Introduction
Colorectal carcinoma (CRC) is the third leading
cause of cancer-associated mortality worldwide (1,2). A total
of ~1 million patients are diagnosed with CRC and ~50% succumb to
this malignancy annually worldwide (3). The development of CRC is a complex
process involving multiple genetic and epigenetic changes. Although
some have already been identified, novel molecules that have been
implicated in CRC carcinogenesis, and may be crucial for the
diagnosis and treatment of CRC, require further investigation.
Therefore, it is imperative to elucidate the mechanisms underlying
CRC tumorigenesis and to identify new molecules involved in its
development and progression.
MicroRNAs (miRNAs) are endogenous non-coding RNAs,
which exert their effects by binding to the 3′untranslated regions
(3′UTRs) of target mRNAs (4,5). miRNAs are involved in various processes
such as gene regulation, apoptosis, hematopoietic development, cell
differentiation and tumorigenesis (4). Additionally, various types of human
cancer, including CRC, have been associated with the dysregulation
of miRNAs (6–8). Accumulating evidence indicates that
miRNAs may serve as potential oncogenes or tumor suppressor genes
in tumorigenesis (9–12).
Shen et al (13) demonstrated that miR-139 inhibits
invasion and metastasis of CRC cells by regulating the type I
insulin-like growth factor receptor. Xu et al (14) observed that miR-503-5p confers drug
resistance by targeting p53 upregulated modulator of apoptosis in
CRC. Recently, Pellatt et al (15) used microarray analysis to demonstrate
that miR-663b was significantly overexpressed in CRC tissues
compared with the normal mucosa. However, the mechanism of action
of miR-663b in CRC remains elusive.
The aim of the present study was to investigate the
expression of miR-663b in CRC cell lines compared with normal
colonic cells, determine its effects on CRC cell proliferation,
migration, invasion and apoptosis in vitro, and elucidate
the underlying mechanism of action, in order to determine whether
miR-663b serves as an oncogene in CRC and identify a potential new
target for CRC diagnosis and treatment.
Materials and methods
Tissue specimens, cell lines and
transfection
A total of 20 paired CRC and adjacent normal tissue
specimens were obtained from the Department of General Surgery,
Union Hospital, Tongji Medical College, Huazhong University of
Science and Technology. The tissue samples were frozen in liquid
nitrogen immediately following surgical removal and stored at −80°C
until used. The study protocol was approved by the Ethics Committee
of Tongji Medical College, Huazhong University of Science and
Technology (IORG no. IORG0003571) and written informed consent was
obtained from each participant. The human CRC HT-29, HCT-116, SW480
and SW620 cell lines, and the normal colonic FHC cell line, were
obtained from American Type Culture Collection. The HT-29 cell line
used in our study has been authenticated using the method of STR
profiling. All cell lines were maintained under the recommended
culture conditions and incubated in a humidified environment with
5% CO2 at 37°C. The gain-of-function study of miR-663b
was conducted with miR-663b mimics (100 nM) and corresponding
negative control (100 nM) on the SW480 cell line. The
loss-of-function assay was conducted with miR-663b inhibitor (100
nM) and corresponding negative control (100 nM) on the HCT-116 cell
line. miR-663b mimics, miR-663b inhibitors and the corresponding
negative control were purchased from Shanghai GenePharma Co., Ltd.
The sequences of the miR-663b mimic and the inhibitor were as
follows: GGUGGCCCGGCCGUGCCUGAGG and CCUCAGGCACGGCCGGGCCACC,
respectively. All assays were performed using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). Subsequent experimentations were performed 48
h-post transfection.
Oligoribonucleotides
All the oligoribonucleotides used in the present
study were purchased from Shanghai GenePharma Co., Ltd. (Table I). The small interfering (si)RNA
targeting human adenomatous polyposis coli 2 (APC2) transcript was
designated as siAPC2.
 | Table I.List of primers. |
Table I.
List of primers.
| Primer |
|
|---|
|
|
|---|
| Name | Sequence
(5′-3′)a |
|---|
| APC2 F |
AAGGTGGAGGTGGTCTTCTGG |
| APC2 R |
GGTGCCGTGGAGGATTTGC |
| U6 F |
CTCGCTTCGGCAGCACA |
| U6 R |
AACGCTTCACGAATTTGCGT |
| GAPDH F |
AAGGTGAAGGTCGGAGTCA |
| GAPDH R |
GGAAGATGGTGATGGGATT |
|
| Primers for
3′UTR cloning |
|
|
| Name | Sequence
(5′-3′)a |
|
| APC2-UTR F |
CGGAGCTCCGTGGTGGCAGCGA |
|
| TGAT |
| APC2-UTR R |
CGCGTCGACAGTGGCGGTCACT |
|
| GTCCAT |
| APC2-MUT F |
GCCTTCTCCATCCCCTGCCGTGG |
|
| GCCGGTGAG |
| APC2-MUT R |
GGCGGTGAGGTGTGGCTCACCG |
|
| GCCCACGG |
RNA extraction and reverse
transcription-quantitative (RT-q)PCR analysis
Total RNA was isolated from the cells using
TRIzol® reagent (Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. RT-qPCR was performed to
evaluate the expression of miR-663b and APC2 mRNA in SW480 and
HCT-116 cells. RNA was reverse transcribed using One Step
PrimeScript miRNA cDNA Synthesis kit (Takara Bio, Inc.). cDNA was
subsequently quantified via qPCR using SYBR Premix Ex Taq (Takara
Bio, Inc.). All PCR reactions were performed using the ABI7500
system (Applied Biosystems; Thermo Fisher Scientific, Inc.). The
thermocycling conditions were as follows: Initial denaturation at
95°C for 5 min; 40 cycles of 95°C for 15 sec; annealing/elongation
at 60°C for 30 sec and a final extension at 72°C for 30 sec. The
relative expression levels of miR-663b and APC2 mRNA were
quantified using the 2−ΔΔCq method (16) and normalized to small nuclear RNA U6
and GAPDH, respectively.
Western blot analysis
Protein was prepared from transfected SW480 and
HCT-116 cells using modified RIPA lysis buffer supplemented with
proteinase inhibitor cocktail (Sangon Biotech Co, Ltd.). Protein
concentrations were measured according to the BCA protein assay kit
(Beyotime Institute of Biotechnology). Equal protein amounts (30-50
µg) were separated in 10% SDS-PAGE gels and transferred onto
polyvinylidene fluoride membranes. The membranes were blocked with
5% non-fat milk for 1 h at room temperature, prior to incubation
overnight at 4°C with the following primary antibodies: Anti-APC2
(cat. no. 12301), anti-c-Myc (cat. no. 9402),
anti-phospho-β-catenin (Ser552; cat. no. 9566),
anti-phospho-β-catenin (Ser675; cat. no. 9567), anti-β-catenin
(cat. no. 9562), anti-cyclin D1 (cat. no. 2922), anti-axin1 (cat.
no. 2087) and anti-β-actin (cat. no. 4970) (all 1:1,000 and all
from Cell Signaling Technology, Inc.). The membranes were
subsequently washed 3 times in 10 ml TBS + 0.2% Tween-20 and
incubated with the corresponding horseradish peroxidase-conjugated
secondary antibody (1:5,000; cat. no. D110291; Sangon Biotech Co.,
Ltd.) for 1 h at room temperature. Secondary antibody binding to
the primary antibody was detected using an enhanced
chemiluminescence system (Pierce; Thermo Fisher Scientific, Inc.).
All experiments were performed in triplicate.
Cell proliferation assay
SW480 and HCT-116 cells seeded at a density of 4,000
cells/well in 96-well plates were transfected with miR-663b
mimic/miR-663b inhibitor or corresponding NC. Following incubation
of the cells for the specified time (1, 2, 3 or 4 days), a Cell
Counting Kit-8 assay was performed according to the manufacturer's
protocol (Dojindo Molecular Technologies, Inc.). The absorbance of
the solution was measured spectrophotometrically at 450 nm with MRX
II absorbance reader (Dynex Technologies). All experiments were
performed in triplicate.
Wound healing assay
The migration of SW480 or HCT-116 cells was assessed
via the wound healing assay. A total of 5×105 cells were
seeded into 6-well plates and cultured in DMEM medium (Sangon
Biotech Co, Ltd.) at 37°C in 5% CO2 until they reached
~100% confluence. Subsequently, artificial wounds were created by
scratching the cell monolayer with a sterile pipette tip. Following
wounding, cells were washed three times with PBS to remove floating
cells and debris, and subsequently incubated in serum-free medium
at 37°C in 5% CO2. Representative images with cells
migrating into the wounds were randomly captured using an inverted
light microscope (magnification, ×40). The experiments were
performed in triplicate.
Transwell invasion assay
Transwell membranes (Corning, Inc.) coated with
Matrigel (BD Biosciences, USA) were used to assay cell invasion
in vitro. A total of 1.0×105 transfected cells
suspended in serum-free medium were added to the upper chamber, and
medium supplemented with 10% FBS (Sangon Biotech Co., Ltd.) was
added to the lower chamber as a chemoattractant. After 24 h of
incubation at 37°C, the invading cells were fixed with 4%
paraformaldehyde for 30 min, followed by staining with 0.1% crystal
violet solution for 20 min, both at room temperature. Subsequently,
each well was captured using an inverted light microscope
(magnification, ×100). Data were obtained from three independent
experiments.
Apoptosis assay
An Annexin V-fluorescein isothiocyanate (FITC)
Apoptosis Detection Kit (Beyotime Institute of Biotechnology) was
used to analyze the cell apoptosis rate according to the
manufacturer's instructions. Following transfection for 72 h, the
cells were harvested and stained in binding buffer with 5 µl of
Annexin V-FITC for 10 min at room temperature. Following incubation
with 5 µl of propidium iodide for 20 min at 4°C, the cell apoptosis
rate was analyzed via flow cytometry (FACScan; BD Biosciences). BD
FACSDiva™ software (version 6.1.3) was used to analyze the flow
cytometry data. The experiments were performed in triplicate.
Dual-luciferase reporter assay
APC2 was predicted to be the target candidate gene
of miR-663b according to TargetScan databases (version 7.2)
(17–21). The 3′UTR fragment of APC2 containing
the putative wild-type (wt) sequence was amplified by PCR. The
amplified product was inserted into the pmirGLO Dual-Luciferase
miRNA Target Expression Vector (Promega Corporation). The
QuikChange Lightning Site-Directed Mutagenesis Kit (Stratagene;
Agilent Technologies, Inc.) was used to construct the miR-663b
binding site mutants according to the manufacturer's protocol.
These constructs were named pmirGLO-APC2-wt and pmirGLO-APC2-mutant
(mut), respectively. SW480 cells were plated into 24-well plates at
a density of ~2×105 cells/well, cultured until they
reached ~70% confluence and co-transfected with either miR-663b
mimic or NC and pmirGLO-APC2-wt or pmirGLO-APC2-mut using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.).
Transfected cells were collected after 48 h of incubation, and the
luciferase activity was measured by a Dual-Luciferase Reporter
Assay System (Promega Corporation) in accordance with the
manufacturer's protocol. The relative luciferase activity,
normalized using Renilla luciferase, was measured 48 h after
transfection. All experiments were performed in triplicate.
Statistical analysis
Experimental data are presented as mean ± standard
deviation. All data were analyzed using one-way ANOVA or Student's
t-test. Multiple comparisons between the groups were performed
using Tukey's post hoc test. SPSS software v.18.0 (SPSS, Inc.) was
used for all data analyses. P<0.05 was considered to indicate a
statistically significant difference.
Results
miR-663b is highly expressed in CRC
tissues and cell lines
To validate the expression of miR-663b in CRC
tissues, the expression level of miR-663b was detected in 20 paired
CRC tissue specimens and adjacent normal tissues. The results
revealed that miR-663b expression was significantly increased in
CRC tissues compared with that in adjacent normal tissues (Fig. 1A). To further investigate the
expression pattern of miR-663b in CRC cells, RT-qPCR was performed
to measure the expression of miR-663b in 4 CRC cell lines and the
normal colonic cell line FHC. It was observed that miR-663b was
markedly upregulated in all 4 CRC cell lines compared with FHC
cells (Fig. 1A). These data suggest
that the abnormal expression of miR-663b may be involved in
tumorigenesis of human CRC. As the expression level of miR-663B was
the lowest in SW480 and highest in HCT-116 among 4 CRC cell lines,
the SW480 and HCT-116 cells were selected for the subsequent
gain/loss-of-function studies and investigation of the underlying
mechanism.
miR-663b promotes CRC cell
proliferation
The overexpression miR-663b in CRC tissues and cells
suggested that miR-663b may serve as an oncogene in CRC. To
investigate the biological function of miR-663b, its effect on the
proliferation of CRC cells was examined using a CCK-8 assay.
miR-663b expression was measured using RT-qPCR to confirm the
transfection efficiency of ectopic miR-663b mimic or inhibitor
(Fig. 1B). It was demonstrated that
ectopic miR-663b expression markedly increased the proliferation of
SW480 cells (Fig. 2A), while
miR-663b knockdown decreased the proliferation of HCT-116 cells at
3 and 4 days after transfection (Fig.
2B).
miR-663b decreases apoptosis of CRC
cells
Following transfection of miR-663b mimic or
inhibitor for 72 h, CRC cell apoptosis was analyzed using flow
cytometry. The apoptotic rate was significantly decreased in the
miR-663b mimic-transfected group compared with the miR-NC group
(Fig. 2C). Additionally, the
apoptotic rate in the miR-663b inhibitor group was increased
compared with that in the corresponding NC group (Fig. 2D). These results confirmed the
anti-apoptotic effect of miR-663b on CRC cells.
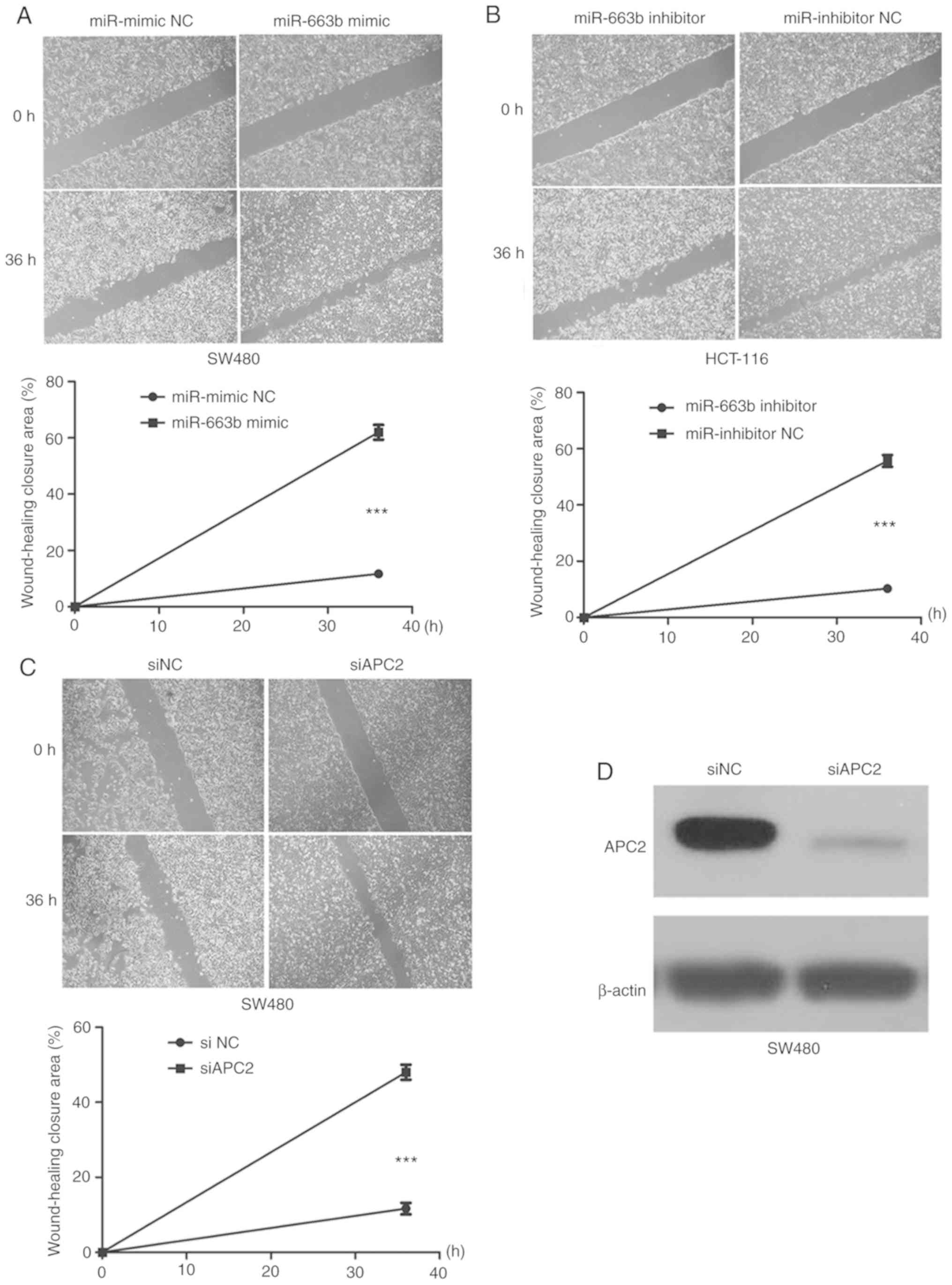
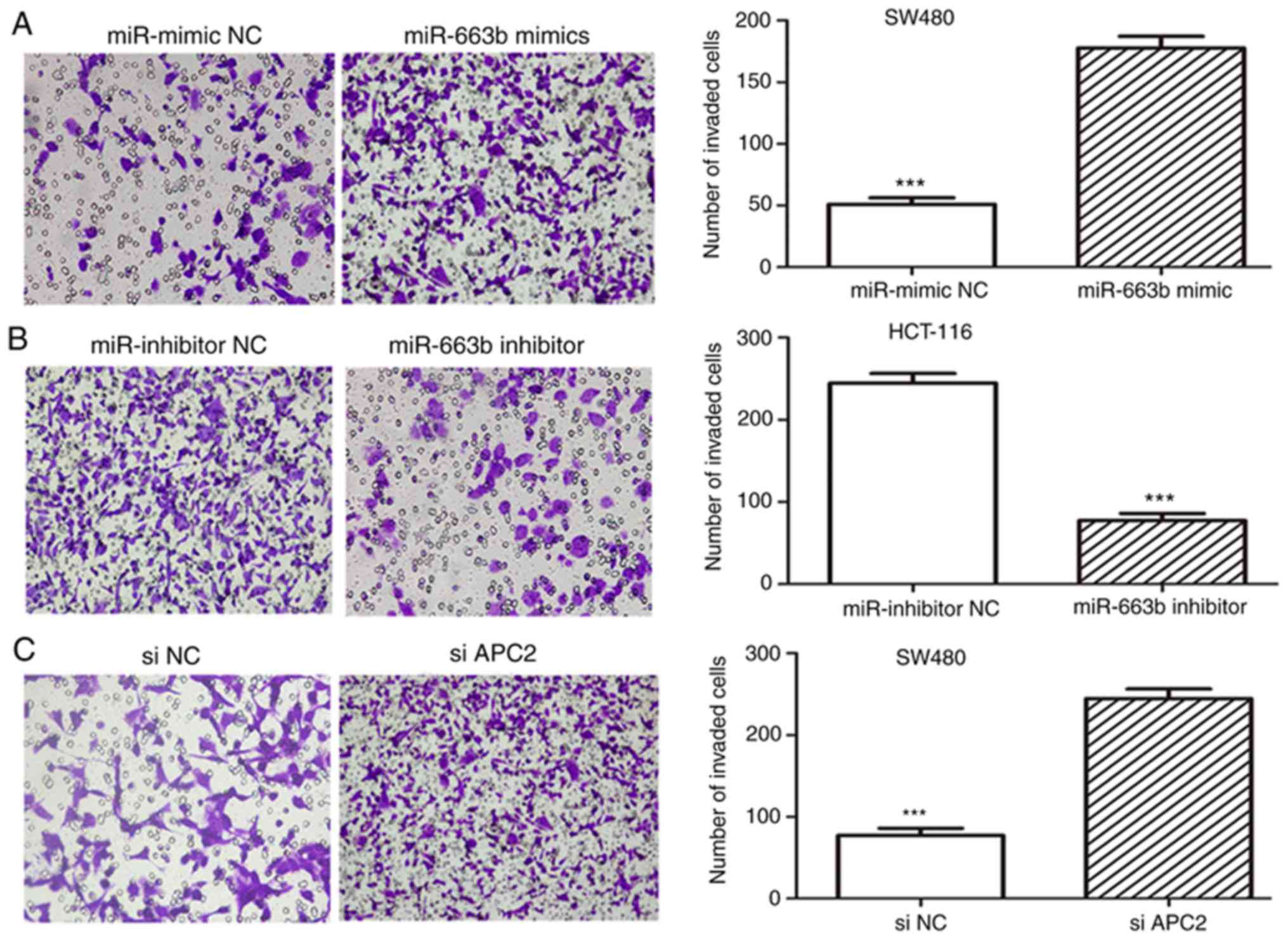
miR-663b promotes the migration and
invasion in CRC cells
To evaluate the role of miR-663b in cancer cell
migration and invasion, wound healing and Transwell assays were
performed. It was observed that the ectopic expression of miR-663b
promoted SW480 cell migration (Fig.
3A), whereas miR-663b inhibitor decreased HCT-116 cell
migration (Fig. 3B). In addition,
the invasion assay suggested that ectopic miR-663b expression
markedly promoted the invasion capacity of SW480 cells (Fig. 4A), while miR-663b knockdown inhibited
invasion of HCT-116 cells (Fig. 4B).
These results indicated that miR-663b may be involved in CRC
progression by promoting cell migration and invasion.
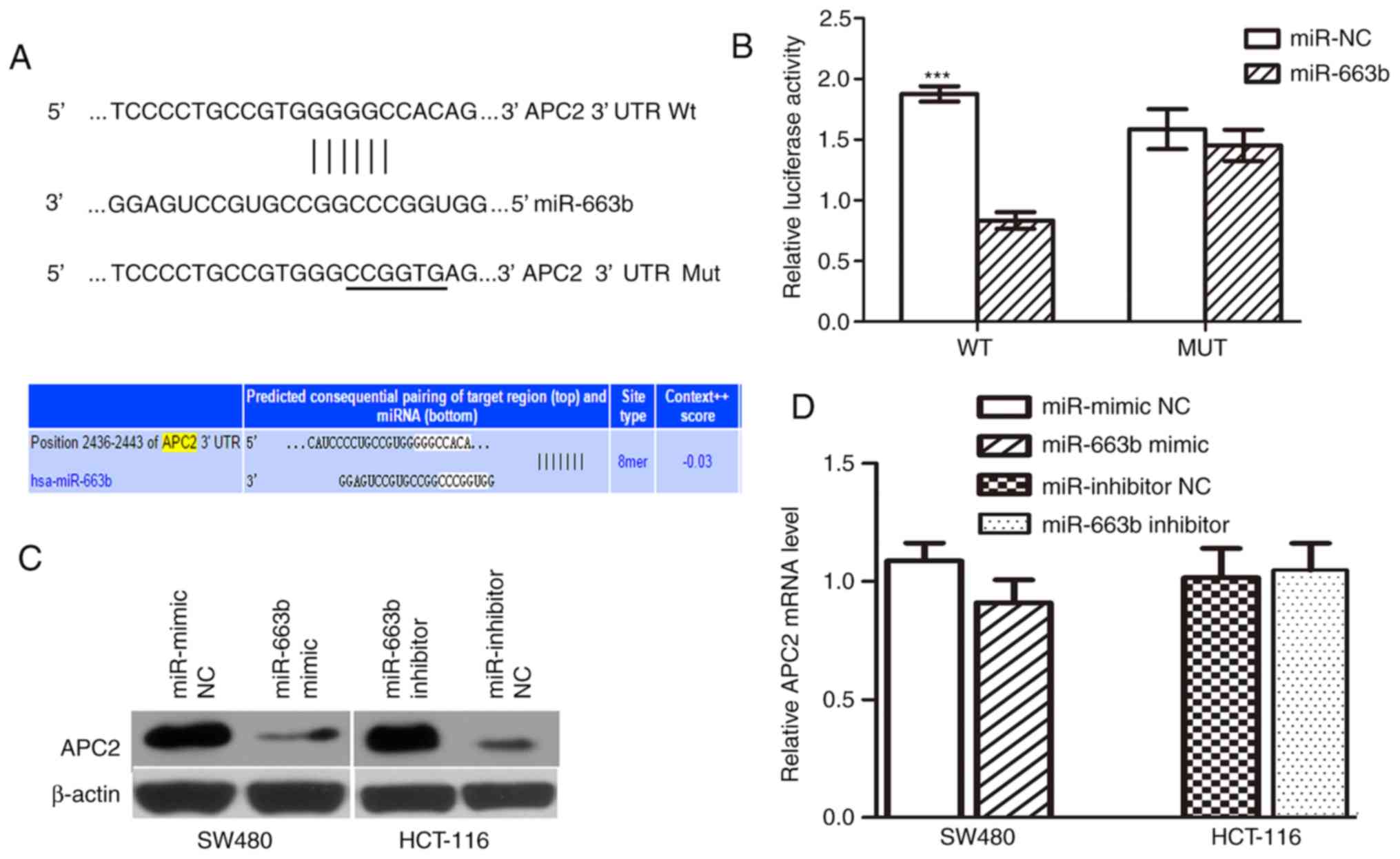
APC2 is a direct functional target of
miR-663b
To elucidate the molecular mechanism by which
miR-663b promotes cancer cell proliferation and invasion, its
target gene was further investigated. APC2, a negative
invasion-associated regulator in the Wnt/β-catenin signaling
pathway, was selected as the candidate target. A seed sequence of
miR-663b was bound to the putative 3′UTR of APC2 (Fig. 5A). First, a dual-luciferase reporter
system was employed to confirm whether APC2 is a target of
miR-663b. PmirGLO-APC2-wt or pmirGLO-APC2-mut was co-transfected
with either miR-NC or miR-663b mimic in SW480 cells. It was
observed that ectopic miR-663b significantly inhibited the firefly
luciferase activity of PmirGLO-APC2-wt but not pmirGLO-APC2-mut
(Fig. 5B), suggesting that APC2 is a
direct target of miR-663b.
In addition, to further validate that APC2 is the
target of miR-663b, western blot analysis and RT-qPCR analysis were
performed to evaluate the effect of miR-663b on endogenous APC2
expression at the protein and mRNA level. The results demonstrated
that the miR-663b inhibited the expression of the APC2 protein in
SW480 cells and that the miR-663b inhibitor upregulated the APC2
level in HCT-116 cells (Fig. 5C).
However, miR-663b overexpression did not decrease the APC2 mRNA
level (P>0.05; Fig. 5D),
indicating that miR-663b downregulates APC2 expression at the
post-transcriptional level. Taken together, these data suggest that
miR-663b may downregulate the expression of APC2 by directly
targeting its 3′UTR.
miR-663b is involved in cancer cell
invasion through activating the Wnt/β-catenin pathway
Further experiments were conducted to explore the
molecular mechanisms by which miR-663b promotes cell invasion by
regulating APC2. The effect of APC2 knockdown on invasion was
investigated. The expression of the APC2 protein was identified to
be markedly downregulated in siAPC2-transfected SW480 cells
(Fig. 3D). The data suggested that
the knockdown of APC2 promoted CRC cell migration and invasion
(Figs. 3C and 4C), eliciting the same effect as miR-663b
overexpression.
Previous evidence has demonstrated that the
Wnt/β-catenin signaling pathway is associated with tumor growth and
invasion (22). Therefore, the
effect of miR-663b on the key molecules of the Wnt/β-catenin
pathway was investigated. It was observed that ectopic miR-663b
expression promoted the expression of MYC proto-oncogene protein
(c-Myc) and cyclin D1 protein in SW480 cells (Fig. 6A). In addition, the ectopic
expression of miR-663b lowered the protein level of phosphorylated
β-catenin. However, there was little effect on the endogenous
expression of axin 1 or total β-catenin protein observed (Fig. 6A).
Collectively, these results suggest that miR-663b
may promote cell migration and invasion in CRC through activating
the Wnt/β-catenin pathway (Fig.
6B).
Discussion
Accumulating evidence indicates that the
dysregulation of miRNAs is involved in various types of diseases
(23,24). The abnormal expression of miR-663b
has been observed in a variety of malignancies: Shu et al
(25) demonstrated that the
knockdown of miR-663b inhibited cell proliferation and promoted
apoptosis in osteosarcoma by regulating tumor protein p73
expression; Du et al (26)
identified that miR-497 and miR-663b levels in the plasma may be
novel biomarkers for bladder cancer; Cai et al (27) suggested that the epigenetic
inhibition of miR-663b by HOTAIR promotes cell proliferation in
pancreatic cancer by upregulating insulin-like growth factor 2;
Wang et al (28) reported
that miR-663b promotes cell proliferation and
epithelial-to-mesenchymal transition by directly targeting SMAD7 in
nasopharyngeal carcinoma; Wang et al (29) demonstrated that pterostilbene
dose-dependently inhibited cell proliferation in human endometrial
cancer by downregulating miR-663b, and BCL2L14 was verified as a
direct target of miR-663b; Liang et al (30) revealed that miR-663b promotes
migration and invasion of nasopharyngeal carcinoma cells through
downregulating TUSC2; and, using a miRNA microarray analysis,
Pellatt et al (15) recently
demonstrated that miR-663b is upregulated in CRC tissues compared
with normal mucosa. However, the mechanism of action of miR-663b in
CRC remains elusive.
In the present study, the expression pattern of
miR-663b in CRC cell lines was first investigated, and it was
identified that miR-663b expression was upregulated in 4 CRC cell
lines compared with FHC cells. This abnormal expression suggests
that miR-663b is likely involved in certain biological properties
of CRC. The function and relative regulatory mechanisms of miR-663b
in CRC tumorigenesis were additionally investigated, and the
results demonstrated that ectopic miR-663b expression promoted cell
proliferation, migration and invasion, and decreased apoptosis
in vitro. It was demonstrated that dysregulation of APC
serves important role in breast cancer cell invasion through WNT
signaling pathway (31). The present
study revealed that the overexpression of microRNA-663b promotes
CRC cell invasion. However, the molecular mechanism in
miR-663b-regulated invasion in CRC remains unclear. Using
bioinformatics analysis, APC2 was predicted to be a functional
target of miR-663b in the present study. To elucidate the molecular
mechanisms by which miR-663b regulates cell invasion, APC2 was
selected as a target candidate for further investigation.
The APC gene is a well-known tumor suppressor that
has been investigated in association with a number of malignancies
(32–34). APC2, an APC homologue located on
chromosome 19p13.3 (35), serves a
key role in several human malignant diseases, such as
retinoblastoma, lymphocytic leukemia and ovarian cancer (36–38).
Previous studies have demonstrated that APC2 interacts with
cytoplasmic β-catenin and negatively regulates the Wnt signaling
pathway (39). Accumulating evidence
suggests that APC2 is involved in the development and progression
of several types of cancer (40).
The Wnt/β-catenin signaling pathway is an important
pathway that is associated with tumor growth and invasion (22). During Wnt signaling, β-catenin is a
key molecule that serves as a co-regulator, cooperating with
transcription factors to regulate gene expression. In the absence
of Wnt signaling, the β-catenin protein is tightly controlled by a
destruction complex that is composed of APC, axin and glycogen
synthetase kinase-3β. This complex then promotes β-catenin protein
phosphorylation, ultimately leading to degradation of β-catenin by
the proteasome system (41). The
results from the present study indicated that the overexpression of
microRNA-663b inhibited the degradation of β-catenin and activated
the expression of downstream genes of β-catenin. To better
demonstrate the conclusions from the present study, phosphorylation
level of β-catenin should be detected at more phosphorylation sites
such as S33, S37 and S45, which are involved in the degradation of
β-catenin. Studies regarding this issue are being conducted by the
present study group. The data from the present study suggested the
involvement of β-catenin phosphorylation regulation, but the
specific mechanism how microRNA-663b regulates β-catenin
phosphorylation requires further investigation. Wnt signaling,
however, inhibits the degradation of the β-catenin protein,
allowing β-catenin to accumulate in the cytoplasm and enter the
nuclei. β-catenin then binds with the T-cell
factor/lymphoid-enhancing factor family molecules to activate Wnt
responsive genes, such as c-myc and cyclin D1 (42). The present study revealed that the
expression of APC2 was downregulated by miR-663b. Therefore, the
phosphorylation of β-catenin by glycogen synthase kinase-3 β
(GSK3β) was decreased. However, unphosphorylated β-catenin protein
was probably upregulated through a separate signaling pathway,
which may explain why the total protein level of β-catenin appeared
to be unchanged between the control and miR-663b groups. The other
pathways through which β-catenin protein was regulated require
further investigation. The results from the present study revealed
that miR-663b inhibited the phosphorylation of β-catenin at
residues S552 and S675, which may suppress downstream target gene
expression. It has been suggested that the phosphorylation of
β-catenin at residues S33, S37 and S45 by GSK3β and casein kinase I
isoform α may inhibit β-catenin activity by degradation of
β-catenin (43). As the expression
of APC2 protein was downregulated by miR-663b, which inhibited
phosphorylation of β-catenin by GSK3β, the degradation of β-catenin
was decreased. Therefore, β-catenin accumulates in cytoplasm and
translocates to the nuclei, where it associates with members of the
T-cell factor/lymphoid-enhancing factor family of transcription
factor to turn on Wnt target genes such as c-myc and cyclin D1.
Consequently, c-Myc and cyclin D1 were upregulated in
miR-663b-overexpressed cells.
The present study demonstrated that APC2 is a
functional target of miR-663b, suggested that the Wnt/β-catenin
signaling pathways are involved in miR-663b-regulated cancer cell
invasion, and that downregulation of APC2 by miR-663b induction may
serve a crucial role in the development and progression of CRC. The
results of the present study also indicated that miR-663b promoted
CRC cell invasion. To further explore whether APC2 was the direct
functional target of miR-663b, invasion assays were performed
following APC2 silencing. The results revealed that miR-663b
promoted CRC cell invasion through directly targeting APC2. A
number of other genes may be involved in miR-663b-regulated cell
invasion, which requires further investigation. Furthermore, the
clinical significance of the dysregulation of miR-663b requires
further investigation.
In summary, the results of the present study
revealed that miR-663b acts as an oncogene by promoting cell
proliferation, migration and invasion by directly targeting the
tumor suppressor gene APC2. It was inferred that miR-663b may serve
a key role in the development and progression of CRC and may
represent a novel therapeutic target for CRC.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
FX and GZ conceived and designed the present study.
FX drafted the initial manuscript. FX and CY performed all the
experiments. WC participated in data analysis and interpretation.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Union Hospital, Tongji Medical College, Huazhong
University of Science and Technology.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J,
Murray T and Thun MJ: Cancer statistics, 2008. CA Cancer J Clin.
58:71–96. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Obuch JC and Ahnen DJ: Colorectal cancer:
Genetics is changing everything. Gastroenterol Clin North Am.
45:459–476. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
DeSantis CE, Lin CC, Mariotto AB, Siegel
RL, Stein KD, Kramer JL, Alteri R, Robbins AS and Jemal A: Cancer
treatment and survivorship statistics, 2014. CA Cancer J Clin.
64:252–271. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moriyama T, Ohuchida K, Mizumoto K, Yu J,
Sato N, Nabae T, Takahata S, Toma H, Nagai E and Tanaka M:
MicroRNA-21 modulates biological functions of pancreatic cancer
cells including their proliferation, invasion, and chemoresistance.
Mol Cancer Ther. 8:1067–1074. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bhaumik D, Scott GK, Schokrpur S, Patil
CK, Campisi J and Benz CC: Expression of microRNA-146 suppresses
NF-kappaB activity with reduction of metastatic potential in breast
cancer cells. Oncogene. 27:5643–5647. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hu G, Chen D, Li X, Yang K, Wang H and Wu
W: miR-133b regulates the MET proto-oncogene and inhibits the
growth of colorectal cancer cells in vitro and in vivo. Cancer Biol
Ther. 10:190–197. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Esquela-Kerscher A and Slack FJ:
Oncomirs-microRNAs with a role in cancer. Nat Rev Cancer.
6:259–269. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang Z, Zhang X, Yang Z, Du H, Wu Z, Gong
J, Yan J and Zheng Q: MiR-145 regulates PAK4 via the MAPK pathway
and exhibits an antitumor effect in human colon cells. Biochem
Biophys Res Commun. 427:444–449. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu L, Cai C, Wang X, Liu M, Li X and Tang
H: MicroRNA-142-3p, a new regulator of RAC1, suppresses the
migration and invasion of hepatocellular carcinoma cells. FEBS
Lett. 585:1322–1330. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu C, Yu J, Yu S, Lavker RM, Cai L, Liu
W, Yang K, He X and Chen S: MicroRNA-21 acts as an oncomir through
multiple targets in human hepatocellular carcinoma. J Hepatol.
53:98–107. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shen K, Liang Q, Xu K, Cui D, Jiang L, Yin
P, Lu Y, Li Q and Liu J: MiR-139 inhibits invasion and metastasis
of colorectal cancer by targeting the type I insulin-like growth
factor receptor. Biochem Pharmacol. 84:320–330. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu K, Chen G, Qiu Y, Yuan Z, Li H, Yuan X,
Sun J, Xu J, Liang X and Yin P: miR-503-5p confers drug resistance
by targeting PUMA in colorectal carcinoma. Oncotarget.
8:21719–21732. 2017.PubMed/NCBI
|
|
15
|
Pellatt DF, Stevens JR, Wolff RK, Mullany
LE, Herrick JS, Samowitz W and Slattery ML: Expression profiles of
miRNA subsets distinguish human colorectal carcinoma and normal
colonic mucosa. Clin Transl Gastroenterol. 7:e1522016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Agarwal V, Bell GW, Nam JW and Bartel DP:
Predicting effective microRNA target sites in mammalian mRNAs.
Elife. 4:2015. View Article : Google Scholar
|
|
18
|
Friedman RC, Farh KK, Burge CB and Bartel
DP: Most mammalian mRNAs are conserved targets of microRNAs. Genome
Res. 19:92–105. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Garcia DM, Baek D, Shin C, Bell GW,
Grimson A and Bartel DP: Weak seed-pairing stability and high
target-site abundance decrease the proficiency of lsy-6 and other
microRNAs. Nat Struct Mol Biol. 18:1139–1146. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Grimson A, Farh KK, Johnston WK,
Garrett-Engele P, Lim LP and Bartel DP: MicroRNA targeting
specificity in mammals: Determinants beyond seed pairing. Mol Cell.
27:91–105. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jeanes A, Gottardi CJ and Yap AS:
Cadherins and cancer: How does cadherin dysfunction promote tumor
progression? Oncogene. 27:6920–6929. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sarver AL, French AJ, Borralho PM,
Thayanithy V, Oberg AL, Silverstein KA, Morlan BW, Riska SM,
Boardman LA, Cunningham JM, et al: Human colon cancer profiles show
differential microRNA expression depending on mismatch repair
status and are characteristic of undifferentiated proliferative
states. BMC Cancer. 9:4012009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schetter AJ, Leung SY, Sohn JJ, Zanetti
KA, Bowman ED, Yanaihara N, Yuen ST, Chan TL, Kwong DL, Au GK, et
al: MicroRNA expression profiles associated with prognosis and
therapeutic outcome in colon adenocarcinoma. JAMA. 299:425–436.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shu Y, Ye W, Gu YL and Sun P: Blockade of
miR-663b inhibits cell proliferation and induces apoptosis in
osteosarcoma via regulating TP73 expression. Bratisl Lek Listy.
119:41–46. 2018.PubMed/NCBI
|
|
26
|
Du M, Shi D, Yuan L, Li P, Chu H, Qin C,
Yin C, Zhang Z and Wang M: Circulating miR-497 and miR-663b in
plasma are potential novel biomarkers for bladder cancer. Sci Rep.
5:104372015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cai H, An Y, Chen X, Sun D, Chen T, Peng
Y, Zhu F, Jiang Y and He X: Epigenetic inhibition of miR-663b by
long non-coding RNA HOTAIR promotes pancreatic cancer cell
proliferation via up-regulation of insulin-like growth factor 2.
Oncotarget. 7:86857–86870. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang M, Jia M and Yuan K: MicroRNA-663b
promotes cell proliferation and epithelial mesenchymal transition
by directly targeting SMAD7 in nasopharyngeal carcinoma. Exp Ther
Med. 16:3129–3134. 2018.PubMed/NCBI
|
|
29
|
Wang YL, Shen Y, Xu JP, Han K, Zhou Y,
Yang S, Yin JY, Min DL and Hu HY: Pterostilbene suppresses human
endometrial cancer cells in vitro by down-regulating miR-663b. Acta
Pharmacol Sin. 38:1394–1400. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liang S, Zhang N, Deng Y, Chen L, Zhang Y,
Zheng Z, Luo W, Lv Z, Li S and Xu T: miR-663b promotes tumor cell
proliferation, migration and invasion in nasopharyngeal carcinoma
through targeting TUSC2. Exp Ther Med. 14:1095–1103. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cho SG: APC downregulated 1 inhibits
breast cancer cell invasion by inhibiting the canonical WNT
signaling pathway. Oncol Lett. 14:4845–4852. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Esteller M, Sparks A, Toyota M,
Sanchez-Cespedes M, Capella G, Peinado MA, Gonzalez S, Tarafa G,
Sidransky D, Meltzer SJ, et al: Analysis of adenomatous polyposis
coli promoter hypermethylation in human cancer. Cancer Res.
60:4366–4371. 2000.PubMed/NCBI
|
|
33
|
Liu M, Cui LH, Li CC and Zhang L:
Association of APC, GSTP1 and SOCS1 promoter methylation with the
risk of hepatocellular carcinoma: A meta-analysis. Eur J Cancer
Prev. 24:470–483. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen Y, Li J, Yu X, Li S, Zhang X, Mo Z
and Hu Y: APC gene hypermethylation and prostate cancer: A
systematic review and meta-analysis. Eur J Hum Genet. 21:929–935.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kinzler KW, Nilbert MC, Su LK, Vogelstein
B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie
D, et al: Identification of FAP locus genes from chromosome 5q21.
Science. 253:661–665. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Beta M, Chitipothu S, Khetan V, Biswas J
and Krishnakumar S: Hypermethylation of adenomatosis polyposis
coli-2 and its tumor suppressor role in retinoblastoma. Curr Eye
Res. 40:719–728. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rahmatpanah FB, Carstens S, Hooshmand SI,
Welsh EC, Sjahputera O, Taylor KH, Bennett LB, Shi H, Davis JW,
Arthur GL, et al: Large-scale analysis of DNA methylation in
chronic lymphocytic leukemia. Epigenomics. 1:39–61. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ying X, Li-ya Q, Feng Z, Yin W and Ji-hong
L: MiR-939 promotes the proliferation of human ovarian cancer cells
by repressing APC2 expression. Biomed Pharmacother. 71:64–69. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nakagawa H, Murata Y, Koyama K, Fujiyama
A, Miyoshi Y, Monden M, Akiyama T and Nakamura Y: Identification of
a brain-specific APC homologue, APCL, and its interaction with
beta-catenin. Cancer Res. 58:5176–5181. 1998.PubMed/NCBI
|
|
40
|
Jarrett CR, Blancato J, Cao T, Bressette
DS, Cepeda M, Young PE, King CR and Byers SW: Human APC2
localization and allelic imbalance. Cancer Res. 61:7978–7984.
2001.PubMed/NCBI
|
|
41
|
Rubinfeld B, Souza B, Albert I, Müller O,
Chamberlain SH, Masiarz FR, Munemitsu S and Polakis P: Association
of the APC gene product with beta-catenin. Science. 262:1731–1734.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Behrens J, von Kries JP, Kühl M, Bruhn L,
Wedlich D, Grosschedl R and Birchmeier W: Functional interaction of
beta-catenin with the transcription factor LEF-1. Nature.
382:638–642. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bustos VH, Ferrarese A, Venerando A, Marin
O, Allende JE and Pinna LA: The first armadillo repeat is involved
in the recognition and regulation of beta-catenin phosphorylation
by protein kinase CK1. Proc Natl Acad Sci USA. 103:19725–19730.
2006. View Article : Google Scholar : PubMed/NCBI
|