Introduction
Uveal melanoma (UM) is the most common primary
malignant tumor of the eye in adults (1). Uveal, as well as cutaneous, melanomas
have origins from the same precursor cell, the melanocyte, which
migrates from the neural crest during embryonic development
(2). UM represents 3–5% of all
melanomas and it arises from proliferating atypical melanocytes
situated in the choroid (85–90%), the ciliary body (5–8%) and the
iris (5–8%) (1). Primary tumor
location has an effect on UM progression: Melanoma originating from
melanocytes in the iris is usually associated with good prognosis,
while choroidal and ciliary ones have a poor prognosis, and in ≤50%
of all cases lead to metastatic disease (3), which most commonly occurs in the liver
(60.5%), lung (24.4%), skin (11%) and bone (8.4%) (1,4).
UM has a median diagnostic age of 62 years, with a
peak between 70 and 79 years (5,6). It has
been recorded to have 30% higher incidence in males; however, to
the best of our knowledge, there is no known reason for this
(7). Based on the Surveillance,
Epidemiology, and End Results database (1973–2009), cases of UMs
affect 5.1 in every million individuals (5). In Europe, the incidence varies between
2 and 8 individuals per million based on latitude according to the
European Cancer Registry (1983–1994) (8). Additionally, UMs are less prevalent in
Asian and black populations (9).
Similarly to other melanomas, the most common risk
factors of UM are fair skin, light eyes, ocular melanocytosis,
dysplastic nevus syndrome and multiple mutations (10–12).
Exposure to UV radiation is a major risk factor for the development
of cutaneous melanoma; however, there is little evidence regarding
its role in UM progression (13).
Since UVA is mainly filtered by the cornea and lens, while UVB and
UVC do not reach the choroid, ‘it is unlikely’ that UV radiation
exposure is responsible for choroidal melanoma (14,15).
The primary tumor is often difficult to diagnose as
a third of the cases are asymptomatic (6). In most cases, UM is manifested through
blurred vision and a variety of other symptoms, such as elevated
intraocular pressure, which causes glaucoma (16). UM is frequently misdiagnosed as
glaucoma, since the latter is one of the potential side effects of
the tumor (16). The median life
expectancy after metastatic growth is 13.4 months with an 8%
survival rate after 2 years (4).
Gene expression profiling is currently used to
classify UMs into two distinct types depending on their ability to
metastasize. Class 1 are tumors with a 1% chance of spreading,
while class 2 are tumors that have a 25.9% chance of forming
secondary tumors (17). Little is
known regarding its molecular pathogenesis, and it has been
considered that a variety of epigenetic alterations occur in the
melanocyte to UM pathway (18). p53
and the retinoblastoma (Rb) signaling pathway are commonly
inhibited, whereas the PI3K/Akt signaling pathway in mostly
activated (18). The present review
discusses these signaling pathways that regulate cell death and the
cell cycle in UM. Increased understanding of these pathways may
lead to the identification of the genetic profile of UM, enable the
design of a personalized targeted therapy for the patient and,
finally, an improved prognosis of patients with UM.
Chromosomal origin of UM
UM is often characterized by multiple chromosomal
aberrations. Abnormalities on chromosomes 1, 3, 6 and 8 have been
observed in 17–61% of UM cases (18). Additionally, these have been
demonstrated to affect the prognosis and development of the tumor
(19). The most common chromosomal
aberrations result in loss of chromosome 3 and 6q, or in the gain
of 6p and 8q (19). Monosomy 3 is
observed in 50% of all tumors, 65% of UMs and in >70% of
metastasizing UMs (20). Chromosome
3 loss results in a >50% reduction in the 5-year survival rate
of patients (19). By contrast,
patients with an intact pair of chromosome 3 have a 5-year survival
rate of 90%, which is reduced to 37% by the loss of one sister
chromatid (20). BRCA1 associated
protein 1 (BAP1) is one of the genes that is mutated in 47% of all
Ums, and, due to its location on chromosome 3, it is important for
the understanding of the disease development (21). This also explains why monosomy 3 is
associated with a poor prognosis. Furthermore, BAP1 is a single
copy gene, which often results in inactivating mutations (21). In UM, this results in an earlier
onset at the age of 30 to 59 years (22). Additionally, mutations in BAP1 have
been associated with an 11% higher risk of secondary malignancies
(22). Other genes located on
chromosome 3 encode PI3K and Ras-association domain family 1
isoform A (RASSF1A), both of which are associated with essential
molecular pathways that are mutated in cancer (23,24).
Other aberrations, including gain of chromosome 8,
loss of chromosome 1 or polysomy 8q, have been associated with a
reduced survival due to various factors, such as exposure to sun
light, oculodermal melanocytosis and dysplastic nevi (25,26).
Some of the genes located on chromosome 1 are associated with
essential molecular pathways by encoding PI3K and Akt, or tumor
suppressor genes (TSGs), such as centromere protein S (27). On the other hand, gain of chromosome
6p has been associated with good prognosis, despite the abundance
of oncogenes (28).
One of the most common chromosomal abnormalities in
UM is rearrangement of chromosome 8q. Copy number variations have
been observed in 79% of UM cases (26). In patients with a normal 8q number
the 5-year survival rate is 93%; however, with the increase of copy
numbers, this rate is reduced to 67 and then 29% (20).
It has been noted that there are two distinct
developmental pathways in UM. Class one exhibits disomy 3 with gain
of chromosome 6p, while class 2 typically exhibits monosomy 3 and a
high metastatic propensity (29,30).
These chromosomal aberrations are present at early stages, while
increased aneuploidy and changes in chromosome 8 are considered to
be associated with later stages (31). While this two-class model is
relatively simplistic, it provides a good basic understanding of
the main chromosomal aberrations and their consequential
effects.
Cell death regulation
One of the hallmarks of cancer is the ability of
cells to evade death signals and proliferate indefinitely (32). Therefore, the regulation of the cell
cycle and the induction of self-mediated cell death, also known as
apoptosis, is vital.
The extrinsic apoptotic signaling pathway is
activated when a death ligand binds to a death receptor on the cell
membrane. These receptors have an intracellular death domain that
recruits adapter proteins, which results in the formation of a
binding site known as death-inducing signaling complex (DISC)
(33). DISC assembles and activates
pro-caspase-8, which initiates apoptosis by cleaving other caspases
(34). Once activated, caspase 3
cleaves the caspase-activated deoxyribonuclease, which begins the
process of DNA degradation (35).
Finally, downstream caspases induce cleavage of protein kinases and
proteins, and break down the cytoskeleton disturbing signaling
pathways, which results in the typical morphological alterations of
apoptosis (36).
The mitochondrial intrinsic pathway is initiated
within the cell due to internal stimuli, such as genetic damage,
hypoxia and oxidative stress (37).
This results in the release of pro-apoptotic molecules, apoptosis
inducing factor mitochondria associated 1, second
mitochondria-derived activator of caspase, diablo IAP-binding
mitochondrial protein, HtrA serine peptidase 2 and cytochrome
c, which initiate apoptosis (38). Subsequently, cytochrome c and
Apaf-1 assemble the apoptosome which activates caspase-9 (38). Alongside caspase-9 activation,
caspase-3 is activated, leading to the same steps as the
aforementioned extrinsic pathway (39). The Bcl-2 family proteins, which are
directly controlled by p53, determine the cell fate through the
balance of pro- and anti-apoptotic molecules (38).
The molecular and genetic makeup of UMs is
considered to be more complicated than the aforementioned
mutations. Therefore, the present review will explore the potential
role of multiple molecular pathways and their role in UM
development and pathogenesis.
G protein subunit αq and G protein subunit
α11
Despite the chromosome abnormalities, most UMs are
considered to be caused by point mutations in G protein α subunits,
specifically in G protein subunit αq (GNAQ) and G protein subunit
α11 (GNA11), regardless of tumor stage or chromosomal constellation
(40). These mutations have similar
effects as mutations in RAS, which are common in a number of other
tumors (41). The G-α subunit is
important due to its involvement in multiple essential cellular
pathways, such as the MAPK (cell proliferation and apoptosis),
PI3K-Akt (growth and homeostasis) and Hippo signaling pathways
(42). GNAQ and GNA11 activate
phospholipase C, which triggers a cascade of events resulting in
the activation of protein kinase C (PKC). PKC then initiates a
phosphorylation cascade, which activates Raf, MEK1/2 and ERK
(43). This process results in the
regulation of cell proliferation and survival (44). It has been hypothesized that these
mutations are early events in the pathogenesis of UM and are
necessary for tumor malignancy (45). On the other hand, mutant GNAQ and
GNA11 are considered to be weak oncogenes and, therefore, cannot
cause damage to melanocytes unless they are already deficient in
the p53 and p16/CDK4/RB signaling pathways (46). Due to the importance of GNAQ and
GNA11 in UM malignancy, the 5-oxo-ETE acid G-protein-coupled
receptor 1 (GPCR) signaling pathway is a potential viable
therapeutic target (42). The
development of inhibitors for specific molecules, such as Gq/11
inhibitor YM-254890 and Arf6-inhibitor NAV-2729, is one of the main
strategies that is currently being investigated (47,48). One
such example is the inhibition of CysLT2R-L129Q, which is
responsible for the constitutive activation of the Gq/11 signaling
pathway in UM (47,49).
RAS interactions
The Ras superfamily consists of small GTPases that
act as switches and modulate a vast array of cell functions by
influencing signaling pathways. They are separated into six
different groups of proteins, and are present in all cell types
(50). One of the subfamilies, also
referred to as RAS, consists of proteins that regulate cell
proliferation. They have downstream effects on signaling pathways
crucial to UM, such as the MAPK/ERK and PI3K/AKT/PTEN signaling
pathways (51). In cancer, RAS is
often mutated which affects a number of these pathways and makes
them less sensitive to apoptosis triggers, thus increasing
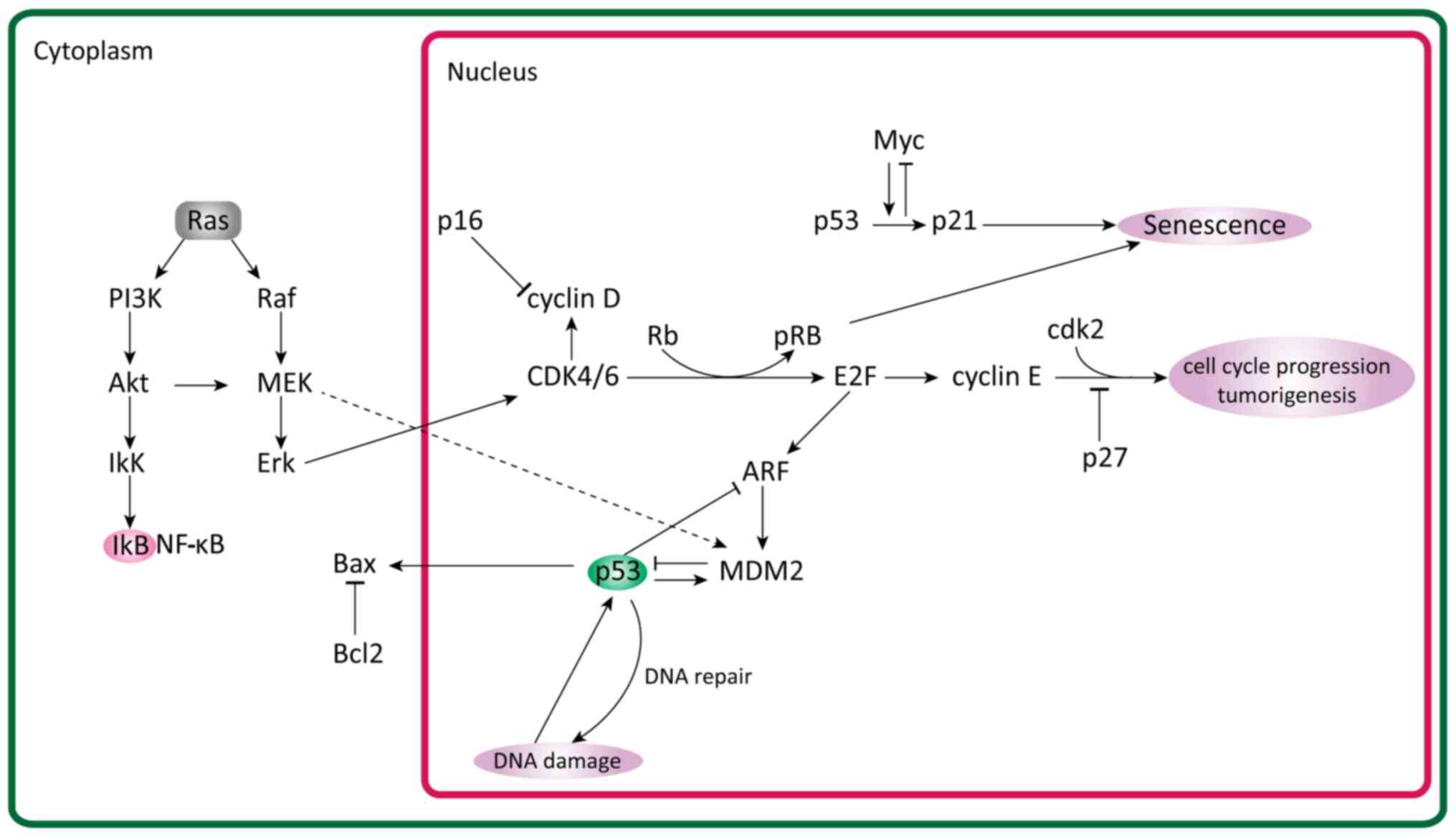
proliferation levels (Fig. 1)
(52).
All three common RAS proteins in humans are highly
conserved in the active regions and often undergo mutations in
codons 12, 13 and 61 (53). The
resulting point mutations lead to preferential binding to GTP over
GDP, which in-turn leads to activation of proliferation pathways
(54). Interestingly, RAS mutations
are not usually associated with UM (55,56).
In general, RAS serve as activating proteins that
remove GDP and allow GTP to bind its target (57). Ras-bound GTP goes on to activate Raf,
which initiates the MAPK/ERK signaling pathway (58). In the case of PI3K, active Ras
directly activates it without an intermediary protein (59).
PI3K/Akt/PTEN signaling pathway
The PI3K/Akt/PTEN is one of the main molecular
pathways involved in cell proliferation. It is mutated in multiple
types of cancer and is constitutively activated in most UMs
(24). RAS directly activates PI3K,
which then goes on to phosphorylate phosphatidylinositol
4,5-bisphosphate to produce phosphatidylinositol (3,4,5)-trisphosphate (PIP3) (60). PIP3 is dephosphorylated by PTEN to
regulate PIP3 levels, which, when elevated, activates Akt.
Subsequently, Akt goes on to phosphorylate a number of signaling
pathways, such as the mTORC1, MDM2 proto-oncogene (MDM2), BAD and
GTPase-activating protein signaling pathways (Fig. 1) (61).
Akt is an anti-apoptotic protein which serves an
important role in cell survival and tumorigenesis (62). It is activated via phosphorylation,
becoming phospho-Akt which inactivates several proteins, including
members of the Bcl-2 family (BAD protein) and caspase-9 (63). Phospho-Akt is involved in the
protection from apoptosis, but also in other cancer development
processes, such as blockage of anti-proliferative signaling,
facilitation of cell replication and angiogenesis (63). Using immunohistochemical testing, it
has been demonstrated that phosphorylation of Akt is associated
with a poor prognosis (62).
PTEN downregulation has also been associated with
various types of cancer, including breast cancer, thyroid cancer,
kidney cancer, endometrial cancer, colorectal cancer and melanoma
(64,65). In UMs, loss of heterozygosity of PTEN
has been observed in 76% of tumors, with 11% being within the PTEN
coding region (66). Loss of
cytoplasmic PTEN in primary UM tumors was associated with shortened
disease-free survival (67). Its
decreased expression can also result in increased aneuploidy and
reduced survival (45).
Overall, these findings suggest that the
PI3K/Akt/PTEN signaling pathway serves a vital role in UM
progression; however, more research needs to be performed to fully
understand its role.
MAPK/ERK signaling pathway
The MAPK/ERK pathway is crucial for mediating
cell-cycle progression. In multiple cancer types, it is
constitutively activated, resulting in proliferation of neoplastic
cells (68,69). It has also been identified to serve
an important role in melanocytic neoplasia (70).
MAPK signaling, similar to the PI3K/Akt signaling
pathway, begins with the activation of RAS, which then recruits RAF
(68). RAFs are a group of kinases
that transduce signals along the MAPK signaling pathway (68). There are three isoforms that are
expressed in humans: A-Raf proto-oncogene serine/threonine kinase,
BRAF and Raf-1 proto-oncogene serine/threonine kinase (cRAF)
(71). cRAF was the first to be
discovered and BRAF has been the most extensively studied due to
its high mutation rate in various types of cancer (72,73).
Activation of BRAF by RAS results in the
phosphorylation of kinases, such as MEK1/2 and ERK1/2, which
induces a multitude of proliferative and survival processes
(74) via the consequent activation
of transcription factors, such as ETS transcription factor ELK1. In
cutaneous melanomas, RAS and BRAF often undergo activating
mutations (75). These also appear
in benign melanocytic naevi and, alongside activation of the MAPK
signaling pathway, constitute early events of melanogenesis
(51,76,77). In
UM, the MAPK signaling pathway is upregulated, which advocates for
the presence of upstream mutations (78). It is also known that mutations in RAS
and BRAF are uncommon in UM (79–81).
Therefore, the constitutive activation is considered to be caused
by mutations in the GNAQ family, which results in the upregulation
of the pathways (82,83).
Yes-associated protein/transcriptional
co-activator with PDZ-binding motif signaling pathway and its
potential for treatment
Yes-associated protein (YAP) and transcriptional
co-activator with PDZ-binding motif (TAZ) modulate regulation of
cell proliferation, migration and survival (84). They are, in turn, negatively
regulated by the Hippo signaling pathway (85), which acts as a tumor suppressor to
limit cell proliferation and organ size regulation (86). As previously mentioned, mutations in
GNAQ and GNA11 are present in >80% of UM cases. This is
essential as GPCRs activate F-actin, which targets YAP/TAZ and,
therefore, could result in the upregulation of the pathway
(87). When this happens, YAP is
activated independently of the Hippo signaling pathway, resulting
in greater resistance to contact inhibition of growth (88–90).
In contrast to MAPK-targeted therapy, which has no
impact on the prognosis of patients with UM, YAP-targeted therapy
is strongly associated with cancer metastasis (91) and shows promise as an ideal target
(91–93). For example, focal adhesion
kinase-targeted therapy reduces YAP levels and counteracts the
effects of the GNAQ/GNA11 mutation (43). This may be effective in treating UMs
that exhibit such mutations (94).
p53
p53 is one of the key apoptotic regulators, which
induces cell cycle arrest and consequently apoptosis. A study by
Liu and Zhou (95) explored the role
of p53 in the development of UM. They observed that p53 expression
and prognosis were negatively associated. The mortality rate
increased with p53 expression. Additionally, inhibition of p53 has
been associated with inhibited invasion in UM (95). Other studies have mentioned that
mutations in the p53 gene are rare and, in fact, most causes of
disruption in the pathways are due to upstream or downstream
mutations (2,96,97). One
such cause could be MDM2 upregulation, which is common in UMs
(98). MDM2 regulates p53 and
reduces its expression (98).
Protection from apoptosis is a major factor in the
metastatic cascade of most types of cancer, including UM. p53 does
not appear to have a significant impact on UM (99). However, a previous study demonstrated
that multiplication of chromosome 8 and c-myc expression are
associated, suggesting that c-myc could be used as a prognostic
factor (11). C-myc alone is
involved in the regulation of cell proliferation and with
p53-dependent mechanisms promotes apoptosis (100). On the other hand, Bcl-2, which is
vital for the intrinsic apoptotic pathway, has been reported to be
upregulated in most UMs (2,101). A strong inverse relationship has
been observed between c-myc (nuclear and cytoplasmic) and Bcl-2,
suggesting that the latter co-operates with c-myc to immortalize UM
cells (79,101).
Brantley and Harbour (2) immunohistochemically analyzed the p53
and retinoblastoma protein (pRb) pathways and found that, in UM
cases, most alterations are due to mutations in other proteins.
CDK and cyclin kinase inhibitors
A fine balance between cyclin kinase inhibitors
(CDKs) and CKIs is required for a normal cell cycle (102). If toxic chemicals, oxidative stress
(reactive oxygen species), ionizing radiation and other factors
induce DNA damage, the cell must repair it and reenter the cell
cycle (102). When the cell fails
to repair the damage, it becomes senescent and the cell cycle is
arrested in G1 phase (diploid DNA) or G2 phase (tetraploid DNA
content). Deregulation of the cell cycle, especially at the G2/M
phase, leads the cell to a more cancerous fate (103).
Cell cycle regulation is achieved through a family
of serine/threonine kinase holoenzyme complexes consisting of
regulatory cyclins that bind to and activate catalytic CDKs. The
cyclins D1, D2, D3 and E are important for the G1-S cell cycle
transition (104). Cyclin A is
involved in DNA synthesis, S-phase completion and preparation for
mitosis, while cyclins B1 and B2 control the onset, sequence of
events and completion of mitosis (105). For example, the cyclin B1/CDK1
complex is a mitotic regulator that is responsible for the
progression of the cell cycle (105).
On the other hand, cyclin-dependent kinase
inhibitors (CDKIs) negatively regulate the kinase activity of the
cyclin-CDK complexes (106). There
are two known families of CDKIs: The cyclin dependent kinase
inhibitor 2A (INK4) family, which includes p16/INK4A, p15/INK4B,
p18/INK4C and p19 (p14)/INK4D, and the cardiac ISL1-interacting
protein (CIP)/calcium and integrin binding 1 (KIP) family, which
includes p21/CIP1, p27/KIP1 and p57/KIP2 (107–109).
Different CKIs have been observed to affect
different CDKs. For example, p15 and p16 inhibit CDK4 and CDK6
respectively, while p21 and p27 act on G1 CDK cyclins and S-phase
CDK2 complexes (110,111). Both p21 and p27 inhibit DNA
replication but through different mechanisms. p21 binds to and
promotes CDK1 and CDK2 with cyclin D activity, while, under certain
conditions, inhibiting CDK4 and CDK6 with cyclin E activity
(112). p27 promotes CDK2 cyclin E
complex and CDK4/6 cyclin D complex formation (113,114).
In uveal and choroidal melanoma cell lines, the
expression levels of p21 and p27 are downregulated resulting in
suppressed p16-CDK interaction (110,115).
This results in more CDK activity, leading to cyclin D and CDKs
phosphorylating pRb, which goes on to release E2F transcription
factor 1 (E2F1) (116). E2F1 is a
transcriptional factor that leads to the expression of numerous
necessary factors for G1 to S phase progression (117). pRb is another one of the vital
molecules in the cell cycle regulation of uveal and choroidal
cells. It is encoded by the Rb gene and serves an important role as
a tumor suppressor (118).
Deregulation and inactivation of p16 and/or overexpression of
cyclin D leads to inactivation of pRb by cyclin-dependent
phosphorylation (119,120). In most UMs, the Rb protein is
constitutively hyperphosphorylated and functionally inactivated
(120). This has been attributed to
cyclin D1 upregulation in 65% of cases and has also been associated
with larger tumor sizes and poor prognosis (63,76,121).
E2F transcription factors are key regulators of cell
division and, among them, E2F1, E2F2 and E2F3a are potent
activators of E2F-responsive genes, but their transcriptional
activity is inhibited by binding to pRb (122,123).
pRb is functionally inactivated at the G1-S transition by cyclin
D-CDK4/CDK6 and cyclin E-CDK2-mediated phosphorylation, thus
enabling E2F transcription factors to activate their target genes
(124).
RASSF1A
RASSF1A is a TSG that is required for death
receptor-dependent apoptosis (80)
and can be found at the 3p21.3 locus. RASSF1A inhibits the
accumulation of cyclin D1 protein without affecting its mRNA levels
(81). This results in suppressed
proliferation via negative regulation of the cell cycle progression
at the G1/S phase transition (125). It has been reported that the
endogenous inactivation of RASSF1A leads to a decrease of p27 which
is a negative cell cycle regulator at the protein level (111,125).
The depletion of this gene in RASSF1A mouse mutants has been noted
as an early event in the senescence of uveal melanocytes and is
considered to contribute to the malignancy of UM (125). RASSF1A is frequently
hypermethylated in UM, which results in its downregulation
(126). Additionally, in 83% of the
cases in a study by Calipel et al (125), the RASSF1A promoter was methylated
which suppressed gene expression. Overall, the downregulation of
RASSF1A is most likely explained by the loss of heterozygosity
typical for UM.
Conclusion
UM is the most common intraocular tumor in adults
and is caused by multiple molecular abnormalities. The most
frequent mutations in GNAQ and GNA11 are considered to be the main
driving events in UM. Due to the involvement of GPCRs in multiple
molecular signaling pathways, the present review explored the
various downstream effects that such mutations could trigger.
Additionally, the potential effects of chromosomal abnormalities,
and how the loss or gain of specific regions could improve or
worsen prognosis, were described. All this information allowed the
authors to pinpoint potential therapeutic targets which could be
used to successfully treat patients with UM. Based on the
understanding of the aforementioned pathways and DNA expression
profiles of UM, prediction models can be produced, and this could
lead to improved prognosis for patients with UM.
Acknowledgements
The authors wish to acknowledge the help provided by
Ms. Heerni Halai (College of Health, Medicine and Life Sciences,
Brunel University, UK).
Funding
No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
PK produced and reviewed the manuscript and figure.
MSK and VA reviewed the manuscript. VA sponsored the publication.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Krantz BA, Dave N, Komatsubara KM, Marr BP
and Carvajal RD: Uveal melanoma: Epidemiology, etiology, and
treatment of primary disease. Clin Ophthalmol. 11:279–289. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brantley MA Jr and Harbour JW:
Deregulation of the Rb and p53 pathways in uveal melanoma. Am J
Pathol. 157:1795–1801. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weber A, Hengge UR, Urbanik D, Markwart A,
Mirmohammadsaegh A, Reichel MB, Wittekind C, Wiedemann P and
Tannapfel A: Absence of mutations of the BRAF gene and constitutive
activation of extracellular-regulated kinase in malignant melanomas
of the uvea. Lab Invest. 83:1771–1776. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kuk D, Shoushtari AN, Barker CA, Panageas
KS, Munhoz RR, Momtaz P, Ariyan CE, Brady MS, Coit DG, Bogatch K,
et al: Prognosis of mucosal, uveal, acral, nonacral cutaneous, and
unknown primary melanoma from the time of first metastasis.
Oncologist. 21:848–854. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Andreoli MT, Mieler WF and Leiderman YI:
Epidemiological trends in uveal melanoma. Br J Ophthalmol.
99:1550–1553. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Damato EM and Damato BE: Detection and
time to treatment of uveal melanoma in the United Kingdom: An
evaluation of 2,384 patients. Ophthalmology. 119:1582–1589. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McLaughlin CC, Wu X, Jemal A, Martin HJ,
Roche LM and Chen VW: Incidence of Noncutaneous Melanomas in the
U.S. Cancer. 103:1000–1007. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Virgili G, Gatta G, Ciccolallo L,
Capocaccia R, Biggeri A, Crocetti E, Lutz JM and Paci E; EUROCARE
Working Group, : Incidence of uveal melanoma in Europe.
Ophthalmology. 114:2309–2315. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hu DN, Yu GP, Mccormick SA, Schneider S
and Finger PT: Population-based incidence of uveal melanoma in
various races and ethnic groups. Am J Ophthalmol. 140:612–617.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nayman T, Bostan C, Logan P and Burnier MN
Jr: Uveal melanoma risk factors: A systematic review of
meta-analyses. Curr Eye Res. 42:1085–1093. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kaliki S, Shields CL and Shields JA: Uveal
melanoma: Estimating prognosis. Indian J Ophthalmol. 63:93–102.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Weis E, Shah CP, Lajous M, Shields JA and
Shields CL: The association of cutaneous and iris nevi with uveal
melanoma: A meta-analysis. Ophthalmology. 116:536–543.e2. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gendron P, Desgarnier MD, Mallet JD and
Rochette PJ: Implication of ultraviolet light in the etiology of
uveal melanoma (Review). Photochem Photobiol. 90:15–21. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mallet JD and Rochette PJ: Themed issue:
Interaction of UV radiation with DNA. Photochem Photobiol Sci.
12:1245–1246. 2013.
|
|
15
|
Mallet JD, Gendron SP, Drigeard Desgarnier
MC and Rochettes PJ: Implication of ultraviolet light in the
etiology of uveal melanoma: A review. Photochem Photobiol.
90:15–21. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shields CL, Materin MA, Shields JA,
Gershenbaum E, Singh sAD and Smith A: Factors associated with
elevated intraocular pressure in eyes with iris melanoma. Br J
Ophthalmol. 85:666–669. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Onken MD, Worley LA, Char DH, Augsburger
JJ, Correa ZM, Nudleman E, Aaberg TM Jr, Altaweel MM, Bardenstein
DS, Finger PT, et al: Collaborative Ocular Oncology Group report
number 1: prospective validation of a multi-gene prognostic assay
in uveal melanoma. Ophthalmology. 119:1596–1603. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Coupland SE, Lake SL, Zeschnigk M and
Damato BE: Molecular pathology of uveal melanoma. Eye (Lond).
27:230–242. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sisley K, Rennie IG, Parsons MA, Jacques
R, Hammond DW, Bell SM, Potter AM and Rees RC: Abnormalities of
chromosomes 3 and 8 in posterior uveal melanoma correlate with
prognosis. Genes Chromosomes Cancer. 19:22–28. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Versluis M, de Lange MJ, van Pelt SI,
Ruivenkamp CA, Kroes WG, Cao J, Jager MJ, Luyten GP and van der
Velden PA: Digital PCR validates 8q dosage as prognostic tool in
uveal melanoma. PLoS One. 10:e01163712015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Harbour JW, Onken MD, Roberson EDO, Duan
S, Cao L, Worley LA, Council ML, Matatall KA, Helms C and Bowcock
AM: Frequent mutation of BAP1 in metastasizing uveal melanomas.
Science. 330:1410–1413. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Laíns I, Bartosch C, Mondim V, Healy B,
Kim IK, Husain D and Miller JW: Second primary neoplasms in
patients with uveal melanoma: A SEER Database Analysis. Am J
Ophthalmol. 165:54–64. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
van der Weyden L and Adams DJ: The
Ras-association domain family (RASSF) members and their role in
human tumourigenesis. Biochim Biophys Acta. 1776:58–85.
2007.PubMed/NCBI
|
|
24
|
Babchia N, Calipel A, Mouriaux F, Faussat
AM and Mascarelli F: The PI3K/Akt and mTOR/P70S6K signaling
pathways in human uveal melanoma cells: Interaction with B-Raf/ERK.
Invest Ophthalmol Vis Sci. 51:421–429. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rodríguez A, Dueñas-Gonzalez A and
Delgado-Pelayo S: Clinical presentation and management of uveal
melanoma. Mol Clin Oncol. 5:675–677. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ewens KG, Kanetsky PA, Richards-yutz J,
Al-Dahmash S, De Luca MC, Bianciotto CG, Shields CL and Ganguly A:
Genomic profile of 320 uveal melanoma cases: Chromosome 8p-loss and
metastatic outcome. Invest Ophthalmol Vis Sci. 54:5721–5729. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
van Gils W, Mensink HW, Kilic E, Vaarwater
J, Verbiest MM, Paridaens D, Luyten GP, de Klein A and Brüggenwirth
HT: Expression of APITD1 is not related to copy number changes of
chromosomal region 1p36 or the prognosis of uveal melanoma. Invest
Ophthalmol Vis Sci. 48:4919–4923. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
van Gils W, Kilic E, Brüggenwirth HT,
Vaarwater J, Verbiest MM, Beverloo B, van Til-Berg ME, Paridaens D,
Luyten GP and de Klein A: Regional deletion and amplification on
chromosome 6 in a uveal melanoma case without abnormalities on
chromosomes 1p, 3 and 8. Melanoma Res. 18:10–15. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Parrella P, Sidransky D and Merbs SL:
Allelotype of posterior uveal melanoma: Implications for a
bifurcated tumor progression pathway. Cancer Res. 59:3032–3037.
1999.PubMed/NCBI
|
|
30
|
Tschentscher F, Hüsing J, Hölter T, Kruse
E, Dresen IG, Jöckel KH, Anastassiou G, Schilling H, Bornfeld N,
Horsthemke B, et al: Tumor classification based on gene expression
profiling shows that uveal melanomas with and without monosomy 3
represent two distinct entities. Cancer Res. 63:2578–2584.
2003.PubMed/NCBI
|
|
31
|
Harbour JW: The genetics of uveal
melanoma: An emerging framework for targeted therapy. Pigment Cell
Melanoma Res. 25:171–181. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim JW, Choi EJ and Joe CO: Activation of
death-inducing signaling complex (DISC) by pro-apoptotic C-terminal
fragment of RIP. Oncogene. 19:4491–4499. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tummers B and Green DR: Caspase-8:
Regulating life and death. Immunol Rev. 277:76–89. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wong RSY: Apoptosis in cancer: From
pathogenesis to treatment. J Exp Clin Cancer Res. 30:872011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang RA, Li QL, Li ZS, Zheng PJ, Zhang HZ,
Huang XF, Chi SM, Yang AG and Cui R: Apoptosis drives cancer cells
proliferate and metastasize. J Cell Mol Med. 17:205–211. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pistritto G, Trisciuoglio D, Ceci C,
Garufi A and D'Orazi G: Apoptosis as anticancer mechanism: Function
and dysfunction of its modulators and targeted therapeutic
strategies. Aging (Albany NY). 8:603–619. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wiman KG: Strategies for therapeutic
targeting of the p53 pathway in cancer. Cell Death Differ.
13:921–926. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Parrish AB, Freel CD and Kornbluth S:
Cellular mechanisms controlling caspase activation and function.
Cold Spring Harb Perspect Biol. 5:52013. View Article : Google Scholar
|
|
40
|
Cancer Genome Atlas Network, . Genomic
Classification of cutaneous melanoma. Cell. 161:1681–1696. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kalinec G, Nazarali AJ, Hermouet S, Xu N
and Gutkind JS: Mutated alpha subunit of the Gq protein induces
malignant transformation in NIH 3T3 cells. Mol Cell Biol.
12:4687–4693. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Urtatiz O and Van Raamsdonk CD: Gnaq and
Gna11 in the endothelin signaling pathway and melanoma. Front
Genet. 7:592016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Croce M, Ferrini S, Pfeffer U and Gangemi
R: Targeted therapy of uveal melanoma: Recent failures and new
perspectives. Cancers (Basel). 11:8462019. View Article : Google Scholar
|
|
44
|
Rozengurt E: Mitogenic signaling pathways
induced by G protein-coupled receptors. J Cell Physiol.
213:589–602. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Landreville S, Agapova OA and Harbour JW:
Emerging insights into the molecular pathogenesis of uveal
melanoma. Future Oncol. 4:629–636. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Van Raamsdonk CD, Bezrookove V, Green G,
Bauer J, Gaugler L, O'Brien JM, Simpson EM, Barsh GS and Bastian
BC: Frequent somatic mutations of GNAQ in uveal melanoma and blue
naevi. Nature. 457:599–602. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ceraudo E, Horioka M, Mattheisen J,
Hitchman TD, Moore AR, Kazmi MA, Chi P, Chen Y, Sakmar TP and Huber
T: Uveal melanoma oncogene CYSLTR2 encodes a constitutively active
GPCR highly biased toward Gq signaling. bioRxiv. Jun 6–2019.(Epub
ahead of print). doi: org/10.1101/663153.
|
|
48
|
Chua V, Lapadula D, Randolph C, Benovic
JL, Wedegaertner PB and Aplin AE: Dysregulated GPCR signaling and
therapeutic options in uveal melanoma. Mol Cancer Res. 15:501–506.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Pandiani C, Béranger GE, Leclerc J,
Ballotti R and Bertolotto C: Focus on cutaneous and uveal melanoma
specificities. Genes Dev. 31:724–743. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zenonos K and Kyprianou K: RAS signaling
pathways, mutations and their role in colorectal cancer. World J
Gastrointest Oncol. 5:97–101. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zuidervaart W, van Nieuwpoort F, Stark M,
Dijkman R, Packer L, Borgstein AM, Pavey S, van der Velden P, Out
C, Jager MJ, et al: Activation of the MAPK pathway is a common
event in uveal melanomas although it rarely occurs through mutation
of BRAF or RAS. Br J Cancer. 92:2032–2038. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Fernández-Medarde A and Santos E: Ras in
cancer and developmental diseases. Genes Cancer. 2:344–358. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Prior IA, Lewis PD and Mattos C: A
comprehensive survey of Ras mutations in cancer. Cancer Res.
72:2457–2467. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Muñoz-Maldonado C, Zimmer Y and Medová M:
A comparative analysis of individual RAS mutations in cancer
biology. Front Oncol. 9:10882019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Mooy CM, Van der Helm MJ, Van der Kwast
TH, De Jong PT, Ruiter DJ and Zwarthoff EC: No N-ras mutations in
human uveal melanoma: The role of ultraviolet light revisited. Br J
Cancer. 64:411–413. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Soparker CN, O'Brien JM and Albert DM:
Investigation of the role of the ras protooncogene point mutation
in human uveal melanomas. Invest Ophthalmol Vis Sci. 34:2203–2209.
1993.PubMed/NCBI
|
|
57
|
Wennerberg K, Rossman KL and Der CJ: The
Ras superfamily at a glance. J Cell Sci. 118:843–846. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kolch W: Meaningful relationships: The
regulation of the Ras/Raf/MEK/ERK pathway by protein interactions.
Biochem J. 351:289–305. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Castellano E and Downward J: RAS
interaction with PI3K: More than just another effector pathway.
Genes Cancer. 2:261–274. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Liu P, Cheng H, Roberts TM and Zhao JJ:
Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev
Drug Discov. 8:627–644. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
O'Donnell JS, Massi D, Teng MW and Mandala
M: PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin
Cancer Biol. 48:91–103. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Saraiva VS, Caissie AL, Segal L, Edelstein
C and Burnier MN Jr: Immunohistochemical expression of phospho-Akt
in uveal melanoma. Melanoma Res. 15:245–250. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Cardone MH, Roy N, Stennicke HR, Salvesen
GS, Franke TF, Stanbridge E, Frisch S and Reed JC: Regulation of
cell death protease caspase-9 by phosphorylation. Science.
282:1318–1321. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Milella M, Falcone I, Conciatori F, Cesta
Incani U, Del Curatolo A, Inzerilli N, Nuzzo CM, Vaccaro V, Vari S,
Cognetti F, et al: PTEN: Multiple functions in human malignant
tumors. Front Oncol. 5:242015. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Chang H, Cai Z and Roberts TM: The
mechanisms underlying PTEN loss in human tumors suggest potential
therapeutic opportunities. Biomolecules. 9:7132019. View Article : Google Scholar
|
|
66
|
Woodman SE: Metastatic uveal melanoma:
Biology and emerging treatments. Cancer J. 18:148–152. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Abdel-Rahman MH, Yang Y, Zhou XP, Craig
EL, Davidorf FH and Eng C: High frequency of submicroscopic
hemizygous deletion is a major mechanism of loss of expression of
PTEN in uveal melanoma. J Clin Oncol. 24:288–295. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Dhillon AS, Hagan S, Rath O and Kolch W:
MAP kinase signalling pathways in cancer. Oncogene. 26:3279–3290.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Burotto M, Chiou VL, Lee JM and Kohn EC:
The MAPK pathway across different malignancies: A new perspective.
Cancer. 120:3446–3456. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Cohen C, Zavala-Pompa A, Sequeira JH,
Shoji M, Sexton DG, Cotsonis G, Cerimele F, Govindarajan B, Macaron
N and Arbiser JL: Mitogen-actived protein kinase activation is an
early event in melanoma progression. Clin Cancer Res. 8:3728–3733.
2002.PubMed/NCBI
|
|
71
|
Leicht DT, Balan V, Kaplun A, Singh-Gupta
V, Kaplun L, Dobson M and Tzivion G: Raf kinases: Function,
regulation and role in human cancer. Biochim Biophys Acta.
1773:1196–1212. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Barras D: BRAF mutation in colorectal
cancer: An update. Biomark Cancer. 7 (Suppl 1):9–12.
2015.PubMed/NCBI
|
|
73
|
Zaman A, Wu W and Bivona TG: Targeting
oncogenic BRAF: Past, present, and future. Cancers (Basel).
11:11972019. View Article : Google Scholar
|
|
74
|
Gaudi S and Messina JL: Molecular bases of
cutaneous and uveal melanomas. Pathol Res Int. 2011:1594212011.
View Article : Google Scholar
|
|
75
|
Glitza IC and Davies MA: Genotyping of
cutaneous melanoma. Chin Clin Oncol. 3:272014.PubMed/NCBI
|
|
76
|
Brose MS, Volpe P, Feldman M, Kumar M,
Rishi I, Gerrero R, Einhorn E, Herlyn M, Minna J, Nicholson A, et
al: Mutations in Human Lung Cancer and Melanoma. Cancer Res.
62:6997–7000. 2002.PubMed/NCBI
|
|
77
|
Shinozaki M, Fujimoto A, Morton DL and
Hoon DS: Incidence of BRAF oncogene mutation and clinical relevance
for primary cutaneous melanomas. Clin Cancer Res. 10:1753–1757.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Harbour JW: Genomic, prognostic, and
cell-signaling advances in uveal melanoma. Am Soc Clin Oncol Educ
book Am Soc Clin Oncol Annu Meet. 2013:388–391. 2013. View Article : Google Scholar
|
|
79
|
Mooy CM, Luyten GP, de Jong PT, Luider TM,
Stijnen T, van de Ham F, van Vroonhoven CC and Bosman FT:
Immunohistochemical and prognostic analysis of apoptosis and
proliferation in uveal melanoma. Am J Pathol. 147:1097–1104.
1995.PubMed/NCBI
|
|
80
|
Merhavi E, Cohen Y, Avraham BC, Frenkel S,
Chowers I, Pe'er J and Goldenberg-Cohen N: Promoter methylation
status of multiple genes in uveal melanoma. Invest Ophthalmol Vis
Sci. 48:4403–4406. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Shivakumar L, Minna J, Sakamaki T, Pestell
R and White MA: The RASSF1A tumor suppressor blocks cell cycle
progression and inhibits cyclin D1 accumulation. Mol Cell Biol.
22:4309–4318. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Kiliç E, Brüggenwirth HT, Verbiest MM,
Zwarthoff EC, Mooy NM, Luyten GP and de Klein A: The RAS-BRAF
kinase pathway is not involved in uveal melanoma. Melanoma Res.
14:203–205. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Helgadottir H and Höiom V: The genetics of
uveal melanoma: Current insights. Appl Clin Genet. 9:147–155. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Liu H, Du S, Lei T, Wang H, He X, Tong R
and Wang Y: Multifaceted regulation and functions of YAP/TAZ in
tumors (Review). Oncol Rep. 40:16–28. 2018.PubMed/NCBI
|
|
85
|
Meng Z, Moroishi T and Guan KL: Mechanisms
of Hippo pathway regulation. Genes Dev. 30:1–17. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Plouffe SW, Hong AW and Guan KL: Disease
implications of the Hippo/YAP pathway. Trends Mol Med. 21:212–222.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Totaro A, Panciera T and Piccolo S:
YAP/TAZ upstream signals and downstream responses. Nat Cell Biol.
20:888–899. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Gumbiner BM and Kim NG: The Hippo-YAP
signaling pathway and contact inhibition of growth. J Cell Sci.
127:709–717. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Field MG and Harbour JW: GNAQ/11 mutations
in uveal melanoma: Is YAP the key to targeted therapy? Cancer Cell.
25:714–715. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Feng X, Degese MS, Iglesias-Bartolome R,
Vaque JP, Molinolo AA, Rodrigues M, Zaidi MR, Ksander BR, Merlino
G, Sodhi A, et al: Hippo-independent activation of YAP by the GNAQ
uveal melanoma oncogene through a trio-regulated rho GTPase
signaling circuitry. Cancer Cell. 25:831–845. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Warren JS, Xiao Y and Lamar JM: YAP/TAZ
activation as a target for treating metastatic Cancer. Cancers
(Basel). 10:102018. View Article : Google Scholar
|
|
92
|
Zanconato F, Battilana G, Cordenonsi M and
Piccolo S: YAP/TAZ as therapeutic targets in cancer. Curr Opin
Pharmacol. 29:26–33. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Moroishi T, Hansen CG and Guan KL: The
emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. 15:73–79.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Feng X, Rigiracciolo D, Lee JS, Yeerna H,
Arang N, Lubrano S, Schlaepfer DD, Tamayo P, Ruppin E and Gutkind
JS: Abstract 968: Targeting FAK inhibits YAP-dependent tumor growth
in uveal melanoma. Cancer Res. 78:9682018.
|
|
95
|
Liu H and Zhou M: Evaluation of p53 gene
expression and prognosis characteristics in uveal melanoma cases.
OncoTargets Ther. 10:3429–3434. 2017. View Article : Google Scholar
|
|
96
|
Hajkova N, Hojny J, Nemejcova K, Dundr P,
Ulrych J, Jirsova K, Glezgova J and Ticha I: Germline mutation in
the TP53 gene in uveal melanoma. Sci Rep. 8:76182018. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Sun Y, Tran BN, Worley LA, Delston RB and
Harbour JW: Functional analysis of the p53 pathway in response to
ionizing radiation in uveal melanoma. Invest Ophthalmol Vis Sci.
46:1561–1564. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Shi D and Gu W: Dual Roles of MDM2 in the
regulation of p53: Ubiquitination dependent and ubiquitination
independent mechanisms of MDM2 tepression of p53 sctivity. Genes
Cancer. 3:240–248. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Hussein MR: The relationships between p53
protein expression and the clinicopathological features in the
uveal melanomas. Cancer Biol Ther. 4:57–59. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Evan GI, Wyllie AH, Gilbert CS, Littlewood
TD, Land H, Brooks M, Waters CM, Penn LZ and Hancock DC: Induction
of apoptosis in fibroblasts by c-myc protein. Cell. 69:119–128.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Schwartz LH, Ferrand R, Boelle PY, Maylin
C and D'Hermies F, Virmont J and D'Hermies F: Lack of correlation
between the location of choroidal melanoma and
ultraviolet-radiation dose distribution. Radiat Res. 147:451–456.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Lim S and Kaldis P: Cdks, cyclins and
CKIs: roles beyond cell cycle regulation. Development.
140:3079–3093, 20132. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Ding L, Cao J, Lin W, Chen H, Xiong X, Ao
H, Yu M, Lin J and Cui Q: The roles of cyclin-dependent kinases in
cell-cycle progression and therapeutic strategies in human breast
cancer. Int J Mol Sci. 21:19602020. View Article : Google Scholar
|
|
104
|
Ando K, Ajchenbaum-Cymbalista F and
Griffin JD: Regulation of G1/S transition by cyclins D2 and D3 in
hematopoietic cells. Proc Natl Acad Sci USA. 90:9571–9575. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Bartkova J, Lukas J, Strauss M and Bartek
J: Cyclin D3: Requirement for G1/S transition and high abundance in
quiescent tissues suggest a dual role in proliferation and
differentiation. Oncogene. 17:1027–1037. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Bonelli P, Tuccillo FM, Borrelli A,
Schiattarella A and Buonaguro FM: CDK/CCN and CDKI alterations for
cancer prognosis and therapeutic predictivity. BioMed Res Int.
2014:3610202014. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Li J, Poi MJ and Tsai MD: Regulatory
mechanisms of tumor suppressor P16(INK4A) and their relevance to
cancer. Biochemistry. 50:5566–5582. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
McConnell BB, Gregory FJ, Stott FJ, Hara E
and Peters G: Induced expression of p16(INK4a) inhibits both CDK4-
and CDK2-associated kinase activity by reassortment of
cyclin-CDK-inhibitor complexes. Mol Cell Biol. 19:1981–1989. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Satyanarayana A and Rudolph KL: p16 and
ARF: Activation of teenage proteins in old age. J Clin Invest.
114:1237–1240. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Macleod KF, Sherry N, Hannon G, Beach D,
Tokino T, Kinzler K, Vogelstein B and Jacks T: p53-dependent and
independent expression of p21 during cell growth, differentiation,
and DNA damage. Genes Dev. 9:935–944. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Mouriaux F, Maurage CA, Labalette P,
Sablonnière B, Malecaze F and Darbon JM: Cyclin-dependent kinase
inhibitory protein expression in human choroidal melanoma tumors.
Invest Ophthalmol Vis Sci. 41:2837–2843. 2000.PubMed/NCBI
|
|
112
|
Abbas T and Dutta A: p21 in cancer:
Intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Abukhdeir AM and Park BH: P21 and p27:
Roles in carcinogenesis and drug resistance. Expert Rev Mol Med.
10:e192008. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Blain SW, Scher HI, Cordon-Cardo C and
Koff A: p27 as a target for cancer therapeutics. Cancer Cell.
3:111–115. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Mouriaux F, Casagrande F, Pillaire MJ,
Manenti S, Malecaze F and Darbon JM: Differential expression of G1
cyclins and cyclin-dependent kinase inhibitors in normal and
transformed melanocytes. Invest Ophthalmol Vis Sci. 39:876–884.
1998.PubMed/NCBI
|
|
116
|
Narasimha AM, Kaulich M, Shapiro GS, Choi
YJ, Sicinski P and Dowdy SF: Cyclin D activates the Rb tumor
suppressor by mono-phosphorylation. eLife. 3:e028722014. View Article : Google Scholar
|
|
117
|
Rayess H, Wang MB and Srivatsan ES:
Cellular senescence and tumor suppressor gene p16. Int J Cancer.
130:1715–1725. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Nagarkatti-Gude N, Wang Y, Ali MJ, Honavar
SG, Jager MJ and Chan CC: Genetics of primary intraocular tumors.
Ocul Immunol Inflamm. 20:244–254. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Bartek J, Bartkova J and Lukas J: The
retinoblastoma protein pathway in cell cycle control and cancer.
Exp Cell Res. 237:1–6. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Brantley MA Jr and Harbour JW:
Inactivation of retinoblastoma protein in uveal melanoma by
phosphorylation of sites in the COOH-terminal region. Cancer Res.
60:4320–4323. 2000.PubMed/NCBI
|
|
121
|
Pollock PM, Harper UL, Hansen KS, Yudt LM,
Stark M, Robbins CM, Moses TY, Hostetter G, Wagner U, Kakareka J,
et al: High frequency of BRAF mutations in nevi. Nat Genet.
33:19–20. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
122
|
Hollern DP, Honeysett J, Cardiff RD and
Andrechek ER: The E2F transcription factors regulate tumor
development and metastasis in a mouse model of metastatic breast
cancer. Mol Cell Biol. 34:3229–3243. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Timmers C, Sharma N, Opavsky R, Maiti B,
Wu L, Wu J, Orringer D, Trikha P, Saavedra HI and Leone G: E2f1,
E2f2, and E2f3 control E2F target expression and cellular
proliferation via a p53-dependent negative feedback loop. Mol Cell
Biol. 27:65–78. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Lundberg AS and Weinberg RA: Functional
inactivation of the retinoblastoma protein requires sequential
modification by at least two distinct cyclin-cdk complexes. Mol
Cell Biol. 18:753–761. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Calipel A, Abonnet V, Nicole O, Mascarelli
F, Coupland SE, Damato B and Mouriaux F: Status of RASSF1A in uveal
melanocytes and melanoma cells. Mol Cancer Res. 9:1187–1198. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Yang ZK, Yang JY, Xu ZZ and Yu WH: DNA
Methylation and Uveal Melanoma. Chin Med J (Engl). 131:845–851.
2018. View Article : Google Scholar : PubMed/NCBI
|