Introduction
Choroid plexus tumors (CPT) have arisen as papillary
neoplasms of choroid plexus in the brain, particularly in children
lateral ventricle or in adult fourth ventricle (1). These tumors originate from the
epithelium of choroid plexus. They can be benign or malignant and
known as choroid plexus papilloma (CPP) or choroid plexus carcinoma
(CPC), respectively (2). In
children, CPP account for 2–3% of all intracranial tumors, with a
median age at diagnosis around 3.5 years old (3) and an annual incidence of 0.3 cases per
million. This neoplasm outnumbers CPC by a factor of 5:1 (4). The tumor is commonly located in the
lateral ventricle, and the main symptom is an elevated intracranial
pressure. Histopathological characteristics observed in CPP are
essentially calcification, hemorrhage, hyalinization, oncocytic
changes, and other presenting symptoms, including headache,
diplopia and ataxia from hydrocephalus (5). Direct mechanical obstruction of
cerebrospinal fluid (CSF) flow, hemorrhage that causes arachnoid
granulation obstruction, and CSF overproduction can stimulate the
development of hydrocephalus, and occasionally lead to CSF leaks
out from the nose (6). Metastasis of
CPP tumors exists; however, extensive metastasis has only been
reported when tumors are malignant (7). Histologically, CPP corresponds to grade
I, according to the World Health Organization (WHO); however
atypical CPP (a-CPP) become more aggressive and reach grade II. It
very rarely transforms into grade III-CPC (4). Although CPPs are commonly benign,
histological analysis is not always predictive of their behavior.
Molecular and genetics analyses may therefore be helpful for
diagnosis and tumor prognosis (7,8).
Increased prevalence of CPC due to R337H mutation of
the germline gene tumor protein p53 (TP53) in southern Brazil was
reported (9). Custodio et al
(9) demonstrated for the first time
that this mutation is responsible for 63% of CPC cases in children,
which indicates a higher incidence of CPC in southern Brazil. This
study including 22 children with CPC and 7 children with CPP was
based on PCR-RFLP and confirmed by sequencing exon 10 of TP53. They
reported that all CPP cases (7/7) were negative for R337H (9). A recent study using whole exome
sequencing demonstrated that homologous recombination deficiency,
amplification of mesenchymal-epithelial transition (MET) and loss
of retinoblastoma susceptibility gene were observed in two cases of
CPC (malignant peripheral nerve sheath tumor sheath tumor)
(10). Conversely with other types
of brain tumor, extensive mutation profiling in CPT cases were not
previously attempted by next generation DNA sequencing (NGS)
methods. However, gene expression profiles using DNA micro-arrays,
Sanger sequencing of TP53 gene, copy number variation and DNA
methylation studies in CPT were attempted (11,12). It
has been reported that mutations in hSNF5/INI1 are associated with
CPCs, and one mutation in the SNF5/INI1 gene in one case of a-CPP
is known (13). However, Mueller
et al (14) have reported
that there is no evidence of hSNF5/INI1 point mutations by SSCP
analysis in CPP. Furthermore, hSNF5/INI1 (SMARCB1) gene serves an
integral role in chromatin remodeling (15). SMARCB1 is an invariant of the SWI/SNF
chromatin-remodeling complex and serves an important role in
transcriptional regulation (16).
Patients with rhabdoid tumors, CPC, epithelioid sarcoma and
peripheral primitive neuroectodermal tumor exhibit a loss of
SMARCB1 expression either by exons duplications or deletions,
although the number of cases with SMARCB1 mutations are limited
(17).
Mutations in ATM gene are associated with breast
cancer and several forms of leukemia and lymphomas (18). However, no association between the
development of brain tumors and ATM gene mutations has been yet
established. A previous study demonstrated an association between
astrocytoma development and familial ataxia-telangiectasia (AT) in
an 8-year-old patient (19). A
previous study using Sanger sequencing reported a compound
heterozygote for two novel mutations: c.3291delC (p.Phe1097fs) at
exon 25 and c.8198A>C (p.Gln2733Pro) at exon 58 in the ATM gene
of patient with AT and with a cerebellar astrocytoma (20). Another study using next-generation
targeted exome sequencing revealed a frameshift mutation p. L2493fs
c.4477_4478CTCT in a patient with astroblastoma, and a missense
mutation p. (Val410Ala) in the ATM gene in a patient with
astrocytoma from the same group (21). To the best of our knowledge,
molecular genetics investigations, particularly ATM gene mutations
in patients with CPP and CPC by DNA sequencing using NGS methods
have not yet been investigated, and the mutation profiling of these
tumors remains unclear. The present study was the first to report
NGS analysis of an a-CPP tumor on Ion Proton using cancer
comprehensive panel genes and identification of a novel and known
mutations.
Materials and methods
The present study was approved by the Institutional
Review Board (IRB) for Bioethics Committee of the King Abdullah
Medical City (IRB no. 14-140) and performed in accordance with the
principles of the Declaration of Helsinki. Informed consent was
obtained from the guardian of the patient prior to the study. The
diagnosis was made following radiological, histopathological and
immunological examinations. Tumors were classified based upon
similarity to the constituent cells of the central nervous system
(CNS), including astrocytes, oligodendrocytes, ependymal and glial
cells, mitotic activity was assessed by counting mitoses in 10
randomly selected high-powered fields at ×40 magnification using
Nikon Eclipse Ci series Upright Light-microscope (Nikon
Corporation). and the proliferation index was assessed by Ki-67
staining as per the WHO grading system (4). Clinicopathological and demographic
characteristics of the patient were obtained and further analyzed
for the present study.
Radiology and histopathological
analysis
A CT scan of the brain was performed using a
multi-slice CT (MSCT), using a 64-detector-row scanner. The use of
CT allowed visualization of detailed images of the soft tissues in
the body in 3D as well as in multiplanar reconstructions. Images
were captured using 5 mm-thick sections on a GE Medical Systems,
light speed VCT, 64-slice multidetector CT (MDCT). High quality
images were processed at low dose performance on Volara™ digital
Data Acquisition System.
The excised tumor was fixed at room temperature in
4% buffered formaldehyde for 12–24 h maximum, then routinely
processed and paraffin embedded. The 4-µm-thick sections were
prepared on clear ground glass microscope slides with ground edges,
and routinely stained using Dako Reagent Management System
(DakoRMS) with hematoxylin for 3 min and eosin for 30 sec at 25°C
each on a Dako Coverstainer (Agilent Technologies). For
immunohistochemistry studies, sections were collected on Citoglas
adhesion microscope slides (Citotest). Briefly, the tissue sections
were deparaffinized with EZ Prep (cat. no. 950-102; Ventana Medical
Systems, Inc.) at 60°C for 1 h. After the antigen retrieval of the
deparaffinized sections using
tris-(hydroxymethyl)-aminomethane-based antigen retrieval reagent
(Ventana Medical Systems, Inc.) at 95–100°C was performed for 30
min by mild cell condition-I protocol using cell conditioning
solution (CC1) Ventana Catalog Number 950–124 and the
immunohistochemistry was performed using the Ventana BenchMark XT
automated stainer (Ventana Medical Systems, Inc.). CONFIRM
anti-Synaptophysin (SP11) rabbit monoclonal antibody (1:20; cat.
no. 790-4407), CONFIRM anti-S100 (1:50; rabbit polyclonal; cat. no.
760-2523), mouse monoclonal β-catenin (1:50; cat. no. 224M-1;
Sigma-Aldrich; Merck KGaA), E-cadherin mouse monoclonal (1:50; cat.
no. 790-4497; Ventana Medical Systems, Inc.) and mouse monoclonal
anti-Ki-67 (1:20; cat. no. KI67-MM1-L-CE; Leica Microsystems, Inc.)
antibodies were used for immunohistochemistry. Following
inactivation of the endogenous peroxidase using a UV-inhibitor for
4 min at 37°C, the primary antibody was added for 16 min at 37°C,
followed by the respective anti-mouse or anti-rabbit secondary
antibody application of HRP Universal Multimer for 8 min, and
detected using the ultraView Universal DAB Detection kit (cat. no.
760-500; Ventana Medical Systems, Inc.) for 38 min. Slides were
counterstained at 25°C with hematoxylin for 8 min and bluing
reagent for 4 min before mounting with cover slips. Following
staining, images were captured on Nikon Eclipse Ci series Upright
Light-microscope using a NIKON Digital Microscope Camera-DS-Ri1,
with NIS Elements imaging software (version 4.0) from Nikon
Corporation. Appropriate positive controls for all of the studied
antibodies were used.
DNA isolation and NGS analysis on Ion
Proton
DNA isolation was performed by Qiagen FFPE kits
(Qiagen Inc.). Briefly, five finely cut 5–10 µm of formalin-fixed
paraffin-embedded sections were deparaffinized using xylene,
cleaned with ethanol to remove xylene, and the pellet was dried at
65°C for 5 min. The pellets were resuspended in buffer ATL, treated
with proteinase K and the later steps were carried out according to
the user manuals. DNA (10 ng) was used for NGS analysis, and DNA
was sequenced using Ion PI v3 chip (Thermo Fisher Scientific, Inc.)
on Ion Proton instrument (22).
Libraries were prepared using Ion AmpliSeq comprehensive cancer
panel (Thermo Fisher Scientific, Inc.) primer pools. Ion AmpliSeq
2.0 library kit, and Ion PI Hi-Q OT2 200 kit, (Thermo Fisher
Scientific, Inc.) was used for libraries and templates preparation,
respectively. Sequencing was done using Ion PI Hi-Q Sequencing 200
kit (Thermo Fisher Scientific, Inc.). Libraries were tagged with
Ion Express barcodes (Thermo Fisher Scientific, Inc.). Following
DNA sequencing, amplicon sequences were aligned to the human
reference genome GRCh37 (hg19) in the target region of
comprehensive cancer panel gens by using the Torrent Suite software
(version 5.10.1.0; Thermo Fisher Scientific, Inc.). Variant call
format (vcf) file was generated by running the Torrent Variant
Caller Plugin v5.2, (Thermo Fisher Scientific, Inc.). Low quality
reads filtering and variant annotations were done on Ion Reporter
software (version 5.10.2.0; Thermo Fisher Scientific, Inc.) for the
annotation. The ‘vcf’ files data were also analyzed using Advaita
Bioinformatics' iVariantGuide (http://www.advaitabio.com/ivariantguide) to determine
for significant variants, associated genes, predicted impacts and
clinical significance.
More details of data analysis and annotation sources
included in Ion Reporter software (Ion Reporter Software 5.6
Publication Number MAN0017204) were obtained, including variant
types, single nucleotide polymorphisms (SNP), insertions and
deletions (Indel), P-value, PolyPhen-2 and coverage analysis. The
coverage parameters were used to avoid false-positive calls that
could result from sequencing coverage not wide enough to provide a
correct genotype. Allele coverage corresponded to the number of
reads supporting the called allele, and the frequency of the allele
observed was annotated as frequency from the raw data. The P-value
was the probability that the variant call made was correct.
P-values closer to 0.0 indicated high confidence in the variant
call. P-values closer to 1.0 represent indicated low confidence in
the variant call. The P-value corresponded to a logarithmic
transformation of the Phred quality score value made by the Variant
caller plugin in Torrent Suite software (version 5.10.1.0; Thermo
Fisher Scientific, Inc.) using an Ion Proton machine. For example,
a variant caller quality score of 20 is associated with a P-value
of 0.01, and a variant caller quality score of 30 is associated
with a P-value of 0.001. Tumor variants with no coverage or low
coverage were designated as non-confident, and the non-confident
variants were not assigned a P-value. These workflows do analysis
reads of a tumor sample and reads against the related reference
sequence. These workflows allowed a statistical evaluation of the
likelihood that the tumor allele may not be present in the normal
sample, and determine a P-value that represented the statistical
confidence of this call. True mutations have been considered based
upon Phred quality score (i.e., -log10 of the probability that the
alternative call is wrong), which was above 20, and mutation with
P-value <0.05 were considered to be significant. The PolyPhen
(Polymorphism Phenotyping; version 2.0; http://genetics.bwh.harvard.edu/pph2) score predicted
the possible impact of an amino acid substitution on the structure
and function of a human protein (23). This score represented the probability
of a substitution to create damage. The PolyPhen score ranged from
0.0 (tolerated damage) to 1.0 (deleterious damage).
Results
Clinical presentation and
radiology
A 3 years old girl was presented with acute subdural
hemorrhage (non-traumatic) with fever, loss of conscious and poor
oral intake. The child has a long head with a circumference of 53
cm. The patient underwent right frontoparietal craniotomy, tumor
excision, admitted into pediatric intensive care unit (PICU) where
she was extubated and into neurosurgery ward. The biopsy specimen
obtained was of tan polypoid formation that consisted in multiple
tissue fragments of rubbery consistency and measuring 5×4×0.5 cm.
The proteins concentration in CSF was high (1,213.9 mg/dl; normal
range, 15–45 mg/dl), the neutrophil count was low and monocyte
count was high. CSF culture showed no growth, ruling out the
possibility of infection. Partial thromboplastin time and
international normalized ratio were normal (13.1 sec and 1.01,
respectively). Blood chemistry and complete blood count results
were normal. Routine radiological and immune-histological
investigations were performed.
The pre-operative computed tomography (CT)
examination revealed a fairly large solid lobulated well-demarcated
intraventricular mass that was isodense to mildly hyperdense (red
arrow; Fig. 1A and B). This lesion
presented homogenous contrast enhancement, which was adjacent to
the right lateral ventricle (Fig.
1A). This lesion measured ~3.7×4.3 cm, exhibited a large
eccentric cystic component (orange arrow), and possessed irregular
or invasive margins with associated mass effect. midline shift
(yellow arrow), perilesional oedema (green arrow) and moderate
supra-tentorial hydrocephalus (white arrow) is also noted (A and
B). Post-operative CT (Fig. 1C)
revealed that, following right craniectomy and extra-axial fluid
collection (green arrow), no evidence of mass lesion could be seen.
Mild supratentorial hydrocephalus was observed (yellow arrow), as
well as evidence of radiopaque shunt marker (red arrow) in the
right parieto-occipital location.
Histopathology and
immunohistochemistry
Histopathology of the papillary lesions stained with
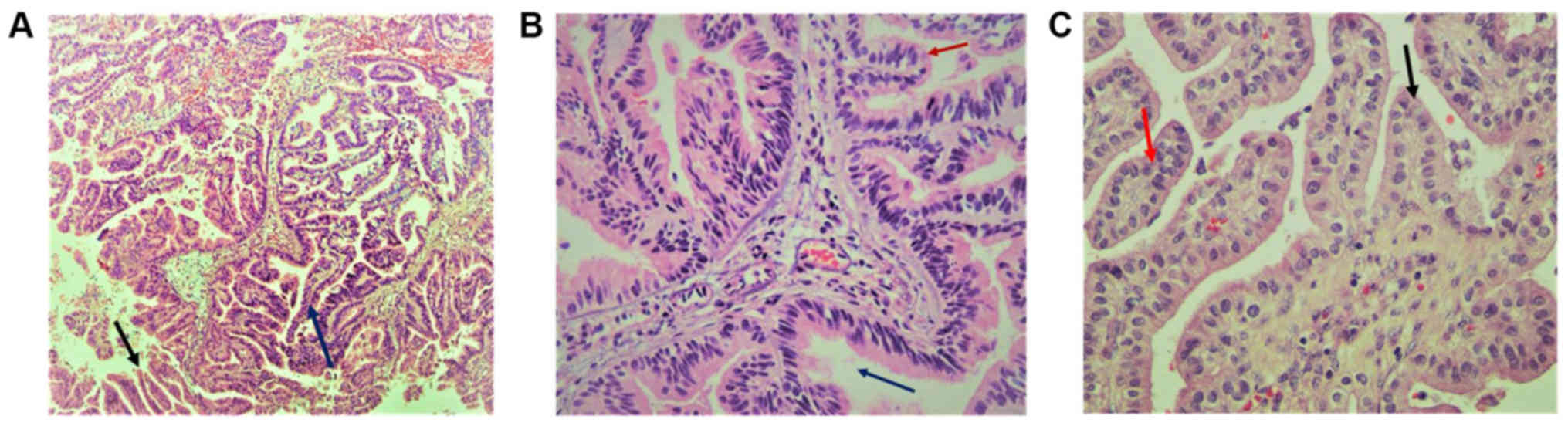
H&E is presented in Fig. 2.
Tumor examination revealed a fibrovascular papillary projection of
choroid plexus that was lined by columnar epithelium with crowding,
elongation and stratification, displaying papillae lined by layers
of cubo-columnar cells having isomorphic nuclei and moderate amount
of cytoplasm delicate papillary fronds (Fig. 2A). In Fig.
2B, H&E stain revealed proliferation of cuboidal cells
arranged in a distinct papillary growth pattern, mitotic figures,
increased mitotic activity and architectural complexity.
Furthermore, cells had increased nucleo-cytoplasmic ratio and
increased mitosis, and the mitotic index was >2 mitoses/10
high-power fields (HPF). H&E slide with isomorphic nuclei and
moderate amount of cytoplasm presented increased mitotic activity
and architectural complexity, typical papillary arrangements,
featuring a single layer of cuboidal or columnar epithelium in a
papillary configuration covering a fibrovascular core and mitotic
cells (Fig. 2C). Areas of cribriform
projections and focal solid growth patterns were also seen
(Fig. 2C). Immunostaining for S-100,
Ki-67 and synaptophysin are presented in Fig. 3. Ki-67 and S-100 were strongly
positive (Fig. 3A and B), and
synaptophysin staining was focally positive (Fig. 3C). Ki-67 proliferation index was
≥85.0%. Immunohistochemistry for E-cadherin and β-catenin is
presented in Fig. 4. The results
revealed that membranes were positively stained for E-cadherin
(Fig. 4A, magnification ×4; Fig. 4B, magnification 20×) and β-catenin
(Fig. 4C, magnification 4×; Fig. 4D, magnification 20×). As presented in
Figs. S1 and S2, immunohistochemical staining for glial
fibrillary acidic protein (GFAP), epithelial membrane antigen
(EMA), vimentin, thyroid transcription factor 1, (TTF-1), P53,
CK-7, and epidermal growth factor receptor (EGFR) were negative in
this a-CPP tumor. The positive controls for these antibodies are
shown in supplementary Fig. S3. The
control tissues used for staining was astrocytoma for GFAP and P53,
lung adenocarcinoma for CK7, papillary thyroid carcinoma for EGFR
and TTF1, myxopapillary ependymoma for EMA and inflammatory
mesenchymal tissue for vimentin staining.
NGS data analysis variant
identification and variant statistics
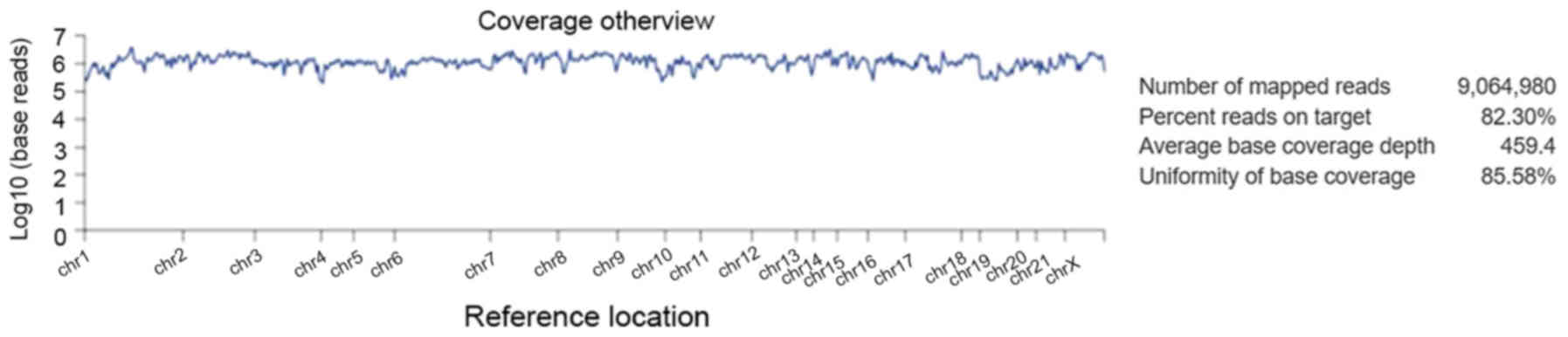
Alignment analysis of the target regions (CCP.
20170413. designed) to the reference genome (Human genome build 19
of hg 19) and sample coverage overview was performed by the Ion
Torrent Suite software v4.4.2 (Fig.
5). Amongst the 9,064,980 reads that were generated for this
sample by Ion PI chip, 83.30% reads were on target, with an average
base coverage depth of 459× and an uniformity of base coverage of
85.58%. Amplicon and target base read coverages of sequencing
demonstrated that out of 16,000 amplicons, 15,992 amplicons were
sequenced with Ion AmpliSeq comprehensive cancer panel primer pool
(Table SI). The 1, 20, 100 and 500×
target base coverage were 99.66, 96.55, 84.35 and 32.99%,
respectively. In addition, the percent end-to-end reads of
amplicons were 92.13% (Table SI).
Initial analysis by Advaita's i-variant software demonstrated that
100% (3,650) variants passed all filters. The variants statistics
distributions according to various filters, including chromosomal
distribution, region in the gene, variant class, effect of variant
on protein, variant impact and clinical significance are presented
in Fig. 6. The chart from panel (A)
represents the relative number of variants located on each
chromosome. The results demonstrated that chromosome 17 possessed
the highest variants (19%) whereas chromosome 21 and X chromosome
had the least variants (0.8%). Panel (B) demonstrated that variant
class represented SNPs at 80.5%, whereas deletions and insertions
represented 8.3 and 3%, respectively. In panel C, variants passed
through the variant impact filter, which gives a prediction of the
disease severity, and the results revealed that 24.9% were of high
impact whereas 44.4% variants had moderate impact on the protein
structure and function. Furthermore, 80.6% were exonic, 12.6% were
intronic and 2% were splice site variants (panel D). In addition,
41.9% were missense variants, 13.3% were synonymous variants and
12.6% were intronic variants (panel E). Clinical significance
represents variants that are associated with clinical outcomes,
including susceptibility to diseases or response to drugs. By
applying the clinical significance filter, the results revealed
that 17.5% were benign variants and 37.8% were pathogenic variants
(panel F).
 | Figure 6.Statistical analysis distribution of
the variants that passed through the filters. The filtration
presented characteristics, including the relative number of
variants located on each chromosome, the variant class, the
substitution types, the insertion and deletion lengths, the variant
effect that gives the changes occurring on the transcript, the
genomic regions impacted by the variant, and the functional
consequences of each variant on the interpretation of the score
severity and on the impact to predict the severity of the disease.
Doughnut charts in panels presented variants that had passed for
each individual filter for (A) Chromosomal distribution, (B)
Variant class, (C) Variant impact on the protein function, (D)
Region in the gene, (E) Variant effect on the protein structure,
and (F) Clinical significance of the variants annotated from the
ClinVar database. |
The summary of all novel missense mutations found in
CPP tumour are presented in Table I.
In this a-CPP tumour, NGS data analysis identified 12 novel
missense mutations by Ion Proton. The gene mutations detected were
as follows: A missense mutation c.5808A>T p. (Leu1936Phe) on
exon 39 of the ATM gene; a c.4214C>A p. (Pro1405His) mutation on
exon 25 of NOTCH1 gene; a c.2439A>T p. (Gln813His) mutation on
exon 21 of SKT36 gene; and a c.2026A>G p. (Arg676Gly) on exon 12
of MAGI1 gene. No exonic mutation in SMARCB1 was found in this
tumor; however, one missense mutation c.2729C>T p. (T910M) was
detected in SMARCA4 gene. Furthermore, novel missense variants were
identified in exon 86 of DST, in exon 8 of RECQL4, in exon 15 of
NUMA1, in exon 17 of THBS1, in exon 17 of MYH11, in exon 9 of MALT1
and in exon 12 of CDH20 (Table I).
In addition, a novel mutation was identified in the exon 39 of ATM
gene locus chr11:108180932. This mutation c.5808A>T (p.
Leu1936Phe) was a missense mutation, and the coding at this
position changed the nucleotide sequence from TTA to TTT. The
allele ratios for A and T were 0.9655 and 0.0345, respectively, and
the frequency of this mutation (A>T) was 3.45%. For this
mutation, the coverage of this variant was 69×, the P-value was
0.00407 and the Phred quality score was 23.909 (Table SII). For each target, frequency,
allele coverage, allele ratio, P-value Phred scores and PolyPhen
scores were determined for these novel mutations (Tables II and SII). The P-value and Phred quality score
were significantly high for all mutations, as revealed by the Ion
Reporter analysis reads of a tumor sample and reads against the
associated reference sequence. Only one novel missense mutation,
the c.2646G>T p. (Gln882His) mutation on gene THBS1, had a
frequency of 80.65% and a Phred quality score of 635.503, which
indicated that this was a germline mutation (Table I). The PolyPhen score of the six
variants viz. DST, RECQL4, NUMA1, THBS1, MYHI1 and SMARCA4 were
high, which suggested that these variants may be pathogenic
variants, whereas NOTCH1 had a PolyPhen score of 0.429, which
indicated that it may be pathogenic. Other variants had low
PolyPhen scores (Table SII).
 | Table I.Novel missense variants observed in
atypical choroid plexus papilloma tumor. |
Table I.
Novel missense variants observed in
atypical choroid plexus papilloma tumor.
| Chromosomal
position | Ref | Observed
allele | Frequency (%) | Gene | Coding | AA change | Phred quality
score | Exon |
|---|
| chr9:139400134 | G | T | 15 | NOTCH1 | c.4214C>A | p.
(Pro1405His) | 47.805 | 25 |
|
chr11:108180932 | A | T | 3.45 | ATM | c.5808A>T | p.
(Leu1936Phe) | 23.909 | 39 |
| chr2:219559286 | A | T | 25 | STK36 | c.2439A>T | p. Gln813His | 36.7001 | 21 |
| chr3:65415336 | T | C | 19.89 | MAGI1 | c.2026A>G | p. Arg676Gly | 625.727 | 12 |
| chr6:56341107 | T | A | 28.71 | DST | c.15347A>T | p. Gln5116Leu | 423.313 | 86 |
| chr8:145740605 | T | G | 11.63 | RECQL4 | c.1412A>C | p. Gln471Pro | 44.974 | 8 |
| chr11:71724390 | G | C | 9.35 | NUMA1 | c.4159C>G | p. Gln1387Glu | 38.5888 | 15 |
| chr15:39884882 | G | T | 80.65 | THBS1 | c.2646G>T | p. Gln882His | 635.503 | 17 |
| chr16:15844106 | C | T | 11.11 | MYH11 | c.1968G>A | p. Met656Ile | 25.4788 | 17 |
| chr18:56383196 | T | G | 4.94 | MALT1 | c.1015T>G | p. Leu339Val | 20.2404 | 9 |
| chr18:59221876 | G | A | 10.99 | CDH20 | c.2354G>A | p. Arg785Gln | 109.339 | 12 |
| chr19:11132513 | C | T | 0.75 | SMARCA4 | c.2729C>T | p. T910M | 22.6552 | 19 |
| Table II.Known missense variants observed in
atypical choroid plexus papilloma tumor. |
Table II.
Known missense variants observed in
atypical choroid plexus papilloma tumor.
| Chromosomal
position | Ref | Observed
allele | Frequency (%) | Gene | Coding | AA change | Phred quality
score | Variant ID | Exon |
|---|
|
chr11:108183167 | A | G | 99.22 | ATM | c.5948A>G | p.
(Asn1983Ser) | 1954.99 | COSM4590264 | 40 |
| chr3:128202753 | G | A | 92.31 | GATA2 | c.967C>T | p. His323Tyr | 813.943 | COSM6854635 | 4 |
| chr4:1808286 | G | A | 93.54 | FGFR3 | c.2044G>A | p. Val682Ile | 4487.31 | COSM6854620 | 16 |
| chrX:53223568 | G | A | 7.49 | KDM5C | c.3791C>T | p. Ala1264Val | 39.1666 | COSM6978062 | 23 |
| chr2:29416572 | T | C | 100 | ALK | c.4381A>G | p. Ile1461Val | 9246.4 | rs1670283 | 29 |
| chr5:112179149 | T | A | 19.78 | APC | c.7858T>A | p. Phe2620Ile | 233.547 | rs587781816 | 16 |
| chr7:55259448 | C | T | 0.84 | EGFR | c.2506C>T | p. R836C | 27.6404 | COSM28604 | 21 |
| chr7:55259493 | G | A | 0.79 | EGFR | c.2551G>A | p. V851I | 30.2797 | COSM12727 | 21 |
| chr7:55259511 | G | A | 1.27 | EGFR | c.2569G>A | p. G857R | 21.9957 | COSM12764 | 21 |
| chr7:55270310 | C | T | 43.99 | EGFR | c.3263C>T | p. Pro1088Leu | 1215.33 | rs771418435 | 27 |
In this tumor, 10 missense mutations identified
(Table II) by Ion Proton have been
previously reported in COSMIC(https://cancer.sanger.ac.uk/cosmic), EXAC browser,
(http://exac.broadinstitute.org/) and
dbSNP databases (https://www.ncbi.nlm.nih.gov/snp/). As described in
Table II, the four known missense
mutations identified in this case were in exon 16 of APC
(c.7858T>A), in exon 16 of FGFR3 (c.2044G>A), in exon 40 of
ATM (c.5948A>G) in exon 29 of ALK (c.4381A>G), in exon 23 of
KDM5C (c.3791C>T) and in exon 4 of GATA2 (c.967C>T). Three
known mutations were detected (c.2506C>T, c.2551G>A and
c.2569G>A) in exon 21 of EGFR, and, one mutation (c.3263C>T)
was also found in exon 27 of EGFR. The quality statistics for these
mutations are presented in Table
SIII. Among these previously reported missense variants, the
ATM, GATA2, FGFR3 and ALK (rs1670283) variants have a frequency of
99.22, 92.31, 93.54 and 100.00% respectively, suggesting that they
may be germline mutations (24,25). All
other variants had low frequencies suggesting those are somatic
mutations (Table II). The PolyPhen
scores of three variants viz. of GATA2 (c.967C>T), FGFR3
(c.2044G>A), KDM5C (c.3791C>T), and of three variants of EGFR
(c.2506C>T; c.2551G>A; and c.2569G>A) were high, which
suggested that they were pathogenic variants; however another EGFR
variant (c.3263C>T) had a low score (Table SIII).
Table III exhibits
the insertions, deletions and nonsense variants that were
identified in the a-CPP tumor tissue. The two novel insertions
found were as follows: The viz. c.1056_1057insG. andc.1056_1057insA
in PARP1 gene, and the c.21904_21905insG in SYNE1 gene. Both caused
frameshift, and the deletion variant in NOTCH1 c.4732_4734delGTG p.
(Val1578delVal) caused a frameshift in reading frame. Two novel
nonsense mutations were also identified in MET gene (c.525T>A;
p. Cys175Ter) and in RNF213 gene (c.6967C>T; p. Gln2323Ter).
Quality statistics of these variants are presented in Table SIV and comprised allele coverage,
allele ratio, P-value, Phred quality score, sequencing coverage and
alleles frequency. The P-value and Phred quality score were
significantly high for all the variants as revealed from the Ion
Reporter analysis reads of a tumor sample and reads against the
associated reference sequence. However, the allele frequency of
PARP1 was low whereas other variants had higher frequencies
(Table SIV), including SYNE1
insertion mutation (p. Phe7302fs) that had a frequency of 100%,
which indicated they may be germline mutations.
| Table III.Novel and known frameshift and
nonsense variants observed in atypical choroid plexus papilloma
tumor. |
Table III.
Novel and known frameshift and
nonsense variants observed in atypical choroid plexus papilloma
tumor.
| Chromosomal
position | Ref | Observed
allele | Frequency (%) | Gene | Coding | AA change | Phred quality
score | Exon |
|---|
| chr9:139399408 | GCACCACCACCA | GCACCACCA | 0.93 | NOTCH1 |
c.4732_4734delGTG | p.
(Val1578delVal) | 112.162 | 26 |
| chr1:226570839 | G | GC, GT | GC=53.44,
T=46.56 | PARP1 | c.1056_1057insG,
c.1056_1057insA | p. Gln353fs, p.
Gln353fs | 8,388.56 | 11 |
| chr6:152540277 | A | AC | 100 | SYNE1 |
c.21904_21905insG | p. Phe7302fs | 3,142.24 | 7 |
| chr7:116339663 | T | A | 8.26 | MET | c.525T>A | p. Cys175Ter | 197.511 | 21 |
| chr17:78319102 | C | T | 12.78 | RNF213 | c.6967C>T | p. Gln2323Ter | 247.344 | 14 |
The present study also identified 15 novel
synonymous mutations in the a-CPP tumor. They were located in exons
11, 7, 21, 14, 12, 16, 13, 10, 24, 46, 19, 21, 1, 4 and 4 of the
genes viz. ABL1, AKT1, PIK3CA, FGFR3, PDGFRA, APC, RET,
SMAD4, NOTCH2, IGF2R, ADGRA2, PRKDC, PAX5, TET1 and IDH2,
respectively (Table IV). As
presented in Table IV, FGFR3,
PDGFRA, RET, IGF2R, ADGRA2, PRKDC and PAX5 synonymous variants had
high frequencies, whereas other synonymous variants were low
frequency variants. The coverage details of these synonymous
mutations are described in Table
SV. In Table V the novel
intronic variants identified in the a-CPP tumor tissue are listed.
The results demonstrated that amongst the 13 novel intronic
variants identified in the present case, the intronic SNPs were in
genes viz. PMS1, FGFR1, ERBB4, RET, FGFR3, SMARCB1, PAX3,
SDHA, LIFR, PDGFRB, MAP3K7, PRKDC and AKT2. In addition, a splice
site mutation at acceptor site (c.132+1G>C) in PMS1 gene, a
variant in 3′-UTR of WT1 gene, and a SNV in 5′-UTR of BRD3 gene
were also identified (Table V).
Quality statistics of these novel intronic variants found in a-CPP
patient are presented in Table SVI
and comprised allele coverage, allele ratio, P-value, coverage and
alleles frequencies. P-values as presented in Table SVI and Phred quality scores as
presented in Table V were
significantly higher for these novel intronic variants.
| Table IV.Novel synonymous variants observed in
atypical choroid plexus papilloma tumor. |
Table IV.
Novel synonymous variants observed in
atypical choroid plexus papilloma tumor.
| Chromosomal
position | Ref | Observed
allele | Frequency (%) | Gene | Coding | AA change | Phred quality
score | Exon |
|---|
| chr3:178952089 | T | C | 4.96 | PIK3CA | c.3144T>C | p.
(His1048His) | 99.3152 | 21 |
| chr4:1807894 | G | A | 100 | FGFR3 | c.1953G>A | p. (Thr651Thr) | 31973.5 | 14 |
| chr4:55141055 | A | G | 100 | PDGFRA | c.1701A>G | p. (Pro567Pro) | 31992 | 12 |
| chr10:43613843 | G | T | 100 | RET | c.2307G>T | p. (Leu769Leu) | 31955.9 | 13 |
| chr1:120469164 | C | A | 3.75 | NOTCH2 | c.3963G>T | p.
(Val1321Val) | 25.14 | 24 |
| chr6:160523656 | G | A | 84.72 | IGF2R | c.6948G>A | p.
(Ala2316Ala) | 1694.61 | 46 |
| chr8:37698673 | C | G | 60.44 | ADGRA2 | c.2817C>G | p.
(Thr3939Thr) | 1510.27 | 19 |
| chr8:48839854 | T | C | 47.76 | PRKDC | c.2319A>G | p. (Leu773Leu) | 8421.16 | 21 |
| chr9:37033987 | C | T | 50.04 | PAX5 | c.42G>A | p. (Arg15Arg) | 6613.8 | 1 |
| chr10:70406386 | C | A | 17.91 | TET1 | c.3900C>A | p.
(Thr1300Thr) | 200.101 | 4 |
| chr15:90631834 | G | A | 6.04 | IDH2 | c.519C>T | p. (His173His) | 33.0969 | 4 |
| Table V.Novel intronic variants observed in
atypical choroid plexus papilloma tumor. |
Table V.
Novel intronic variants observed in
atypical choroid plexus papilloma tumor.
| Chromosomal
position | Ref | Observed
allele | Frequency (%) | Gene | Coding | Phred quality
score |
|---|
| chr2:212522461 | A | C | 46.69 | ERBB4 |
c.1946+18T>G | 1,140.64 |
| chr10:43613960 | C | T | 6.34 | RET |
c.2392+32C>T | 22.0765 |
| chr8:38272057 | A | C | 30.43 | FGFR1 |
c.2141+20T>G | 45.0631 |
| chr4:1808524 | G | A | 7.47 | FGFR3 |
c.2169-32G>A | 277.191 |
| chr2:190656668 | G | C | 7.65 |
PMS1a | c.132+1G>C | 221.546 |
| chr2:190717340 | A | C | C=6.77 | PMS1 | c.700-41A>C | 53.9539 |
| chr2:223158437 | CTC | C | 14.29 | PAX3 |
c.586+449GAG>G | 94.7574 |
| chr5:233504 | T | C | 5.56 | SDHA | c.896-88T>C | 24.9682 |
| chr5:38493661 | T | A | 28.86 | LIFR |
c.2065+47A>T | 1,786.08 |
| chr5:149499530 | A | T | A=24.24, | PDGFRB |
c.2698+45T>A | 207.989 |
| chr6:91228938 | AGAA | A | 6.35 | MAP3K7 |
c.1524+39TTCT>T | 24.0052 |
| chr8:48847670 | CA | AC | 4.79 | PRKDC |
c.1448-52TG>GT | 47.8405 |
| chr9:136918606 | G | A | 19.05 | BRD3,
LOC100130548b | c.-7C>T | 20.7564 |
| chr11:32410514 | T | C | 19.58 |
WT1c | c.1644A>G | 353.703 |
| chr19:40744051 | C | A | 8.16 | AKT2 | c.709-53G>T | 38.5654 |
| chr22:24167632 | T | C | 27.78 | SMARCB1 | c.986+30T>C | 83.9641 |
Discussion
The a-CPP grade II tumor is situated between the CPP
and CPC tumors (11). A-CPP tumors
were previously considered to be a separate pathologic subgroup
that exhibits an intermediate degree of mitotic activity and
overall survival rate. The intermediate position of a-CPP between
CPP and CPC was supported by the clinical data, such as the
presence of metastases at diagnosis, event-free survival rates, and
increases in Ki-67 proliferation marker and the p53 tumor
suppressor protein across the three histological subtypes (from CPP
to a-CPP and then CPC) (26).
Histopathological examination of this tumor revealed increased
mitotic index of 2 mitoses/10 HPF and cribriform morphology, which
confirmed that, according to the diagnostic criteria of WHO
classification of CNS tumors, the present case was an a-CPP
(27). Extensive metastasis from
CPPs can take place into the subarachnoid space when the tumors are
malignant (28). In low-grade CPP,
the unique papillary configuration is a distinct feature, whereas
in CPC the papillary features are unclear and this ill-defined
growth pattern sometimes makes the diagnosis challenging (29). The fibrovascular papillary
projections of CPP are lined by cuboidal to columnar epithelium
compared with normal choroid plexus tissue, which has orderly
cobblestone appearance (1,5). CPP tumours commonly exhibit higher
nuclear-to-cytoplasmic ratio and nuclear hyperchromasia. This was
the first case of a-CPP reported in Saudi Arabia. However, a case
of CPP was reported by Jamjoom et al (30).
Most of the CPP cases have strong immunoreactivity
to S-100 protein, whereas glial fibrillary acidic protein (GFAP)
are expressed weakly and only focally in a few tumors (31). In the present study, no EMA staining
was observed, although it has been reported to be commonly positive
in CPP cases (32). In the present
study, GFAP and EGFR staining was negative, whereas synaptophysin
was focally positive. GFAP, synaptophysin, transthyretin and S100
protein stainings were reported to be more variable in CPP cases
(33). However, pan-cytokeratin and
vimentin are commonly positively expressed in CPP tumours (1,33). It
has been reported that most CPPs express cytokeratins, vimentin and
podoplanin, and are also usually positive for E-cadherin (4,34). The
loss of E-cadherin expression would increase cell proliferation
migration and invasion (35,36). Although E-cadherin is present on the
basolateral surface of the majority of CPPs, decreased expression
in atypical papillomas and CPCs has been reported; however, the
present study demonstrated positive staining for E-cadherin
(37,38).
The novelty of the mutations identified in the
present study (Table I) were
verified by COSMIC and ExAc databases. Several molecular pathways
associated with CPC have been previously reported and allow
distinction of CPC from CPTs (11,39,40).
Mutations in TP53 were demonstrated to be associated with increased
tumor aggressiveness and worsen survival outcome in CPCs; however,
no mutation in TP53 was reported in a-CPP and it is in agreement
with the results from the present study (11,39–41). No
mutation profiling in CPP cases has been previously attempted by
NGS methods; however, it was done in numerous types of brain tumor
and used to distinguish these tumors from their mutational profiles
(42,43). For example, the distinction between
adamantinomatous craniopharyngiomas and papillary
craniopharyngiomas was based upon the presence of mutations in the
genes catenin-β 1 (CTNNB1) and B-Raf proto-oncogene,
serine/threonine kinase following whole exome sequencing (44). To the best of our knowledge,
mutations in the genes NOTCH1, ATM, STK36, MAGI1, GATA2, FGFR3,
DST, RECQL4, NUMA1, THBS1, MYH11, MALT1, CDH20 and KDM5C have been
reported in CPP tumors. STK36 is a serine/threonine protein kinase
that serves a crucial role in the sonic hedgehog pathway by
regulating the activity of the GLI transcription factors and that
is essential for postnatal development, which may be mediated by
CSF homeostatis or ciliary function regulation (45). Overproduction of CSF, growth
retardation, hydrocephalus development and early mortality were
reported in STK36 knockout mice (45). CSF overproduction also worsens the
risk of developing hydrocephalus (6). The patient from the present study also
presented hydrocephalous and CSF accumulation. This indicates that
STK36 mutations may serve a crucial role in choroid plexus tumor
progression. However, further investigation is required to
understand this phenomenon. Shajani-Yi et al (46) reported that 28% patients with
glioblastoma present mutations in TP53, following NGS using Cancer
HotSpot Pane, and also identified mutations in clinically
actionable genes, including IDH1, IDH2, CDKN2A and PIK3CA (46). In the present study we have also
found a known missense mutation in exon 5 of TP53, mutation in
c.419C>T p. (T140I COSM43742), with a p. value of 0.05396, Phred
quality score of 12.6795 and frequency of 1.36% (not shown in
results). A missense mutation in the exon 10 of IDH2 was also
observed in this tumor. This mutation in c.1261G>A (p. Gly421Ser
COSM3420710) had a frequency of 5.06%, a P-value of 0.01377 and a
Phred quality score 18.6103 (not shown in results). These two
mutations have Phred score <20, and were subsequently not
included in the analyses or Tables. In addition, the tumor from the
present study presented a novel synonymous variant in IDH2. The
a-CPP tumor assessed in the present study harbored mutations only
in the EGFR, a driver gene for glioma, activation and
autophosphorylation domains, whereas other driver genes for various
gliomas, such as TP53, PTEN, NF1 and IDH1, did not have any
mutations (47,48). The present study was the first to
reveal a complete mutational profile of a-CPP tumor by NGS
technology on an Ion Proton platform and using 409 tumor suppressor
and oncogenes targeted for mutations. It has been reported that
glioblastoma tumors frequently overexpress the
mesenchymal-epithelial transition (MET) proto-oncogene, of which
expression correlates with tumor grade. In addition, MET signaling
confers resistance to radiotherapy, and various MET inhibition
strategies are being developed to treat GBM tumors (49). A novel nonsense mutation c.525T>A
p. (Cys175Ter) in a-CPP tumor was reported in the present
study.
The present study identified a novel intronic
variant in hSNF5/INI1 (SMARCB1) gene. No exonic mutation was found
in SMARCB1; however, one missense mutation was detected in SMARCA4
gene. Hasselblatt et al (2011) reported that a nonsense
mutation inactivates SMARCA4 in an atypical teratoid/rhabdoid tumor
with intact SMARCB1 (INI1) expression (50). To the best of our knowledge, the
present study is the first to report that SMARCA4 mutation is
associated with a-CPP. For the first time in brain tumors, four
missense variants in ATM, GATA2, FGFR3 and KDM5C and one deletion
variant in NOTCH1 were identified in the present study. The ATM
missense mutation [c.5948A>G; p. (Asn1983Ser)] identified in the
present study was of germline origin, according to ClinVar database
by ‘Ambry Genetics’ with the variation ID No. 140790. This mutation
was also found in SNP database with rs # 659243, and in a patient
with familial history gastrointestinal cancer; however, to the best
of our knowledge, this mutation has not been reported in patients
with brain cancer (24). This ATM
mutation [c.5948A>G; p. (Asn1983Ser)] is adjacent to the Ser
1981 and Ser 1985, which is a part of the focal adhesion targeting
(FAT) domain that is required for ATM autophosphorylation (51,52).
However, this mutation was reported to be benign with a FATHMM
prediction score 0.01 (53). The
mutation reported in the a-CPP case from the present study in exon
16 of the FGFR3 gene [COSM6854620 c.2044G>A p. (Val682Ile)] had
also been reported previously in large intestine (25), with a FATHMM prediction score of
0.97, which suggests that this mutation might be pathogenic
(25). However, this mutation has
not yet been reported in brain tumors.
The formation of various types of tumor in humans is
modulated by NOTCH signaling pathway, which has been reported to
serve a crucial role in the development and formation of normal
choroid plexus (54). Insertion of G
and A in PARP1 gene (c.1056_1057insG and c.1056_1057insA) causes a
frameshift in the reading frame and a termination at 35th coding
position from this insertion. In addition, insertion of G
(c.21904_21905insG) in PARP1 causes a frameshift in the reading
frame and a termination codon at 3rd coding position from insertion
(55). PARP enzymes are involved in
DNA damage repair (56). Mutations
in SYNE1 (ARCA1) gene are known to cause autosomal recessive
cerebellar ataxia type-1 (57), and
patients with CPP are known to have symptoms of ataxia (5). Previous studies reported that SYNE1 and
STK36 mutations are present in GBM cases (58). In addition, deletion variant in
NOTCH1 [c.4732_4734delGTG p. (Val1578delVal)], which causes a
frameshift in reading frame, was also observed in hematopoietic and
lymphoid tissues, but this variant has not previously reported in
GBM (59). Mutations in NOTCH1 are
associated with altered drug sensitivity, such as to tyrosine
kinase inhibitors (60).
Frameshift-deletion in exon 26 of NOTCH1 has been reported in
hematopoietic and lymphoid skin, salivary gland and thymus
(61). KDM5C, also known as
‘Jumonji. AT-rich interactive domain 1c’ JARID1C, codes for a
histone demethylase, and mutations in this gene are associated with
mental retardation and microcephaly (62). This suggests that alteration in
chromatin remodeling might serves a role in CPP. It is therefore
crucial to further investigate choroid plexus tumor to better
understand the role of chromatin remodeling, since this type of
tumor exhibits mutations in enzymes responsible for chromatin
remodeling, including PARP, SMARCA4 and ATM. A mutation in GATA2
gene [c.967C>T p. (His323Tyr)] has been previously identified in
large intestine with a FATHMM prediction score of 1.00 (25). To the best of our knowledge, the
missense mutation detected in the GATA2 gene in the present study
has not yet been reported in GBM tumors. GATA2 is a hematopoietic
factor that has been implicated in hematopoietic malignancies, and
this transcription factor has been implicated in prostate
tumorigenesis (63,64).
ATM kinase is an important tumor suppressor, and its
activation causes the phosphorylation of numerous targets,
including proteins involved in cell cycle arrest, DNA repair and
apoptosis (65). Blake et al
(66) have demonstrated that
inactivation of the ATMIN/ATM pathway protects against glioblastoma
pathogenesis. Eich et al (67) revealed that ATM and ATR inhibition
sensitizes cancer cells to temozolomide (TMZ) and to other
anticancer compounds. In addition, several studies also reported
that ATM- and ATR (ataxia telangiectasia and Rad3 related)-mutated
cells are sensitive to TMZ mediated cytotoxicity (68), Inhibition of ATM-AMPK enhances
TMZ-induced cytotoxicity in inherently TMZ-sensitive glioma cells,
and ATM kinase inhibition also sensitizes p53 mutant glioma cells
to ionizing radiation (69,70). The novel mutation revealed in the
present study was located on chromosome 11, in the exon 39 of ATM
gene. In addition, amongst patients with prostate cancer in AT
heterozygotes, a higher proportion ATM exon 39 polymorphism
(G5557A) was reported (71). Copy
number analysis by molecular inversion probe method revealed focal
chromosomal gains in the region of chromosome 11, which is where
ATM gene locus is present (31).
Ion AmpliSeq comprehensive cancer panel consists of
16,000 primers supplied in 4 tubes (aliquots), which targets 409
genes. This panel targets the exons of numerous tumor suppressor
genes and oncogenes that are frequently mutated in various types of
cancer. The mutations detected in the present study with the Ion
Proton system presented high accuracy and high depth of coverage,
which allowed to reliably detect low frequency mutations with high
confidence. Individual allele coverage for this novel ATM mutation
A>T are in forward strands, the sequencing reads for the A
allele are 1,026 and 904, respectively, and for the T allele, the
sequencing reads are 45 and 24 respectively. This variant coverage
was 1999, mutation frequency was 3.45%, allele frequency was
0.034517 and P-value was 0.00407. Instead of using whole exome
sequencing, cancer panels analysis also became a common practice on
Ion Proton. A previous study analyzed circulating tumor DNA in
non-small cell lung cancer by Ion Proton (22). This instrument offers the advantage
of pooling more samples using the barcodes, which is what the
authors of the aforementioned study did by using Ion AmpliSeq Colon
and Lung Cancer Research Panel v2 on Ion Proton. Pooled samples
sequencing using Ion PI chip enables a highest throughput up to 15
Gb data, with >60–80 million reads passing filter.
It is crucial to obtain well-calibrated quality
scores, since SNP and genotype calling at a specific position in
the genome depends on the base calls and the per-base quality
scores of the reads overlapping the position. Coverage values
represent the read depth (DP), which represents the number of reads
passing quality control that are used to calculate the genotype at
a specific site and in a specific sample with higher values for DP,
generally leading to more accurate genotype calls (72). In the present study, the Phred
quality score of the novel ATM variant was 23.909. A score in this
range corresponds to a <0.5% error rate in base calling. These
results suggested that this novel mutation may be observable in
more CPP tumors. The Polyphen score indicates whether a
substitution is damaging, and predicts the possible impact of an
amino acid substitution on the structure and function of a human
protein. Variants with Polyphen scores in the range of 0.0–0.15 are
predicted to be benign, with the score of 0.15–1.0 potentially
having a damaging effect, and 0.85–1.0 score established as
damaging (23). The novel ATM
variant revealed in the present study had a Polyphen score of
0.097, which suggested that it may be benign.
Numerous ATM mutants possess the ability to
autophosphorylate on Ser-1981 (73).
This activation by the autophosphorylation on Ser1981 leads to
dissociation of the inactive ATM dimer (or higher-order multimer)
into single protein molecules with kinase activity (51). Most missense mutations occur in the
C-terminal part of the amino acid 1966. Mutations of the N-terminal
part are therefore less likely to result in loss of kinase activity
(74). The novel mutation found in
this study was Leu1936Phe that was between the autophosphorylation
sites viz, Ser1893 and Ser1981 (73). This result suggested that the kinase
activity of ATM protein may not be affected by this mutation, since
the kinase domain in ATM was situated between the amino acids 2712
and 2962. Also FRAP/ATM/TRRAP (FAT) domain is not affected because
it is at region 1960 to 2566 amino acids (51).
In conclusion, the present study identified a novel
ATM mutation and 11 novel mutations in an a-CPP tumor, which had
not yet been reported in any databases. Since these novel mutations
have not been previously implicated in choroid plexus tumors, the
findings from the present study may reveal a genetic signature that
could be useful to distinguish choroid plexus tumors between CPP,
a-CPP and CPC. In addition, the present study was the first
reported case of a-CPP from Saudi Arabia. In this tumor, EGFR
mutations were detected in the kinase and autophosphorylation
domains of EGFR. The presence of mutations in this driver gene may
indicate the transformation of a a-CPP grade II tumor to a CPC
grade III tumor. Furthermore, the present study revealed mutations
in KDM5C, PARP, SMARCA4 and ATM genes, which suggested that they
may serve crucial role in chromatin remodeling, and may serve in
understanding the progression of the benign tumor to malignant
tumor. The results from this study may help the development of
targeted therapies for this pediatric tumor, which had not been
previously due to the lack of mutational information. This
investigation may allow understanding this rare tumor at a
molecular level.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to Mr. Mohammed Bader
Al-Hamad (Histopathology Division, Al-Noor Specialist Hospital,
Makkah) for his help in histopathology and immunology work. The
authors would also like to acknowledge Mr. Udaya Raja (Integrated
Gulf Biosystems, Riyadh) for his help with Ion Reporter 5.6
software.
Funding
This study was supported by the National plan for
Science, Technology and Innovation (MAARIFAH) King Abdul Aziz City
for Science and Technology that awarded Dr MM. Taher (grant no.
12-MED 2961-10).
Availability of data and materials
The datasets generated during the current study are
available from the corresponding author on reasonable request.
Authors' contributions
THN and AB collected the data. MS, ZA and AAH
provided formal analysis of the results. Data curation was done by
RAJ and HA. The study was investigated by GD and MA. The
methodology of the current study was designed by WME, GD and MMT.
The original draft of the manuscript was written by MMT and MS.
Ethics approval and consent to
participate
This study was approved by the Institutional Review
Board for Bioethics Committee of King Abdullah Medical City,
Makkah, Kingdom of Saudi Arabia (IRB no. 14-140), and performed in
accordance with the principles of the Declaration of Helsinki.
Informed consent was obtained from the guardian of the patient
prior to the study.
Patient consent for publication
Not applicable.
Competing interests
All authors agreed with the contents of this
manuscript and declare that they have no competing interests.
Glossary
Abbreviations
Abbreviations:
|
a-CPP
|
atypical choroid plexus papilloma
|
|
AT
|
ataxia-telangiectasia
|
|
CNS
|
central nervous system
|
|
CNV
|
copy number variation
|
|
CSF
|
cerebrospinal fluid
|
|
HPF
|
high-power fields
|
|
NGS
|
next generation DNA sequencing
|
|
PICU
|
pediatric intensive care unit
|
|
PolyPhen
|
polymorphism phenotyping
|
|
SNP
|
single nucleotide polymorphisms
|
|
vcf
|
variant call format
|
References
|
1
|
Safaee M, Oh MC, Bloch O, Sun MZ, Kaur G,
Auguste KI, Tihan T and Parsa AT: Choroid plexus papillomas:
Advances in molecular biology and understanding of tumorigenesis.
Neuro Oncol. 15:255–267. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ellenbogen RG, Winston KR and Kupsky WJ:
Tumors of the choroid plexus in children. Neurosurgery. 25:327–335.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rickert CH and Paulus W: Tumors of the
choroid plexus. Microsc Res Tech. 52:104–111. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Louis DN, Perry A, Reifenberger G, von
Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD,
Kleihues P and Ellison DW: The 2016 World Health Organization
Classification of tumors of the central nervous system: A summary.
Acta Neuropathol. 131:803–820. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jaiswal S, Vij M, Mehrotra A, Kumar B,
Nair A, Jaiswal AK, Behari S and Jain VK: Choroid plexus tumors: A
clinico-pathological and neuro-radiological study of 23 cases.
Asian J Neurosurg. 8:29–35. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lechanoine F, Zemmoura I and Velut S:
Treating cerebrospinal fluid rhinorrhea without dura repair: A case
report of posterior fossa choroid plexus papilloma and review of
the literature. World Neurosurg. 108:990.e1–990.e9. 2017.
View Article : Google Scholar
|
|
7
|
Wolff JE, Sajedi M, Coppes MJ, Anderson RA
and Egeler RM: Radiation therapy and survival in choroid plexus
carcinoma. Lancet. 353:21261999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Park SH, Won J, Kim SI, Lee Y, Park CK,
Kim SK and Choi SH: Molecular testing of brain tumor. J Pathol
Transl Med. 51:205–223. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Custodio G, Taques GR, Figueiredo BC,
Gugelmin ES, Oliveira Figueiredo MM, Watanabe F, Pontarolo R, Lalli
E and Torres LF: Increased incidence of choroid plexus carcinoma
due to the germline TP53 R337H mutation in southern Brazil. PLoS
One. 6:e180152011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Østrup O, Nysom K, Scheie D, Schmidt AY,
Mathiasen R, Hjalgrim LL, Olsen TE, Skjøth-Rasmussen J, Henriksen
BM, Nielsen FC, et al: Importance of comprehensive molecular
profiling for clinical outcome in children with recurrent cancer.
Front Pediatr. 6:1142018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Merino DM, Shlien A, Villani A, Pienkowska
M, Mack S, Ramaswamy V, Shih D, Tatevossian R, Novokmet A, Choufani
S, et al: Molecular characterization of choroid plexus tumors
reveals novel clinically relevant subgroups. Clin Cancer Res.
21:184–192. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hasselblatt M, Böhm C, Tatenhorst L, Dinh
V, Newrzella D, Keyvani K, Jeibmann A, Buerger H, Rickert CH and
Paulus W: Identification of novel diagnostic markers for choroid
plexus tumors: A microarray-based approach. Am J Surg Pathol.
30:66–74. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sevenet N, Sheridan E, Amram D, Schneider
P, Handgretinger R and Delattre O: Constitutional mutations of the
hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet.
65:1342–1348. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mueller W, Eum JH, Lass U, Paulus W,
Sarkar C, Bruck W and von Deimling A: No evidence of hSNF5/INI1
point mutations in choroid plexus papilloma. Neuropathol Appl
Neurobiol. 30:304–307. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Phelan ML, Sif S, Narlikar GJ and Kingston
RE: Reconstitution of a core chromatin remodeling complex from
SWI/SNF subunits. Mol Cell. 3:247–253. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reisman D, Glaros S and Thompson EA: The
SWI/SNF complex and cancer. Oncogene. 28:1653–1668. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kreiger PA, Judkins AR, Russo PA, Biegel
JA, Lestini BJ, Assanasen C and Pawel BR: Loss of INI1 expression
defines a unique subset of pediatric undifferentiated soft tissue
sarcomas. Mod Pathol. 22:142–150. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
de Jong MM, Nolte IM, Meerman GJ, van der
Graaf WT, Oosterwijk JC, Kleibeuker JH, Schaapveld M and de Vries
EG: Genes other than BRCA1 and BRCA2 involved in breast cancer
susceptibility. J Med Genet. 39:225–242. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Miyagi K, Mukawa J, Kinjo N, Horikawa K,
Mekaru S, Nakasone S, Koga H, Higa Y and Naito M: Astrocytoma
linked to familial ataxia-telangiectasia. Acta Neurochir (Wien).
135:87–92. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Piane M, Molinaro A, Soresina A, Costa S,
Maffeis M, Germani A, Pinelli L, Meschini R, Plebani A, Chessa L
and Micheli R: Novel compound heterozygous mutations in a child
with Ataxia-Telangiectasia showing unrelated cerebellar disorders.
J Neurol Sci. 371:48–53. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cryan JB, Haidar S, Ramkissoon LA, Bi WL,
Knoff DS, Schultz N, Abedalthagafi M, Brown L, Wen PY, Reardon DA,
et al: Clinical multiplexed exome sequencing distinguishes adult
oligodendroglial neoplasms from astrocytic and mixed lineage
gliomas. Oncotarget. 5:8083–8092. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pécuchet N, Zonta E, Didelot A, Combe P,
Thibault C, Gibault L, Lours C, Rozenholc Y, Taly V, Laurent-Puig
P, et al: Base-position error rate analysis of next-generation
sequencing applied to circulating tumor DNA in non-small cell lung
cancer: A prospective study. PLoS Med. 13:e10021992016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Han CM, Hwang Y, Kim CK and Oh JH: Genetic
profile analysis of a patient with metachronous gastric cancer with
a family history of gastrointestinal cancers. Korean J Helicobacter
Upper Gastrointest Res. 17:218–223. 2017. View Article : Google Scholar
|
|
25
|
Chakrabarty S, Varghese VK, Sahu P,
Jayaram P, Shivakumar BM, Pai CG and Satyamoorthy K: Targeted
sequencing-based analyses of candidate gene variants in ulcerative
colitis-associated colorectal neoplasia. Br J Cancer. 117:136–143.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wrede B, Hasselblatt M, Peters O, Thall
PF, Kutluk T, Moghrabi A, Mahajan A, Rutkowski S, Diez B, Wang X,
et al: Atypical choroid plexus papilloma: Clinical experience in
the CPT-SIOP-2000 study. J Neurooncol. 95:383–392. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Adesina AM: Intraoperative consultation in
the diagnosis of pediatric brain tumors. Arch Pathol Lab Med.
129:1653–1660. 2005.PubMed/NCBI
|
|
28
|
Uff CE, Galloway M and Bradford R:
Metastatic atypical choroid plexus papilloma: A case report. J
Neurooncol. 82:69–74. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Singh A, Vermani S, Sharma S and Chand K:
Papillary meningioma: A rare but distinct variant of malignant
meningioma. Diagn Pathol. 2:32007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jamjoom AA, Sharab MA, Jamjoom AB and
Satti MB: Rapid evolution of a choroid plexus papilloma in an
infant. Br J Neurosurg. 23:324–325. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Japp AS, Gessi M, Messing-Jünger M,
Denkhaus D, Zur Mühlen A, Wolff JE, Hartung S, Kordes U,
Klein-Hitpass L and Pietsch T: High-resolution genomic analysis
does not qualify atypical plexus papilloma as a separate entity
among choroid plexus tumors. J Neuropathol Exp Neurol. 74:110–120.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Matsushima T, Inoue T, Takeshita I, Fukui
M, Iwaki T and Kitamoto T: Choroid plexus papillomas: An
immunohistochemical study with particular reference to the
coexpression of prealbumin. Neurosurgery. 23:384–389. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sameshima T, Tanikawa R, Sugimura T, Izumi
N, Seki T, Maeda T, Tsuboi T, Hashimoto M, Kimura T and Nabeshima
K: Choroid plexus papilloma originating in the sella turcica-case
report. Neurol Med Chir (Tokyo). 50:144–146. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Figarella-Branger D, Lepidi H, Poncet C,
Gambarelli D, Bianco N, Rougon G and Pellissier JF: Differential
expression of cell adhesion molecules (CAM). neural CAM and
epithelial cadherin in ependymomas and choroid plexus tumors. Acta
Neuropathol. 89:248–257. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim SA, Inamura K, Yamauchi M, Nishihara
R, Mima K, Sukawa Y, Li T, Yasunari M, Morikawa T, Fitzgerald KC,
et al: Loss of CDH1 (E-cadherin) expression is associated with
infiltrative tumour growth and lymph node metastasis. Br J Cancer.
114:199–206. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gumbiner BM: Regulation of
cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol.
6:622–634. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Losi-Guembarovski R, Kuasne H,
Guembarovski AL, Rainho CA and Cólus IM: DNA methylation patterns
of the CDH1. RARB. and SFN genes in choroid plexus tumors. Cancer
Genet Cytogenet. 179:140–145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hirohashi S and Kanai Y: Cell adhesion
system and human cancer morphogenesis. Cancer Sci. 94:575–581.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lv SQ, Song YC, Xu JP, Shu HF, Zhou Z, An
N, Huang QL and Yang H: A novel TP53 somatic mutation involved in
the pathogenesis of pediatric choroid plexus carcinoma. Med Sci
Monit. 18:CS37–CS41. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tabori U, Shlien A, Baskin B, Levitt S,
Ray P, Alon N, Hawkins C, Bouffet E, Pienkowska M, Lafay-Cousin L,
et al: TP53 alterations determine clinical subgroups and survival
of patients with choroid plexus tumors. J Clin Oncol. 28:1995–2001.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Vital A, Bringuier PP, Huang H, San Galli
F, Rivel J, Ansoborlo S, Cazauran JM, Taillandier L, Kleihues P and
Ohgaki H: Astrocytomas and choroid plexus tumours in two families
with identical p53 germline mutations. J Neuropathol Exp Neurol.
57:1061–1069. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bettegowda C, Agrawal N, Jiao Y, Wang Y,
Wood LD, Rodriguez FJ, Hruban RH, Gallia GL, Binder ZA, Riggins CJ,
et al: Exomic sequencing of four rare central nervous system tumor
types. Oncotarget. 4:572–583. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Synhaeve NE, van den Bent MJ, French PJ,
Dinjens WNM, Atmodimedjo PN, Kros JM, Verdijk R, Dirven CMF and
Dubbink HJ: Clinical evaluation of a dedicated next generation
sequencing panel for routine glioma diagnostics. Acta Neuropathol
Commun. 6:1262018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Brastianos PK, Taylor-Weiner A, Manley PE,
Jones RT, Dias-Santagata D, Thorner AR, Lawrence MS, Rodriguez FJ,
Bernardo LA, Schubert L, et al: Exome sequencing identifies BRAF
mutations in papillary craniopharyngiomas. Nat Genet. 46:161–165.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Merchant M, Evangelista M, Luoh SM, Frantz
GD, Chalasani S, Carano RA, van Hoy M, Ramirez J, Ogasawara AK,
McFarland LM, et al: Loss of the serine/threonine kinase fused
results in postnatal growth defects and lethality due to
progressive hydrocephalus. Mol Cell Biol. 25:7054–7068. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shajani-Yi Z, de Abreu FB, Peterson JD and
Tsongalis GJ: Frequency of somatic TP53 mutations in combination
with known pathogenic mutations in colon adenocarcinoma, non-small
cell lung carcinoma and gliomas as identified by next-generation
sequencing. Neoplasia. 20:256–262. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Parsons DW, Jones S, Zhang X, Lin JC,
Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et
al: An integrated genomic analysis of human glioblastoma
multiforme. Science. 321:1807–1812. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
The Cancer Genome Atlas Research Network,
. Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Rath PB, Lal O, Ajala Y, Li Y, Xia S, Kim
J and Laterra J: In vivo c-Met pathway inhibition depletes human
glioma xenografts of tumor-propagating stem-like cells. Transl
Oncol. 6:104–111. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hasselblatt M, Gesk S, Oyen F, Rossi S,
Viscardi E, Giangaspero F, Giannini C, Judkins AR, Frühwald MC,
Obser T, et al: Nonsense mutation and inactivation of SMARCA4
(BRG1) in an atypical teratoid/rhabdoid tumor showing retained
SMARCB1 (INI1) expression. Am J Surg Pathol. 35:933–935. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bakkenist CJ and Kastan MB: DNA damage
activates ATM through intermolecular autophosphorylation and dimer
dissociation. Nature. 421:499–506. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kozlov SV, Graham ME, Jakob B, Tobias F,
Kijas AW, Tanuji M, Chen P, Robinson PJ, Taucher-Scholz G, Suzuki
K, et al: Autophosphorylation and ATM activation: Additional sites
add to the complexity. J Biol Chem. 286:9107–9119. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hirsch P, Zhang Y, Tang R, Joulin J,
Boutroux H, Pronier E, Moatti H, Flandrin P, Marzac C, Bories D, et
al: Genetic hierarchy and temporal variegation in the clonal
history of acute myeloid leukaemia. Nat Commun. 7:124752016.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Beschorner R, Waidelich J, Trautmann K,
Psaras T and Schittenhelm J: Notch receptors in human choroid
plexus tumors. Histol Histopathol. 28:1055–1063. 2013.PubMed/NCBI
|
|
55
|
Ricks TK, Chiu HJ, Ison G, Kim G, McKee
AE, Kluetz P and Pazdur R: Successes and challenges of PARP
inhibitors in cancer therapy. Front Oncol. 5:2222015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ray Chaudhuri A and Nussenzweig A: The
multifaceted roles of PARP1 in DNA repair and chromatin
remodelling. Nat Rev Mol Cell Biol. 18:610–621. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Gros-Louis F, Dupré N, Dion P, Fox MA,
Laurent S, Verreault S, Sanes JR, Bouchard JP and Rouleau GA:
Mutations in SYNE1 lead to a newly discovered form of autosomal
recessive cerebellar ataxia. Nat Genet. 39:80–85. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Masica DL and Karchin R: Correlation of
somatic mutation and expression identifies genes important in human
glioblastoma progression and survival. Cancer Res. 71:4550–4561.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Weng AP, Ferrando AA, Lee W, Morris JP IV,
Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT and Aster
JC: Activating mutations of NOTCH1 in human T cell acute
lymphoblastic leukemia. Science. 306:269–271. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Espinoza I and Miele L: Notch inhibitors
for cancer treatment. Pharmacol Ther. 139:95–110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Lee SY, Kumano K, Masuda S, Hangaishi A,
Takita J, Nakazaki K, Kurokawa M, Hayashi Y, Ogawa S and Chiba S:
Mutations of the Notch1 gene in T-cell acute lymphoblastic
leukemia: Analysis in adults and children. Leukemia. 19:1841–1843.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sheardown S, Norris D, Fisher A and
Brockdorff N: The mouse Smcx gene exhibits developmental and tissue
specific variation in degree of escape from X inactivation. Hum Mol
Genet. 5:1355–1360. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wlodarski MW, Collin M and Horwitzd MS:
GATA2 deficiency and related myeloid neoplasms. Semin Hematol.
54:81–86. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Rodriguez-Bravo V, Carceles-Cordon M,
Hoshida Y, Cordon-Cardo C, Galsky MD and Domingo-Domenech J: The
role of GATA2 in lethal prostate cancer aggressiveness. Nat Rev
Urol. 14:38–48. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kastan MB and Lim DS: The many substrates
and functions of ATM. Nat Rev Mol Cell Biol. 1:179–186. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Blake SM, Stricker SH, Halavach H, Poetsch
AR, Cresswell G, Kelly G, Kanu N, Marino S, Luscombe NM, Pollard SM
and Behrens A: Inactivation of the ATMIN/ATM pathway protects
against glioblastoma formation. Elife. 5:e087112016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Eich M, Roos WP, Nikolova T and Kaina B:
Contribution of ATM and ATR to the resistance of glioblastoma and
malignant melanoma cells to the methylating anticancer drug
temozolomide. Mol Cancer Ther. 12:2529–2540. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Maréchal A and Zou L: DNA damage sensing
by the ATM and ATR kinases. Cold Spring Harb Perspect Biol.
5:a0127162013. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Biddlestone-Thorpe L, Sajjad M, Rosenberg
E, Beckta JM, Valerie NC, Tokarz M, Adams BR, Wagner AF, Khalil A,
Gilfor D, et al: ATM kinase inhibition preferentially sensitizes
p53 mutant glioma to ionizing radiation. Clin Cancer Res.
19:3189–3200. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zou Y, Wang Q, Li B, Xie B and Wang W:
Temozolomide induces autophagy via ATM-AMPK-ULK1 pathways in
glioma. Mol Med Rep. 10:411–416. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Janin N, Andrieu N, Ossian K, Laugé A,
Croquette MF, Griscelli C, Debré M, Bressac-de-Paillerets B, Aurias
A and Stoppa-Lyonnet D: Breast cancer risk in ataxia telangiectasia
(AT) heterozygotes: Haplotype study in French AT families. Br J
Cancer. 80:1042–1045. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Carson AR, Smith EN, Matsui H, Brækkan SK,
Jepsen K, Hansen JB and Frazer KA: Effective filtering strategies
to improve data quality from population-based whole exome
sequencing studies. BMC Bioinformatics. 15:1252014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Khalil HS, Tummala H and Zhelev N: ATM in
focus: A damage sensor and cancer target. Biodiscov. 5:12012.
|
|
74
|
Lavina MF, Scotta S, Guevena N, Kozlova S,
Penga C and Chena P: Functional consequences of sequence
alterations in the ATM gene. DNA Repair (Amst). 3:1197–1205. 2004.
View Article : Google Scholar : PubMed/NCBI
|