Introduction
Renal cell carcinoma (RCC) originally develops in
the renal epithelium and accounts for >90% of kidney cancer
cases (1). In total, >30% of
patients with RCC are diagnosed with locally advanced and
metastatic disease (2). Among RCC
cases clear cell RCC (CCRCC) is a typical histologic type,
accounting for 80-90% of cases (3).
Although surgical resection can effectively resolve CCRCC, 20-40%
of patients still develop metastasis or recurrence following
surgery (4). In addition, CCRCC has
the worst prognosis among the common epithelial kidney tumors
(5). Recently, significant progress
has been made in the current understanding of CCRCC development;
for example, the von Hippel-Lindau tumor suppressor gene is
inactivated frequently by genetic alteration (6). However, the molecular mechanisms
underlying CCRCC pathogenesis remain unknown. Therefore, the aim of
the present study was to screen for effective and potential
pathogenic biomarkers of CCRCC.
Recently, microarrays and RNA-sequencing on
high-throughput platforms and public databases, such as Gene
Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA), have
emerged as novel and potential methods to examine significant
genetic alternations in carcinogenesis. These methods have been
applied to identify promising biomarkers for the diagnosis and
prognosis of several types of human cancer, including CCRCC. Song
et al (7) used a total of six
GEO datasets with gene expression profiles (GEPs) of CCRCC and
identified 129 up- and 123 downregulated genes, which were
associated with signal transduction, metabolism and immune system
pathways. Another previous study used the GSE53000 dataset to
identify 533 up- and 642 downregulated differentially expressed
genes (DEGs). A total of 10 genes were identified as hub genes of
CCRCC, namely DNA topoisomerase II α (TOP2A), matrix
metalloproteinase 9 (MMP-9), albumin, cyclin-dependent kinase 1,
MYC, vascular endothelial growth factor A (VEGFA), calcium sensing
receptor (CASR), protein tyrosine phosphatase receptor type C,
prostaglandin-endoperoxide synthase 2 and endothelial growth factor
receptor. A total of six CCRCC-related modules were identified in
co-expression networks, which were associated with metabolic
processes, immunoreaction, cell cycle regulation, ion transport and
angiogenesis (8). Furthermore, based
on weighted gene co-expression network analysis, Chen et al
(9) identified 29 hub genes that
exhibited a significant positive correlation with CCRCC progression
at four stages of CCRCC. These genes could also serve as
prognostic, recurrence or progression biomarkers of CCRCC.
However, the use of different experimental
platforms, limited sample sizes and inappropriate analysis methods
lead to outliers and inconsistent results. Therefore, unbiased and
integrated approaches are necessary. The robust rank aggregation
(RRA) method can identify genes exhibiting a better-than-expected
performance according to the null hypothesis of uncorrelated inputs
and robust regarding the result errors and noise. This method can
reduce noise compared to signal while integrating data information
of different platforms, which can make the research results more
reliable (10). The RRA method
integrates multiple gene expression datasets well, which improves
high-throughput characterization of novel tumor genes with regard
to their molecular mechanisms. This method was recently reported in
studies focusing on several types of cancer, included gastric
(11), bladder (12), non-small cell lung (13) and ovarian cancer (14). However, to the best of the authors'
knowledge, no study to date has used this integration approach to
investigate CCRCC.
In the present study, an integrated bioinformatics
analysis was conducted to identify promising biomarkers for the
diagnosis and prognosis of CCRCC. According to the GEPs of GEO
datasets, 980 markedly robust DEGs (including 429 up- and 551
downregulated) were identified using the RRA method. Functional
biological analysis suggested that these DEGs exhibited a marked
enrichment in cancer-related biological pathways and functions. A
protein-protein interaction (PPI) network was constructed using the
Search Tool for the Retrieval of Interacting Genes/Proteins
(STRING) database. The Cytoscape MCODE plug-in helped perform
module analysis of the whole network. In total, six hub genes,
C-X-C motif chemokine ligand 12 (CXCL12), bradykinin receptor B2
(BDKRB2), adenylate cyclase 7 (ADYC7) CASR, kininogen 1 (KNG1) and
lysophosphatidic acid receptor 5 (LPAR5) were identified and
verified in TCGA database. Moreover, it was identified that CASR
exhibited marked association with CCRCC prognosis, as suggested by
the survival analysis.
To the best of the authors' knowledge, the present
study constituted the first time the RRA algorithm was used to
investigate CCRC. Moreover, key genes were identified through
bioinformatics methods, including analysis associated with clinical
pathology and prognosis survival. The accuracy of the
bioinformatics results of the present study was further confirmed
by experimental verification methods. In conclusion, the present
study provided new insight and promising potential biomarkers for
CCRCC diagnosis and prognosis.
Materials and methods
CCRCC GEPs and data processing
The GEPs of CCRCC GSE16441 (15), GSE36895 (16), GSE40435 (17), GSE46699 (18), GSE66270 (19) and GSE71963 (20) were downloaded from the GEO database
(https://www.ncbi.nlm.nih.gov/geo/).
The screening criteria were as follows: i) CCRCC; ii) samples
containing tumor and paired normal tissues; iii) array-based gene
expression profiling as the study type; iv) Homo sapiens as the
organism; and v) sample size >20. Supplementary information on
the aforementioned six GEO datasets is listed in detail in Table SI. If multiple probes responded to
any given gene, the median expression level of the gene was
regarded as the final expression. Finally, quantile normalization
was performed to standardize the gene expression levels (Fig. S1).
DEG screening in GEO datasets
In order to investigate DEGs in every GEO dataset,
the limma package v.3.42.2 (21) in
R language was used to compare tumor and normal tissues with the
cut-off set at |log2FC|>1 and modified P<0.05,
where FC indicates fold change. All gene list results, which were
categorized according to log2(FC) in every dataset, were
preserved for later analysis.
RRA method
To find consistent and robust DEGs among the GEO
datasets, the RRA method was used, which can identify genes with a
better-than-expected performance on the basis of the null
hypothesis of uncorrelated inputs and is robust to result errors
and noise. RRA was carried out using the R package RobustRankAggreg
v.1.1 (10). A
|log2FC|>1 and corrected P<0.05 was considered to
indicate a statistically significant difference for robust DEGs. A
comprehensive list of up- and downregulated DEGs used for further
analysis is presented in Table
SII.
Gene ontology (GO) term and kyoto
encyclopedia of genes and genomes (KEGG) pathway enrichment
analysis
With the aim of examining the important biological
function of the identified DEGs, the Database for Annotation,
Visualization and Integrated Discovery (DAVID; version 6.8;
http://david.ncifcrf.gov/) was used to
perform enrichment analysis on GO terms (22). The GO terms included molecular
function (MF), biological process (BP) and cellular component (CC)
with a false discovery rate cut-off value <0.05. The KEGG
Orthology Based Annotation System (KOBAS) online analysis database
(version 3.0; http://kobas.cbi.pku.edu.cn/) (23) was used for KEGG pathway enrichment
analysis of DEGs, with a corrected P<0.05.
PPI network building and module
analysis
A PPI network was constructed using STRING (version
10.5; http://string-db.org/) (24). The interaction parameter was set as a
maximum confidence of 0.90. Cytoscape software (version 3.6.1;
http://www.cytoscape.org/) was used to visualize
and analyze the PPI network. The Cytoscape MCODE plug-in v.1.6
(25), which can automatically
identify the molecular complexes in a large PPI network, was used
to screen for key modules from the whole network with parameters
set to default (false degree cut-off, 2; K-Core, 2; haircut, true;
node score cut-off, 0.2; fluf, false; maximum depth from seed,
100).
Identification of hub genes
The cytoHubba v.0.1 (26) plug-in was used to select potential
hub genes from the identified DEGs. The Maximal Clique Centrality
(MCC) as well as Maximum Neighborhood Component (MNC) analysis was
used to identify the hub genes, with the shared hub genes screened
using a Venn diagram. The two aforementioned plug-ins were derived
from Cytoscape MCODE plug-in v.1.6.
TCGA-based validation and prognostic
analysis
The fragments per kilobase per million value was
used to determine up- or downregulation of GEPs of CCRCC from TCGA
database (https://cancergenome.nih.gov/) helped validate the DEG
results obtained from 72 pairs of tumor and normal tissues. The
expression heat map and violin plots were used to represent tumors
and normal samples exhibiting different expression patterns. The
Gene Expression Profiling Interactive Analysis (GEPIA) online
analysis tool (http://gepia.cancer-pku.cn/) (27) was used to analyze the prognosis and
stage of hub genes. Pearson's correlation coefficient (R) was
calculated to evaluate the association among these hub genes.
P<0.05 was considered to indicate a statistically significant
difference.
Tissue sample collection
All tissue samples were obtained from The Shengjing
Hospital of China Medical University, including 20 pairs of CCRCC
and matched normal tissue. Patients who underwent curative surgery
from July 2003 to September 2007 without receiving any
pre-operative chemotherapy, radiotherapy or targeted therapy prior
to resection, were included in the present study. Patients with
metastases were excluded. CCRCC diagnosis was confirmed by a
pathologist using histochemical examination of tumor biopsies.
Pathological grading of the tumor samples was based on the 7th TNM
staging (28). Tissue samples were
frozen in liquid nitrogen immediately after surgical resection and
stored at −80°C before further use. Informed consent forms were
signed by all patients prior to sample collection. The present
study was approved by The Research Ethics Committee of China
Medical University (approval no. 2015PS44K).
RNA isolation and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from CCRCC and normal tissue
using TRIzol® (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. Total RNA was
solubilized in RNase-free water, and the concentration and purity
of the samples were determined using a NanoDrop™ 2000
spectrophotometer (Thermo Fisher Scientific, Inc.). RT was
performed using a reverse transcription cDNA kit (Thermo First
Strand cDNA Synthesis kit (Thermo Fisher Scientific, Inc.) with the
following conditions: 42°C for 60 min, 70°C for 5 min, then 4°C
preservation. SYBR® Green Fast qPCR Mix (High ROX)
(Servicebio) was used for RT-qPCR with the StepOne Plus PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.). Thermocycling
conditions were as follows: Pre-denaturation at 95°C for 10 min,
followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. GAPDH
expression was used for normalization. The quantification was
performed using the 2−ΔΔCq method (29). The primer sequences were as follows:
KNG1-primer-forward (F), 5′-TGTGGATGCTGCTCTGAAGAAATA-3′;
KNG1-primer-reverse (R), 5′-GTCAGAGCCAACCGTCTTAGTG-3′;
CASR-primer-F, 5′-CTCTACGATTGCTGTGGTGGGA-3′; CASR-primer-R,
5′-CTGCTGGAGGAGGCATAACTGA-3′; GAPDH-primer-F,
5′-GGAAGCTTGTCATCAATGGAAATC-3′; and GAPDH-primer-R,
5′-TGATGACCCTTTTGGCTCCC-3′. All PCRs were performed in
triplicate.
Statistical analysis
Statistical analysis was carried out using R
software (version 3.6.1 http://www.r-project.org/). A paired t-test was
employed to compare gene expression patterns in CCRCC and normal
tissues from TCGA database, and for RT-qPCR experimental
validation. The log-rank test was applied to compare the outcomes
of Kaplan-Meier analysis between high and low-risk patients.
Wilcoxon test was used to compare two independent non-parametric
samples, while the Kruskal-Wallis test was used for multiple
independent non-parametric samples. The Pearson correlation
analysis was used to study the expression correlation between two
genes. A two-tailed P<0.05 was considered to indicate a
statistically significant difference.
Results
DEG identification using CCRCC GEO
datasets
A multistep analysis was conducted based on selected
GEO datasets. Significant DEGs and their biological characteristics
were identified using integrated bioinformatics analysis (Fig. 1). Since paired tumor and normal
tissues from the same patient should ideally be compared to find
genetic alterations (30), only GEO
datasets with paired tumor and normal controls were selected. In
brief, there were a total of six GEO datasets with GEPs of CCRCC,
including 235 paired tumor and normal tissues.
Following processing and normalization of gene
expression data (Fig. S1), DEGs
were screened in each GEO dataset with a cut-off value of a
corrected P<0.05 and |log2FC|>1. Overall, the
GSE16441 dataset included 3,138 DEGs (1,484 up- and 1,654
downregulated), the GSE36895 dataset 1,609 (724 up- and 885
downregulated), the GSE40435 1,108 (498 up- and 610 downregulated),
the GSE46699 1,056 (516 up- and 540 downregulated), the GSE66270
4,474 (2,334 up- and 2,140 downregulated) and the GSE71963 1,844
(799 up- and 1,045 downregulated). Volcano plots of DEGs in each
dataset are presented in Fig.
2A.
DEG selection using the RRA
method
After screening DEGs in each dataset, all 6 gene
lists were sorted according to log2FC. The RRA method
was then used to compare the ranked gene lists. In brief, this
method was based on the hypothesis of randomly arranged genes in
each dataset. The higher the rank of a gene in all DEGs results,
the smaller the P-value calculated by the RRA method, and the
larger the possibility of robust DEGs. Based on this method, a
total of 980 robust DEGs were obtained (429 up- and 551
downregulated; Table SII). A gene
expression heatmap of the top 15 up- and downregulated DEGs is
presented in Fig. 2B.
Functional enrichment analysis of
DEGs
Next, the present study aimed to examine the
biological functions associated with the aforementioned DEGs. DAVID
(Table SIII) was used to conduct GO
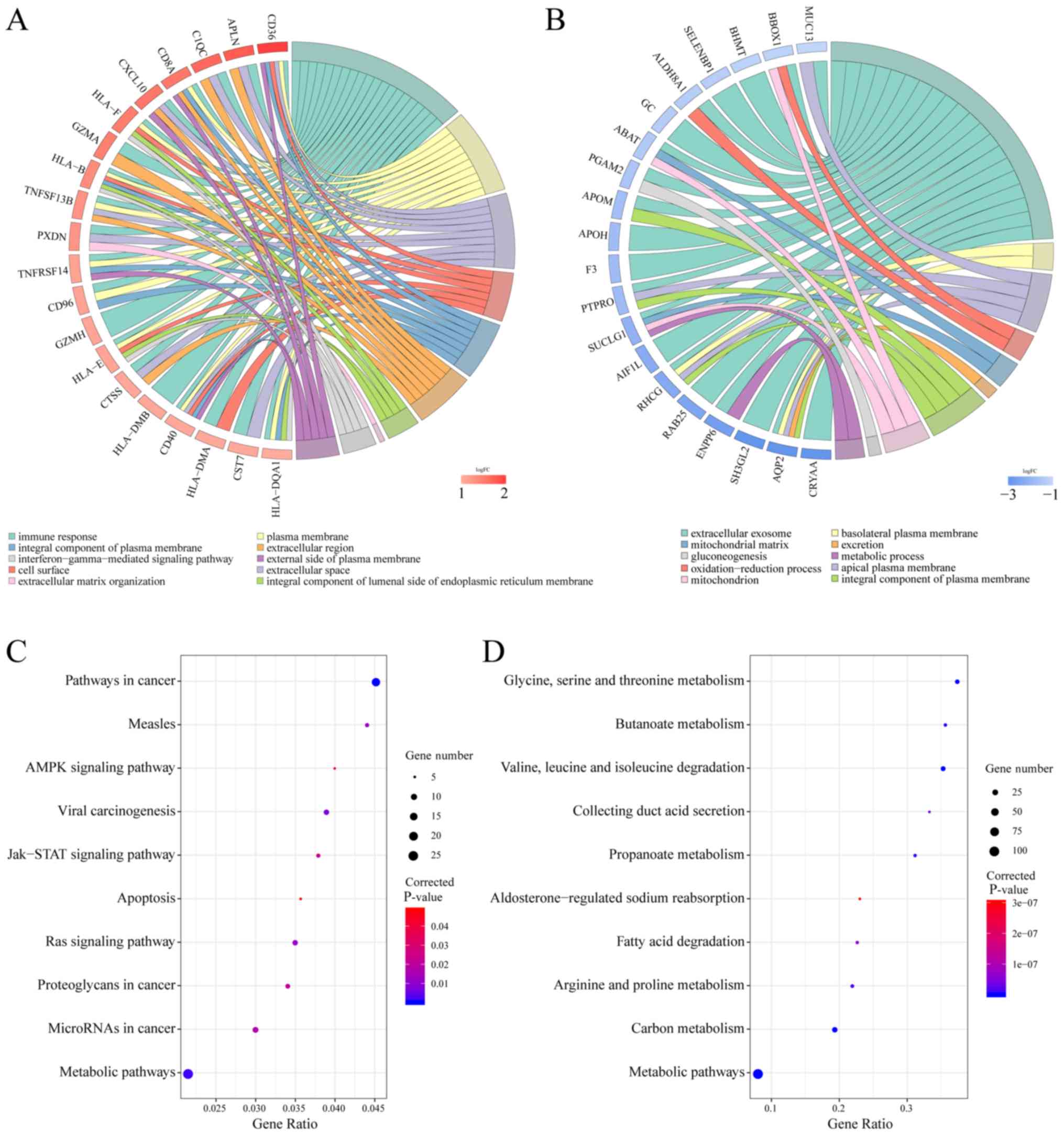
term enrichment analysis of integrated DEGs. The GO terms included
BP, MF and CC. With regard to MF, upregulated DEGs were enriched in
‘peptide antigen binding’, ‘MHC class II receptor activity’ and
‘receptor binding’ (Table SIII).
These genes were also clearly enriched in ‘immune response’,
‘interferon-gamma-mediated signaling pathway’ and BP ‘inflammatory
response’ (Table SIII). With regard
to CC, upregulated genes were enriched mainly in ‘plasma membrane’,
‘extracellular space’ and ‘cell surface’ (Fig. 3A). In addition, downregulated DEGs
were enriched in ‘oxidoreductase activity’, ‘anion:anion antiporter
activity’ and ‘pyridoxal phosphate binding’ in the MF group
(Table SIII). With regard to BP,
these genes were enriched in ‘oxidation-reduction process’,
‘gluconeogenesis’ and ‘metabolic process’. With regard to CC, these
genes were mainly enriched in ‘extracellular exosome’ and
‘basolateral plasma membrane’ (Fig.
3B).
Using KEGG pathway enrichment analysis based on
KOBAS, upregulated genes exhibited an enrichment in ‘apoptosis)’,
‘AMPK signalling pathway’, as well as ‘Jak-STAT signalling pathway’
(Fig. 3C). In addition, these genes
were closely associated with various signaling pathways, such as
the HIF-1, PI3K-Akt, Rap1, NF-κB and p53 (Table SIII). The downregulated DEGs were
mainly associated with metabolism, such as ‘carbon metabolism’,
‘metabolic pathways’, ‘valine, leucine, isoleucine degradation’, as
well as ‘glycine, serine and threonine metabolism’ (Fig. 3D). According to these results, these
DEGs exhibited a marked enrichment in cancer-related biological
function.
PPI network construction and module
analysis
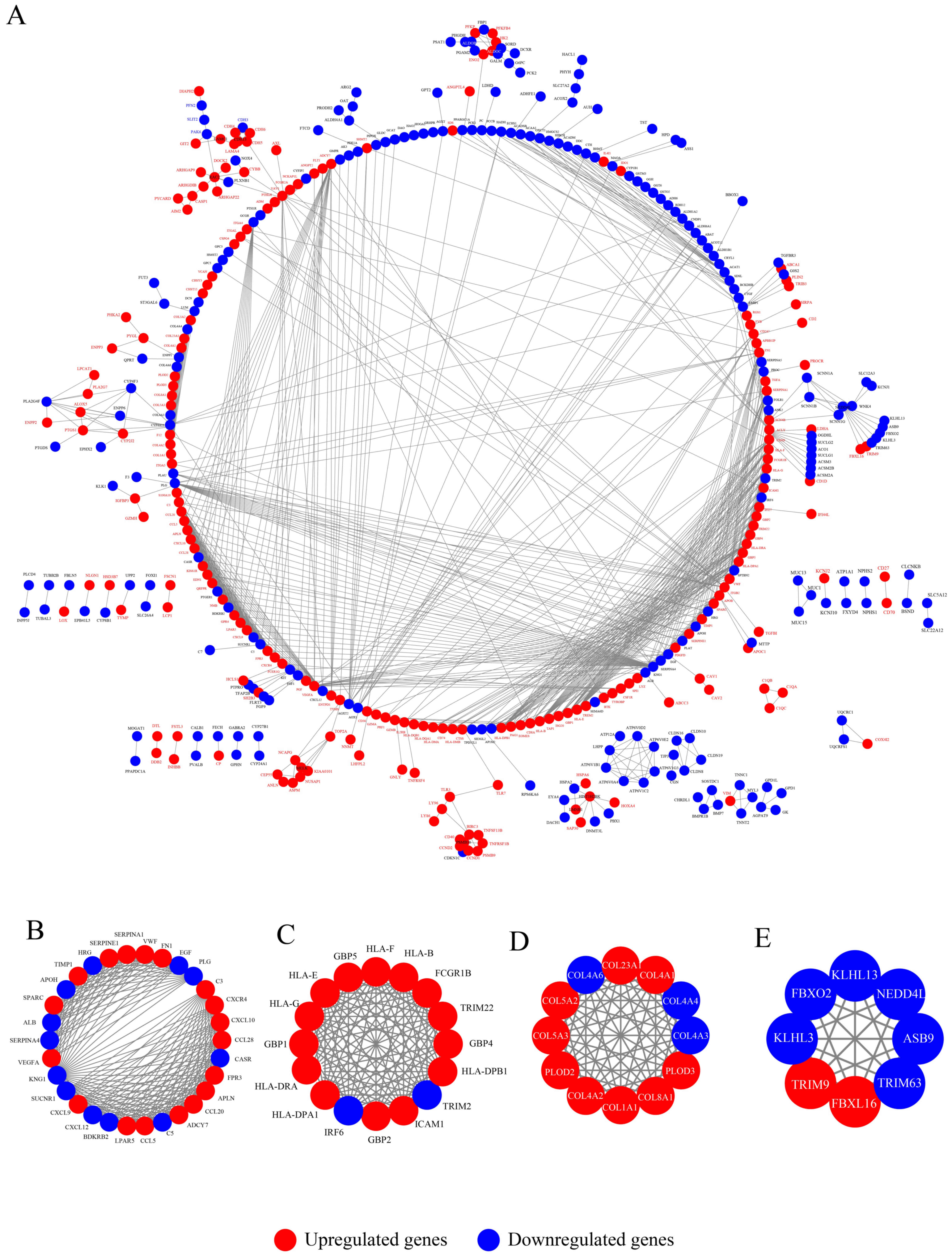
The STRING database was used to construct a PPI
network with a high confidence score ≥0.90, which included 1,154
edges and 429 nodes (205 up- and 224 downregulated genes; Table SIV), as presented in (Fig. 4A; Table
SV).
MCODE was used to identify four significant modules
from the whole PPI network (Fig.
4B-E), with an enrichment of DEGs in module 1 in ‘complement
and coagulation cascades’, ‘chemokine signaling pathway’ and
‘cytokine-cytokine receptor interaction’. An apparent enrichment of
genes in module 2 was observed in ‘cell adhesion molecules (CAMs)’,
‘antigen processing and presentation’ and ‘phagosome’. An apparent
enrichment of genes in module 3 was observed in ‘protein digestion
and absorption’, ‘ECM-receptor interaction’ and ‘focal adhesion’.
Finally, an apparent enrichment of genes was observed in module 4
in ‘ubiquitin mediated proteolysis’, ‘aldosterone-regulated sodium
reabsorption’ and ‘protein processing in endoplasmic reticulum’
(Table SVI).
Hub gene selection from the entire PPI
network
Using the cytoHubba plug-in of Cytoscape software,
hub genes were identified according to the MCC and MNC algorithms.
A total of six mutual hub genes were found using the two methods,
as demonstrated by a Venn diagram: CXCL12, BDKRB2, ADCY7, CASR,
KNG1 and LPAR5 (Fig. 5A; Table I). Therefore, these significant hub
genes could be promising and potential biomarkers, as well as
diagnostic and prognostic targets for CCRCC.
 | Figure 5.TCGA database validation and
prognostic significance of hub genes. (A) Venn diagram of shared
hub genes, as determined using the MCC and MNC methods. (B) Gene
expression heat map of six hub genes in TCGA database. (C) Violin
plot of six hub genes in TCGA database. (D) Kaplan-Meier survival
analysis according to CASR gene expression. (E) Gene expression of
BDKRB2, CASR and KNG1 at different clear cell renal cell carcinoma
stages. TCGA, The Cancer Genome Atlas; ADCY7, adenylate cyclase 7;
LPAR5, lysophosphatidic acid receptor 5; CXCL12, C-X-C motif
chemokine ligand 12; BDKRB2, bradykinin receptor B2; CASR,
calcium-sensing receptor; KNG1, kininogen 1; HR, hazard ratio; MCC,
Maximal Clique Centrality; MNC, Maximum Neighborhood Component; FC,
fold-change; TPM, transcript per million. |
 | Table I.Hub genes identified from the Maximal
Clique Centrality and Maximum Neighborhood Component methods using
Venn diagram. |
Table I.
Hub genes identified from the Maximal
Clique Centrality and Maximum Neighborhood Component methods using
Venn diagram.
| Gene | Entrez | Full gene name |
Log2FC | Expression |
|---|
| ADCY7 | 113 | Adenylate cyclase
7 | 1.030452496 | Upregulated |
| LPAR5 | 57121 | Lysophosphatidic
acid receptor 5 | 1.194101348 | Upregulated |
| CXCL12 | 6387 | C-X-C motif
chemokine ligand 12 | −1.430728025 | Downregulated |
| BDKRB2 | 624 | Bradykinin receptor
B2 | −1.115489479 | Downregulated |
| CASR | 846 | Calcium-sensing
receptor | −1.704317596 | Downregulated |
| KNG1 | 3827 | Kininogen 1 | −5.140318003 | Downregulated |
TCGA database validation and
prognostic significance of hub genes
TCGA database was used to validate integrated DEG
results based on the RNA sequencing data. First, the GEPs of the
aforementioned six hub genes helped obtain 72 pairs of CCRCC and
normal samples from TCGA database. These six hub genes were also
found to be significantly differentially expressed in the TCGA
database (P<0.001), which was consistent with the GEO dataset
results (Fig. 5B and C).
The GEPIA online analysis tool was used to examine
the prognosis of the aforementioned hub genes using the GEPs from
TCGA database. Only CASR exhibited a marked association with the
overall survival (OS) of patients with CCRCC (log-rank P=0.027;
Fig. 5D). Furthermore, BDKRB2
(P=0.0125), CASR (P=0.00433) and KNG1 (P=0.0305) were
differentially expressed at different stages of CCRCC (Fig. 5E).
To examine the association among these six hub
genes, Pearson's correlation coefficient was used (Fig. S2). KNG1 positively correlated with
CASR (R=0.92) and CXCL12 (R=0.51). However, KNG1 was negatively
correlated with ADCY7 (R=−0.53) and LPAR5 (R=−0.55).
Moreover, in order to find clinical use of the hub
genes, correlation analysis based on TCGA data and clinical
information was performed (Fig.
S3). Gene expression of LPAR5 (P=0.019), BDKRB2 (P<0.001)
and CASR (P=0.016) was associated with sex. In addition, BDKRB2
(P=0.016) and CASR (P=0.002) were associated with T stage. LPAR5
was associated with N stage (P=0.004) and M stage (P=0.009). ADCY7
(P=0.010) and CASR (P=0.006) were closely associated with M stage.
Finally, LPAR5 (P=0.039) and CASR (P=0.009) were associated with
histological grade.
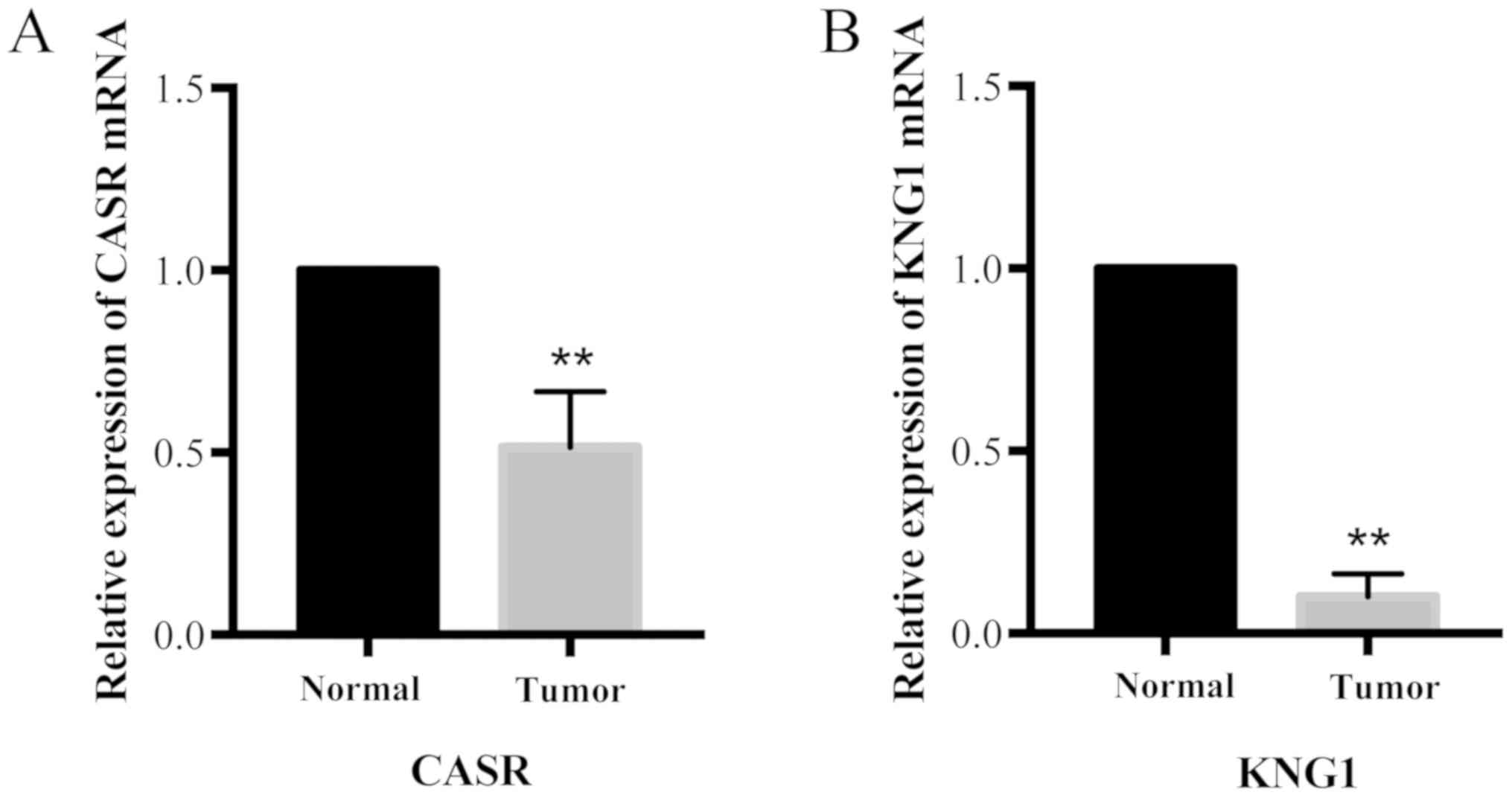
Experimental validation of hub genes
by RT-qPCR
In order to validate the results of identified hub
genes, experimental validation was performed using RT-qPCR in 20
pairs of CCRCC and matched normal tissues (n=40). The clinical
information of patients with CCRCC in the validation experiment is
presented in Table II. In the
validation experiments, the two hub genes with the most significant
changes, CASR and KNG1, were selected. The expressions of both
genes were downregulated in tumor tissues, compared with matched
normal tissues (P<0.01; Fig. 6),
suggesting that the results obtained using bioinformatics analysis
were consistent with experimental validation.
| Table II.Clinical information of patients with
clear cell renal cell carcinoma in the validation experiment. |
Table II.
Clinical information of patients with
clear cell renal cell carcinoma in the validation experiment.
| Clinicopathological
parameter | Alive, n=16, n
(%) | Dead, n=4, n
(%) | Total, n=20, n
(%) |
|---|
| Sex |
|
|
|
|
Female | 10 (62.5) | 0 (0) | 10 (50) |
|
Male | 6 (37.5) | 4 (100) | 10 (50) |
| Age, mean (SD) | 56.8 (9.6) | 58.2 (17.8) | 57 (11.1) |
| Age, median
(range) | 56.5 (44–76) | 56 (42–79) | 56.5 (42–79) |
| Stagea |
|
|
|
| Stage
I | 9 (56.2) | 3 (75) | 12 (60) |
| Stage
II | 3 (18.8) | 0 (0) | 3 (15) |
| Stage
III | 3 (18.8) | 1 (25) | 4 (20) |
| Stage
IV | 1 (6.2) | 0 (0) | 1 (5) |
Discussion
In the present study, a comprehensive bioinformatics
analysis was conducted for the identification of robust DEGs that
could become potential biomarkers of CCRCC. According to GEPs of
GEO datasets, the RRA method was used to identify 980 markedly
robust DEGs (429 upregulated genes and 551 downregulated).
Functional biological analysis showed that these DEGs were clearly
enriched in cancer-related biological functions and pathways. The
STRING database was used for the construction of the PPI network.
The Cytoscape MCODE plug-in was used to analyze modules from the
entire network. In total, six hub genes were identified, including
CXCL12, BDKRB2, ADCY7, CASR, KNG1 and LPAR5, which were verified in
the TCGA database. Moreover, survival analysis suggested that CASR
could affect the prognosis of patients with CCRCC.
Based on microarrays, RNA-sequencing technology and
public databases, such as GEO, several gene expression studies on
human cancer have been conducted in the last decade, including
CCRCC. Chen et al (9) used
two GEO microarray datasets of 101 tumor and 95 normal kidney
samples to identify 2,425 DEGs (1,259 up- and 1,166 downregulated).
In addition, the sva R package (31) was used to eliminate batch effects
between datasets GSE36895 and GSE53757. The weighted gene
co-expression network and module preservation analysis were carried
out to identify 11 co-expressed gene modules. According to the
functional enrichment analysis, the patho-module BPs were involved
in biological pathways and processes associated with cell cycle and
division. A total of 29 hub genes were finally identified, which
exhibited a stronger association with CCRCC progression (9). In another previous CCRCC study, Song
et al (7) used four GEO
datasets containing 244 matched primary tumor and adjacent normal
tissues. DEGs were identified in each dataset, and 424 aberrantly
expressed mRNAs that were shared among these datasets were
identified. Following integration with TCGA data, 252 shared genes
with aberrant expression in both the GEO and TCGA datasets were
identified. Similarly, 330 up- and 545 downregulated DEGs were
found to overlap in 3 GEPs from GEO CCRCC datasets (32). A total of 8 DEGs were identified as
biomarkers, including VEGFA, peroxisome proliferator activated
receptor α, cyclin D1, fms related receptor tyrosine kinase 1,
CXCL12, fibronectin 1, decorin and erb-b2 receptor tyrosine kinase
4.
However, due to the heterogeneity of individuals,
paired tumor and normal tissues from the same patient should
ideally be compared to find genetic alterations (30). Thus, only GEO datasets with paired
tumor and normal tissues were selected in the present study. In
addition, the methods used in the aforementioned studies were not
suitable for various datasets, different platforms or limited
sample sizes. By contrast, the RRA method used in the present study
was suitable for the comparison of ranked gene lists, based on a
hypothesis of randomly ordered genes. The higher the rank of a gene
in all experiments, the smaller the P-value, and the larger the
likelihood that the gene is a DEG. This method has been
increasingly used recently in different types of human cancer. In
one study of non-small cell lung cancer, Ni et al (13) analyzed four GEO datasets using the
RRA method for gene integration, and identified 249 DEGs (113 up-
and 136 downregulated); the 5 hub genes with the strongest
connectivity were identified as CCNB1, TOP2A, ubiquitin conjugating
enzyme E2 C (UBE2C), CCNA2 and kinesin family member 20A. Gao et
al (12) identified 343 DEGs
(111 upregulated and 232 downregulated) using the RRA method on
five GEO datasets of bladder cancer to identify 10 hub genes:
CCNB1, VEGFA, actin α 2, TOP2A, aurora kinase (AURK) B, cell
division cycle 20, AURKA, centrosomal protein 55 (CEP55), CCNB2 and
UBE2C. In a gastric cancer study, Liu et al (11) merged nine GEO datasets and used the
RRA method. A total of 9 hub genes that could affect the
pathogenesis of gastric cancer were identified, namely TOP2A,
cyclin-dependent kinase inhibitor 3, collagen type I α 1 chain
(COL1A1), NDC80 kinetochore complex component, TPX2 microtubule
nucleation factor, collagen type III α 1 chain, CEP55, collagen
type I α 2 chain and tissue inhibitor of metallopeptidase 1. Other
previous studies, such as one focusing on ovarian cancer (14) and hepatocellular carcinoma (33) have also used this method.
The integrated bioinformatics analysis identified
six important hub genes, CXCL12, ADCY7, BDKRB2, LPAR5, CASR and
KNG1. CXCL12 is a member of the C-X-C motif chemokine subfamily.
CXCL12/C-X-C motif chemokine receptor 4 signaling is known to
affect tumor growth; therefore, targeting this pathway could serve
as a new treatment strategy against cancer (34). CXCL12 exhibited a markedly higher
expression in lung metastases, as compared with paired primary
lesions of colorectal cancer (35).
BDKRB2 has also been detected in numerous types of human cancer.
Wei et al (36) screened
potential biomarkers of the chemoresistant ovarian carcinoma,
including six essential genes (phosphoinositide-3-kinase regulatory
subunit 5, phosphatase and tensin homolog, BDKRB2, MAPK3,
sphingosine-1-phosphate receptor 3 and nuclear cap binding protein
subunit 2). In addition, microRNA (miRNA)-129-1-3p could target
BDKRB2 in gastric cancer, thereby suppressing cell migration
(37). The expression of ADCY7 was
shown to be inversely correlated with the OS of patients with acute
myeloid leukemia (38). CASR
promoted bone metastasis in RCC (39) and was also identified as a hub gene
in CCRCC by co-expression analysis (8). Overexpression of KNG1 promoted
apoptosis and G1 phase cell cycle arrest, whilst
suppressing the PI3K/Akt pathway in glioma cells (40). This gene was also reported in thyroid
(41) and colorectal cancer
(42), as well as tongue squamous
cell carcinoma (43). LPAR5 was
identified as a hub gene in thyroid cancer (41). LPAR5 was downregulated in primary
nasopharyngeal carcinoma tissues, which, in turn, promoted
lysophosphatidic acid-induced cell migration (44).
In the present study, 20 pairs of CCRCC and matched
normal tissues were used for validation experiments. The results
obtained from the experimental validation were consistent with the
results obtained from the bioinformatics analysis. The sex
distribution, age range as well as tumor stages observed in the
patient recruited in the present study were similar to the clinical
information obtained from public databases. Most of the datasets
originated from Europe and America, and only one dataset was from
Japan. However, patients with CCRCC with different geographical
backgrounds also have different molecular biology backgrounds
(45). Therefore, future studies
will need to include datasets from other regional backgrounds,
including for instance, the Chinese population.
In addition, future research of RCC should be
focused on new methods or gene models. By literature searching, for
example, a recent trend of developing computational models to
identify some important RNAs (non-coding RNAs or miRNAs) related to
disease was increasingly reported (46). New models have been established and
implemented to identify novel miRNA-disease associations (47). For example, Chen et al
(48) presented a novel model of
Inductive Matrix Completion for miRNA-Disease Association
prediction, with the aim of predicting unknown miRNA-disease
associations based on known associations between integrated miRNA
and disease similarity. Moreover, a matrix decomposition and
heterogeneous graph inference calculation model for miRNA disease
association prediction, which identifies new miRNA disease
associations by integrating predictive association probabilities
obtained from matrix decomposition through sparse learning methods,
miRNA functional similarity, as well as other theories, was
developed (49). Another previous
study also developed a method of Laplacian Regularized Least
Squares for lncRNA-Disease Association in the semi-supervised
learning framework (50). The
aforementioned studies may provide insight into the
characterization of disease-related biomarkers.
To the best of the authors' knowledge, the present
study is the first to use the RRA algorithm in the study of CCRCC.
In addition, bioinformatics analysis was used to identify key genes
in the development and progression of CCRCC. However, there are
several limitations to the present study. Firstly, a larger sample
size needs to be validated to obtain reliable results in further
study. Moreover, novel and effective experimental methods and
computational models need to be designed. Lastly, further research
on hub genes needs to be carried out to examine molecular function
or biological behavior in the occurrence and development of
CCRCC.
In conclusion, GEO datasets were used in the present
study to screen for robust DEGs that could be involved in CCRCC
carcinogenesis. An integrated bioinformatics analysis was also
performed. The present study identified reliable molecular
biomarkers for the screening, diagnosis and prognosis of CCRCC,
which may also serve as new therapeutic targets.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by The National
Science and Technology Major Project of the Ministry of Science and
Technology of China (grant no. 2017ZX09304025), The Science and
Technology Plan Project of Liaoning Province (grant no. 2016007010)
and The General Projects of Liaoning Province Colleges and
Universities (grant no. LFWK201706).
Availability of data and materials
All data generated or analyzed during the current
study are included in this published article.
Authors' contributions
HC and LX conceived and designed the present study.
HC and LX performed the analysis. ZL, KZH, XFC and BFL contributed
to the literature search and did the initial analysis of the GEO
data. HC and XJQ wrote the initial draft of the manuscript. XJQ and
YPL assisted with the study design, obtained funding and also
revised the manuscript critically for important intellectual
content.. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Informed consent forms were signed by all patients
prior to sample collection. The present study was approved by The
Research Ethics Committee of China Medical University (approval no.
2015PS44K).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
RCC
|
renal cell carcinoma
|
|
CCRCC
|
clear cell renal cell carcinoma
|
|
GEP
|
gene expression profile
|
|
GEO
|
Gene Expression Omnibus
|
|
DEGs
|
differentially expressed genes
|
|
RRA
|
robust rank aggregation
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
GO
|
Gene Ontology
|
|
PPI
|
protein-protein interaction
|
|
TCGA
|
The Cancer Genome Atlas
|
|
FC
|
fold-change
|
|
DAVID
|
Database for Annotation, Visualization
and Integrated Discovery
|
|
MF
|
molecular function
|
|
BP
|
biological process
|
|
CC
|
cellular component
|
|
MCC
|
Maximal Clique Centrality
|
|
MNC
|
Maximum Neighborhood Component
|
References
|
1
|
Hsieh JJ, Purdue MP, Signoretti S, Swanton
C, Albiges L, Schmidinger M, Heng DY, Larkin J and Ficarra V: Renal
cell carcinoma. Nat Rev Dis Primers. 3:170092017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
Statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ljungberg B, Bensalah K, Canfield S,
Dabestani S, Hofmann F, Hora M, Kuczyk MA, Lam T, Marconi L,
Merseburger AS, et al: EAU guidelines on renal cell carcinoma: 2014
update. Eur Urol. 67:913–924. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tosco L, Van Poppel H, Frea B, Gregoraci G
and Joniau S: Survival and impact of clinical prognostic factors in
surgically treated metastatic renal cell carcinoma. Eur Urol.
63:646–652. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Grignon DJ and Che M: Clear cell renal
cell carcinoma. Clin Lab Med. 25:305–316. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim HS, Kim JH, Jang HJ, Han B and Zang
DY: Clinicopathologic significance of VHL gene alteration in
clear-cell renal cell carcinoma: An updated meta-analysis and
review. Int J Mol Sci. 19(pii): E25292018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Song E, Song W, Ren M, Xing L, Ni W, Li Y,
Gong M, Zhao M, Ma X, Zhang X and An R: Identification of potential
crucial genes associated with carcinogenesis of clear cell renal
cell carcinoma. J Cell Biochem. 119:5163–5174. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yuan L, Zeng G, Chen L, Wang G and Wang X,
Cao X, Lu M, Liu X, Qian G, Xiao Y and Wang X: Identification of
key genes and pathways in human clear cell renal cell carcinoma
(ccRCC) by co-expression analysis. Int J Biol Sci. 14:266–279.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen L, Yuan L, Qian K, Qian G, Zhu Y, Wu
CL, Dan HC, Xiao Y and Wang X: Identification of biomarkers
associated with pathological stage and prognosis of clear cell
renal cell carcinoma by co-expression network analysis. Front
Physiol. 9:3992018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kolde R, Laur S, Adler P and Vilo J:
Robust rank aggregation for gene list integration and
meta-analysis. Bioinformatics. 28:573–580. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu X, Wu J, Zhang D, Bing Z, Tian J, Ni
M, Zhang X, Meng Z and Liu S: Identification of potential key genes
associated with the pathogenesis and prognosis of gastric cancer
based on integrated bioinformatics analysis. Front Genet.
9:2652018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gao X, Chen Y, Chen M, Wang S, Wen X and
Zhang S: Identification of key candidate genes and biological
pathways in bladder cancer. PeerJ. 6:e60362018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ni M, Liu X, Wu J, Zhang D, Tian J, Wang
T, Liu S, Meng Z, Wang K, Duan X, et al: Identification of
candidate biomarkers correlated with the pathogenesis and prognosis
of non-small cell lung cancer via integrated bioinformatics
analysis. Front Genet. 9:4692018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang X, Zhu S, Li L, Zhang L, Xian S, Wang
Y and Cheng Y: Identification of differentially expressed genes and
signaling pathways in ovarian cancer by integrated bioinformatics
analysis. Onco Targets Ther. 11:1457–1474. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu H, Brannon AR, Reddy AR, Alexe G,
Seiler MW, Arreola A, Oza JH, Yao M, Juan D, Liou LS, et al:
Identifying mRNA targets of microRNA dysregulated in cancer: With
application to clear cell renal cell carcinoma. BMC Syst Biol.
4:512010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Peña-Llopis S, Vega-Rubin-de-Celis S, Liao
A, Leng N, Pavía-Jiménez A, Wang S, Yamasaki T, Zhrebker L,
Sivanand S, Spence P, et al: BAP1 loss defines a new class of renal
cell carcinoma. Nat Genet. 44:751–759. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wozniak MB, Le Calvez-Kelm F,
Abedi-Ardekani B, Byrnes G, Durand G, Carreira C, Michelon J,
Janout V, Holcatova I, Foretova L, et al: Integrative genome-wide
gene expression profiling of clear cell renal cell carcinoma in
Czech Republic and in the United States. PLoS One. 8:e578862013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Eckel-Passow JE, Serie DJ, Bot BM, Joseph
RW, Cheville JC and Parker AS: ANKS1B is a smoking-related
molecular alteration in clear cell renal cell carcinoma. BMC Urol.
14:142014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wotschofsky Z, Gummlich L, Liep J, Stephan
C, Kilic E, Jung K, Billaud JN and Meyer HA: Integrated microRNA
and mRNA signature associated with the transition from the locally
confined to the metastasized clear cell renal cell carcinoma
exemplified by miR-146-5p. PLoS One. 11:e01487462016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Takahashi M, Tsukamoto Y, Kai T, Tokunaga
A, Nakada C, Hijiya N, Uchida T, Daa T, Nomura T, Sato F, et al:
Downregulation of WDR20 due to loss of 14q is involved in the
malignant transformation of clear cell renal cell carcinoma. Cancer
Sci. 107:417–423. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar
|
|
23
|
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong
S, Kong L, Gao G, Li CY and Wei L: KOBAS 2.0: A web server for
annotation and identification of enriched pathways and diseases.
Nucleic Acids Res. 39:(Web Server Issue). W316–W322. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Szklarczyk D, Morris JH, Cook H, Kuhn M,
Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al:
The STRING database in 2017: Quality-controlled protein-protein
association networks, made broadly accessible. Nucleic Acids Res.
45(D1): D362–D368. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bader GD and Hogue CW: An automated method
for finding molecular complexes in large protein interaction
networks. BMC Bioinformatics. 4:22003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chin CH, Chen SH, Wu HH, Ho CW, Ko MT and
Lin CY: cytoHubba: Identifying hub objects and sub-networks from
complex interactome. BMC Syst Biol. 8 (Suppl 4):S112014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45((W1)):
W98–W102. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Greene FL, Gospodarowicz MK and Wittekend
C: American Joint Committee On Cancer (AJCC) cancer staging manual.
(7th). Philadelphia. (Springer). 2009.PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ge S, Xia X, Ding C, Zhen B, Zhou Q, Feng
J, Yuan J, Chen R, Li Y, Ge Z, et al: A proteomic landscape of
diffuse-type gastric cancer. Nat Commun. 9:10122018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Leek JT and Storey JD: Capturing
heterogeneity in gene expression studies by surrogate variable
analysis. PLoS Genet. 3:1724–1735. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu F, Wu S and Gou X: Identification of
biomarkers and potential molecular mechanisms of clear cell renal
cell carcinoma. Neoplasma. 65:242–252. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang Y, Lu Q, Shao X, Mo B, Nie X, Liu W,
Chen X, Tang Y, Deng Y and Yan J: Development of A three-gene
prognostic signature for hepatitis B virus associated
hepatocellular carcinoma based on integrated transcriptomic
analysis. J Cancer. 11:989–2002. 2018.PubMed/NCBI
|
|
34
|
Zhou Y, Cao HB, Li WJ and Zhao L: The
CXCL12 (SDF-1)/CXCR4 chemokine axis: Oncogenic properties,
molecular targeting, and synthetic and natural product CXCR4
inhibitors for cancer therapy. Chin J Nat Med. 16:801–810.
2018.PubMed/NCBI
|
|
35
|
Wang M, Yang X, Wei M and Wang Z: The role
of CXCL12 axis in lung metastasis of colorectal cancer. J Cancer.
9:3898–3903. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wei S, Wang Y, Xu H and Kuang Y: Screening
of potential biomarkers for chemoresistant ovarian carcinoma with
miRNA expression profiling data by bioinformatics approach. Oncol
Lett. 10:2427–2431. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang D, Luo L and Guo J: miR-129-1-3p
inhibits cell migration by targeting BDKRB2 in gastric cancer. Med
Oncol. 31:982014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li C, Xie J, Lu Z, Chen C, Yin Y, Zhan R,
Fang Y, Hu X and Zhang CC: ADCY7 supports development of acute
myeloid leukemia. Biochem Biophys Res Commun. 465:47–52. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Frees S, Breuksch I, Haber T, Bauer HK,
Chavez-Munoz C, Raven P, Moskalev I, D Costa N, Tan Z, Daugaard M,
et al: Calcium-sensing receptor (CaSR) promotes development of bone
metastasis in renal cell carcinoma. Oncotarget. 9:15766–15779.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xu J, Fang J, Cheng Z, Fan L, Hu W, Zhou F
and Shen H: Overexpression of the Kininogen-1 inhibits
proliferation and induces apoptosis of glioma cells. J Exp Clin
Cancer Res. 37:1802018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tang J, Kong D, Cui Q, Wang K, Zhang D,
Yuan Q, Liao X, Gong Y and Wu G: Bioinformatic analysis and
identification of potential prognostic microRNAs and mRNAs in
thyroid cancer. PeerJ. 6:e46742018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yu J, Huang Y, Lin C, Li X, Fang X, Zhong
C, Yuan Y and Zheng S: Identification of kininogen 1 as a serum
protein marker of colorectal adenoma in patients with a family
history of colorectal cancer. J Cancer. 9:540–547. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang H, Liu J, Fu X and Yang A:
Identification of key genes and pathways in tongue squamous cell
carcinoma using bioinformatics analysis. Med Sci Monit.
23:5924–5932. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yap LF, Velapasamy S, Lee HM, Thavaraj S,
Rajadurai P, Wei W, Vrzalikova K, Ibrahim MH, Khoo AS, Tsao SW, et
al: Down-regulation of LPA receptor 5 contributes to aberrant LPA
signalling in EBV-associated nasopharyngeal carcinoma. J Pathol.
235:456–465. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Znaor A, Lortet-Tieulent J, Laversanne M,
Jemal A and Bray F: International variations and trends in renal
cell carcinoma incidence and mortality. Eur Urol. 67:519–530. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lopez R, Wang R and Seelig G: A molecular
multi-gene classifier for disease diagnostics. Nat Chem.
10:746–754. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chen X, Xie D, Zhao Q and You ZH:
MicroRNAs and complex diseases: From experimental results to
computational models. Brief Bioinform. 20:515–539. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen X, Wang L, Qu J, Guan NN and Li JQ:
Predicting miRNA-disease association based on inductive matrix
completion. Bioinformatics. 34:4256–4265. 2018.PubMed/NCBI
|
|
49
|
Chen X, Yin J, Qu J and Huang L: MDHGI:
Matrix decomposition and heterogeneous Graph inference for
miRNA-disease association prediction. PLoS Comput Biol.
14:e10064182018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chen X and Yan GY: Novel human
lncRNA-disease association inference based on lncRNA expression
profiles. Bioinformatics. 29:2617–2624. 2013. View Article : Google Scholar : PubMed/NCBI
|