Introduction
Nasopharyngeal carcinoma (NPC) is the most prevalent
malignancy in southern China and Southeast Asia. In total, ~129,000
new cases are reported annually worldwide, with >70% reported in
South China and Southeast Asia (1).
Its pathogenesis is associated with three primary etiological
factors; Epstein-Barr virus infection, genetic susceptibility and
environmental condition (2).
However, the pathophysiological mechanism underlying NPC
progression is yet to be elucidated.
Previous studies have predominantly focused on the
function of specific genes expressed in NPC instead of the
molecular pathogenesis of the disease (1,2).
Notably, >98% of human genes are non-protein coding, and the
expression of these genes generates non-coding RNAs (ncRNAs)
(3). There are multiple families of
ncRNA, including ribosomal (r), short interfering (si), micro (mi),
circular (circ) and long non-coding (lnc)RNA (4–6). The
latter refers to ncRNAs >200 nucleotides in length (4,5).
Previous studies have reported that lncRNAs are important
regulators of numerous biological processes, including tumor
progression (7,8). miRNAs are a class of small ncRNAs of
~22 nucleotides, which serve as key regulators of multiple
disease-associated processes (9).
Unlike linear RNA, circRNAs exhibit a covalently closed continuous
loop, and serve as an miRNA sponge to regulate transcription
(10). A number of studies have
indicated that ncRNAs play important roles in transcriptional
regulation by forming regulatory networks and subsequently
interacting with their respective target genes (4–7).
lncRNAs and miRNAs have become increasingly
associated with the progression of NPC (11); however, the majority of studies have
solely focused on the role of single or small groups of these
molecules. Ma et al (12)
reported that lncRNA HOX transcript antisense RNA contributes
towards the tumorigenesis of NPC via the upregulation of fatty acid
synthase. Moreover, Zhang et al (13) discovered that miRNA-200c acts as an
oncogene in NPC, by regulating the phosphatase and tensin homolog
genes. To the best of our knowledge, a limited number of studies to
date have investigated the association between circRNA expression
and NPC, and no data are currently available concerning circRNAs
and their target genes in NPC. Shuai et al (14) indicated that circRNA_0000285 may be
used as a novel biomarker for NPC radiosensitivity. As the
initiation and progression of different types of tumor is a
multi-gene, multi-step process, research is typically focused on
gene regulatory networks at a genome-wide level.
The purpose of the present study was to
comprehensively analyze the expression profiles of lncRNAs,
circRNAs and mRNAs in NPC, and their inter-regulatory molecular
mechanisms. It was also aimed to identify the target genes of
differentially expressed (DE) lncRNAs and circRNAs, and the DE
genes (DEGs) within key signaling pathways influencing NPC
progression, in order to elucidate the regulatory network in NPC.
First, the present study compared the transcriptome profiling of
lncRNAs, circRNAs and mRNAs between NPC and chronic nasopharyngitis
(CNP) tissues, using microarray technology at the whole genome
level. Subsequently, integrated bioinformatics analysis was
performed on the three microarray datasets. The results of the
present study may help to clarify the associations between lncRNAs,
circRNAs and miRNAs (and their target genes), and elucidate the
notable regulatory networks involved in the molecular pathogenesis
of NPC.
Materials and methods
Specimens
A total of 42 human nasopharyngeal tissue samples
were collected from 42 patients (30 men and 12 women) during
nasopharyngeal biopsy between August 2013 and October 2014, at
Zhongshan Hospital (Ximen, China). The tissue samples included 21
cases of primary NPC and 21 cases of CNP from patients suspected of
having cancer. All specimens were confirmed by histopathological
examination. Patients did not receive chemoradiotherapy and
biotherapy prior to biopsy. The 21 patients with primary NPC
comprised 15 men and six women (age range, 20–69 years; median,
45.5 years), and the 21 patients with CNP comprised 16 men and five
women (age range, 21–60 years; median, 43.2 years).
All tissues were immediately stored at −80°C
following biopsy, prior to subsequent RNA extraction. Tumor tissues
were isolated via micro-dissection and specimens containing >70%
tumor cells were further analyzed. In total, six pairs of NPC and
CNP specimens among the 42 collected tissues were selected for
lncRNA, mRNA and circRNA expression microarray, while all 42
specimens were used for reverse transcription-quantitative
(RT-q)PCR. These specimens were used to evaluate differences in the
expression levels between NPC and CNP tissues. The present study
was approved by the Medical Ethics Committee of Zhongshan Hospital,
Xiamen University (Fujian, China) and written informed consent was
obtained from all patients prior to the study start.
RNA extraction and quality
control
Total RNA was extracted from each tissue sample
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) and purified using the RNeasy Mini kit (Qiagen
GmbH), according to the manufacturers' protocol. The quantity and
quality of the RNA were determined using a NanoDrop ND-1000
spectrophotometer (NanoDrop Technologies; Thermo Fisher Scientific,
Inc.) and the RNA integrity was assessed via electrophoresis on a
denaturing agarose gel.
RNA labeling, lncRNA and mRNA
microarray
Sample labeling and microarray hybridization were
performed using a modified version of the Agilent One-Color
Microarray-Based Gene Expression Analysis protocol (Agilent
Technologies, Inc.). The rRNA was removed from the total RNA sample
using the Ribo-Zero™ rRNA Removal kit (Epicentre; Illumina, Inc.)
and mRNA was purified using the mRNA-ONLY™ Eukaryotic mRNA
Isolation kit (Epicentre; Illumina, Inc.). Subsequently, each
sample was amplified and transcribed into fluorescent cRNA along
the entire length of the transcripts, using a random priming
method. The labeled cRNAs were purified using the RNeasy Mini kit
(Qiagen GmbH), and the concentration and specific activity of the
labeled cRNAs [pmol cyanine (Cy)3/µg cRNA] were measured using
NanoDrop ND-1000 (NanoDrop Technologies; Thermo Fisher Scientific,
Inc.). A total of 0.6 µg of each labeled cRNA was fragmented via
the addition of 5 µl 10X blocking agent and 1 µl 25X fragmentation
buffer. The mixture was heated at 60°C for 30 min, and 25 µl 2X GEx
hybridization buffer (Agilent Technologies, Inc.) was added to
dilute the labeled cRNA. A total of 40 µl hybridization solution
was dispensed into a gasket slide and placed into the Human LncRNA
Array (version 3.0; 8×60 K; Arraystar, Inc.), which contained
30,586 lncRNAs and 26,109 mRNAs. The slides were incubated for 17 h
at 65°C in an Agilent Microarray hybridization oven. The hybridized
arrays were washed with Gene Expression Wash Buffer (Agilent
Technologies, Inc.) and subsequently fixed with 3.7%
paraformaldehyde for 15 min at room temperature prior to being
scanned using the Agilent DNA Microarray Scanner System (G2505C;
Agilent Technologies, Inc.).
Agilent Feature Extraction software (version
11.0.1.1; Agilent Technologies, Inc.) was used to analyze the
acquired array images. Quantile normalization and subsequent data
processing were performed using the GeneSpring GX software package
(version 11.5.1; Agilent Technologies, Inc.). Following quantile
normalization of the raw data, lncRNAs and mRNAs that were flagged
as Present or Marginal (‘All Targets Value’), in ≥6 out of 12
samples, were selected for further analyses. Significantly DE
lncRNAs and mRNAs (fold-change ≥2.0; P≤0.05) between the two groups
were identified via volcano plot filtering. Hierarchical clustering
was performed using the Agilent GeneSpring GX software (version
11.5.1; Agilent Technologies, Inc.). Gene Ontology (GO; http://geneontology.org) and Kyoto Encyclopedia of
Genes and Genomes (KEGG; http://www.genome.jp/kegg) pathway analyses were
performed using the standard enrichment computation method for DE
mRNAs. GO analysis was performed in order to characterize genes and
gene products in terms of cellular component, molecular function
and biological process. KEGG pathway analysis was performed to
identify the signaling pathways in which DE mRNAs underwent
significant enrichment, and thus predict the underlying biological
functions of the DEGs. P<0.05 and the false discovery rate
denoted the significance of the GO term enrichment and the
biological pathways. The computational data analysis was performed
by Kangchen BioTech Co., Ltd.
RNA labeling and circRNA
microarray
Sample labeling and Arraystar Human circRNA Array
hybridization (Arraystar, Inc.) were performed according to the
manufacturer's protocol. Briefly, circRNAs were treated with RNase
R (Epicentre; Illumina, Inc.) to remove the linear RNA. Each sample
was subsequently amplified and transcribed into fluorescent cRNA
using the Arraystar Super RNA Labeling kit (Arraystar, Inc.) and a
random priming method. The labeled cRNAs were purified using the
RNeasy Mini kit (Qiagen GmbH) and the concentration and specific
activity of the labeled cRNAs (pmol Cy3/µg cRNA) was determined
using NanoDrop ND-1000 (NanoDrop Technologies; Thermo Fisher
Scientific, Inc.). A total of 1 µg of each labeled cRNA was
fragmented using 5 µl 10X blocking agent and 1 µl 25X fragmentation
buffer. The sample was heated at 60°C for 30 min, and 25 µl 2X
hybridization buffer (Agilent Technologies, Inc.) was added to
dilute the labeled cRNAs. A total of 50 µl of hybridization
solution was dispensed into a gasket slide and assembled into the
Arraystar Human CircRNA Microarray slide (Arraystar, Inc.). The
slides were incubated for 17 h at 65°C in an Agilent hybridization
oven. The hybridized arrays were washed with Gene Expression Wash
Buffer (Agilent Technologies, Inc.) and subsequently fixed in 3.7%
paraformaldehyde for 15 min at room temperature, prior to being
scanned using the Agilent Microarray Scanner System (Agilent
Technologies, Inc.).
The scanned images were imported into Agilent
Feature Extraction software (version 11.0.1.1; Agilent
Technologies, Inc.) for raw data extraction. Quantile normalization
of the raw data and subsequent data processing were performed using
the R software package (version 3.28.0; http://bioconductor.org/packages/edgeR). Low-intensity
filtering was performed and circRNAs that were flagged as Present
or Marginal (‘All Targets Value’) in ≥6 out of 12 samples were
retained for further analyses. The fold-change between the groups
for each circRNA was computed to allow for comparisons between two
groups of profile differences (such as cancer vs. inflammation).
circRNAs with a fold-change ≥1.5 and P≤0.05 were selected as
significantly DE. The analysis outputs were filtered and the DE
circRNAs were ranked according to their fold-change and P-value
using Microsoft Excel's Data/Sort & Filter functionalities
(Microsoft Corporation). The computational data analysis was
performed by Kangchen BioTech Co., Ltd.
RT-qPCR validation
Randomly selected DE lncRNAs, mRNAs and circRNAs
were evaluated using RT-qPCR. The specific primer sequences for 12
lncRNAs, eight mRNAs and four circRNAs were designed using Primer
(version 5.0; Premier Biosoft, Inc.) and are presented in Table SI. The total RNA (1.5 µg) was
reverse transcribed into cDNA using the PrimeScript™ RT Reagent kit
(Takara Bio, Inc.), according to the manufacturer's protocol. qPCR
was performed on a total reaction volume of 10 µl, comprised of 5
µl 2X Master Mix (Arraystar, Inc.), 0.5 µl each of the PCR forward
and reverse primers (10 µM), 2 µl DNA and 2 µl double-distilled
water. The following thermocycling conditions were used for
RT-qPCR: An initial denaturation step of 10 min at 95°C, followed
by 40 cycles of 95°C for 10 sec and 60°C for 1 min. All experiments
were performed in triplicate. For RT-qPCR validation analysis, all
42 samples were normalized to GAPDH. The fold-change in expression
was calculated using the 2−ΔΔCq method (15).
Identification of the nearby coding
genes of DE lncRNAs
A nearby coding gene is defined as a coding
transcript <300 kb between the DE lncRNA and the neighboring
coding mRNA. In the present study, genomic coordinate analysis of
DE lncRNAs was performed alongside computational analysis of lncRNA
and mRNA microarray data. The NPC-associated DE lncRNAs and their
neighboring coding genes were annotated, and genomic coordinates of
the lncRNAs, and the association between an lncRNA and its nearby
coding gene were also detailed. Additionally, nearby coding genes
of DE lncRNAs in NPC were obtained following lncRNA classification,
subgroup analysis and genomic coordinate determination.
Predictive analysis of potential
targets of DE circRNAs
Overlap analysis was performed via three steps.
First, the circRNA-miRNA interaction was predicted with Arraystar's
proprietary miRNA target prediction software (version 1.0) using
datasets retrieved from the TargetScan and miRanda databases, and
the DE circRNAs within all the comparisons were annotated in detail
using the circRNA-miRNA interaction information. A total of five
target miRNAs for each DE circRNA were subsequently identified
according to the number of conservative miRNA binding sites.
Subsequently, the candidate target mRNAs for the selected target
miRNAs of DE circRNAs were analyzed using Overlap software (version
1.0; Kangchen BioTech Co., Ltd.), based on three miRNA databases
(miRanda, miRDB and TargetScan). The intersection of the mRNAs
between the aforementioned candidate mRNAs, and the mRNAs in the
lncRNA and mRNA microarray data, was determined using Venny
software (version 2.1; http://bioinfogp.cnb.csic.es/tools/venny/index.html).
Following the determination of DE circRNAs and their
corresponding target genes, the circRNA-miRNA-mRNA regulatory
network was constructed and visualized using Cytoscape software
(version 3.7.1; http://www.cytoscape.org).
Statistical analysis
The differences in lncRNA, circRNA and mRNA
expression levels between NPC and CNP tissues (from the microarray
and RT-q-PCR data) were analyzed using the paired Student's t-test,
according to their fold-change. Fisher's exact test was used for GO
and pathway analyses. P<0.05 was considered to indicate a
statistically significant difference. For the microarray analysis,
the false discovery rate was calculated to correct the P-value.
Results
Profiles of DE lncRNAs and mRNAs
Among all the lncRNA and mRNA probes in the
microarray, 2.80% (856/30,586) lncRNAs (425 upregulated and 431
downregulated) and 2.94% (767/26,109) mRNAs (426 upregulated and
341 downregulated) were significantly DE between the two groups
(Tables SII and SIII). The top 20 most significantly DE
lncRNAs consisted of more upregulated lncRNAs compared with
downregulated lncRNAs (ratio, 16:4). Furthermore, uc004ebm.1
(fold-change, 38.478134) was the most significantly upregulated
lncRNA, while ENST00000572818 was the most significantly
downregulated lncRNA (fold-change, 12.04174). Of the 20 most
significantly DE mRNAs; the number of upregulated and downregulated
mRNAs were equal (ratio, 10:10). Furthermore, SPRR2E (fold-change,
63.81733) was the most significantly downregulated mRNA, while
PCDH10 (fold-change, 18.465176) was the most significantly
upregulated mRNA. Differences in mRNA expression between NPC and
CNP tissues are presented using a volcano plot, scatter plot and
clustering heat-map (Fig. 1A-C,
respectively). In addition, pathway analysis revealed that 31
significantly enriched signaling pathways (18 upregulated and 13
downregulated) corresponded to DE mRNAs in this microarray
(Tables I, II, SIV and
SV).
 | Table I.Key signaling pathways associated with
upregulated DEGs. |
Table I.
Key signaling pathways associated with
upregulated DEGs.
| Pathway ID | Definition | DEGs |
|---|
| hsa04060 | Cytokine-cytokine
receptor interaction-Homo sapiens (human) | CCL2, CCL4, CCR8,
CXCL10, CXCL2, CXCL3, CXCL6, CXCR6, EGFR, FAS, GHR, IFNG, IL12A,
IL15, IL22RA2, IL23A, LIFR, TNFRSF11B, TNFSF10, TNFSF18 |
| hsa05164 | Influenza A-Homo
sapiens (human) | CCL2, CXCL10,
EIF2AK2, FAS, IFIH1, IFNG, IL12A, MX1, OAS1, OAS2, RSAD2, STAT1,
TMPRSS13, TNFSF10 |
| hsa05162 | Measles-Homo
sapiens (human) | EIF2AK2, FAS,
IFIH1, IFNG, IL12A, MX1, OAS1, OAS2, STAT1, TNFSF10 |
| hsa00260 | Glycine, serine and
threonine metabolism-Homo sapiens (human) | CHDH, GATM, PIPOX,
PSAT1, SDS |
| hsa04630 | Jak-STAT signaling
pathway-Homo sapiens (human) | GHR, IFNG, IL12A,
IL13RA2, IL15, IL22RA2, IL23A, LIFR, SPRY2, STAT1 |
| hsa05160 | Hepatitis C-Homo
sapiens (human) | CLDN1, EGFR,
EIF2AK2, IFIT1, OAS1, OAS2, PPP2R2B, PPP2R2C, STAT1 |
| hsa05168 | Herpes simplex
infection-Homo sapiens (human) | CCL2, EIF2AK2, FAS,
IFIH1, IFIT1, IFNG, IL12A, IL15, OAS1, OAS2, STAT1 |
| hsa05142 | Chagas disease
(American trypanosomiasis)-Homo sapiens (human) | C1QB, CCL2, FAS,
IFNG, IL12A, PPP2R2B, PPP2R2C |
| hsa04940 | Type I diabetes
mellitus-Homo sapiens (human) | FAS, GAD1, IFNG,
IL12A |
| hsa05144 | Malaria-Homo
sapiens (human) | CCL2, IFNG, IL12A,
KLRK1 |
| hsa05412 | Arrhythmogenic
right ventricular cardiomyopathy (ARVC)-Homo sapiens
(human) | CACNA2D1, DSG2,
DSP, ITGAV, ITGB8 |
| hsa04062 | Chemokine signaling
pathway-Homo sapiens (human) | CCL2, CCL4, CCR8,
CXCL10, CXCL2, CXCL3, CXCL6, CXCR6, STAT1 |
| hsa04080 | Neuroactive
ligand-receptor interaction-Homo sapiens (human) | C3AR1, CHRNA7,
CHRNB4, EDN1, GABRE, GAL, GHR, GRIN2A, GZMA, LGR5, LHCGR, PPYR1,
SSTR2 |
| hsa04066 | HIF-1 signaling
pathway-Homo sapiens (human) | ANGPT2, EDN1, EGFR,
IFNG, NOX1, TF |
| hsa05143 | African
trypanosomiasis-Homo sapiens (human) | FAS, IFNG,
IL12A |
| hsa05410 | Hypertrophic
cardiomyopathy (HCM)-Homo sapiens (human) | CACNA2D1, EDN1,
ITGAV, ITGB8, TPM1 |
| hsa04512 | ECM-receptor
interaction-Homo sapiens (human) | COL4A5, ITGAV,
ITGB8, LAMA3, LAMB3 |
| hsa05132 | Salmonella
infection-Homo sapiens (human) | CCL4, CXCL2, CXCL3,
DYNC1I1, IFNG |
| Table II.Key signaling pathways associated
with downregulated DEGs. |
Table II.
Key signaling pathways associated
with downregulated DEGs.
| Pathway ID | Definition | DEGs |
|---|
| hsa04640 | Hematopoietic cell
lineage-Homo sapiens (human) | CD19, CD1C, CD22,
CD37, CR1, CR2, FCER2, MME, MS4A1 |
| hsa04080 | Neuroactive
ligand-receptor interaction-Homo sapiens (human) | ADRA2A, CNR2, CTSG,
EDN3, GALR2, GPR77, GRM5, HTR2A, LEP, MC1R, P2RX5, PRSS1, PRSS3,
S1PR4, SCT, TAC4 |
| hsa04662 | B cell receptor
signaling pathway-Homo sapiens (human) | CD19, CD22, CD72,
CD79A, CD79B, CR2, VAV3 |
| hsa05217 | Basal cell
carcinoma - Homo sapiens (human) | GLI1, PTCH1, TCF7,
WNT16, WNT9A |
| hsa05340 | Primary
immunodeficiency-Homo sapiens (human) | CD19, CD40LG,
CD79A, TNFRSF13C |
| hsa04340 | Hedgehog signaling
pathway-Homo sapiens (human) | GLI1, PTCH1, WNT16,
WNT9A |
| hsa04064 | NF-kappa B
signaling pathway-Homo sapiens (human) | CCL19, CCL21,
CD40LG, CXCL12, TNFRSF13C |
| hsa00920 | Sulfur
metabolism-Homo sapiens (human) | SULT2B1, SUOX |
| hsa04614 | Renin-angiotensin
system-Homo sapiens (human) | CTSG, MME |
| hsa04060 | Cytokine-cytokine
receptor interaction-Homo sapiens (human) | AMHR2, CCL17,
CCL19, CCL21, CD40LG, CXCL12, LEP, TNFRSF10D, TNFRSF13C |
| hsa04062 | Chemokine signaling
pathway-Homo sapiens (human) | CCL17, CCL19,
CCL21, CXCL12, GNG7, RASGRP2, VAV3 |
| hsa04672 | Intestinal immune
network for IgA production-Homo sapiens (human) | CD40LG, CXCL12,
TNFRSF13C |
| hsa05030 | Cocaine
addiction-Homo sapiens (human) | CDK5R1, DLG4,
FOSB |
Profiles of DE circRNAs
In the circRNA microarray, 0.96% of all probes (31
circRNAs, 18 upregulated and 13 downregulated) were significantly
DE between the NPC and CNP samples (Tables III and SVI). These comprised two circRNAs
(upregulated); hsa_circRNA_100160 (circRNA identifier) and
hsa_circRNA_100989, with >6-fold-change differences in
expression. The variation of circRNAs between NPC and CNP tissues
is presented in a volcano plot, scatter plot and clustering heat
map (Fig. 2A-C, respectively).
| Table III.Differentially expressed circRNAs and
their miRNA binding sites. |
Table III.
Differentially expressed circRNAs and
their miRNA binding sites.
| Differentially
expressed circRNAs | miRNA binding
sites |
|---|
|
|
|---|
| circRNA ID | Regulation | Chromosome | MRE1 | MRE2 | MRE3 | MRE4 | MRE5 |
|---|
|
hsa_circRNA_100160 | Up | chr 1 |
hsa-miR-193b-5p |
hsa-miR-518a-5p | hsa-miR-527 | hsa-miR-1264 | hsa-miR-584-3p |
|
hsa_circRNA_100181 | Up | chr1 | hsa-miR-223-5p |
hsa-miR-148a-3p |
hsa-miR-148b-3p | hsa-miR-152-3p |
hsa-miR-146b-5p |
|
hsa_circRNA_100386 | Up | chr1 |
hsa-miR-193a-5p | hsa-miR-149-3p |
hsa-miR-1301-3p | hsa-miR-139-3p | hsa-miR-494-5p |
|
hsa_circRNA_100491 | Up | chr1 | hsa-miR-223-3p | hsa-miR-26a-5p | hsa-miR-26b-5p | hsa-miR-212-5p | hsa-miR-663a |
|
hsa_circRNA_100989 | Up | chr11 | hsa-miR-30b-3p | hsa-miR-889-5p | hsa-miR-766-3p | hsa-miR-509-3p | hsa-miR-608 |
|
hsa_circRNA_101252 | Up | chr13 | hsa-miR-762 | hsa-miR-654-3p |
hsa-miR-1301-3p | hsa-miR-15a-3p | hsa-miR-145-5p |
|
hsa_circRNA_101728 | Up | chr16 |
hsa-miR-518c-5p |
hsa-miR-514a-5p | hsa-miR-762 | hsa-miR-105-5p | hsa-miR-585-5p |
|
hsa_circRNA_101965 | Up | chr17 |
hsa-miR-519d-5p | hsa-miR-489-3p |
hsa-miR-376c-5p |
hsa-miR-376b-5p | hsa-miR-298 |
|
hsa_circRNA_102539 | Up | chr19 | hsa-miR-224-3p | hsa-miR-370-3p | hsa-miR-452-5p | hsa-miR-506-3p | hsa-miR-522-3p |
|
hsa_circRNA_102682 | Up | chr2 | hsa-miR-383-3p | hsa-miR-486-3p | hsa-miR-541-3p | hsa-miR-557 | hsa-miR-18a-5p |
|
hsa_circRNA_102817 | Up | chr2 | hsa-miR-331-5p | hsa-miR-298 | hsa-miR-296-3p |
hsa-miR-30c-1-3p | hsa-miR-588 |
|
hsa_circRNA_103578 | Up | chr4 | hsa-miR-149-5p | hsa-miR-650 |
hsa-miR-548a-3p |
hsa-miR-148b-5p | hsa-miR-589-3p |
|
hsa_circRNA_103964 | Up | chr5 | hsa-miR-512-3p | hsa-miR-215-3p | hsa-miR-492 | hsa-miR-105-5p |
hsa-miR-200a-3p |
|
hsa_circRNA_103965 | Up | chr5 | hsa-miR-512-3p | hsa-miR-215-3p | hsa-miR-492 | hsa-miR-105-5p | hsa-miR-377-3p |
|
hsa_circRNA_104204 | Up | chr6 | hsa-miR-619-5p | hsa-miR-370-3p | hsa-miR-448 | hsa-miR-18b-5p | hsa-miR-18a-5p |
|
hsa_circRNA_104244 | Up | chr6 | hsa-miR-892b | hsa-miR-149-5p |
hsa-miR-130b-5p |
hsa-miR-1271-3p | hsa-miR-432-3p |
|
hsa_circRNA_104405 | Up | chr7 | hsa-miR-122-5p | hsa-miR-205-5p | hsa-miR-136-5p | hsa-miR-214-3p | hsa-miR-138-5p |
|
hsa_circRNA_104589 | Up | chr8 | hsa-miR-511-5p |
hsa-miR-548d-5p |
hsa-miR-548b-5p |
hsa-miR-548c-5p | hsa-miR-658 |
|
hsa_circRNA_000250 | Down | chr18 |
hsa-miR-181c-5p |
hsa-miR-181b-5p |
hsa-miR-181d-5p | hsa-miR-224-3p |
hsa-miR-181a-5p |
|
hsa_circRNA_000526 | Down | chr10 |
hsa-miR-92a-2-5p | hsa-miR-488-5p | hsa-miR-542-3p | hsa-miR-525-5p |
hsa-miR-193b-5p |
|
hsa_circRNA_001430 | Down | chr5 |
hsa-miR-92a-2-5p | hsa-miR-491-5p | hsa-miR-16-5p | hsa-let-7g-5p |
hsa-miR-193a-5p |
|
hsa_circRNA_100044 | Down | chr1 | hsa-miR-629-3p | hsa-miR-486-3p | hsa-miR-134-3p | hsa-miR-877-3p | hsa-miR-377-5p |
|
hsa_circRNA_100499 | Down | chr1 | hsa-miR-504-3p | hsa-miR-21-5p | hsa-miR-337-3p |
hsa-miR-642a-5p |
hsa-miR-16-2-3p |
|
hsa_circRNA_101969 | Down | chr17 | hsa-miR-18a-3p |
hsa-miR-519a-5p |
hsa-miR-519b-5p |
hsa-miR-519c-5p |
hsa-miR-518e-5p |
|
hsa_circRNA_102062 | Down | chr17 | hsa-miR-18a-5p | hsa-miR-485-5p | hsa-miR-150-3p | hsa-miR-433-5p | hsa-miR-18b-5p |
|
hsa_circRNA_102113 | Down | chr17 |
hsa-miR-106b-3p | hsa-miR-545-5p | hsa-miR-766-5p | hsa-miR-660-3p | hsa-miR-625-5p |
|
hsa_circRNA_102224 | Down | chr17 | hsa-miR-17-3p |
hsa-miR-520g-3p | hsa-miR-422a | hsa-miR-520h | hsa-miR-545-3p |
|
hsa_circRNA_102226 | Down | chr17 | hsa-miR-17-3p | hsa-miR-802 | hsa-miR-495-3p | hsa-miR-143-5p |
hsa-miR-516b-5p |
|
hsa_circRNA_102535 | Down | chr19 | hsa-miR-221-5p | hsa-miR-431-5p | hsa-miR-602 | hsa-miR-662 | hsa-miR-661 |
|
hsa_circRNA_103992 | Down | chr5 |
hsa-miR-1301-3p |
hsa-miR-130b-5p | hsa-miR-204-3p |
hsa-miR-29b-1-5p | hsa-miR-877-3p |
|
hsa_circRNA_104652 | Down | chr8 | hsa-miR-25-3p | hsa-miR-512-3p | hsa-miR-134-5p | hsa-miR-134-3p | hsa-let-7i-5p |
Validation of selected DE lncRNAs,
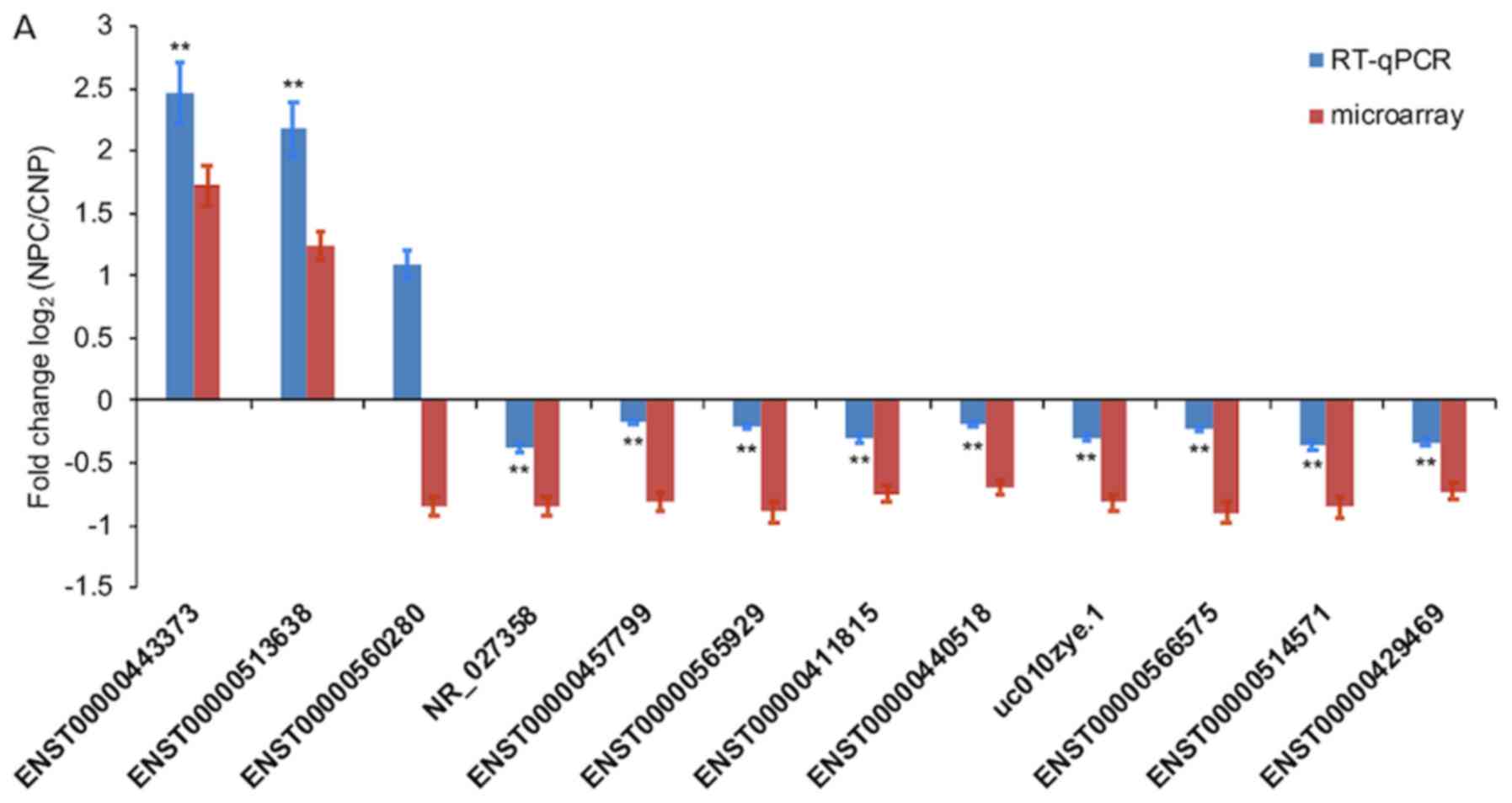
mRNAs and circRNAs using RT-qPCR
Of the 12 selected DE lncRNAs (NR_027358,
ENST00000566575, ENST00000560280, ENST00000411815, ENST00000565929,
ENST00000457799, ENST00000514571, ENST00000440518, uc010zye.1,
ENST00000443373, ENST00000513638 and ENST00000429469), only
ENST00000560280 exhibited significantly opposing microarray
expression patterns, which was compared with those resulting from
RT-q-PCR analysis. Changes in the expression levels of the other 11
lncRNAs were also verified by RT-q-PCR (Fig. 3A). Of the 8 selected DE mRNAs
(SHISA8, XAF1, RORA, MEF2C, TXLNB, MCM2, STAT1 and caspase 3), the
expression levels of all except MCM2 exhibited the same trends of
up- and downregulation as the microarray data, which indicated a
significant difference between NPC and CNP tissues, following
RT-qPCR (Fig. 3B). The changes in
expression of the four selected DE circRNAs (hsa_circRNA_104204,
hsa_circRNA_101252, hsa_circRNA_100160 and hsa_circRNA_001430) were
consistent with the results of microarray analysis (Fig. 3C).
Prediction of potential targets of DE
lncRNAs
Following bioinformatics and genomic coordinate
analyses, the NPC-associated nearby coding genes of DE lncRNAs were
listed in the relevant tables (Tables
IV, V, SII, SVII
and SVIII), including their
association and corresponding gene accession number, gene name,
protein name, gene strand, gene start location and gene end
location. In summary, 420 NPC-associated nearby coding genes,
corresponding with DE lncRNAs, were accurately defined and
comprised 196 upregulated and 224 downregulated genes.
| Table IV.Nearby coding genes for long
intergenic non-coding RNAs. |
Table IV.
Nearby coding genes for long
intergenic non-coding RNAs.
| Sequence name | Gene symbol | Genomic
location | GR | Nearby gene |
|---|
| AW833912 |
| Chr 3:
172556888-172557496 | D | NM_018098 |
| BF108976 |
| Chr 2:
192068946-192069452 | D | NM_007315 |
| BG953017R |
| Chr 4:
184736396-184736595 | U |
ENST00000296741 |
|
ENST00000400353 | AP000569.8 | Chr 21:
35303517-35343487 | U | NM_001001132 |
|
ENST00000411844 | KIAA0664L3 | Chr 16:
3171560031717339 | U |
ENST00000389202 |
|
ENST00000412797 | RP11-70P17.1 | Chr 1:
25907968-25916847 | D | NM_024037 |
|
ENST00000413991 | AC073257.2 | Chr 2:
121300484-121301902 | U | NM_005270 |
|
ENST00000420672 | AC009948.5 | Chr 2:
179278665-179295551 | U | NM_145739 |
|
ENST00000425214 | CCDC144B | Chr 17:
18494172-18507053 | U | NM_016078 |
|
ENST00000431729 | RP11-191N8.2 | Chr 1:
222001007-222014008 | D | NM_144729 |
|
ENST00000434893 | GUSBP11 | Chr 22:
23995356-24029101 | U | NM_013378 |
|
ENST00000438082 | RP11-57C13.6 | Chr 10:
89367741-89419036 | D | NM_004670 |
|
ENST00000439051 | RP11-57C13.6 | Chr 10:
89367768-89419036 | D | NM_004670 |
|
ENST00000439472 | TTTY10 | Chr Y:
22669139-22680293 | U | NM_001039567 |
|
ENST00000440357 | RP4-738P15.1 | Chr 20:
25124000-25129876 | D |
ENST00000480798 |
|
ENST00000441287 | AC011193.1 | Chr 17:
32806352-32806976 | U | NM_002982 |
|
ENST00000442583 | CCDC144B | Chr 17:
18491592-18509704 | U | NM_016078 |
|
ENST00000449023 | SRGAP3-AS4 | Chr 3:
9298442-9299191 | U | NM_014850 |
|
ENST00000457217 | RP11-222A5.1 | Chr 1:
175846478-175849604 | U | NM_003285 |
|
ENST00000483245 | RP11-202A13.1 | Chr 3:
133774099-133776492 | D | NM_001063 |
|
ENST00000486295 | EGFEM1P | Chr 3:
168538977-168547319 | D |
ENST00000264674 |
|
ENST00000514571 | CTC-454M9.1 | Chr 5:
88261691-88464485 | U | NM_001193347 |
|
ENST00000520323 | CTB-11I22.2 | Chr 5:
158654722-158672135 | U | NM_024007 |
|
ENST00000520840 | RP11-875O11.3 | Chr 8:
22928889-22932001 | U |
ENST00000312584 |
|
ENST00000536112 | RP11-81H14.2 | Chr 12:
68825634-68826434 | D | NM_000619 |
|
ENST00000537192 | RP11-1038A11.3 | Chr 12:
5399645-5487520 | D | NM_002527 |
|
ENST00000538430 | RP11-1038A11.1 | Chr 12:
5497754-5515817 | D | NM_002527 |
|
ENST00000539404 | RP11-81H14.2 | Chr 12:
68726727-68797580 | D | NM_000619 |
|
ENST00000541707 | RP11-81H14.2 | Chr 12:
68726667-68729561 | D | NM_000619 |
|
ENST00000544591 | RP11-291B21.2 | Chr 12:
10705961-10710648 | U | NM_007333 |
|
ENST00000544842 | RP11-319E16.2 | Chr 12:
5425126-5428513 | D | NM_002527 |
|
ENST00000546086 | RP11-81H14.2 | Chr 12:
68727032-68835996 | D | NM_000619 |
|
ENST00000546968 | RP11-44N21.1 | Chr 14:
105561527-105565341 | U | NM_138790 |
|
ENST00000548846 | RP3-473L9.4 | Chr 12:
111834638-111841111 | D | NM_001136538 |
|
ENST00000549710 | RP11-498M15.1 | Chr 12:
72102950-72104154 | D | NM_003667 |
|
ENST00000552154 | RP11-554D14.7 | Chr 12:
108226634-108228807 | D |
ENST00000342331 |
|
ENST00000556624 | RP11-219E7.1 | Chr 14:
21252046-21252452 | D |
ENST00000298687 |
|
ENST00000558147 | LINC00277 | Chr 15;
69373189-69383734 | U |
ENST00000310673 |
|
ENST00000558419 | CTD-2008A1.1 | Chr 15:
45118737-45119292 | D | NM_003104 |
|
ENST00000559914 | LINC00277 | Chr 15:
69365277-69367206 | U |
ENST00000310673 |
|
ENST00000561384 | CTD-2008A1.2 | Chr 15:
45119397-45176892 | U | NM_003104 |
|
ENST00000562834 | RP3-523K23.2 | Chr 6:
54807964-54809897 | U | NM_001010872 |
|
ENST00000563852 | RP11-506G7.1 | Chr 17:
41020507-41025481 | D | NM_007299 |
|
ENST00000564832 | RP11-531A24.3 | Chr 8:
73859384-73862680 | D | NM_001243237 |
|
ENST00000566575 | CTA-250D10.23 | Chr 22:
42318026-42319104 | D | NM_001207020 |
|
ENST00000568337 | RP11-160C18.2 | Chr 15:
79021382-79026298 | U | NM_000750 |
|
ENST00000569215 | RP11-609N14.1 | Chr 16:
10445296-10446609 | D | NM_001134407 |
|
ENST00000569655 | RP11-143K11.1 | Chr 17:
71171621-71172772 | U | NM_001050 |
|
ENST00000569892 | RP11-114H24.3 | Chr 15:
78246416-78255996 | U | NM_015162 |
|
ENST00000575693 | LA16c-325D7.2 | Chr 16:
2916348-2917619 | U | NM_024507 |
|
ENST00000577807 | RP11-599B13.3 | Chr 17:
7959542-7960939 | D | NM_001039131 |
| HMlincRNA791− | HMlincRNA791 | Chr 18:
52298998-52308760 | U | NM_001143829 |
| HMlincRNA963+ | HMlincRNA963 | Chr 3:
168554930-168560248 | D |
ENST00000264674 |
| NR_024475 | LOC100216001 | Chr 10:
4692376-4720262 | U | NM_001353 |
| NR_026878 | FOXD2-AS1 | Chr 1:
47897806-47900313 | D |
ENST00000337817 |
| NR_027994 | NHEG1 | Chr 6:
137303295-137314368 | U | NM_181310 |
| NR_038293 | LOC100507173 | Chr 6:
27661813-27678001 | D |
ENST00000331442 |
| NR_040109 | LOC100505495 | Chr 19:
41960073-42006554 | U | NM_006890 |
| TCONS_00001315 | XLOC_000595 | Chr 1:
227976987-227979782 | D | NM_003395 |
| TCONS_00001451 | XLOC_000781 | Chr 1:
35081179-35083207 | U | NM_005268 |
| TCONS_00005258 | XLOC_002368 | Chr 2:
160780449-160792478 | U | NM_001007267 |
| TCONS_00005268 | XLOC_002383 | Chr 2:
169197716-169198115 | U | NM_203463 |
| TCONS_00006514 | XLOC_003131 | Chr 3:
54048256-54065456 | D | NM_018397 |
| TCONS_00008529 | XLOC_004016 | Chr 4:
90459366-90472707 | U |
ENST00000420646 |
| TCONS_00009933 | XLOC_004361 | Chr 5:
42922835-42924839 | U | NM_000163 |
| TCONS_00010742 | XLOC_004475 | Chr 5:
92906525-92909378 | D | NM_005654 |
| TCONS_00011633 | XLOC_005123 | Chr 6:
1489677-1490173 | U | NM_033260 |
| TCONS_00011758 | XLOC_005220 | Chr 6:
27677988-27680876 | D |
ENST00000331442 |
| TCONS_00012442 | XLOC_005214 | Chr 6:
26674955-26677930 | U | NM_001732 |
| TCONS_00012443 | XLOC_005214 | Chr 6:
26675224-26688063 | U | NM_001732 |
| TCONS_00014617 | XLOC_006712 | Chr 8:
11500332-11506826 | U | NM_001715 |
| TCONS_00014681 | XLOC_006779 | Chr 8:
39891375-39891902 | U | NM_001464 |
| TCONS_00017282 | XLOC_008100 | Chr X:
2484083-2488088 | U | NM_001141919 |
| TCONS_00017293 | XLOC_008116 | Chr X:
13405670-13437996 | U | NM_001167890 |
| TCONS_00018417 | XLOC_008704 | Chr 10:
4790106-4806336 | U | NM_001353 |
| TCONS_00021032 | XLOC_009637 | Chr 12:
7491433-7494514 | U | NM_031491 |
| TCONS_00021064 | XLOC_009662 | Chr 12:
10725616-10727581 | U | NM_007333 |
| TCONS_00029036 | XLOC_013955 | Chr 21:
44232379-44237997 | U | NM_001001568 |
| TCONS_00029753 | XLOC_014147 | Chr 22:
18848963-18851914 | U | NM_017414 |
| TCONS_00029855 | XLOC_014297 | Chr 22:
19543858-19552723 | D | NM_001178010 |
| uc001yfd.1 | BX247990 | Chr 14:
96181819-96223116 | D | NM_001252507 |
| uc002ebp.1 | TRIM72 | Chr 16:
31237192-31237830 | D |
ENST00000389202 |
| uc002iby.2 | LOC388387 | Chr 17:
41026690-41050751 | D | NM_001158 |
| uc002nbr.3 | UCA1 | Chr 19:
15939756-15946230 | D |
ENST00000344824 |
| uc002zbk.2 | BC041455 | Chr 21:
44019513-44035168 | U | NM_001001568 |
| .uc002zob.1 | GGT3P | Chr 22:
18761201-18792992 | D | NM_017414 |
| uc003fif.1 | AK127557 | Chr 3:
172308502-172312373 | D |
ENST00000241261 |
| uc003ihb.3 | BC042378 | Chr 4:
134114523-134115760 | U |
ENST00000264360 |
| uc003qhh.4 | NHEG1 | Chr 6:
137303295-137314368 | U | NM_181310 |
| uc010jbc.2 | FLJ42709 | Chr 5:
92877577-92916738 | U | NM_005654 |
| uc010vdm.1 | RRN3P2 | Chr 16:
29086162-29107582 | U | NM_001178098 |
| Table V.Nearby coding genes for enhancer long
non-coding RNAs. |
Table V.
Nearby coding genes for enhancer long
non-coding RNAs.
| Sequence name | Gene symbol | Genomic
location | GR | Nearby gene |
|---|
|
ENST00000400353 | AP000569.8 | Chr 21:
35303517-35343487 | U | NM_001001132 |
|
ENST00000411844 | KIAA0664L3 | Chr 16:
31715600-31717339 | U |
ENST00000389202 |
|
ENST00000412797 | RP11-70P17.1 | Chr 1:
25907968-25916847 | D | NM_024037 |
|
ENST00000431729 | RP11-191N8.2 | Chr 1:
222001007-222014008 | D | NM_144729 |
|
ENST00000440357 | RP4-738P15.1 | Chr 20:
25124000-25129876 | D |
ENST00000480798 |
|
ENST00000440357 | RP4-738P15.1 | Chr 20:
25124000-25129876 | U | NM_021067 |
|
ENST00000441287 | AC011193.1 | Chr 17:
32806352-32806976 | U | NM_002982 |
| NR_024475 | LOC100216001 | Chr 10:
4692376-4720262 | U | NM_001353 |
| NR_026878 | FOXD2-AS1 | Chr 1:
47897806-47900313 | D |
ENST00000337817 |
| NR_027994 | NHEG1 | Chr 6:
137303295-137314368 | U | NM_181310 |
| uc003qhh.4 | NHEG1 | Chr 6:
137303295-137314368 | U | NM_181310 |
Prediction of potential targets within
significant pathway
In total, 31 significantly enriched signaling
pathways (18 associated with upregulated and 13 associated with
downregulated genes) were obtained via pathway analysis. Among the
31 signaling pathways corresponding to DE mRNAs, each included a
different number of DEGs associated with the pathway identifier.
The most significant enrichment pathway was upregulated and
included 20 targeted genes associated with ‘cytokine-cytokine
receptor interaction-Homo sapiens (human)’. The least
significantly enriched pathways were downregulated and only
included two target genes; the target genes, SULT2B1 and SUOX, were
involved in ‘Sulfur Metabolism-Homo sapiens (human)’, while
the CTSG and MME genes were identified within the
‘Renin-angiotensin system-Homo sapiens (human)’. In summary,
221 potential target genes are closely associated with 31 key
signaling pathways in NPC (Tables I
and II).
Prediction of potential target miRNAs
of DE circRNAs
All DE circRNAs were annotated in detail with their
respective circRNA-miRNA interaction network information (Table SVI). A total of five miRNA response
elements for each DE circRNA were predicted from the results of the
circRNA microarray (Table
III).
Prediction of potential targets of DE
circRNAs
In combination with the lncRNA, mRNA and circRNA
microarray datasets, the original results of the overlap analysis
were exported (Table SIX). Upon
induction, 23 DE circRNAs (16 upregulated and 7 downregulated) and
their associated target genes (37 miRNAs and 50 mRNAs) were
selected. A regulatory network, including 37 circRNA-miRNA
interactions and 50 miRNA-mRNA interactions, was then constructed
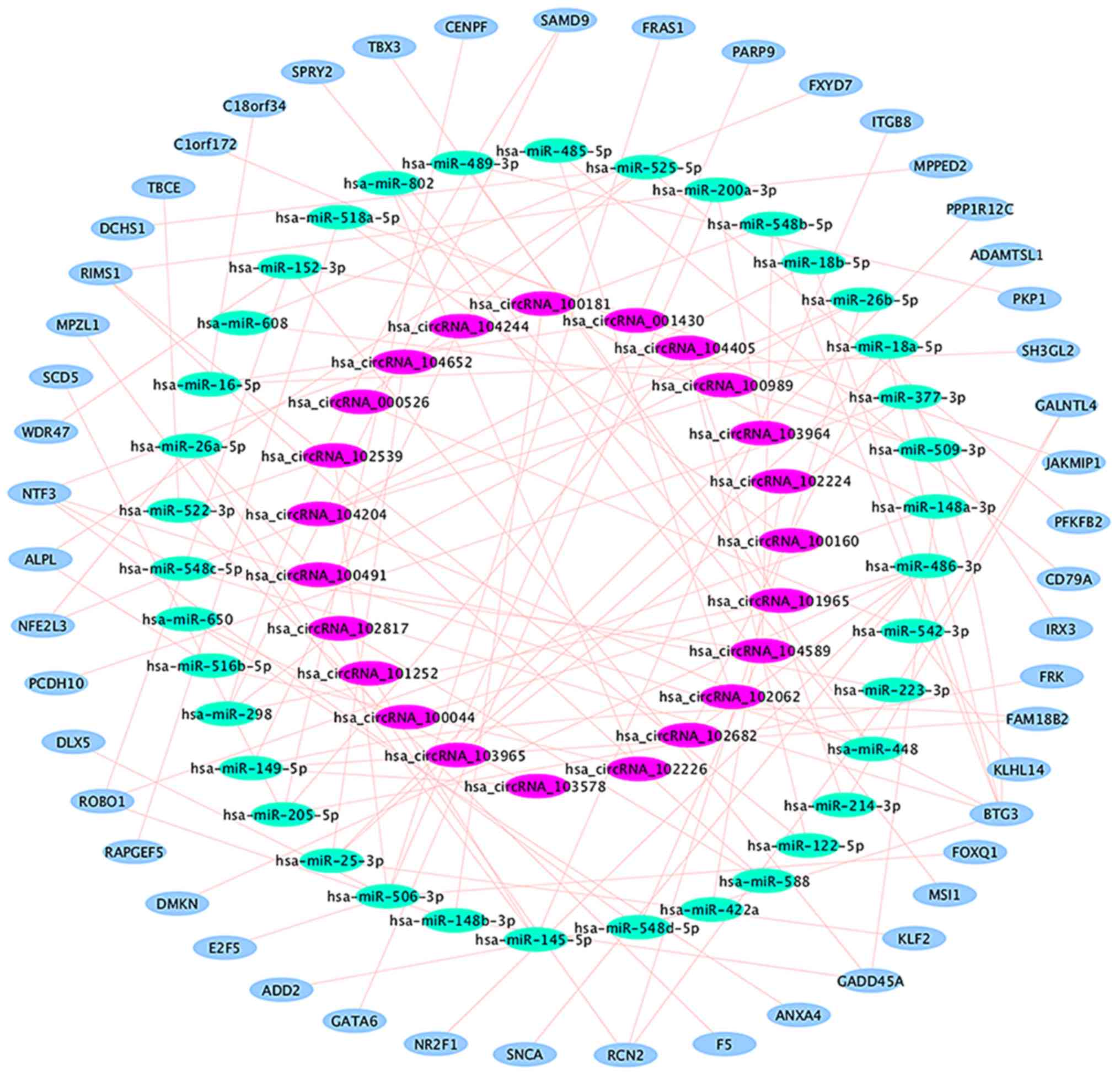
(Table VI and Fig. 4), in which 14 circRNA-miRNA-mRNA
regulatory modules were identified (Fig.
5). According to the data, there is no one-to-one
correspondence between circRNA and its target genes. For example,
hsa_circRNA_104405 is associated with 2 target miRNAs
(hsa-miR-122-5p and hsa-miR-205-5p), while hsa-miR-122 is
associated with 1 target mRNA (RIMS1) and hsa-miR-205-5p is
associated with 3 target mRNAs (CENPF, FRK and SCD5).
| Table VI.Regulatory network components of
differentially expressed circRNAs in nasopharyngeal carcinoma. |
Table VI.
Regulatory network components of
differentially expressed circRNAs in nasopharyngeal carcinoma.
| circRNA
identifier | Regulation | miRNA | mRNA |
|---|
|
hsa_circRNA_100181 | Up |
hsa-miR-148a-3p | GADD45A, ROBO1 |
|
|
|
hsa-miR-148b-3p | GADD45A, ROBO1 |
|
|
| hsa-miR-152-3p | GADD45A, ROBO1,
WDR47 |
|
hsa_circRNA_104405 | Up | hsa-miR-122-5p | RIMS1 |
|
|
| hsa-miR-205-5p | CENPF, FRK,
SCD5 |
|
|
| hsa-miR-214-3p | GALNTL4 |
|
hsa_circRNA_102682 | Up | hsa-miR-486-3p | SNCA |
|
|
| hsa-miR-18a-5p | BTG3 |
|
hsa_circRNA_102539 | Up | hsa-miR-506-3p | DLX5, E2F5, FOXQ1,
FRAS1, PARP9 |
|
|
| hsa-miR-522-3p | F5, TBCE |
|
hsa_circRNA_103578 | Up | hsa-miR-149-5p | FAM18B2 |
|
|
| hsa-miR-650 | ANXA4 |
|
hsa_circRNA_101252 | Up | hsa-miR-145-5p | ITGB8, MPZL1 |
|
hsa_circRNA_101965 | Up | hsa-miR-489-3p | PKP1 |
|
|
| hsa-miR-298 | SAMD9 |
|
hsa_circRNA_103965 | Up | hsa-miR-377-3p | IRX3, PCDH10 |
|
hsa_circRNA_104204 | Up | hsa-miR-448 | BTG3, NTF3, SPRY2,
TBX3 |
|
|
| hsa-miR-18b-5p | BTG3 |
|
|
| hsa-miR-18a-5p | BTG3 |
|
hsa_circRNA_102817 | Up | hsa-miR-298 | SAMD9 |
|
|
| hsa-miR-588 | RIMS1 |
|
hsa_circRNA_104244 | Up | hsa-miR-149-5p | FAM18B2 |
|
hsa_circRNA_103964 | Up |
hsa-miR-200a-3p | GATA6, MPPED2,
RIMS1 |
|
hsa_circRNA_100989 | Up | hsa-miR-509-3p | C1orf172 |
|
|
| hsa-miR-608 | FXYD7 |
|
hsa_circRNA_100160 | Up |
hsa-miR-518a-5p | JAKMIP1,
RAPGEF5 |
|
hsa_circRNA_100491 | Up | hsa-miR-223-3p | GALNTL4, RCN2 |
|
|
| hsa-miR-26a-5p | NFE2L3, RCN2 |
|
|
| hsa-miR-26b-5p | NFE2L3, RCN2 |
|
hsa_circRNA_104589 | Up |
hsa-miR-548d-5p | BTG3, NTF3 |
|
|
|
hsa-miR-548b-5p | BTG3, NTF3 |
|
|
|
hsa-miR-548c-5p | BTG3, NTF3 |
|
hsa_circRNA_100044 | Down | hsa-miR-486-3p | DMKN, NR2F1 |
|
hsa_circRNA_104652 | Down | hsa-miR-25-3p | ADAMTSL1, KLF2,
PPP1R12C |
|
hsa_circRNA_000526 | Down | hsa-miR-542-3p | KLHL14 |
|
|
| hsa-miR-525-5p | ALPL, DCHS1,
PFKFB2 |
|
hsa_circRNA_001430 | Down | hsa-miR-16-5p | C18orf34,
SH3GL2 |
|
hsa_circRNA_102062 | Down | hsa-miR-485-5p | CD79A |
|
hsa_circRNA_102224 | Down | hsa-miR-422a | ADD2 |
|
hsa_circRNA_102226 | Down | hsa-miR-802 | MSI1 |
|
|
|
hsa-miR-516b-5p | ALPL |
Discussion
NPC is a type of head and neck cancer with a high
incidence and poor overall survival rate, particularly in the
endemic regions of Southeast Asia (4). Although a clear understanding of its
etiology is yet to be determined, NPC is widely suspected to be the
result of both genetic susceptibility, exposure to certain
environmental factors or Epstein-Barr virus infection (1,2).
Genome-wide association and regulatory ncRNA studies may improve
understanding of the etiological and essential molecular mechanisms
underpinning NPC progression (14,16,17).
The non-coding regions of the human genome have been
closely associated with the biological processes of disease
(6). Furthermore, it has been
demonstrated that lncRNAs, miRNAs and circRNAs all regulate the
physiological and pathological processes of numerous types of
cancer, and that these regulatory ncRNAs can affect the functions
of their target mRNAs (11,17–19). It
has been reported that ncRNA molecules influence tumorigenesis and
tumor progression by forming regulatory networks with their target
genes (10), which corresponds with
the multi-gene and multi-step regulation of tumor development.
Certain studies have investigated the ncRNA
regulatory networks that influence the occurrence and development
of various types of tumor, including NPC (10,11,13,20).
Therefore, in order to delineate an NPC-specific regulatory gene
network containing ncRNAs in >10,000 human genes, the
comprehensive identification of NPC-associated DE ncRNAs and their
targets represents the initial step in establishing this network.
To the best of our knowledge, the present study is the first to
simultaneously screen and predict the possible target genes of DE
ncRNAs (lncRNAs, miRNAs and circRNAs) using three sets of
high-throughput microarray data based on transcriptome profiling of
NPC tissues. The results constitute a foundation for subsequent
comprehensive studies into the regulatory network behind the
molecular pathogenesis of NPC.
In the present study, >100 DE lncRNAs and mRNAs
were identified in NPC tissues. Subsequently, the NPC-associated
nearby coding genes that may represent targets of DE lncRNAs were
predicted via bioinformatics analysis, and their associations
between DE lncRNAs, nearby coding genes and genome coordinates were
also evaluated. Pathway analysis was conducted to determine the
biological function of the selected DE mRNAs in NPC pathogenesis,
and to predict the essential genes regulating various
NPC-associated signaling pathways. Additional target genes of DE
lncRNAs were identified from 31 significantly enriched signaling
pathways associated with NPC. The results suggest that aberrantly
expressed lncRNAs may influence NPC development and progression
through certain mechanisms, such as the interaction between a DE
lncRNA and its adjacent protein-coding gene, or via interaction
with its target gene in the corresponding signaling pathway. Thus,
lncRNA-mRNA networks may serve an important role in the
transcriptional regulation of NPC.
Pathway analysis demonstrated that 31 signaling
pathways were associated with DEGs, including 18 pathways
associated with upregulated, and 13 associated with downregulated
genes; three of these pathways (‘cytokine-cytokine receptor
interaction’, ‘chemokine signaling pathway’ and ‘neuroactive
ligand-receptor interaction’) were simultaneously associated with
upregulated and downregulated signaling pathways. Of the 74 target
genes associated with the above three pathways, 14 genes (LEP,
CCL17, CCL19, CCL21, CXCL12, CCL2, CCL4, CCR8, CXCL10, CXCL2,
CXCL3, CXCL6, CXCR6 and GHR) were separately involved in ≥2 of
these pathways. The functions of these target genes were associated
with the following biological processes: ‘Signal transduction’,
‘cell adhesion and migration’, ‘cell proliferation’, ‘inflammatory
cell infiltration’, ‘angiogenesis’ and ‘immunoregulation’.
Additionally, various pathways and target genes were associated
with the development and progression of several other human cancer
types (21–23). The present results indicate that the
three aforementioned pathways, and 14 identified genes, may
represent key regulators of NPC tumorigenesis.
Additionally, multiple DE circRNAs associated with
NPC, and their target miRNAs/mRNAs, were investigated alongside
their corresponding association and genome mapping. However, thus
far, no detailed reports are available on the association between
circRNAs and their targets in NPC. The present study identified
regulatory circRNA-miRNA-mRNA networks in NPC, which contained
different modules consisting of relevant target genes. The current
results indicate that aberrantly expressed circRNAs may influence
different pathophysiological mechanisms of NPC via interaction with
miRNAs and mRNAs, and also that the circRNA-miRNA-mRNA motifs serve
a key regulatory function in NPC. Taken together, the present data
indicate that lncRNAs do not serve an isolated role, but target the
mRNAs of various other genes, and influence other associated genes
involved in the tumorigenesis and progression of NPC, by forming
regulatory networks.
In the circRNA-miRNA-mRNA network, 50 mRNAs were
identified as the final functional genes. According to the National
Center for Biotechnology Information gene database, the functions
of these target genes were associated with the following
physiological and pathological mechanisms: ‘Environmental stress’,
‘cell motility and migration’, ‘cytoskeleton’, ‘antiproliferative
activity’, ‘regulation of voltage-gated calcium channels’, ‘cell
proliferation and apoptosis’, ‘desmosome formation’, ‘annexin’, ‘B
lymphocyte antigen receptor’ ‘bimodal regulator of epidermal growth
factor receptor and mitogen-activated protein kinase signaling’,
‘extracellular matrix protein’ and ‘chromosome segregation’.
Notably, previous studies have revealed that certain identified
target genes are associated with cancer cell proliferation and
metastasis (24,25), and thus serve important roles in NPC
development and progression (26–28).
Overall, the present study simultaneously identified
DE lncRNAs, circRNAs and mRNAs between NPC and CNP tissues via the
integrated analysis of three transcriptome profiling datasets.
Furthermore, potential target genes for these DE ncRNAs, and key
signaling pathways associated with NPC, were identified using
bioinformatics analysis. Finally, possible regulatory networks
comprised of different modules in NPC were predicted and
constructed. The present study serves to evaluate the association
between these genes and NPC at the RNA transcriptome level. It also
provides novel information to elucidate the molecular pathogenesis
of NPC from a networking perspective. In further studies, the
biological functions of these regulatory networks in NPC should be
verified.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by the Natural Science
Fund of Fujian Province, China (grant no. 2017J01374).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YFD designed the study. YFD and CSY revised the
manuscript. DNZ, CSY and QQY performed the research, collected and
analyzed the data, and wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was ethically approved by the
Medical Ethics Committee of Zhongshan Hospital, Xiamen University
(Xiamen, China), and written informed consent was obtained from all
subjects prior to the study start.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Tsang CM, Lui VWY, Bruce JP, Pugh TJ and
Lo KW: Translational genomics of nasopharyngeal cancer. Semin
Cancer Biol. Sep 12–2019.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen YP, Chan ATC, Le QT, Blanchard P, Sun
Y and Ma J: Nasopharyngeal carcinoma. Lancet. 394:64–80. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wilusz JE and Sharp PA: A circuitous route
to noncoding RNA. Science. 340:440–441. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wu J and Hann SS: Functions and roles of
long-non-coding RNAs in human nasopharyngeal carcinoma. Cell
Physiol Biochem. 45:1191–1204. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Anastasiadou E, Jacob LS and Slack FJ:
Non-coding RNA networks in cancer. Nat Rev Cancer. 18:5–18. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li LJ, Leng RX, Fan YG, Pan HF and Ye DQ:
Translation of noncoding RNAs: Focus on lncRNAs, pri-miRNAs, and
circRNAs. Exp Cell Res. 361:1–8. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bhan A, Soleimani M and Mandal SS: Long
noncoding RNA and cancer: A new paradigm. Cancer Res. 77:3965–3981.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang QQ and Deng YF: Long non-coding RNAs
as novel biomarkers and therapeutic targets in head and neck
cancers. Int J Clin Exp Pathol. 7:1286–1292. 2014.PubMed/NCBI
|
|
9
|
Anfossi S, Fu X, Nagvekar R and Calin GA:
MicroRNAs, regulatory messengers inside and outside cancer cells.
Adv Exp Med Biol. 1056:87–108. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zang J, Lu D and Xu A: The interaction of
circRNAs and RNA binding proteins: An important part of circRNA
maintenance and function. J Neurosci Res. 98:87–97. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gong Z, Yang Q, Zeng Z, Zhang W, Li X, Zu
X, Deng H, Chen P, Liao Q, Xiang B, et al: An integrative
transcriptomic analysis reveals p53 regulated miRNA, mRNA, and
lncRNA networks in nasopharyngeal carcinoma. Tumour Biol.
37:3683–3695. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma DD, Yuan LL and Lin LQ: lncRNA HOTAIR
contributes to the tumorigenesis of nasopharyngeal carcinoma via
up-regulating FASN. Eur Rev Med Pharmacol Sci. 21:5143–5152.
2017.PubMed/NCBI
|
|
13
|
Zhang ZZ, Cao HC, Huang DL, Chen XF, Wan J
and Zhang W: MicroRNA-200c plays an oncogenic role in
nasopharyngeal carcinoma by targeting PTEN. Tumour Biol.
39:10104283177036552017.PubMed/NCBI
|
|
14
|
Shuai M, Hong J, Huang D, Zhang X and Tian
Y: Upregulation of circRNA_0000285 serves as a prognostic biomarker
for nasopharyngeal carcinoma and is involved in radiosensitivity.
Oncol Lett. 16:6495–6501. 2018.PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Paul P, Deka H, Malakar AK, Halder B and
Chakraborty S: Nasopharyngeal carcinoma: Understanding its
molecular biology at a fine scale. Eur J Cancer Prev. 27:33–41.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nicolas FE: Role of ncRNAs in development,
diagnosis and treatment of human cancer. Recent Pat Anticancer Drug
Discov. 12:128–135. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kristensen LS, Hansen TB, Venø MT and
Kjems J: Circular RNAs in cancer: Opportunities and challenges in
the field. Oncogene. 37:555–565. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Du WW, Zhang C, Yang W, Yong T, Awan FM
and Yang BB: Identifying and characterizing circRNA-protein
interaction. Theranostics. 7:4183–4191. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu M, Zhu K, Qian X and Li W:
Identification of miRNA/mRNA-negative regulation pairs in
nasopharyngeal carcinoma. Med Sci Monit. 22:2215–2234. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang H, Liu J, Fu X and Yang A:
Identification of key genes and pathways in tongue squamous cell
carcinoma using bioinformatics analysis. Med Sci Monit.
23:5924–5932. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lim SY, Yuzhalin AE, Gordon-Weeks AN and
Muschel RJ: Targeting the CCL2-CCR2 signaling axis in cancer
metastasis. Oncotarget. 7:28697–28710. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fang ZQ, Zang WD, Chen R, Ye BW, Wang XW,
Yi SH, Chen W, He F and Ye G: Gene expression profile and
enrichment pathways in different stages of bladder cancer. Genet
Mol Res. 12:1479–1489. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Okai I, Wang L, Gong L, Arko-Boham B, Hao
L, Zhou X, Qi X, Hu J and Shao S: Overexpression of JAKMIP1
associates with Wnt/β-catenin pathway activation and promotes
cancer cell proliferation in vitro. Biomed Pharmacother.
67:228–234. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hu X, Zhao Y, Wei L, Zhu B, Song D, Wang
J, Yu L and Wu J: CCDC178 promotes hepatocellular carcinoma
metastasis through modulation of anoikis. Oncogene. 36:4047–4059.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Alajez NM, Lenarduzzi M, Ito E, Hui AB,
Shi W, Bruce J, Yue S, Huang SH, Xu W, Waldron J, et al: MiR-218
suppresses nasopharyngeal cancer progression through downregulation
of survivin and the SLIT2-ROBO1 pathway. Cancer Res. 71:2381–2391.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ying J, Li H, Seng TJ, Langford C,
Srivastava G, Tsao SW, Putti T, Murray P, Chan AT and Tao Q:
Functional epigenetics identifies a protocadherin PCDH10 as a
candidate tumor suppressor for nasopharyngeal, esophageal and
multiple other carcinomas with frequent methylation. Oncogene.
25:1070–1080. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cao JY, Liu L, Chen SP, Zhang X, Mi YJ,
Liu ZG, Li MZ, Zhang H, Qian CN, Shao JY, et al: Prognostic
significance and therapeutic implications of centromere protein F
expression in human nasopharyngeal carcinoma. Mol Cancer.
9:2372010. View Article : Google Scholar : PubMed/NCBI
|