Introduction
Pancreatic cancer is a highly malignant tumor of the
digestive system. In the United States, in 2018, an estimated
56,770 cases of pancreatic cancer occurred, and 45,750 patients
succumbed to the disease (1).
Pancreatic cancer is one of the most fatal types of cancer, for its
mortality rates closely parallels its incidence rates (2). Most patients have obvious symptoms,
particularly in the advanced stage of pancreatic cancer (2). Gemcitabine is a widely used drug for
the treatment of pancreatic cancer, to improve the surgical success
ratio. However, its serious side effects attenuate its efficacy,
and cause harm to patients (3).
Therefore, it is crucial to identify an efficacious drug, with
fewer serious side effects, for the treatment of pancreatic
cancer.
Ganoderma lucidum is a Traditional Chinese
Medicine. Several biological activities and pharmacological
functions of polysaccharides, proteins, and triterpenoids, found in
Ganoderma lucidum, have been reported, including the defense
of tumors, management of blood sugar levels, modulation of
immunity, and protection of hepatocytes (4). In our previous study a proteoglycan,
Fudan-Yueyang Ganoderma lucidum (FYGL) was extracted from the
Ganoderma lucidum fruiting body (5). FYGL is composed of both proteins and
polysaccharides, and its molecular weight is 2.6×105.
FYGL is an amphiphilic hyperbranched proteoglycan consisting of
hydrophilic polysaccharides (4 saccharide units) and lipophilic
protein moieties (17 amino acids), with a saccharide:protein ratio
of 77:17 (6). Our previous study
demonstrated that, FYGL could efficiently and safely manage blood
sugar levels in vivo, and affect multiple signaling pathways
for insulin regulation (5,7,8).
Oxidative stress is crucial for both anticancer
effects and cancer development (9).
Excessive reactive oxygen species (ROS) can damage the cancer cell
structures, including lipids, proteins, and DNA, resulting in
cancer cell death, apoptosis and autophagy (10,11).
Furthermore, high levels of ROS, and the resultant oxidative
damage, can decrease the mitochondrial membrane potential (MMP),
leading to cancer cell death (12,13).
However, the production of ROS by mammalian mitochondria has been
associated with multiple pathologies, including neurodegenerative
diseases (14), diabetes (15), cancer (16) and premature aging (17). Therefore, the regulation of ROS at a
suitable concentration in the body is crucial for maintaining
health. In addition, the autophagy of cancer cells is a catabolic
pathway for supporting metabolism in response to starvation, and
for removing damaged proteins and organelles in response to stress
(18). Autophagy maintains
mitochondrial metabolic function, which is beneficial for the
growth of aggressive cancers (19);
however, pharmacological inhibition of autophagy can cause the
accumulation of defective mitochondria, and consequently lead to
metabolomic issues (20). Cell
autophagy is essential for pancreatic cancer activity and it has
been found that inhibition of autophagy in cancer cells through
genetically and pharmacologically increasing ROS; therefore,
damaging DNA, is an alternative method of inducing apoptosis in
pancreatic cancer cells (21,22).
In the present study, the anti-pancreatic cancer
ability of FYGL was investigated and to compare its selectivity in
3 different pancreatic cancer cell lines (PANC-1, BxPC-3 and Mia
PaCa-2), as well as in the HepG2 hepatic cell line. In addition,
the effect of FYGL on cell migration, colony formation, ROS
production, and cell autophagy were also investigated. The results
indicated that FYGL was effective at inducing apoptosis in the
PANC-1 cells, by increasing ROS and inhibiting autophagy.
Materials and methods
Cell lines and reagents
FYGL was extracted from Ganderma lucidum in
our laboratory (5). The human
pancreatic cancer cell line, PANC-1, and the human liver cancer
cell line, HepG2, were obtained from Procell Life Science &
Technology Co., Ltd., while the human pancreatic cancer cell line,
Mia PaCa-2, was donated by Professor Wuli Yang (Fudan University,
Shanghai, China). The human pancreatic cancer cell line, BxPC-3,
was donated by Doctor Yiqun Ma (Zhongshan Hospital, Shanghai,
China). The PANC-1, Mia PaCa-2 and HepG2 cells were cultured in
DMEM (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10%
FBS (Gibco; Thermo Fisher Scientific, Inc.), 1%
penicillin/streptomycin and 1% L-glutamine (Thermo Fisher
Scientific, Inc.). The BxPC-3 cells were cultured in RPMI-1640
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS,
1% penicillin/streptomycin and 1% L-glutamine. Cells were incubated
at 37°C in a humidified atmosphere with 5% CO2. The
following antibodies: Caspase-3 (dilution 1:1,000; cat. no. 9662S),
cleaved-caspase-3 (dilution 1:1,000; cat. no. 9661S), Bcl-2
(dilution 1:1,000; cat. no. 4223S), P62 (dilution 1:1,000; cat. no.
39749S), LC3A/B (dilution 1:1,000; cat. no. 12741S), β-actin
(dilution 1:1,000; cat. no. 4970S), GAPDH (dilution 1:1,000; cat.
no. 5174S), and HRP-conjugated secondary anti-rabbit (dilution
1:2,000; cat. no. 7074S) were all purchased from Cell Signaling
Inc.
Cell viability assay
The cells were seeded in 96-well plates at 37°C in a
humidified atmosphere with 5% CO2, at a density of
5×104 cells/well. After 24 h, the cells in each well
were treated with FYGL at a concentration range of 0–1,000 µg/ml,
and gemcitabine (Eli Lilly and Company) as a positive control at a
concentration range of 5–20 µM for 24 and 48 h, then, 10 µl Cell
Counting Kit-8 (CCK-8) reagent (Shanghai Yeasen Biotechnology Co.,
Ltd.) was added into each well. The absorbance was measured at 450
nm, using a microplate reader (Cytation 3; BioTek Instruments,
Inc.), 1–4 h later.
Confocal microscopy analysis
The cells were seeded in cell culture dishes (Wuxi
NEST Biotechnology Co., Ltd.) at a density of 1×105
cells/well at 37°C in a humidified atmosphere with 5%
CO2. After treatment with 0 (control) and 150 µg/ml
FYGL, the cells were fixed with 4% paraformaldehyde (PFA) for 10
min at 37°C, then permeabilized with 0.3% Triton X-100 (Sinopharm
Chemical Reagent Co., Ltd.). FYGL was stained with a green
fluorescent agent, fluorescein isothiocyanate (FITC), and the cell
nucleus and cytoskeleton were stained with a blue fluorescent
agent, 4′,6-diamidino-2-phenylindole (DAPI), and a red fluorescent
agent, rhodamine-labeled phalloidin (all from Shanghai Yeasen
Biotech Co., Ltd.), respectively for 10 min at 37°C. Images were
obtained using a laser scanning confocal microscope (magnification,
×100; LSCM; Nikon C2+; Nikon Corporation).
Wound healing assay
The cells were seeded (5×105 cells/well)
in 6-well plates at 37°C in a humidified atmosphere with 5%
CO2, and the cell monolayer was scratched with a pipette
tip when the cells grew to a single layer. Following which, the
cells were treated with FYGL at a concentration range of 0–1,000
µg/ml in DMEM medium with 3% FBS for 48 h. Cell images were
obtained using an inverted optical microscope (Nikon ECLIPSE Ts2;
Nikon Corporation) to observe the migration of the cells across the
wound (magnification, ×4). ImageJ software (v1.51j8; National
Institutes of Health) was used for digital analysis of the wound
healing area.
Colony viability assay
The cells were seeded in 6-well culture plates at
37°C in a humidified atmosphere with 5% CO2, at a
density of 200 cells/well and incubated for 24 h. Then, the cells
in each well were treated with FYGL at a concentration range of
0–1,000 µg/ml for 12 days. All colonies were fixed with 4% PFA for
10 min at 37°C, and washed with PBS (Sangon Biotech Co., Ltd.)
subsequently, the cells in each well were stained with a purple
dye, giemsa (Shanghai Yeasen Biotechnology Co., Ltd.) for 10 min at
37°C. Cell images were obtained using an optical camera
(magnification, ×1; Sony α6400; Sony Corporation).
Cell apoptosis assay
The percentage of apoptotic cells was determined
using an Annexin V-FITC/PI kit (Shanghai Yeasen Biotechnology, Co.,
Ltd.). Cells were seeded in 6-well plates, at a density of
5×105 cells/well. After incubation for 24 h, the cells
were treated with FYGL at a concentration range of 0–500 µg/ml for
24 h and harvested using 0.25% trypsin (Sangon Biotech Co., Ltd.),
then the cells were incubated with 5 µl Annexin V-FITC and 10 µl PI
working solution for 30 min at 37°C. Finally, the stained cells
were analyzed using flow cytometry (Beckman Coulter, Inc.), and the
data were analyzed using FlowJo software (version 10.4; BD
Biosciences).
Measurement of intracellular ROS
levels
The fluorescent agent,
2,7-dichlorodi-hydrofluorescein diacetate (DCFH-DA; Shanghai Yeasen
Biotech Co., Ltd.) was used to investigate ROS production. The
cells were seeded in 6-well plates at 37°C in a humidified
atmosphere with 5% CO2, at a density of 5×105
cells/well. After incubation for 24 h, the cells were treated with
FYGL at a concentration range of 0–500 µg/ml for 24 h. Following
which, the cells were washed with PBS and added to 1 ml serum-free
medium with 10 µl DCFH-DA per well. After incubation for 30 min at
37°C, the level of ROS in the cells were identified using DCFH, and
analyzed using flow cytometry (Beckman Coulter, Inc.), and the data
were analyzed using FlowJo software (version 10.4; BD
Biosciences).
Analysis of mitochondrial membrane
potential
The cells were seeded in cell culture dishes at 37°C
in a humidified atmosphere with 5% CO2, at a density of
1×105 cells/well and incubated for 24 h, then, the cells
were treated with FYGL at a concentration range of 0–1,000 µg/ml
for 24 h. The medium was removed and the cells were washed with
PBS, then stained with 10 µl rhodamine 123 (Rh-123; a green
fluorescent agent; 5 mg/ml; Shanghai Yeasen Biotech Co. Ltd.) for
30 min in the dark at 37°C, to detect the mitochondrial membrane
potential. Following which, the cells were fixed with 4% PFA for 10
min at 37°C, and permeabilized with 0.3% Triton X-100.
Subsequently, the cell nucleus and cytoskeleton were stained blue
with DAPI and red with rhodamine-labeled phalloidin for 10 min at
37°C, respectively. Images were taken by LSCM (magnification, ×60;
Nikon C2+, Nikon Corporation).
Detection of autophagy
The cells were seeded in cell culture dishes at 37°C
in a humidified atmosphere with 5% CO2, at a density of
1×105 cells/well then, treated with FYGL at a
concentration range of 0–1,000 µg/ml for 24 h. After washing with
PBS three times, the cells were incubated with a Cyto-ID autophagy
detection kit (Enzo Life Sciences, Inc.) for 30 min at 37°C, which
including a mitochondrial staining reagent Cyto-ID and nuclear
staining reagent Hoechst 33258, subsequently were washed with PBS
and fixed with 4% PFA for 15 min at 37°C. Images were obtained
using LSCM (magnification, ×60; Nikon C2+; Nikon Corporation).
Western blot analysis
Total protein was lysed from the cells using a RIPA
containing phenylmethylsulfonyl fluoride, then the concentration
was measured using a BCA protein assay kit (all from Shanghai
Yeasen Biotech Co., Ltd.). Following which, the lysed proteins were
separated using a 10–15% SDS-PAGE and transferred to PVDF membranes
(Beyotime Institute of Biotechnology). The membranes were blocked
with a buffer containing 10 mM Tris-HCl, (pH 7.4), 150 mM NaCl,
0.1% Tween-20, and 5% skimmed milk (Beyotime Institute of
Biotechnology), then incubated with the primary antibodies for 12 h
at 4°C, followed by incubation with the HRP-conjugated secondary
antibody for 1 h at 25°C. The antibodies were detected using a
super enhanced chemiluminescence detection reagent (Shanghai Yeasen
Biotech Co., Ltd.) and a chemiluminescent imaging system (Bio-Rad
ChemiDoc MP; Bio-Rad Laboratories, Inc.). GAPDH and β-actin were
used as the loading controls.
Imaging mCherry-green fluorescent
protein-LC3B (mCherry-GFP-LC3) fusion protein
The adenovirus expressing (Ad)-mCherry-GFP-LC3
fusion protein (Beyotime Institute of Biotechnology) was used to
investigate the binding of autophagosomes and lysosomes. The PANC-1
cell line was transfected with ad-mCherry-GFP-LC3 adenovirus, at 20
multiplicity of infection for 24 h at 37°C. Then, the culture
medium was removed and fresh DMEM with FYGL at a concentration
range of 0–500 µg/ml was added. After 24 h, the cells were detected
with LSCM (magnification, ×60; Nikon C2+; Nikon Corporation).
Statistical analysis
The results are presented as the mean ± standard
deviation (n=3). SPSS software (v20.0; IBM Corp.) was used for
statistical analyses. ImageJ software (v1.51j8; National Institutes
of Health) was used for digital analysis of the western blots.
One-way ANOVA was performed to compare the mean values among
multiple groups followed by Tukey's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Inhibition of pancreatic cancer cell
viability by FYGL
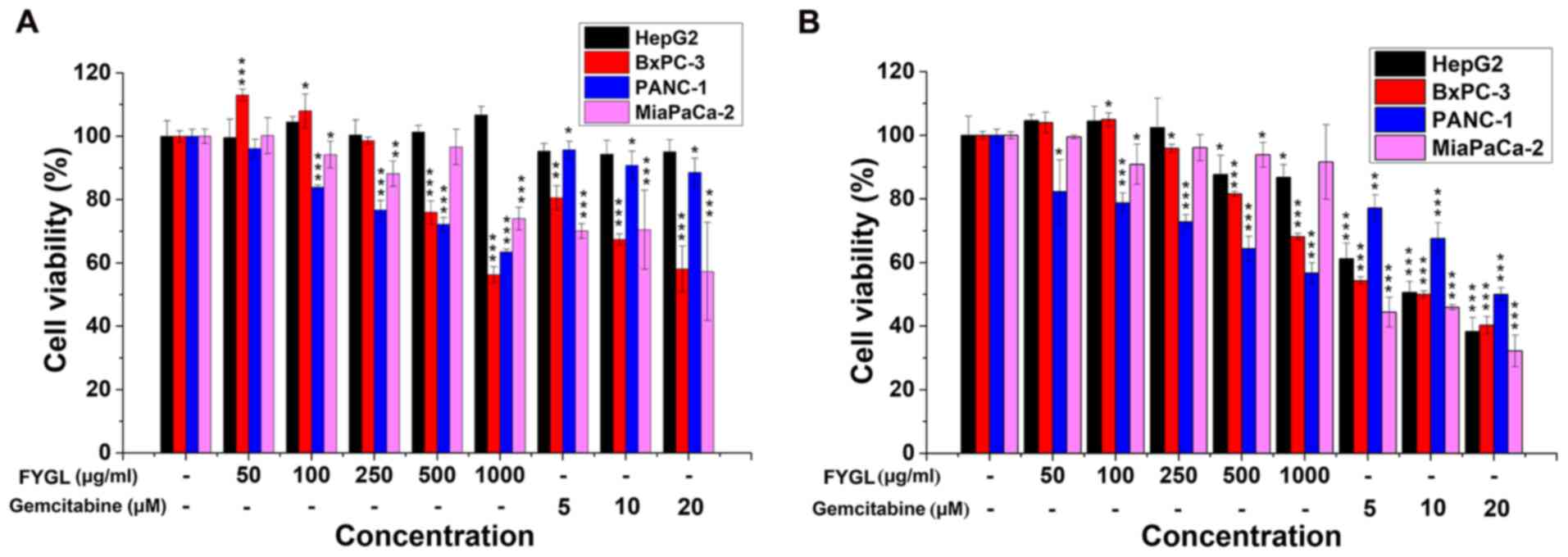
To investigate the effects of FYGL on the viability
of 2 different cancer cell lines, three pancreatic cancer cell
lines, PANC-1, BxPC-3, and Mia PaCa-2, and one hepatic cell line,
HepG2, were treated with FYGL, at a concentration range of 50–1,000
µg/ml, with gemcitabine, as a positive control, at a concentration
range of 5–20 µM. All the cell lines were treated for 24 (Fig. 1A) and 48 h (Fig. 1B), then the viability was
investigated using a CCK-8 assay. The results indicated that FYGL
had different effects on cell viability in the 2 different cancer
cell lines. The viability of PANC-1 and BxPC-3 cells was markedly
decreased by the treatment of FYGL in a dose dependent manner at
both 24 and 48 h, comparable with the cells treated with the
positive agent, gemcitabine. The viability of the Mia PaCa-2 cells
was not affected by FYGL treatment in a dose dependent manner, both
at 24 and 48 h. As well as in HepG2 cells, however, gemcitabine was
markedly toxic for all the cell lines following treatment for 48 h.
The results, therefore, indicated that FYGL was moderate, safe, and
selective for the given cell lines.
FYGL absorption in cells
To understand the functional mechanism of FYGL in
the cells, the PANC-1 and Mia PaCa-2 cell lines were selected for
further research. First, the absorption of FYGL into the cells was
investigated. The FYGL, cell nucleus, and cytoskeleton were all
labeled with fluorescent agents namely: FITC (green), DAPI (blue),
and rhodamine-labeled phalloidin (red). As shown in Fig. 2, the green fluorescence from FYGL
overlapped with the red fluorescence from the cytoskeleton.
Furthermore, some green fluorescent areas were found between the
red and the blue fluorescent areas (from the nucleus labelled with
DAPI), indicating that FYGL was encapsulated by vesicles and
dispersed into the cytoplasm. Some green fluorescent areas were
found to be outside of the cells, indicating not all of the FYGL
was absorbed into the cells; however, most of the FYGL was absorbed
in the cells.
Effects of FYGL on cell migration and
colony formation
Cell migration in cell treated with FYGL was
determined using a wound healing assay. Both PANC-1 and Mia PaCa-2
cells were treated with FYGL for 48 h. The cell migration of
PANC-1 cells into the wound area was halted by the increased
concentrations of FYGL (Fig. 3A).
The wound healing percentage of PANC-1 cells significantly
decreased at 250, 500 and 1,000 µg/ml concentrations of FYGL
compared with that in the control group (Fig. 3B). However, the wound healing
percentage of Mia PaCa-2 cells increased at 250 µg/ml
concentrations of FYGL and decreased at 1,000 µg/ml concentrations
of FYGL, but no significant differences were observed compared with
that in the control group, which indicated that cell migration was
not affected by FYGL in the Mia PaCa-2 cells (Fig. 3C).
Fig. 3D and E showed
the colony formation of the PANC-1 and Mia PaCa-2 cells treated
with different concentrations of FYGL. A total of 200 cells, in
each well, were incubated with FYGL for 12 days. The colony
formation of the PANC-1 cells was low at higher concentrations,
indicating sensitivity to FYGL treatment, while there was a high
number of colonies with the Mia PaCa-2 cell line indicating
insensitivity to FYGL treatment.
Effects of FYGL on apoptosis of the
pancreatic cancer cells
Apoptosis of cancer cells is a critical index for an
anticancer drug (23), and is
typically investigated using an Annexin V-FITC/PI kit and measured
using flow cytometry (24). The
apoptotic rate of the PANC-1 cells increased to 23%, as the
concentration of FYGL increased to 500 µg/ml (Fig. 4A), however, the apoptotic rate of Mia
PaCa-2 cells was unaffected (Fig.
4B).
Furthermore, western blot analysis was used to
analyze the expression levels of key proteins for example,
caspase-3, Bcl-2 and cleaved-caspase-3, which are involved in
apoptosis. The production of cleaved caspase-3 from caspase-3
promotes cell apoptosis, while the expression level of Bcl-2
prevents the cells from undergoing apoptosis (25,26).
Fig. 5A shows that the protein
expression levels of caspase-3 and Bcl-2 in the PANC-1 cells
significantly decreased at 250, 500 and 1,000 µg/ml concentrations
of FYGL compared with that in the control group, while Fig. 5B shows that the protein expression
level of cleaved-caspase-3 in the PANC-1 cells was significantly
increased, at 500 and 1,000 µg/ml concentrations of FYGL, compared
with that in the control group. The results indicated that cell
apoptosis in the PANC-1 cells was induced by FYGL.
However, Fig. 5C and
D demonstrated that the protein expression levels of caspase-3
in the Mia PaCa-2 cells decreased at 50 and 100 µg/ml
concentrations of FYGL and increased at 500 and 1,000 µg/ml
concentrations of FYGL, but no significant differences were
observed compared with that in the control group. The protein
expression levels of Bcl-2 in the Mia PaCa-2 cells increased at 50,
100 and 500 µg/ml concentrations of FYGL, but no significant
differences were observed compared with that in the control group.
The protein expression levels of cleaved-caspase-3 in the Mia
PaCa-2 cells increased at 50 µg/ml concentrations of FYGL and
decreased at 250, 500 and 1,000 µg/ml concentrations of FYGL, but
no significant differences were observed compared with that in the
control group. These results indicated that FYGL does not induce
Mia PaCa-2 cell apoptosis.
FYGL effects on oxidative stress and
MMP
The fluorescent probe, DCFH-DA was used to detect
ROS in the cells using flow cytometry. Fig. 6A showed that the ROS level in the
PANC-1 cells significantly increased as the concentration of FYGL
increased from 0 to 500 µg/ml; however, there was no increase in
the Mia PaCa-2 cells (Fig. 6B). The
decrease of MMP has been associated with the increase of ROS, and
with cell apoptosis (11,27).
Therefore, in the subsequent experiments, the green
fluorescent agent, Rh-123 was used to detect the MMP in the PANC-1
and Mia PaCa-2 cell lines, treated with FYGL for 24 h and analyzed
using confocal microscopy. Fig. 7A and
B showed the fluorescent images of the PANC-1 and Mia PaCa-2
cell lines, respectively; wherein blue, green, and red represent
the cell nucleus (DAPI), MMP (Rh-23), and cytoskeleton
(rhodamine-labeled phalloidin), respectively. The MMP was found to
be deceased in the PANC-1 cells, as the concentrations of FYGL
increased; however, there was no marked change in the Mia PaCa-2
cells, which were consistent with the results from the ROS
experiments. The results indicated that FYGL could selectively
induce the production of ROS, leading to the reduction of the MMP
and apoptosis in the PANC-1 cells line.
Effects of FYGL on autophagy of the
pancreatic cancer cells
The cell autophagy process occurs from autophagosome
formation, along with organelle damage, to the movement of
autophagosomes into lysosomes for the recycling of damaged
organelles (18). The
polyubiquitinated protein, LC3-I/II, and the polyubiquitin-binding
protein, P62, are autophagic effector proteins. During the cell
autophagy process, LC3-I is covalently bound to
phosphatidylethanolamine (PE) to form LC3-II, which then localizes
on the autophagosome membrane (28).
Once the autophagy process has been completed, the P62 protein is
degraded, and the autophagosomes enter the lysosomes (29). Fig. 8A
showed that the autophagosomes, labeled with the green fluorescent
agent, cyto-ID, increased, as the concentration of FYGL increased,
in the PANC-1 cells treated with FYGL for 24 h, suggesting that
FYGL promoted autophagosome formation in the PANC-1 cell line. FYGL
had almost no effect on the fluorescence intensity in the Mia
PaCa-2 cell line, indicating that FYGL did not induce autophagosome
formation in the Mia PaCa-2 cell line (Fig. 8B).
Fig. 9A and B showed
the protein expression level of LC3-I/II and P62 in the PANC-1 and
Mia PaCa-2 cells, respectively, treated with different
concentrations of FYGL for 24 h. In the PANC-1 cells, the cascade
protein ratio of LC3-I/II significantly decreased at 50, 100, 250,
500 and 1,000 µg/ml concentrations of FYGL compared with that in
the control group further indicating that there was an increase in
the number of autophagosomes in the PANC-1 cells. However, it was
found that in the PANC-1 cells treated with different
concentrations of FYGL, the relative protein expression level of
P62 was significantly increased, at 500 and 1,000 µg/ml compared
with that in the control group, indicating that the cell autophagy
process was halted, and not completed in the signaling pathway. In
the Mia PaCa-2 cell line, the ratio of LC3-I/II decreased at 50,
250, 500 and 1,000 µg/ml concentrations of FYGL and increased at
100 µg/ml concentrations of FYGL, but no significant differences
were observed compared with that in the control group. The protein
expression levels of P62 in the Mia PaCa-2 cells increased at 500
and 1,000 µg/ml concentrations of FYGL, but no significant
differences were observed compared with that in the control group.
These results indicated that FYGL had almost no effect on
the autophagy of Mia PaCa-2 cells.
In addition, the fluorescent adenovirus,
ad-mCherry-GFP-LC3 was used to confirm that the autophagosomes
enter the lysosomes. When the ad-mCherry-GFP-LC3 adenovirus, bound
to the autophagosomes, are present in the lysosomes of cells, the
GFP in the fusion protein is quenched. Fig. 9C shows the fluorescence images of the
PANC-1 cells infected with Ad-mCherry-GFP-LC3 and treated with (500
µg/ml) or without (control) FYGL for 24 h. It was found that the
fluorescent intensity of GFP in FYGL-treated cells was markedly
higher compared with that in the control cells, suggesting that
FYGL prevented autophagosomes from entering the lysosomes.
Discussion
The present study demonstrated that FYGL could
promote cell apoptosis in the PANC-1 cell line, but not in the Mia
PaCa-2 cell line. The Bcl-2 protein is one of the most important
anti-apoptotic proteins, and prevents the loss of MMP (26). ROS can damage lipids, proteins, DNA,
and organelles, leading to cell apoptosis (12). Numerous studies (30–34) have
found that drugs, such as Berberine, BML-275 and Resveratrol could
induce apoptosis of the PANC-1 and MiaPaCa-2 cell lines via ROS
production, without selectivity, indicating that ROS has the same
sensitivity for both the PANC-1 and Mia PaCa-2 cell lines treated
with those drugs; therefore, we hypothesized that a similar
mechanism was involved following treatment with FYGL.
Western blot analysis showed that the protein expression level of
the Bcl-2 protein was decreased in the PANC-1 cell line treated
with FYGL. In addition, the ROS was also increased, as a result,
the caspase-3/cleaved-caspase-3 proteins were activated, which led
to a reduction in MMP and ultimately, PANC-1 cell apoptosis;
however, this did not occur in the Mia PaCa-2 cell line.
Furthermore, ROS also plays a crucial role in the
activation of cell autophagy (18,35).
Cell autophagy can provide nutrients and energy for cancer cells,
promote the development of cancer cells, and leads to cancer cell
resistance to treatment. The excessive accumulation of ROS can
induce cell autophagy (36). In the
present study, the ratio of LC3-I/II was decreased in PANC-1 cells,
indicating an increase in the number of autophagosomes. However,
the protein expression level of P62 was increased in the PANC-1
cells treated with 500 and 1,000 µg/ml FYGL compared with that in
the control cells, indicating an inhibition of autophagy. We
hypothesized that the failure of the autophagy process resulted
from the inhibition of autophagosomes from entering the lysosomes.
A schematic of FYGL killing pancreatic cancer cells was proposed,
as shown in Fig. 10. First, FYGL
was absorbed into the cells probably through the process of
micropinocytosis (37). Then, FYGL
decreased MMP and induced ROS production through inhibiting the
expression of Bcl-2 protein. The increase of ROS production damaged
organelles and proteins, which activated both the cell autophagy
and cell apoptosis pathways. More autophagosomes were synthesized
to wrap damaged organelles, consuming lots of LC3-I protein and PE
to synthesize LC3-II protein, which is an important part of the
autophagosome membrane. However, FYGL halts the downstream process
of autophagy by inhibiting the fusion of autophagosomes and
lysosomes, which caused the upregulation of P62 proteins and
increased the production of ROS, as well as induced metabolic
deficiency. The increase of ROS and the blockage of autophagy
promoted apoptosis in the PANC-1 cells in the present study.
 | Figure 10.The proposed mechanism of FYGL
killing the PANC-1 cancer cells. FYGL inhibits Bcl-2 protein
expression and, selectively, induces the increase in ROS, leading
to the decrease of caspase-3/cleaved-caspase-3, and simultaneously
halts the downstream process of autophagy by inhibiting the fusion
of autophagosomes to lysosomes, leading to cell metabolic
deficiency. Both processes result in cancer cell apoptosis. Green
arrow, increase; red arrow, decrease; FYGL, Fudan-Yueyang-Ganoderma
lucidum; PE, phosphatidylethanolamine; ROS, reactive oxygen
species. |
In addition, Boya et al (22) reported that autophagy inhibition lead
to the accumulation of defective mitochondria and an increase in
ROS production, while Humpton et al (38) found that KRAS-NIX-mediated mitophagy
was a novel driver of glycolysis and redox activities. Future
studies will investigate whether the energy metabolic processes of
pancreatic cancer cells are forced to switch under the treatment of
FYGL.
In pancreatic cells, macropinocytosis drives the
internalization of extracellular proteins and saccharides, and
thereafter degradation in lysosomes, to produce amino acids and
sugars (39,40). When nutrients, such as amino acids,
are plentiful, the mTORC1 signaling pathway could inactivate
lysosomes to inhibit autophagy (41,42).
FYGL endocytosed in the PANC-1 cells might be degraded into amino
acids and sugars, potentially inhibiting cell autophagy.
A limitation of the present study was that in
vitro experiments alone are not sufficient to validate the
inhibitory effects of FYGL on tumors. Therefore, future studies
should include in vivo experiments. However, the present
study conclusively demonstrated that, FYGL could selectively kill
pancreatic cancer cells through regulation of ROS and the
simultaneous inhibition of autophagy in the following processes:
FYGL promoted PANC-1 cell apoptosis by inducing ROS, and the
increase of ROS increased the number of autophagosomes. On the
other hand, FYGL halts the downstream process of autophagy by
inhibiting the fusion of autophagosomes and lysosomes. This
increased the accumulation of defective mitochondria and the
production of ROS, as well as induced metabolic deficiency. Both
processes promoted apoptosis in the PANC-1 cells. The results from
the present study suggested that FYGL could be used as a potential
agent for the treatment of pancreatic cancer.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural
Science Foundation of China (grant nos. 21374022 and 81374032); The
Scientific National Major Scientific and Technological Special
Project for ‘Significant New Drugs Development’ (grant no.
2017ZX09301006) and the Shanghai Science and Technology Innovation
Action Plan ‘Science and Technology Support Project in Biomedical
Science’ (grant no. 17401902700).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Author's contributions
XW and PZ designed and performed the experiments. LJ
and JL performed part of the western blotting experiments. HL, YP
and SY performed the experiments involving cell culture and cell
viability. ZZ, YH, HY and YT contributed to the data analysis. XW
wrote the manuscript. HY and PZ reviewed and revised the manuscript
for important intellectual information. PZ conceived the study and
was responsible for the revision of the manuscript and final
decision to submit the article for publication. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2019. CA Cancer J Clin. 69:7–34. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kamisawa T, Wood LD, Itoi T and Takaori K:
Pancreatic cancer. Lancet. 388:73–85. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Burris HR III, Moore MJ, Andersen J, Green
MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo
AM, Tarassoff P, et al: Improvements in survival and clinical
benefit with gemcitabine as first-line therapy for patients with
advanced pancreas cancer: A randomized trial. J Clin Oncol.
15:2403–2413. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shiao MS: Natural products of the
medicinal fungus Ganoderma lucidum: Occurrence, biological
activities, and pharmacological functions. Chem Rec. 3:172–180.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Teng BS, Wang CD, Yang HJ, Wu JS, Zhang D,
Zheng M, Fan ZH, Pan D and Zhou P: A protein tyrosine phosphatase
1B activity inhibitor from the fruiting bodies of Ganoderma
lucidum (Fr.) Karst and its hypoglycemic potency on
streptozotocin-induced type 2 diabetic mice. J Agric Food Chem.
59:6492–6500. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pan D, Wang L, Chen C, Hu B and Zhou P:
Isolation and characterization of a hyperbranched proteoglycan from
Ganoderma lucidum for anti-diabetes. Carbohyd Polym.
117:106–114. 2015. View Article : Google Scholar
|
|
7
|
Pan D, Zhang D, Wu J, Chen C, Xu Z, Yang H
and Zhou P: Antidiabetic, antihyperlipidemic and antioxidant
activities of a novel proteoglycan from Ganoderma lucidum
fruiting bodies on db/db mice and the possible mechanism. PLoS One.
8:e683322013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Teng BS, Wang CD, Zhang D, Wu JS, Pan D,
Pan LF, Yang HJ and Zhou P: Hypoglycemic effect and mechanism of a
proteoglycan from ganoderma lucidum on
streptozotocin-induced type 2 diabetic rats. Eur Rev Med Pharmacol
Sci. 16:166–175. 2012.PubMed/NCBI
|
|
9
|
Gorrini C, Harris IS and Mak TW:
Modulation of oxidative stress as an anticancer strategy. Nat Rev
Drug Discov. 12:931–947. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen Y, McMillan-Ward E, Kong J, Israels
SJ and Gibson SB: Oxidative stress induces autophagic cell death
independent of apoptosis in transformed and cancer cells. Cell
Death Differ. 15:171–182. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Khan M, Ding C, Rasul A, Yi F, Li T, Gao
H, Gao R, Zhong L, Zhang K, Fang X and Ma T: Isoalantolactone
induces reactive oxygen species mediated apoptosis in pancreatic
carcinoma PANC-1 cells. Int J Biol Sci. 8:533–547. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schumacker PT: Reactive oxygen species in
cancer cells: Live by the sword, die by the sword. Cancer Cell.
10:175–176. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li PF, Dietz R and von Harsdorf R: p53
regulates mitochondrial membrane potential through reactive oxygen
species and induces cytochrome c-independent apoptosis blocked by
Bcl-2. EMBO J. 18:6027–6036. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
de Vries HE, Witte M, Hondius D,
Rozemuller AJ, Drukarch B, Hoozemans J and van Horssen J:
Nrf2-induced antioxidant protection: A promising target to
counteract ROS-mediated damage in neurodegenerative disease? Free
Radic Biol Med. 45:1375–1383. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hur J, Sullivan KA, Schuyler AD, Hong Y,
Pande M, States DJ, Jagadish HV and Feldman EL: Literature-based
discovery of diabetes- and ROS-related targets. BMC Med Genomics.
3:492010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pelicano H, Carney D and Huang P: ROS
stress in cancer cells and therapeutic implications. Drug Resist
Updat. 7:97–110. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liochev SI: Reactive oxygen species and
the free radical theory of aging. Free Radic Biol Med. 60:1–4.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mizushima N: Autophagy: Process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Degenhardt K, Mathew R, Beaudoin B, Bray
K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation, and tumorigenesis. Cancer Cell. 10:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guo JY, Chen HY, Mathew R, Fan J,
Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM,
Karantza V, et al: Activated Ras requires autophagy to maintain
oxidative metabolism and tumorigenesis. Genes Dev. 25:460–470.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang S, Wang X, Contino G, Liesa M, Sahin
E, Ying H, Bause A, Li Y, Stommel JM, Dell'Antonio G, et al:
Pancreatic cancers require autophagy for tumor growth. Genes Dev.
25:717–729. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Boya P, Gonzalez-Polo RA, Casares N,
Perfettini JL, Dessen P, Larochette N, Metivier D, Meley D,
Souquere S, Yoshimori T, et al: Inhibition of macroautophagy
triggers apoptosis. Mol Cell Biol. 25:1025–1040. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
El-Khattouti A, Selimovic D, Haikel Y and
Hassan M: Crosstalk between apoptosis and autophagy: Molecular
mechanisms and therapeutic strategies in cancer. J Cell Death.
6:37–55. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen S, Cheng AC, Wang MS and Peng X:
Detection of apoptosis induced by new type gosling viral enteritis
virus in vitro through fluorescein annexin V-FITC/PI double
labeling. World J Gastroenterol. 14:2174–2178. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Budihardjo I, Oliver H, Lutter M, Luo X
and Wang X: Biochemical pathways of caspase activation during
apoptosis. Annu Rev Cell Dev Biol. 15:269–290. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by
Bcl-2: Release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Poupel F, Aghaei M, Movahedian A, Jafari
SM and Shahrestanaki MK: Dihydroartemisinin induces apoptosis in
human bladder cancer cell lines through reactive oxygen species,
mitochondrial membrane potential, and cytochrome c pathway. Int J
Prev Med. 8:782017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pankiv S, Clausen TH, Lamark T, Brech A,
Bruun JA, Outzen H, Øvervatn A, Bjørkøy G and Johansen T:
p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem.
282:24131–24145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Park SH, Sung JH, Kim EJ and Chung N:
Berberine induces apoptosis via ROS generation in PANC-1 and
MIA-PaCa2 pancreatic cell lines. Braz J Med Biol Res. 48:111–119.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Duong HQ, Hwang JS, Kim HJ, Seong YS and
Bae I: BML-275, an AMPK inhibitor, induces DNA damage, G2/M arrest
and apoptosis in human pancreatic cancer cells. Int J Oncol.
41:2227–2236. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Messner MC and Cabot MC: Cytotoxic
responses to N-(4-hydroxyphenyl)retinamide in human pancreatic
cancer cells. Cancer Chemother Pharmacol. 68:477–487. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vaquero EC, Edderkaoui M, Pandol SJ,
Gukovsky I and Gukovskaya AS: Reactive oxygen species produced by
NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J
Biol Chem. 279:34643–34654. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cheng L, Yan B, Chen K, Jiang Z, Zhou C,
Cao J, Qian W, Li J, Sun L, Ma J, et al: Resveratrol-induced
downregulation of NAF-1 enhances the sensitivity of pancreatic
cancer cells to gemcitabine via the ROS/Nrf2 signaling pathways.
Oxid Med Cell Longev. 2018:94820182018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Scherz-Shouval R, Shvets E, Fass E, Shorer
H, Gil L and Elazar Z: Reactive oxygen species are essential for
autophagy and specifically regulate the activity of Atg4. EMBO J.
26:1749–1760. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Scherz-Shouval R and Elazar Z: ROS,
mitochondria and the regulation of autophagy. Trends Cell Biol.
17:422–427. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang Z, Wu F, Yang H and Zhou P:
Endocytosis mechanism of a novel proteoglycan, extracted from
Ganoderma lucidum, in HepG2 cells. Rsc Adv. 7:41779–41786.
2017. View Article : Google Scholar
|
|
38
|
Humpton TJ, Alagesan B, DeNicola GM, Lu D,
Yordanov GN, Leonhardt CS, Yao MA, Alagesan P, Zaatari MN, Park Y,
et al: Oncogenic KRAS induces NIX-mediated mitophagy to promote
pancreatic cancer. Cancer Discov. 9:1268–1287. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang Y and Commisso C: Macropinocytosis
in cancer: A complex signaling network. Trends Cancer. 5:332–334.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Commisso C, Davidson SM, Soydaner-Azeloglu
RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin
JA, Thompson CB, et al: Macropinocytosis of protein is an amino
acid supply route in Ras-transformed cells. Nature. 497:633–637.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Florey O and Overholtzer M:
Macropinocytosis and autophagy crosstalk in nutrient scavenging.
Philos Trans R Soc Lond B Biol Sci. 374:201801542019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Palm W, Park Y, Wright K, Pavlova NN,
Tuveson DA and Thompson CB: The utilization of extracellular
proteins as nutrients is suppressed by mTORC1. Cell. 162:259–270.
2015. View Article : Google Scholar : PubMed/NCBI
|