression of cervical cancer, GO functional
and KEGG pathway enrichment analyses were performed using the DAVID
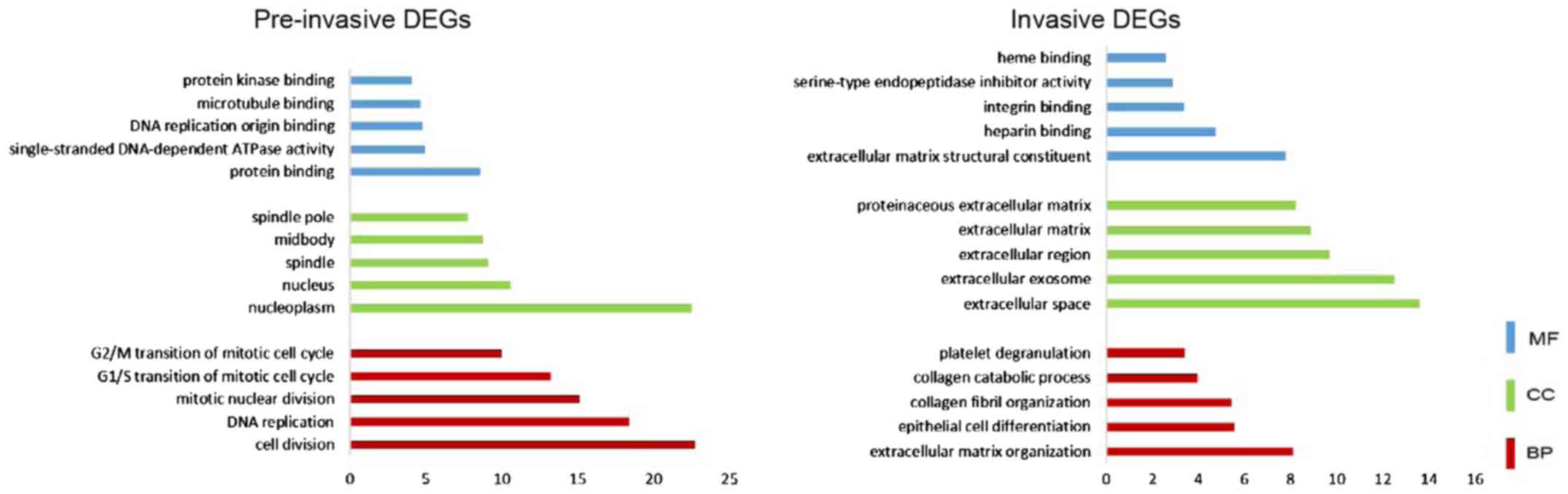
database. As shown in Fig. 3, for
pre-invasive DEGs, ‘nucleoplasm’, ‘nucleus’, ‘spindle’, and
‘midbody’ were enriched from the CC category; enriched BP terms
included ‘cell division’, ‘DNA replication’, ‘cell cycle’ and
‘transcription regulation’; enriched MF terms included ‘protein
binding’, ‘single-stranded DNA-dependent ATPase activity’, ‘DNA
replication origin binding’ and ‘microtubule binding’. Based on
KEGG pathway enrichment analysis (Fig.
4), the pre-invasive DEGs were significantly associated with
the cell cycle, DNA replication and p53 signaling pathways.
For the invasive DEGs, CC terms were mainly enriched
in ‘extracellular space’, ‘extracellular exosome’, ‘extracellular
region’ and ‘extracellular matrix’; BP terms included
‘extracellular matrix organization’, ‘epithelial cell
differentiation’ and ‘collagen fibril organization’; the identified
MF terms included ‘extracellular matrix structural constituent’,
‘heparin binding’ and ‘integrin binding’. The significantly
enriched KEGG pathways included amoebiasis, focal adhesion,
ECM-receptor interaction and platelet activation.
PPI network construction
The screened DEGs were used to construct PPI
networks. CytoHubba was used to rank nodes by their network
features, select the top 10 genes from each methods, and eliminate
duplicate genes. From the pre-invasive DEGs, we screened 23 hub
genes, while from the invasive DEGs, we screened 21 hub genes.
Finally, all the hub genes were summarized and imported into STRING
software to construct the PPI network., there were 25 nodes and 128
edges in the network. The hub genes are listed in Table I, among which BUB1B, MAD2L1, CHEK1,
CCNB1, CCNB2, CDC20, CDC6, CCNA2 and PCNA were associated with the
cell cycle, RFC3, RFC4, FEN1 and PCNA were associated with DNA
replication, and PIK3CA, VEGFA, ITGA1, PTK2, ITGB1, ACTN1, FN1,
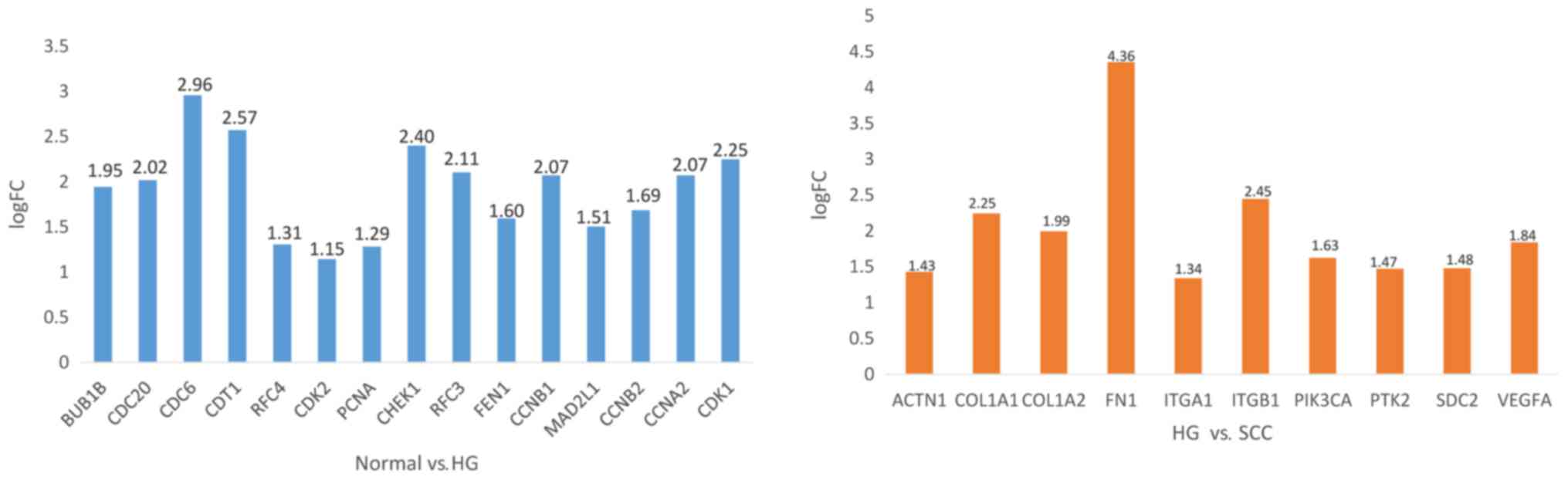
COL1A1 and COL1A2 were associated with focal adhesion (Table II). As shown Fig. 4, the expression of CDC6, CDT1, CHEK1
were significantly increased from normal tissue to SIL, while FN1,
ITGB1 were significantly increased from SIL to cancer.
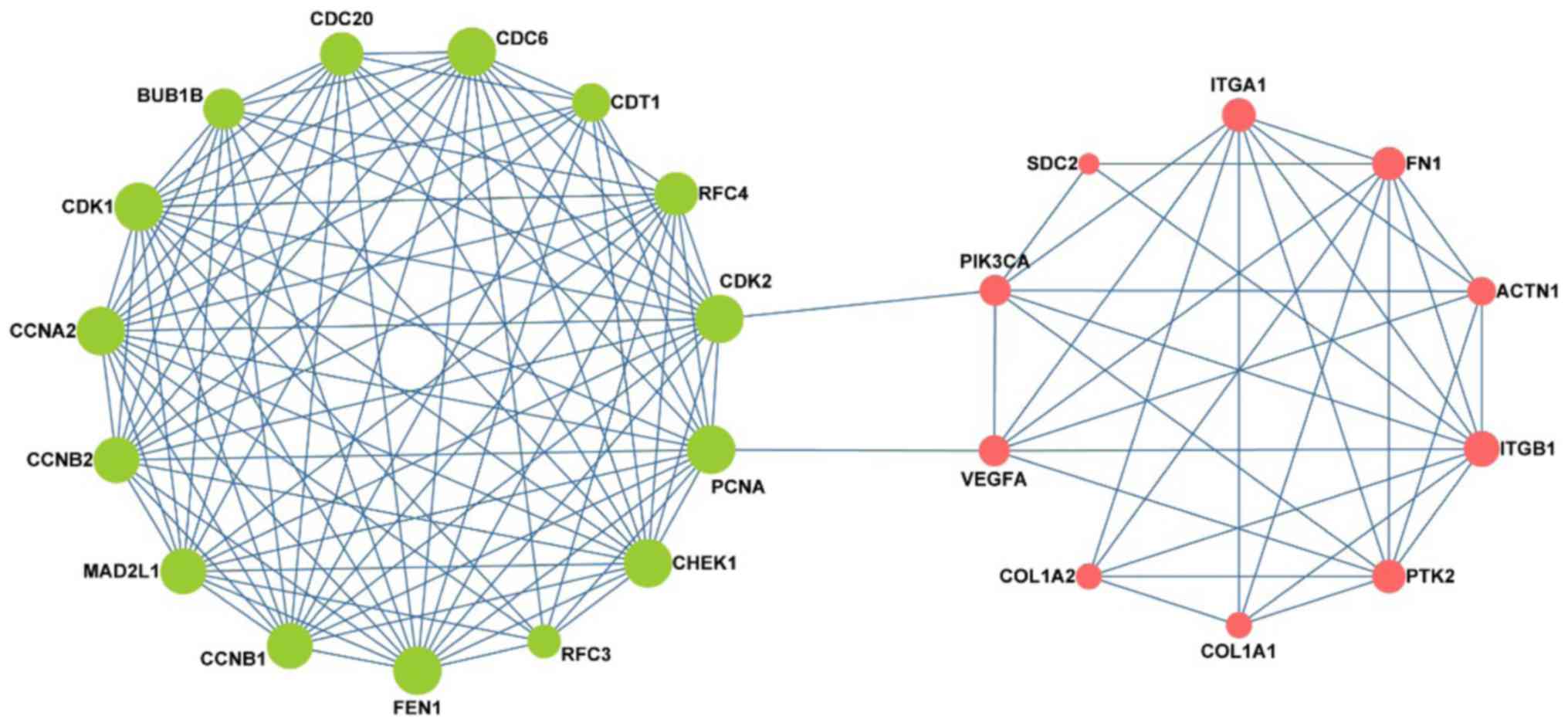
Interestingly, as shown in Fig. 5 the
network consisted of two clusters: The left network was composed of
pre-invasive DEGs, while the right was composed of invasive DEGs;
moreover, these two parts were connected by 4 key nodes, including
PCNA, CDK2, VEGFA and PIK3CA. Notably, the left network was
predominantly associated with the cell cycle and DNA replication,
while the right was mainly associated with focal adhesion.
 | Table I.Whole hub genes screened by
Cytoscape. |
Table I.
Whole hub genes screened by
Cytoscape.
| Pre-invasive hub
genes | Invasive hub
genes |
|---|
| BUB1B, CDC20,
CDC6 | ACTN1, COL1A1,
COL1A2 |
| CDT1, RFC4, CDK2 | FN1, ITGA1,
ITGB1 |
| PCNA, CHEK1,
RFC3 | PIK3CA, PTK2,
SDC2 |
| FEN1, CCNB1,
MAD2L1 | VEGFA |
| Table II.Kyoto Encyclopedia of Genes and
Genomes pathway analysis of differentially expressed hub genes
associated with cervical cancer. |
Table II.
Kyoto Encyclopedia of Genes and
Genomes pathway analysis of differentially expressed hub genes
associated with cervical cancer.
| Pathway ID | Name | Gene count | FDR | Genes |
|---|
| 4110 | Cell cycle | 12 |
2.38×10−17 | BUB1B, CCNA2, CCNB1,
CCNB2, CDC20, CDC6, CHEK1, MAD2L1, PCNA |
| 3030 | DNA replication | 6 |
8.80×10−10 | FEN1, PCNA, RFC3,
RFC4 |
| 4510 | Focal adhesion | 9 |
8.80×10−10 | ACTN1, COL1A1,
COL1A2, FN1, ITGA1, ITGB1, PIK3CA, PTK2, VEGFA |
Discussion
Malignant transformation in tumor progression is
caused by a series of genetic alterations. To better understand the
genetic alterations occurring during cervical cancer progression,
bioinformatics methods were used to extract data from the GSE7803
and GSE63514 gene expression profiles. In this study, we identified
127 DEGs between normal squamous cervical epithelium and HSIL,
while 99 DEGs were identified between HSIL and invasive SCC of the
cervix. Functional analysis demonstrated that these DEGs were
mainly involved in the cell cycle, DNA replication, p53 signaling
and focal adhesion pathways.
From the PPI network constructed from the DEGs, we
found that the network was composed of two clusters. Notably, the
left cluster consisted of pre-invasive DEGs, suggesting that these
genes were involved in the progression to HSIL. GO term analysis
revealed that the pre-invasive DEGs were mainly involved in cell
division, DNA replication, the cell cycle and transcription
regulation, among which BUB1B, MAD2L1, CHEK1, CCNB1, CCNB2, CDC20,
CDC6, CCNA2 and PCNA were involved in the cell cycle, whereas RFC3,
RFC4, FEN1 and PCNA were involved in DNA replication.
DNA replication is a key process for cell
proliferation; however, the abnormal proliferation of tumor cells
may be characterized by irregularities in pathways involved in DNA
replication, cell cycle, apoptosis resistance and metabolic
capacity, with significant implications in tumorigenesis. The cell
cycle is a series of events leading to DNA division and replication
to produce two daughter cells. Enhanced cell proliferation capacity
is the hallmark of cancer. We observed that the biological
processes of DNA replication and cell cycle transition were
significantly increased in cervical cancer tissues. To maintain a
hyperproliferative state, cervical cancer cells upregulate a group
of genes that control multiple steps of DNA replication (12). The mitotic spindle checkpoint Bub1 is
involved in monitoring the assembly of the mitotic spindle, which
ensures the accurate segregation of sister chromatids during
mitosis (13). Bub1 was found to be
mutated in human cancers, such as colorectal cancer, which is
characterized by chromosomal instability and increased aneuploidy
(14). The cyclin proteins CCNA2 and
CCNB1 and their associated kinases CHEK1 and CDK1 were
significantly upregulated in cervical cancer tissue; these proteins
promote cell cycle transition from the G1 to the S phase, and from
the G2 to the M phase. Furthermore, PCNA was also found to be
upregulated in cervical cancer tissues (12). CDC20 is upregulated in HSIL as well as
SCC of the uterine cervix (15).
Replication factor C (RFC) is important for DNA replication and
cell cycle control (16). RFC3 and
RFC4 were reported to promote tumor cell proliferation, and the
high expression of RFC3 was associated with poor prognosis in a
variety of cancers (17,18).
The right cluster consisted of invasive DEGs, which
were involved in biological processes such as ECM organization,
epithelial cell differentiation and collagen fibril organization,
suggesting that these genes were involved in the progression of
SCC. This is in accord with established paradigm that the
dysfunction of cell proliferation and cell cycle regulation is the
primary cause of tumor development (19). Among the identified DEGs, PIK3CA,
VEGFA, ITGA1, PTK2, ITGB1, ACTN1, FN1, COL1A1, COL1A2 and SDC2 were
associated with focal adhesion. Focal adhesions are large
macromolecular assemblies through which mechanical force and
regulatory signals are transmitted between the ECM and interacting
cells. Focal adhesion kinase (FAK) is the key enzyme in regulating
the formation of focal adhesions, and a key regulator of survival,
proliferation, migration and invasion, which endows cells with
higher motility (20). Indeed, FAK
overexpression has been identified in aggressive cervical cancer
(21). FAK was recently established
as a cardinal controller of cell migration, particularly during
tumor metastasis (22). In human
cervical cancer samples, the high expression or phosphorylation of
FAK is associated with an aggressive phenotype (23). Overall, FAK is crucial for cervical
cancer metastasis.
Co-expressed genes are a group of genes with similar
expression profiles that are often involved in parallel biological
processes. By constructing a PPI network from the DEGs, we found
that the two clusters were connected by 4 key genes, namely PCNA,
CDK2, VEGFA and PIK3CA.
From the GO analysis, it was observed that most
pre-invasive DEGs were enriched in the nucleoplasm, cell division
and protein binding, while most invasive DEGs were enriched in the
extracellular space, ECM organization and structural
constituents.
Proliferating cell nuclear antigen (PCNA) is
reported as an important marker of the progression of tumors, which
acts as a central coordinator of DNA transactions by providing
interaction surface for factors involved in DNA replication,
repair, chromatin dynamics and cell cycle regulation (24). In a systematic review by Lv et
al, PCNA upregulation was found to be significantly associated
with poor 5-year survival, advanced disease stage and higher WHO
grade in cervical cancer, suggesting that PCNA may be a useful
prognostic and diagnostic biomarker in cervical cancer (25). Kim et al considered PCNA to be
a biomarker to reflect cellular proliferation, and PCNA protein
immunostaining enhanced the diagnostic accuracy for HSIL,
indicating that PCNA may act as a key gene mediating the
progression from HSIL to cervical cancer (26).
Vascular endothelial growth factor A (VEGFA) is a
significant biomarker that elicits tumor angiogenesis, a BP crucial
for primary tumor growth and metastasis. The overexpression of
VEGFA is associated with poor survival in a variety of cancers,
such as lung, colorectal and cervical cancer (27–29),
suggesting that VEGFA is significantly involved in cervical
tumorigenesis. Combined with the KEGG pathway analysis of the hub
genes, which indicated that PCNA was involved in the cell cycle and
DNA replication, and VEGFA in focal adhesion, we may infer that the
effect of PCNA upregulation on the cell cycle promoted the
connection between cells and the ECM by focal adhesions, thus
activating extracellular angiogenesis, which promoted the
transition from HSIL to cervical cancer.
Cyclin-dependent kinases (CDKs) play key roles in
cell proliferation, and have attracted considerable attention in
the study of tumor growth. CDK2 is a member of the CDK family,
which associates with cyclin A or cyclin E, and is considered to be
essential in the cell cycle, driving cells through the S phase by
binding with cyclin A (30).
PIK3CA is a part of the PI3K/AKT/mTOR pathway, a
pathway that is disrupted in several types of cancer with high
frequency and is involved in the regulation of cell growth,
proliferation, differentiation, glucose metabolism, protein
synthesis and apoptosis (31).
Somatic mutations in PIK3CA have been detected in a variety of
human malignant solid tumors, including cervical cancer (32). Chung et al performed
whole-exome sequencing in 15 paired cervical adenocarcinoma and
peripheral leukocyte DNA samples, and identified specific PIK3CA
aberrations in cervical cancer (33).
In addition, Cui et al (34)
analyzed PIK3CA mutations in CIN3 lesions and cervical carcinomas,
and identified somatic mutations in 8.15% of cervical carcinomas,
whereas there were no mutations in CIN3 cases, suggesting that
genetic alterations of PIK3CA are late events during cervical
carcinogenesis. Hence, we hypothesized that the upregulation of
CDK2 promoted the cell cycle and DNA replication in cervical
epithelial cells, leading to the development of HSIL, which,
following PIK3CA mutation and focal adhesion dysregulation,
ultimately developed into cervical cancer.
In summary, a comprehensive bioinformatics analysis
of DEGs that may be involved in cervical cancer development is
provided by the present study. Furthermore, a series of useful
targets for the future study of biomarkers and molecular mechanisms
were identified. Further molecular biological experiments, however,
are required to confirm the role of the identified genes in
cervical cancer.
Acknowledgements
Not applicable.
Funding
This study was supported by the Zhongnan Hospital of
Wuhan University Science, Technology and Innovation Seed Fund
(grant no. znpy2016040).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
KW and YY performed the experiments and wrote the
paper. FL, WW and YC analyzed the data. WZ designed the study and
reviewed the manuscript. All authors discussed the results and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vogelstein B and Kinzler KW: Cancer genes
and the pathways they control. Nat Med. 10:789–799. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wu NL, Huang DY, Tsou HN, Lin YC and Lin
WW: Syk mediates IL-17-induced CCL20 expression by targeting
Act1-dependent k63-linked ubiquitination of TRAF6. J Invest
Dermatol. 135:490–498. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kulasingam V and Diamandis EP: Strategies
for discovering novel cancer biomarkers through utilization of
emerging technologies. Nat Clin Pract Oncol. 5:588–599. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu SF, Qian WY, Zhang JW, Yang YB, Liu Y,
Dong Y, Zhang ZB, Zhu YP and Feng YJ: Network motifs in the
transcriptional regulation network of cervical carcinoma cells
respond to EGF. Arch Gynecol Obstet. 287:771–777. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Luo Y, Wu Y, Peng Y, Liu X, Bie J and Li
S: Systematic analysis to identify a key role of CDK1 in mediating
gene interaction networks in cervical cancer development. Ir J Med
Sci. 185:231–239. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jiao X, Sherman BT, Huang da W, Stephens
R, Baseler MW, Lane HC and Lempicki RA: DAVID-WS: A stateful web
service to facilitate gene/protein list analysis. Bioinformatics.
28:1805–1806. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gene Ontology Consortium: The Gene
Ontology (GO) project in 2006. Nucleic Acids Res. 34:(Database
Issue):. D322–D326. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chin CH, Chen SH, Wu HH, Ho CW, Ko MT and
Lin CY: CytoHubba: Identifying hub objects and sub-networks from
complex interactome. BMC Syst Biol. 8(Suppl 4): S112014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cheng J, Lu X, Wang J, Zhang H, Duan P and
Li C: Interactome analysis of gene expression profiles of cervical
cancer reveals dysregulated mitotic gene clusters. Am J Transl Res.
9:3048–3059. 2017.PubMed/NCBI
|
|
13
|
Cahill DP, Lengauer C, Yu J, Riggins GJ,
Willson JK, Markowitz SD, Kinzler KW and Vogelstein B: Mutations of
mitotic checkpoint genes in human cancers. Nature. 392:300–303.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ru HY, Chen RL, Lu WC and Chen JH: Hbub1
defects in leukemia and lymphoma cells. Oncogene. 21:4673–4679.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim Y, Choi JW, Lee JH and Kim YS: MAD2
and CDC20 are upregulated in high-grade squamous intraepithelial
lesions and squamous cell carcinomas of the uterine cervix. Int J
Gynecol Pathol. 33:517–523. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Masuda Y, Suzuki M, Piao J, Gu Y,
Tsurimoto T and Kamiya K: Dynamics of human replication factors in
the elongation phase of DNA replication. Nucleic Acids Res.
35:6904–6916. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lockwood WW, Thu KL, Lin L, Pikor LA,
Chari R, Lam WL and Beer DG: Integrative genomics identified RFC3
as an amplified candidate oncogene in esophageal adenocarcinoma.
Clin Cancer Res. 18:1936–1946. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Arai M, Kondoh N, Imazeki N, Hada A,
Hatsuse K, Matsubara O and Yamamoto M: The knockdown of endogenous
replication factor C4 decreases the growth and enhances the
chemosensitivity of hepatocellular carcinoma cells. Liver Int.
29:55–62. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Perez R, Wu N, Klipfel AA and Beart RW Jr:
A better cell cycle target for gene therapy of colorectal cancer:
Cyclin G. J Gastrointest Surg. 7:884–889. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
van Nimwegen MJ and van de Water B: Focal
adhesion kinase: A potential target in cancer therapy. Biochem
Pharmacol. 73:597–609. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oktay MH, Oktay K, Hamele-Bena D, Buyuk A
and Koss LG: Focal adhesion kinase as a marker of malignant
phenotype in breast and cervical carcinomas. Hum Pathol.
34:240–245. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fong YC, Liu SC, Huang CY, Li TM, Hsu SF,
Kao ST, Tsai FJ, Chen WC, Chen CY and Tang CH: Osteopontin
increases lung cancer cells migration via activation of the
alphavbeta3 integrin/FAK/Akt and NF-kappaB-dependent pathway. Lung
Cancer. 64:263–270. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Moon HS, Park WI, Choi EA, Chung HW and
Kim SC: The expression and tyrosine phosphorylation of
E-cadherin/catenin adhesion complex, and focal adhesion kinase in
invasive cervical carcinomas. Int J Gynecol Cancer. 13:640–646.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Srinivasan M and Jewell SD: Quantitative
estimation of PCNA, c-myc, EGFR and TGF-alpha in oral submucous
fibrosis-an immunohistochemical study. Oral Oncol. 37:461–467.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lv Q, Zhang J, Yi Y, Huang Y, Wang Y, Wang
Y and Zhang W: Proliferating cell nuclear antigen has an
association with prognosis and risks factors of cancer patients: A
systematic review. Mol Neurobiol. 53:6209–6217. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim TH, Han JH, Shin E, Noh JH, Kim HS and
Song YS: Clinical implication of p16, Ki-67, and proliferating cell
nuclear antigen expression in cervical neoplasia: Improvement of
diagnostic accuracy for high-grade squamous intraepithelial lesion
and prediction of resection margin involvement on conization
specime. J Cancer Prev. 20:70–77. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gkiozos I, Tsagouli S, Charpidou A, Grapsa
D, Kainis E, Gratziou C and Syrigos K: Levels of vascular
endothelial growth factor in serum and pleural fluid are
independent predictors of survival in advanced non-small cell lung
cancer: Results of a prospective study. Anticancer Res.
35:1129–1137. 2015.PubMed/NCBI
|
|
28
|
Zhang J, Liu J, Zhu C, He J, Chen J, Liang
Y, Yang F, Wu X and Ma X: Prognostic role of vascular endothelial
growth factor in cervical cancer: A meta-analysis. Oncotarget.
8:24797–24803. 2017.PubMed/NCBI
|
|
29
|
Tsai HL, Yang IP, Lin CH, Chai CY, Huang
YH, Chen CF, Hou MF, Kuo CH, Juo SH and Wang JY: Predictive value
of vascular endothelial growth factor overexpression in early
relapse of colorectal cancer patients after curative resection. Int
J Colorectal Dis. 28:415–424. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tian RQ, Wang XH, Hou LJ, Jia WH, Yang Q,
Li YX, Liu M, Li X and Tang H: MicroRNA-372 is down-regulated and
targets cyclin-dependent kinase 2 (CDK2) and cyclin A1 in human
cervical cancer, which may contribute to tumorigene. J Biol Chem.
286:25556–25563. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Koncar RF, Feldman R, Bahassi EM and
Hashemi Sadraei N: Comparative molecular profiling of HPV-induced
squamous cell carcinomas. Cancer Med. 6:1673–1685. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
McIntyre JB, Wu JS, Craighead PS, Phan T,
Köbel M, Lees-Miller SP, Ghatage P, Magliocco AM and Doll CM:
PIK3CA mutational status and overall survival in patients with
cervical cancer treated with radical chemoradiotherapy. Gynecol
Oncol. 128:409–414. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chung TK, van Hummelen P, Chan PK, Cheung
TH, Yim SF, Yu MY, Ducar MD, Thorner AR, MacConaill LE, Doran G, et
al: Genomic aberrations in cervical adenocarcinomas in Hong Kong
Chinese women. Int J Cancer. 137:776–783. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cui B, Zheng B, Zhang X, Stendahl U,
Andersson S and Wallin KL: Mutation of PIK3CA: Possible risk factor
for cervical carcinogenesis in older women. Int J Oncol.
34:409–416. 2009.PubMed/NCBI
|