Introduction
High risk human papillomaviruses (HR HPV) are ~55 nm
non-enveloped circular DNA viruses causing anogenital and
oropharyngeal cancers (1).
Carcinogenic properties of HR HPV are mainly due to the continuous
expression of E6 and E7 oncoproteins. Indeed, the most described
effects of these two oncoproteins are to bind and abrogate the
functions of the tumor suppressor proteins p53 and pRb respectively
(2–4). This leads to altered cell cycle
regulation with increased cell proliferation rate, immortalization,
chromosomal instability driving malignant transformation of
infected cells (5). However, E6 and
E7 interact with many other proteins and particularly with
epigenetic enzymes. Among them, DNMT1 is a DNA methyltransferase
maintaining methylation on CpG dinucleotide during cell division.
This epigenetic mark generally drives chromatin compaction, thus
repressing genes under its dependence. Interestingly, E6 and E7
expression are also regulated by epigenetic mechanisms, and
methylation level on viral promoter is higher in HPV-associated
cancers than in precancerous lesions or in normal tissues (6). Recently, studies revealed that E6 and
E7 expression was downregulated following treatment of HPV positive
cancer cell lines by 5aza-2′-deoxycytidine (5azadC), a DNA methyl
transferase (DNMT) inhibitor also named decitabine (7–9). The
downregulation of E6 was partly due to the upregulation of miR-375
(8,9), known to target the early HPV16
transcripts (10). Indeed, E6
expression was only partially restored by miR-375 inhibitor in
5azadC-treated cells (9). This
suggests that other mechanisms are involved in E6 downregulation
following 5azadC treatment. In fact, although expression of
transcription factors known to bind to early viral promoter (SP1,
AP1, YY1 and NF1) is not modified by 5azadC treatment, not all
transcription factor expression has been assessed. This is why the
role of T-box transcription factor 2 (TBX2), another transcription
factor binding viral promoter, has been evaluated.
In 2013, it has been shown that the HPV16 LCR could
be repressed, at least in vitro, by TBX2, a member of the
T-Box protein family (11). This
family include transcription factors involved in embryonic
development and encoded by highly conserved genes among vertebrates
(12). T-Box factors recognize and
bind the core sequence GGTGTGA, also known as the T-element
(13). Although the HPV16 LCR lacks
the canonical T-element, TBX2 is able to repress the p97 activity
by binding a sequence located between nt 7564 and nt 7756 (11). Conversely to other T-Box family
members, TBX2 has repressive activities thanks to the presence of a
strong repression domain located in its C-terminal region.
The role of TBX2 in tumorigenesis remains
controversial because of both anti- and pro-tumorigenic activities.
On the one hand, TBX2 is able to bypass senescence and activate
cell proliferation by repressing p14ARF and p21 in
different cancer models (12). TBX2
expression was significantly increased in prostate cancers compared
to healthy adjacent tissues (14),
suggesting an oncogenic activity. On the other hand, TBX2 inhibits
cell cycle progression in lung adenocarcinoma (15) and its expression is decreased in lung
cancers compared to normal tissue (16). Furthermore, TBX2 methylation has been
associated with a poor prognosis in chronic lymphocytic leukemia
(17), endometrial cancer (18) and bladder cancer (19). Thus, TBX2 may also exert an
anti-tumor activity depending on the context and cellular origin of
cancers.
In the present study, the role of TBX2 was
investigated to determine whether, in addition to miR-375, it could
induce E6 repression following 5azadC treatment of HPV16 cervical
cancer cells.
Materials and methods
Cell lines and cell culture
MCF-7 cells [mammary cancer cells, American Type
Culture Collection (ATCC)], SiHa cells (cervical cancer cells,
ATCC) and U-2 OS cells (bone osteosarcoma cells, ATCC) were
cultured in Dulbecco's modified Eagle's medium (DMEM); Ca Ski cells
(cervical cancer cells, ATCC) were grown in Roswell Park Memorial
Institute (RPMI) medium while C-33 A and HeLa cells (both cervical
cancer cells, ATCC) were cultured in Eagle's minimum essential
medium (EMEM). All media were supplemented with 10% (v/v) fetal
bovine serum (FBS, Lonza) but not with antibiotics. Cells were
cultured at 37°C in a 5% CO2 humidified incubator.
Drug treatment
SiHa and Ca Ski cells were treated by 5-aza-
2′-deoxycytidine (5azadC) (Epigentek). This demethylating agent was
dissolved in DMSO at 220 mM and then diluted in appropriate medium
at 0.25 or 5.0 µM to treat SiHa cells (10,000 cells/cm2)
and Ca Ski cells (10,000 cells/cm2) for 24, 48, 72 and
96 h. The treatment medium was renewed every day. Untreated HeLa,
C-33 A and MCF-7 cells were harvested as controls for western
blotting studies.
U-2 OS, SiHa and Ca Ski cells were transfected as
described below.
Plasmid transfection and luciferase
assay
U-2 OS cells were plated in 96-well microplates at a
density of 15,000 cells per well. They were transfected for 24 h
with either pT-REx-DEST30/empty or pT-REx-DEST30/TBX2-3XFlag plus a
mixture of pGL3-Luc-16LCR (obtained by cloning the HPV16 DNA
sequence from nt 7135 to nt 105 into the pGL3 plasmid) and
pRenilla (Promega) with JetPEI® transfection
reagent (Polyplus-transfection) according to the manufacturer's
recommendations. Cells were lysed by 1X Lysis reagent of
Dual-Luciferase Reporter Assay System kit (Promega). The luciferase
assay reagent was mixed with lysates and the luminescence was
recorded using the TECAN Infinite 200 Pro instrument.
Unfortunately, luciferase activity could not have been normalized
by Renilla signals because TBX2 strongly repressed the CMV-driven
vector pRenilla, as already documented by Schneider and
collaborators (11).
SiHa and Ca Ski cells were plated in 6-well plates
at a density of 350,000 cells per well and transfected with
pT-REx-DEST30/empty or pT-REx-DEST30/TBX2-3XFlag using
JetPEI® transfection reagent according to the
manufacturer's recommendations. At 48 h after plasmid transfection,
cells were harvested for checking overexpression efficiency and
subsequent analyses via RTqPCR and western-blotting.
5azadC and TBX2 combinatory
treatment
SiHa and Ca Ski cells were seeded at 10,000
cells/cm2 in 6-well plates and treated with 5azadC at
0.25 µM for 72 h. The treatment medium was renewed every day.
Twenty-four hours after the beginning of 5azadC treatment, cells
were transfected with pT-REx-DEST30/empty or
pT-REx-DEST30/TBX2-3XFlag using JetPEI® transfection
reagent according to the manufacturer's recommendations. Cells were
then cultured for 48 h and harvested for E6 expression analysis by
RT-qPCR.
Transfection of siRNA
MCF-7 cells were transfected with 20 nM of siRNA
targeting TBX2 (5′-GGA-GCU-GUG-GGA- CCA-GUU-CTT-3′) or control
siRNA (SR-CL000-005; Eurogentec) using Lipofectamine 2000 (Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions (ratio siRNA/Lipofectamine of 1:3). Forty-eight hours
after transfection, cells were harvested directly in Ribozol™
solution for RNA extraction or scrapped, centrifuged and lysed in
RIPA solution for protein extraction.
RNA extraction and reverse
transcription
Total cellular RNAs were isolated by
RiboZol™-chloroform method (VWR). Briefly, cells were lysed in 500
µl of RiboZol and 100 µl of chloroform were added. After a
centrifugation at 12,000 g (15 min, 4°C), aqueous phase was
harvested and incubated 10 min with 500 µl of isopropanol. Then,
total RNAs were pelleted by centrifugation at 12,000 g, (10
min, 4°C), washed with cold ethanol and dissolved in molecular
biology grade water. cDNAs were synthetized using 500 ng of total
RNA with the Maxima First Strand cDNA Synthesis kit (Thermo Fisher
Scientific, Inc.) according to the manufacturer's
recommendations.
RT-qPCR
Primers were synthetized by Eurogentec. Real-time
quantitative PCR was performed using SYBR Green real time PCR
master mix (Life Technologies) in the ABI 7500 Real-Time PCR System
(Applied Biosystems). The cDNAs were amplified using the following
cycling parameters: 95°C for 5 min followed by 40 cycles of 95°C
for 30 sec and 60°C for 1 min. Transcript levels for E6 and TBX2
were measured with GAGAACTGCAATGTTTCAGGACC forward and
TGTATAGTTGTTTGCAGCTCTGTGC reverse primers for E6,
CTCTGACAAGCACGGCTTCA forward and TGTCGTTGGCTCGCACTATG reverse
primers for TBX2 and normalized with β2M mRNA level measured with
GATGAGTATGCCGTGTG forward and CAATCCAAATGCGGCATCT reverse primers
using the 2−ΔΔCt method (20).
Western blotting
Following different conditions, cells were harvested
by scraping in PBS and stored in dry pellets at −80°C until use.
Proteins were extracted with radio immunoprecipitation assay (RIPA)
lysis buffer [50 mM Tris/HCl, pH 7.4, 150 mM NaCl, 1% (v/w) Nonidet
P-40, 0.5% (w/w) Na deoxycholate, 1 mM EDTA, 30 µg/ml protease
inhibitor] (Roche Diagnostics). After sonication, protein
concentrations were determined using the Bio-Rad Protein assay
(Bio-Rad) according to the manufacturer's recommendations. Each
sample was resolved on 12% SDS-PAGE gels and then transferred to
Hybond® polyvinylidene difluoride (PVDF) membranes (GE
Healthcare). The membranes were blocked with 5% nonfat milk
overnight at 4°C under constant shaking and then incubated for 2 h
at room temperature under constant agitation with primary
antibodies: Anti-E6HPV16, 1/1,000 (2E-3F8; Euromedex,
Souffelweyersheim, France); DO7 anti-p53, 1/2,000 (554298, BD
Biosciences, Le Pont de Claix, France); 6B6 anti-p21, 1/2,000
(554228; BD Biosciences); AC15 anti-β-actin, 1/20,000 (A1978;
Sigma-Aldrich); C-17 anti-TBX2, 1/500 (sc-17880). After several
washes, membranes were incubated with goat anti-mouse, goat
anti-rabbit (BD Pharmingen) or rabbit anti-goat (Agilent)
immunoglobulin antibodies conjugated with horseradish-peroxidase.
The immune complexes were revealed using an enhanced
chemiluminescence detection system with Pierce ECL2 western
blotting substrate (Thermo Fisher Scientific, Inc.) using ChemiDoc
XRS+. The band densities were normalized against the β-actin
internal control and analyzed by Image Lab software (Bio-Rad).
Statistical analysis
A two-tailed unpaired Student's t-test was used to
assess differences between two groups. In each case non-treated
cells served as reference control. Two-way ANOVA followed by Levene
test were used to analyze differences in E6 expression following
combinatory treatments. All data were obtained from at least three
independent experiments or as specified for each figure and are
presented as mean ± standard deviation (SD). P<0.05 was
considered to indicate a statistically significant difference.
Results
5azadC treatment induces HPV16 E6
downregulation and p53 and p21 upregulation
As we previously published, 5azadC treatment of SiHa
cells induced a time-dependent decrease of E6 expression at both
mRNA and protein levels (Fig. 1A-C).
The effect of 5azadC on E6 repression was observed starting 24 h
after treatment, with a maximal decrease in transcripts reaching
60% at 96 h of treatment (Fig. 1C),
a result consistent with the decreased expression of E6 observed at
the protein level (Fig. 1A and B).
In contrast, the effect of 5azadC on E6 RNA expression did not
appear dependent on the concentration used (0.25 or 5 µM) to treat
SiHa cells. At the protein levels, 5azadC at 5 µM seemed to cause
sooner and more prominent decrease in E6 expression. As expected,
the decreased level of E6 was accompanied by an increased
expression of p53 and an up-regulation of the cyclin dependent
kinase inhibitor p21.
E6 expression was also investigated in Ca Ski cells
treated with 0.25 and 5 µM of 5azadC up to 96 h (Fig. 1D-F). As in SiHa cells, E6 expression
was downregulated at both protein and mRNA levels. The
downregulation of E6 in Ca Ski cells was also accompanied by the
restoration of p53 and p21 expression.
5azadC treatment induces TBX2 mRNA
expression
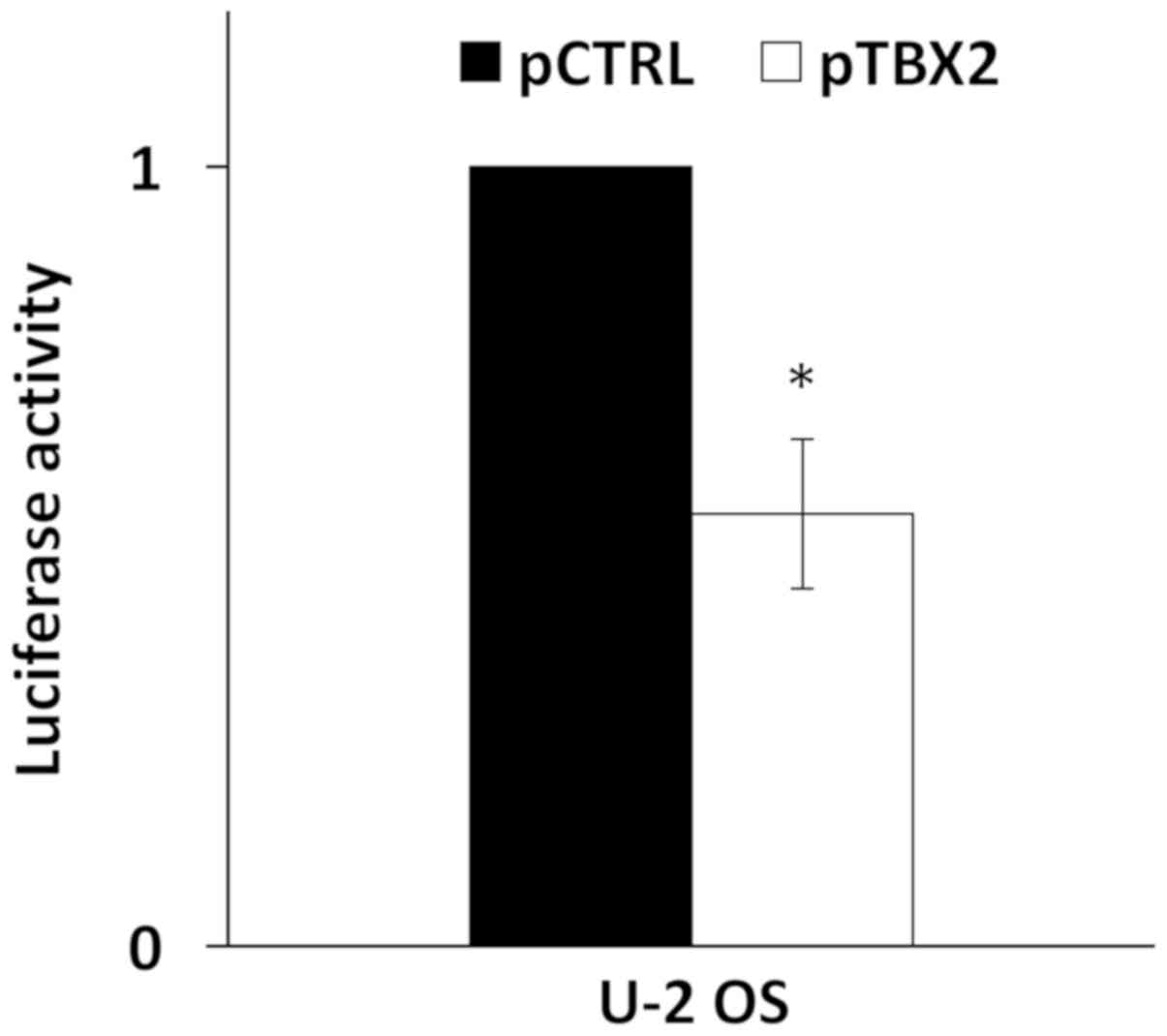
A recent report has shown that TBX2 was able to bind
and repress the HPV16 LCR (11). To
confirm this, the pT-REx-DEST30/TBX2-3XFlag was co-transfected with
pGL3-Luc-16LCR plasmid in U-2 OS cells and the p97 promoter
activity was measured. As shown in Fig.
2, the overexpression of TBX2 induced a reduction of the
relative luciferase activity of 40% compared to cells transfected
with the empty vector. TBX2 transfection efficiency in U-2 OS cells
was assessed by western blotting (Fig.
S1).
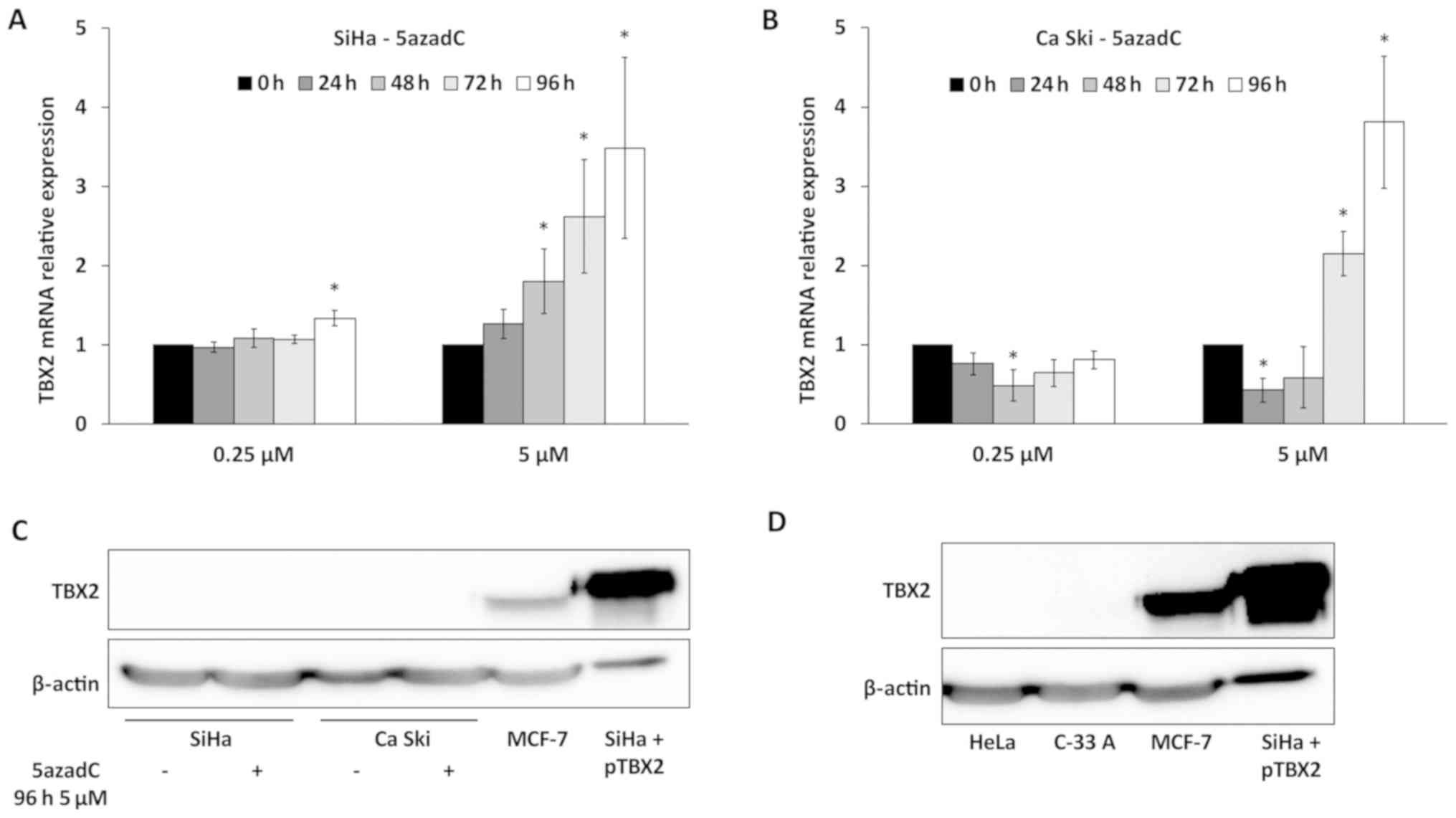
Then, the expression of TBX2 was studied in
5azadC-treated SiHa and Ca Ski cells. In a first set of
experiments, the specificity of TBX2 primers and anti-TBX2 antibody
was confirmed in MCF-7 cells, known to express TBX2, treated by
TBX2 siRNA (Fig. S1). As shown in
Fig. 3A, a slight increase of TBX2
RNA level was observed in SiHa cells treated with 0.25 µM of 5azadC
at 96 h. In Ca Ski cells, no variation of TBX2 expression was
observed with the same treatment (Fig.
3B). In contrast, a clear increase of TBX2 RNA was observed in
both cell lines for the highest concentration (5 µM) of 5azadC,
especially at 96 h, reaching 3.5 and 3.8-fold in SiHa and Ca Ski
cells respectively (Fig. 3A and B).
It is interesting to note that the pattern of variation of E6 mRNA
(Fig. 1C and F) was not inversely
related to that of TBX2.
The endogenous expression of TBX2 was
then analyzed by western blotting
The TBX2 protein was not detected in SiHa and Ca Ski
cells either treated or not with the highest concentration of
5azadC during 96 h (Fig. 3C).
Furthermore, TBX2 was neither detected in Hela (HPV18 positive)
cells and C-33 A (HPV negative) cervical cancer cell lines
(Fig. 3D). In contrast, a strong
signal was observed in MCF-7 cells (a positive control for TBX2
expression) or in SiHa cells transfected with
pT-REx-DEST30/TBX2-3XFlag (Fig. 3C and
D). It is noteworthy that TBX2 mRNA expression is 700 and
90-fold more important in MCF-7 compared to Ca Ski and SiHa cells,
respectively (not shown). Thus, these data indicate that TBX2 is
not expressed or at a very low level in cervical cancer cell lines
compared to MCF-7 cells.
Ectopic TBX2 expression does not
repress endogenous E6 expression
Then, we investigated whether ectopic TBX2
expression could downregulate endogenous E6 expression in SiHa and
Ca Ski cells transfected with pT-REx-DEST30/TBX2- 3XFlag. As shown
in Fig. 4A, TBX2 protein was readily
detected in transfected SiHa and Ca Ski cells. But any changes were
observed in E6 protein (Fig. 4A) and
mRNA (Fig. 4B) expression following
ectopic TBX2 expression. In keeping with this observation, TBX2
unlikely downregulates endogenous E6 expression in cervical cancer
cell lines.
TBX2 overexpression and 5azadC
combinatory treatment does not enhance E6 repression
In order to rule out the possibility that TBX2 was
involved in E6 repression following 5azadC treatment, TBX2
overexpression and 5azadC combinatory treatment were conducted.
TBX2 overexpression has been confirmed by RT-qPCR in cells treated
or not with 5azadC (Fig. S1).
Fig. 4C shows that E6
RNA relative expression was significantly decreased following
5azadC treatment in SiHa (P=0.03) and Ca Ski (P=0.001) cells. In
contrast, no difference in E6 RNA relative expression was observed
between the pT-REx-DEST30/TBX2-3XFlag-(pTBX2) and the
pT-REx-DEST30/empty-(pCTRL) transfected cells, whether they were
treated by 5azadC or not. Thus, overexpression of TBX2 did not
enhance 5azadC-induced E6 repression.
Discussion
In this study, we confirmed that the treatment of
HPV16 positive cancer cell lines with 5azadC leads to the
downregulation of E6 at both mRNA and protein levels (7–9). As
expected, a p53 and p21 restoration was observed in treated cells
confirming the functional loss of E6. Interestingly, the treatment
of the tongue HPV16-positive cancer cell line UPCI:SCC090 with
5azadC also leads to the decrease of E6 and to the increase of p53
and p21 expression (Fig. S2). These
results, in line with those obtained by Stich et al with two
other head and neck cancer-derived cell lines (UM-SCC-47 and
UM-SCC-104) (8), suggest that the
effect of 5azadC treatment on E6 repression is independent of the
tumor origin. Since 5azadC (decitabine) is already used to treat
myelodysplastic syndromes (21) and
acute myeloid leukemia (22) its
usefulness in the treatment of HPV-associated cancers probably
deserves to be addressed. In this line, Biktasova et al
recently reported that the treatment of patients presenting
HPV-positive head and neck cancers with 5-azacytidine, a 5azadC
analogue, induced E6 and E7 RNA repression in their tumors
accompanied by reactivation of the p53 pathway and apoptosis
induction (23). This is the first
study presenting the potential effects of a demethylating treatment
against human HPV+ tumors in a clinical trial.
In SiHa cells, the effect of 5azadC on E6 RNA
repression was not dependent on the concentration used since E6
downregulation was similar whatever the concentration was (0.25 µM
or 5 µM) at each time point over 96 h. Similarly, Stich et
al showed no dose effect of 5azadC treatment on E6*I/E7 mRNA
downregulation even with a concentration as low as 100 nM (8). In contrast, a clear time-dependent
effect of 5azadC treatment was observed on E6 downregulation with a
maximum achieved at 96 h. This may reflect the 5azadC mechanism of
action that requires successive cell divisions to passively
demethylate DNA leading to the progressive upregulation of
factor(s) involved in direct or indirect E6 repression. However,
such a time dependent effect was not reproduced in Ca Ski cells
treated with 5 µM of 5azadC. At this concentration, a cut-off
effect was observed starting at 24 h of treatment, an observation
consistent with a more sensitive phenotype of Ca Ski cells
regarding 5azadC effects on E6 downregulation.
In order to highlight the mechanism involved in E6
downregulation following 5azadC treatment, we explored the role of
the transcription factor TBX2. Indeed, Schneider and collaborators
have recently proposed that TBX2 might decrease the HPV gene
expression through LCR inhibition (11). Furthermore, they mapped the minimal
sequence required for TBX2 inhibition from nt 7564 to nt 7756 on
the LCR. A first series of experiments permitted to confirm in an
in vitro assay that the overexpression of TBX2 downregulated
HPV16 LCR activity.
In both SiHa and Ca Ski cells, TBX2 mRNA expression
was up-regulated following 5azadC exposure in a time and dose
dependent manner. However, this regulation pattern differed
substantially from the one observed for E6. This was especially
true for the lowest concentration of 5azadC that had weak effects
on TBX2 mRNA upregulation while E6 mRNA was clearly downregulated.
Surprisingly, the TBX2 protein was undetectable in SiHa and Ca Ski
cells as well as in two other cervical cancer cell lines infected
by HPV18 (HeLa cells) or not infected by HPV (C-33 A cells). In
contrast TBX2 protein was clearly detected in MCF-7 cells and in
SiHa cells transfected with pT-REx-DEST30/TBX2-3XFlag that served
as positive controls. While we can hypothesize that TBX2 protein is
not expressed in cervical cancer we cannot rule out that its
expression is very low in cervical cancer cells (under the limit of
detection of the western blotting assay). Indeed, Schneider et
al reported only a very faint band for TBX2 in their western
blotting experiments performed with SiHa cell protein extracts. In
the present study, TBX2 protein was not detected even after the
treatment of cervical cancer cell lines with 5 µM of 5azadC during
96 h, whereas the relative expression of the corresponding mRNA was
increased more than 3-fold. Thus, we provide no evidence that the
downregulation of E6 expression following 5azadC treatment is
mediated through an increased expression of endogenous TBX2.
The increased expression of TBX2 mRNA transcripts
without effective translation could be explained by the fact that
Ca Ski and SiHa cells expressed at extremely low level TBX2 mRNA
(700-fold less in Ca Ski and 90-fold less in SiHa cells), compared
to TBX2 mRNA levels in MCF-7 cells as determined by RT-qPCR (data
not shown). These results are in line with the Human Protein Atlas
data, that report no TBX2 mRNA expression in SiHa cells (0.0 TPM)
while TBX2 is expressed at 62.0 TPM in MCF-7 cells). Another
explanation could be the inhibition of translation due to mRNA
sequestration in the nucleus, or due to miRNA interference.
Because TBX2 protein was shown to inhibit the
activity of a cloned HPV16 LCR in overexpression experiments
[(11) and present study), we
wondered to assess whether it could also repress the endogenous
HPV16 LCR in Ca Ski and SiHa cells. While TBX2 was readily detected
in both transfected cell lines, no variation in E6 expression at
both protein and mRNA expression was evidenced. Similarly, TBX2
overexpression in the presence of 5azadC did not enhance E6
repression that is solely due to 5azadC. These observations again
reinforce the idea that TBX2 is unlikely involved in the regulation
of E6 expression in both cell lines.
A limitation of the present study is the lack of
TBX2 target assessment. No specific TBX2 target is described in
cervical cancer cell models, in the literature. This is why TBX2
targets described in cell lines derived from other tissues [p21
(24), p14/p16 (25–27), p27
(28), PTEN (29) and NDRG1 (30)] were tested. However, none of these
targets was significantly repressed following TBX2 transfection.
There is no clear explanation to this observation, but it has to be
noted that p21, p27, p14/p16 are already deregulated by HPV
oncogenes in cervical cancer cells.
Epigenetic regulation of HPV16 oncogene expression
is very likely since the use of a DNA demethylating agent leads to
E6 mRNA and protein repression. Indirect effects linked to the
re-expression of miR-375, that targets HPV16 early transcripts has
been well documented (8,9). By contrast, the involvement of TBX2 in
E6 repression is very unlikely in SiHa and Ca Ski cells treated by
5azadC even if this transcription factor may exert inhibitory
effect on HPV16 LCR cloned upstream a reporter gene. In particular,
combinatory experiments conducted in the present study permitted to
confirmed that TBX2 was not involved in E6 repression even in a
context of demethylated DNA. Whether the structure of chromatin
affect the accessibility of TBX2 to occupy its binding site in the
native HPV16 LCR remains a challenging question.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Schneider
(University Medical Center of Johannes Gutenberg University Mainz,
Mainz, Germany) for providing us with the
pT-REx-DEST30/TBX2-3XFlag, Dr Caroline Demeret (Pasteur Institute,
Paris, France) for providing us with pGL3-Luc-16LCR and Dr F
Monnien (Centre Hospitalier Universitaire de Besançon, Besançon,
France) for aiding in the statistical analysis of the present
study. The authors would also like to thank Julie Durel and Anne
Peigney (both, Université Bourgogne Franche-Comté, Université de
Franche Comté, Besançon, France) for excellent technical
assistance.
Funding
The present study was supported by research grants
from La Ligue Contre le Cancer (grant no. CCIR-GE) and the Conseil
Régional de Franche-Comté (grant no. 2016Y7570-2016Y7571). J.
Perrard and A. Morel were recipients of a predoctoral scholarship
from the Conseil Régional de Franche-Comté and K. Meznad was the
recipient of a predoctoral scholarship from Ministère de
l'Enseignement Supérieur et de la Recherche Scientifique.
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
JP, AM, ChM, VD, CC and JLP conceived and designed
the experiments. JP, KM, CeM and PPB performed the experiments. JP,
AB, SF, VD, DG and CC analyzed the data. JP, AB, DG, ChM and JLP
wrote the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Doorbar J, Egawa N, Griffin H, Kranjec C
and Murakami I: Human papillomavirus molecular biology and disease
association. Rev Med Virol. 25 (Suppl 1):S2–S23. 2015. View Article : Google Scholar
|
|
2
|
Scheffner M, Werness BA, Huibregtse JM,
Levine AJ and Howley PM: The E6 oncoprotein encoded by human
papillomavirus types 16 and 18 promotes the degradation of p53.
Cell. 63:1129–1136. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Münger K, Scheffner M, Huibregtse JM and
Howley PM: Interactions of HPV E6 and E7 oncoproteins with tumour
suppressor gene products. Cancer Surv. 12:197–217. 1992.PubMed/NCBI
|
|
4
|
Huh K, Zhou X, Hayakawa H, Cho JY,
Libermann TA, Jin J, Harper JW and Munger K: Human papillomavirus
type 16 E7 oncoprotein associates with the cullin 2 ubiquitin
ligase complex, which contributes to degradation of the
retinoblastoma tumor suppressor. J Virol. 81:9737–9747. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
zur Hausen H: Human papillomaviruses in
the pathogenesis of anogenital cancer. Virology. 184:9–13. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jacquin E, Baraquin A, Ramanah R,
Carcopino X, Morel A, Valmary-Degano S, Bravo IG, de Sanjosé S,
Riethmuller D, Mougin C and Prétet JL: Methylation of human
papillomavirus Type 16 CpG sites at E2-binding site 1 (E2BS1),
E2BS2, and the Sp1-binding site in cervical cancer samples as
determined by high-resolution melting analysis-PCR. J Clin
Microbiol. 51:3207–3215. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang C, Deng Z, Pan X, Uehara T, Suzuki M
and Xie M: Effects of methylation status of CpG sites within the
HPV16 long control region on HPV16-positive head and neck cancer
cells. PLoS One. 10:e01412452015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stich M, Ganss L, Puschhof J, Prigge ES,
Reuschenbach M, Guiterrez A, Vinokurova S and von Knebel Doeberitz
M: 5-aza-2′-deoxycytidine (DAC) treatment downregulates the HPV E6
and E7 oncogene expression and blocks neoplastic growth of
HPV-associated cancer cells. Oncotarget. 8:52104–52117.
2016.PubMed/NCBI
|
|
9
|
Morel A, Baguet A, Perrard J, Demeret C,
Jacquin E, Guenat D, Mougin C and Prétet JL: 5azadC treatment
upregulates miR-375 level and represses HPV16 E6 expression.
Oncotarget. 8:46163–46176. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jung HM, Phillips BL and Chan EK: miR-375
activates p21 and suppresses telomerase activity by coordinately
regulating HPV E6/E7, E6AP, CIP2A, and 14-3-3ζ. Mol Cancer.
13:802014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schneider MA, Scheffer KD, Bund T,
Boukhallouk F, Lambert C, Cotarelo C, Pflugfelder GO, Florin L and
Spoden GA: The transcription factors TBX2 and TBX3 interact with
human papillomavirus 16 (HPV16) L2 and repress the long control
region of HPVs. J Virol. 87:4461–4474. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wansleben S, Peres J, Hare S, Goding CR
and Prince S: T-box transcription factors in cancer biology.
Biochim Biophys Acta. 1846:380–391. 2014.PubMed/NCBI
|
|
13
|
Abrahams A, Parker MI and Prince S: The
T-box transcription factor Tbx2: Its role in development and
possible implication in cancer. IUBMB Life. 62:92–102.
2010.PubMed/NCBI
|
|
14
|
Du WL, Fang Q, Chen Y, Teng JW, Xiao YS,
Xie P, Jin B and Wang JQ: Effect of silencing the T-Box
transcription factor TBX2 in prostate cancer PC3 and LNCaP cells.
Mol Med Rep. 16:6050–6058. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Khalil A, Dekmak B, Boulos F, Kantrowitz
J, Spira A, Fujimoto J, Kadara H, El-Hachem N and Nemer G:
Transcriptomic alterations in lung adenocarcinoma unveil new
mechanisms targeted by the TBX2 subfamily of tumor suppressor
genes. Front Oncol. 8:4822018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Khalil AA, Sivakumar S, Lucas FAS,
McDowell T, Lang W, Tabata K, Fujimoto J, Yatabe Y, Spira A, Scheet
P, et al: TBX2 subfamily suppression in lung cancer pathogenesis: A
high-potential marker for early detection. Oncotarget.
8:68230–68241. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rani L, Mathur N, Gupta R, Gogia A, Kaur
G, Dhanjal JK, Sundar D, Kumar L and Sharma A: Genome-wide DNA
methylation profiling integrated with gene expression profiling
identifiesPAX9as a novel prognostic marker in chronic lymphocytic
leukemia. Clin Epigenetics. 9:572017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Farkas SA, Sorbe BG and Nilsson TK:
Epigenetic changes as prognostic predictors in endometrial
carcinomas. Epigenetics. 12:19–26. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kandimalla R, van Tilborg AA, Kompier LC,
Stumpel DJ, Stam RW, Bangma CH and Zwarthoff EC: Genome-wide
analysis of CpG island methylation in bladder cancer identified
TBX2, TBX3, GATA2, and ZIC4 as pTa-specific prognostic markers. Eur
Urol. 61:1245–1256. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gangat N, Patnaik MM and Tefferi A:
Myelodysplastic syndromes: Contemporary review and how we treat. Am
J Hematol. 91:76–89. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nieto M, Demolis P, Béhanzin E, Moreau A,
Hudson I, Flores B, Stemplewski H, Salmonson T, Gisselbrecht C,
Bowen D and Pignatti F: The european medicines agency review of
decitabine (Dacogen) for the treatment of adult patients with acute
myeloid leukemia: Summary of the scientific assessment of the
committee for medicinal products for human use. Oncologist.
21:692–700. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Biktasova A, Hajek M, Sewell A, Gary C,
Bellinger G, Deshpande HA, Bhatia A, Burtness B, Judson B, Mehra S,
et al: Demethylation therapy as a targeted treatment for human
papillomavirus-associated head and neck cancer. Clin Cancer Res.
23:7276–7287. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Prince S, Carreira S, Vance KW, Abrahams A
and Goding CR: Tbx2 directly represses the expression of the
p21(WAF1) cyclin-dependent kinase inhibitor. Cancer Res.
64:1669–1674. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jacobs JJ, Keblusek P, Robanus-Maandag E,
Kristel P, Lingbeek M, Nederlof PM, van Welsem T, van de Vijver MJ,
Koh EY, Daley GQ and van Lohuizen M: Senescence bypass screen
identifies TBX2, which represses Cdkn2a (p19(ARF)) and is amplified
in a subset of human breast cancers. Nat Genet. 26:291–299. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vance KW, Carreira S, Brosch G and Goding
CR: Tbx2 is overexpressed and plays an important role in
maintaining proliferation and suppression of senescence in
melanomas. Cancer Res. 65:2260–2268. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Harrelson Z, Kelly RG, Goldin SN,
Gibson-Brown JJ, Bollag RJ, Silver LM and Papaioannou VE: Tbx2 is
essential for patterning the atrioventricular canal and for
morphogenesis of the outflow tract during heart development.
Development. 131:5041–5052. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lüdtke TH, Rudat C, Wojahn I, Weiss AC,
Kleppa MJ, Kurz J, Farin HF, Moon A, Christoffels VM and Kispert A:
Tbx2 and Tbx3 act downstream of shh to maintain canonical wnt
signaling during branching morphogenesis of the murine lung. Dev
Cell. 39:239–253. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhu B, Zhang M, Williams EM, Keller C,
Mansoor A and Davie JK: TBX2 represses PTEN in rhabdomyosarcoma and
skeletal muscle. Oncogene. 35:4212–4224. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Redmond KL, Crawford NT, Farmer H, D'Costa
ZC, O'Brien GJ, Buckley NE, Kennedy RD, Johnston PG, Harkin DP and
Mullan PB: T-box 2 represses NDRG1 through an EGR1-dependent
mechanism to drive the proliferation of breast cancer cells.
Oncogene. 29:3252–3262. 2010. View Article : Google Scholar : PubMed/NCBI
|