Introduction
Colorectal cancer (CRC) is one of the most prevalent
malignancies worldwide, and according to the global cancer
statistics in 2012, it has high rates of morbidity (9.7%) and
mortality (8.5%) (1,2). The occurrence of CRC is the result of
the interaction of multiple factors, and its pathology causes
include unhealthy lifestyle, high-fat and high-protein diet,
carcinogenic environment, and chromosome instability (3–5).
Although there has been advances made in the diagnostic and
treatment strategies for CRC treatment, the survival rate from this
type of cancer remains low due to the complex pathology of the
disease (6).
At present, treatment for CRC mainly consists of
surgery supplemented by chemotherapy (5-fluorouracil, oxaliplatin,
irinotecan and capecitabine), which can achieve satisfactory
therapeutic effects (7–9). However, a lack of effective treatment
methods remain for patients with advanced-stage CRC tumors
(7). Chemotherapy is one method
that is currently in use for patients with cancer, including CRC
(10). Therefore, discovering
novel agents and analyzing their mechanism of action in relation to
the occurrence and development of CRC has important implications
for the development of novel therapies (11).
To increase the therapeutic efficacy of chemotherapy
in colorectal cancer, the search for downstream effectors is
required. In particular, capecitabine is typically administered as
a combinational chemotherapy (with surgery) or as a monotherapy for
CRC treatment (9). It
preferentially generates 5-flurouracil (5-FU) which represents a
well-established treatment method (12). Due to the acquisition of
chemotherapy resistance, capecitabine treatment frequently fails in
CRC (13,14). Therefore, interrogation of new
effectors associated with its treatment is required to potentially
discover novel therapeutic targets.
The receptor activator of nuclear factor-κB (RANK)
and RANK ligand (RANKL) are two important components of the
RANK/RANKL pathway (15). RANK and
RANKL are both members of the TNF superfamily of receptors and is
regulated by osteoprotegerin (OPG), which is a decoy receptor for
RANKL that prevents its binding to its receptor RANK (16). This pathway exerts its function in
differentiating microfold cells by recruiting TNF
receptor-associated factor adaptor proteins and activating
downstream pathways, such as the NF-κB and EMT pathways (17–19).
Several studies have reported dysregulation and function of this
pathway in a number of cancers. RANK and c-Met-regulated pathways
have been reported to enhance metastatic colonization in prostate
cancer (20). In addition, the
RANKL/RANK/MMP-1 pathway was found to contribute to the metastasis
of breast and prostate cancer cells (21). RANK/RANKL can also be exploited as
a target for the treatment of myeloma and solid tumors, such as
prostate and breast cancer (22).
In tissues from liver (23),
stomach (24), breast (25), thyroid (26), prostate (27) and pancreatic cancers (28), high levels RANK expression were
previously detected, suggesting that RANK may be involved in the
occurrence and development of malignant tumors. Therefore, RANK may
apply a multitude of mechanisms to facilitate the tumorigenic
process. Although the role of RANK in a variety of malignant tumors
has been previously studied, its possible oncogenic role in CRC and
its significance in occurrence remain unclear, where there are few
reports. These reports only mentioned that RANK promotes CRC
migration and invasion, and it may be related to the relapse risk
of stage II CRC (29,30).
In the present study, the possible oncogenic
effectors associated with capecitabine in CRC were investigated.
The expression levels of components in the RANK/RANKL pathway in
CRC tissues and cell lines were measured, where the roles of
capecitabine and the RANK/RANKL pathway were investigated. Finally,
the potential effects of capecitabine on the RANK/RANKL pathway in
CRC cells were also studied.
Materials and methods
Tissue samples
In the present study, 50 pairs of CRC (>1.5 cm
away from the negative tumor margin) and adjacent normal tissues
were collected from January to December 2019 and stored at −80°C.
Western blotting was applied to detect the expression levels of
RANK, RANKL and OPG. These tissue samples were procured after
surgical excisions of tissues from patients with CRC at the Taizhou
Hospital of Zhejiang Province affiliated to Wenzhou Medical
University (Linhai, China). Patients had a pathological diagnosis
of CRC according to the Chinese Protocol of Diagnosis and Treatment
of Colorectal Cancer (2020 edition) (31) and did not undergo any pre-operative
treatment, and each patient had provided appropriately signed
consent forms. The clinicopathological features of patients were
listed in the Table SI, and the
average age was 61.16±7.46 years and the proportion of male
patients was 60%. The Ethics Committee of Taizhou Hospital of
Zhejiang Province affiliated to Wenzhou Medical University reviewed
and approved the present study (approval no. ZXC2019005; Linhai,
China).
Patient inclusion and exclusion
criteria
The inclusion criteria were: i) Age ≥40 years and
≤80 years; ii) colorectal adenocarcinoma was confirmed by
colonoscopy and biopsy before surgery; iii) no tumor metastasis or
implantation in adjacent organs was found in preoperative CT
imaging evaluation; and iv) obtained the patients' informed consent
and they were willing to cooperate. The exclusion criteria were: i)
Age <40 years; ii) cases of emergency operation due to acute
intestinal obstruction, perforation or bleeding; iii) combined with
malignant tumors of other organs; iv) CT examination confirmed that
the tumor invaded adjacent organs or distant metastasis, which made
it impossible to perform radical resection; v) contraindications of
laparoscopic surgery; and vi) unwilling to cooperate and poor
compliance.
Cell lines and treatment
CRC cell lines SW480, SW620, LOVO, HT29 and HCT116
and normal control cells (colonic epithelial cell NCM460) were from
American Type Culture Collection. They were cultured in DMEM
(Nanjing KeyGen Biotech Co., Ltd.) supplemented with 10% FBS
(Gibco; Thermo Fisher Scientific, Inc.) and 1% antibiotics (100
µg/ml streptomycin and 100 U/ml penicillin; Invitrogen; Thermo
Fisher Scientific, Inc.). These cells were maintained in a
humidified atmosphere under 5% CO2 at 37°C for
subsequent assays. Different concentrations of capecitabine (25,
50, 100, 200 and 400 µM; Shanghai Roche Pharmaceuticals Co., Ltd.)
were applied to the CRC cells (HT29, SW480, SW620, LOVO and HCT116)
for 48 h at 37°C.
Cell transfection
CRC cells (HT29, SW480, SW620, LOVO and HCT116) were
divided into the si-RNA negative control (NC; Guangzhou RiboBio
Co., Ltd.), si-RANK, pcDNA3.1 vector (Thermo Fisher Scientific,
Inc.) and pcDNA3.1-RANK groups. pcDNA3.1-RANK was constructed as
follows: Firstly, primers were designed according to the complete
sequence of RANK gene in the GenBank (https://www.ncbi.nlm.nih.gov/genbank/; sense,
5′-GGCTGGCTACCACTGGAACT-3′ and antisense, 5′-TCCTGTAGTAAACGCCGAAGA
−3′). Then, the restriction endonucleases BamHI and
XhoI (Promega Corporation) were added to the 5′ ends of the
sense and antisense primers, respectively. The DNA was extracted
from HT29 cells with DNA Extraction Reagent (cat. no. P1012;
Beijing Solarbio Science & Technology Co., Ltd.), and then the
RANK gene was amplified by PCR with 2X Taq plus MasterMix (cat. no.
BL553B; Biosharp; Beijing Labgic Co., Ltd.). The thermocycling
conditions were as follows: Initial denaturation at 94°C for 2 min;
followed by denaturation at 94°C for 30 sec, annealing at 60°C for
30 sec and extension at 72°C for 1 min for 30 cycles; and extension
at 72°C for 5 min. A 1% agarose gel was prepared, the amplified DNA
was electrophoresed, and the gel dyed with ethidium bromide was
observed under an ultraviolet lamp. Afterwards, the PCR recovered
product and pcDNA3.1(+) vector were purified by double enzyme
digestion with BamHI and XhoI. Afterwards, T4 DNA
ligase (Promega Corporation) was added to the centrifuge tube and
ligated overnight in a 16°C water bath. The ligation product was
transformed into competent DH5α Escherichia coli and grown
on the selective LB solid medium (containing 100 µg/ml ampicillin;
Gibco; Thermo Fisher Scientific, Inc.). The recombinant plasmids of
the positive clones were extracted, digested with BamHI and
XhoI, and then subjected to agarose gel electrophoresis for
further sequencing and identification. The si-NC and pcDNA3.1
vector groups served as control groups. Cells were transfected with
50 nM si-NC, si-RANK, pcDNA3.1 vector or pcDNA3.1-RANK using
Lipofectamine® 2000 (Gibco; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocols. The si-NC (sense,
5′-UAGCGACUAAACACAUCAAUU −3′ and antisense,
5′-UUAUCGCUGAUUUGUGUAGUU −3′), si-RANK (sense,
5′-CCAAGGAGGCCCAGGCUUAUU −3′ and antisense,
5′-UAAGCCUGGGCCUCCUUGGUU −3′), pcDNA3.1 and pcDNA3.1-RANK vectors
were transfected at 37°C for 48 h. Afterwards, the cells were
cultured at 37°C for 24 h before they were used for subsequent
experiments.
RNA isolation and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from CRC cells (HT29, SW480,
SW620, LOVO and HCT116) using a UNlQ-10 Column Trizol Total RNA
Isolation Kit (cat. no. B511321-0100; Sangon Biotech Co., Ltd.).
The RNA samples were quantified and then reverse-transcribed using
Reverse Transcriptase M-MLV (cat. no. 28025013; Thermo Fisher
Scientific, Inc.). The conditions were as follows: 37°C for 10 min,
42°C for 50 min and 95°C for 5 min. qPCR was performed using TB
Green™ Premix Ex Taq™ II (Tli RNaseH Plus; Takara Bio, Inc.). The
following primer sequences were used in the present study: RANK
forward, 5′-AGCATTGTTAGAGCCTGTGG-3′ and reverse,
5′-CAGACGTGGCAGGACTAAGG-3′ and GAPDH forward,
5′-CCATGGGGAAGGTGAAGGTC-3′ and reverse, 5′-AGTGATGGCATGGACTGTGG-3′.
The thermocycling conditions were as follows: Initial denaturation
at 95°C for 3 min, followed by each step of denaturation at 95°C
for 10 sec and annealing extension at 58°C for 30 sec, for 35
cycles. The 2−ΔΔCq method was applied to calculate the
relative RANK mRNA expression level (32), and GAPDH was used as the reference
gene.
MTT assay
Cell viability was evaluated using MTT as previously
described (33). A total of
1×104 HT29 cells were seeded into 96-well culture plates
and subsequently grown overnight at 37°C. Next, 10 µl MTT at a
concentration of 5 mg/ml was added into each well and incubated at
37°C for a further 4 h. DMSO was used to dissolve the formazan
crystals. The absorbance at 540 nm in each well was then measured
using a Multiplate reader (Tecan Group, Ltd.), which was normalized
to the absorbance values of untreated cells at the corresponding
time points (12, 24, 36, 48 and 60 h).
Cell Counting Kit-8 (CCK-8) assay
Referring to the protocol of the CCK-8 (cat. no.
C0038; Beyotime Institute of Biotechnology), NCM460, SW480, SW620,
LOVO, HT29 and HCT116 cells (2×104 cells/well) were
inoculated into 96-well plates. Afterwards, 20 µl CCK-8 solution
and 100 µl DMEM (with 10% FBS) were added and cells were incubated
for 1 h. The absorbance values at 450 nm were measured by a
microplate reader (Tecan Group, Ltd.).
EdU staining
Cell proliferation was measured using an EdU assay
kit (Guangzhou RiboBio Co., Ltd). Briefly, stably transfected HT29
cells (2×103 cells/well) were seeded into 96-well
culture dishes. After cell culture overnight, 10 nM EdU was
supplemented and cells were cultured for a further 12 h at 37°C.
They were then washed with PBS, fixed in 4% paraformaldehyde for 30
min at 37°C, and stained with Apollo® Fluorescent dye
solution (cat. no. C10310-3; Guangzhou RiboBio Co., Ltd.) for 30
min at 37°C. The ApolloR Fluorescent dye solution was
discarded, and then 0.1 ml 0.5% Triton X-100 was added for 10 min
at room temperature. Afterwards, the TritonX-100 was discarded, and
0.1 ml 1 mg/ml DAPI solution (cat. no. C0060; Beijing Solarbio
Science & Technology Co., Ltd.) was added for 5 min at 37°C. In
total, three randomly selected fields were imaged using a
fluorescence microscope (Olympus IX73; Olympus Corporation) and
cells exhibiting blue fluorescence were defined as positive and
counted using the ImageJ software v1.8 (National Institutes of
Health). The ratio of the positive cell number/total cell number is
a measure of the relative EdU incorporation rate.
Transwell assay
The 24-well Millicell uncoated chambers were used
for the measurement of cell migration. At 2 days post-transfection,
cancer cells (3×105 cells) grown in serum-free DMEM were
placed into the upper compartment (8-µm pore size; EMD Millipore)
whereas the lower compartment was filled with 500 µl DMEM and 15%
FBS. In total, 24 h later at room temperature, non-migratory cells
were discarded, whilst migratory cells were fixed using cold 98%
ethanol at 37°C for 10 min and stained with 0.1% Crystal Violet at
37°C for 20 min. At the end of the experiment, five visual fields
were randomly selected, and then stained cells were counted using
an inverted light microscope (Nikon Corporation). Relative cell
migration (%)=(migrated cell number of treatment group/migrated
cell number of DMSO group) ×100%.
Wound healing assay
Wound healing assays were performed also for the
assessment of cell migration. Cells were plated into six-well
plates (5×104 cells per well) with DMEM. A sterilized
5-µl plastic micropipette was then used to make a scratch on the
surfaces of cell monolayers at 90% confluency. The cells were
incubated under standard conditions (5% CO2; 37°C; DMEM;
1% antibiotics) for 24 h and then washed three times with PBS. The
wound regions at different time points (0 and 24 h) were imaged and
photographed using an IX71 inverted light microscope (Olympus
Corporation). Cell migration rate=(cell migration distance of
treatment group/cell migration distance of control group) ×100%,
and cell migration distance referred to the distance that cells
migrated between 0 and 24 h.
Western blotting
Expression levels of proteins in the RANK/RANKL
pathway and in the epithelial-to-mesenchymal transition (EMT)
process of CRC cells (HT29, SW480, SW620, LOVO and HCT116) were
analyzed by western blotting. In the present study, RIPA buffer
(cat. no. P0013B; Beyotime Institute of Biotechnology) was used to
obstain cell protein lysates. First, the cells were trypsinized and
then centrifuged at 134 × g at 37°C for 5 min. Next, the
supernatant was removed and the cell pellet was washed three times
with cold PBS. The cell lysates were produced by the addition of
100 µl RIPA buffer and incubated on ice for 1 h. The samples were
sonicated (80 W ultrasonic power; 2 sec working time and 4 sec
interval time) for 3 min on ice and centrifuged at 12,000 × g for
12 min at 4°C. A BCA protein assay kit (Abcam) was used to
determine the protein concentration according to the manufacturer's
protocols. In total, 25 µg protein was loaded into each lane of a
10% Mini-PROTEAN® TGX Stain-Free™ Protein gel with
electrophoresis buffer (25 mM Tris, 0.192 M glycine, 0.1% SDS) and
run for 0.5 h at 200 V. After the proteins were separated, they
were transferred onto Trans-Blot® Turbo™ Mini PVDF
membranes and blocked in 1% BSA (Abcam) for 3 h at 37°C. The
membranes were then incubated with relevant the primary antibodies
(RANK, dilution, 1:1,000; cat. no. ab200369; RANKL, dilution,
1:1,000; cat. no. ab9957; OPG, dilution, 1:1,000; cat. no. ab73400;
E-cadherin, dilution, 1:1,000; ab40772; vimentin, dilution,
1:1,000; ab92547; N-cadherin, dilution, 1:1,000; ab18203; GAPDH,
dilution, 1:2,000; ab9485; Abcam) overnight at 4°C. The next day,
TBS-T (5% blocking buffer, 0.1% Tween-20) was used to wash the
membrane six times and then HRP-conjugated secondary antibody
(dilution, 1:2,000; cat. no. ab6721; Abcam) was applied for 1 h at
37°C. At the end of this incubation, the membranes were washed and
proteins visualized using Enhanced ECL Chemiluminescence Substrate
Kit (cat. no. 36222ES60; Shanghai Yeasen Biotechnology Co., Ltd.)
and then employing the ChemiDoc imager (Bio-Rad Laboratories,
Inc.). The exposure time was determined according to the brightness
of the visible bands after adding the luminescent reagent, and a
film was exposed. Image J v1.8.0 software (National Institutes of
Health) was used to quantify the gray values of the protein bands.
The gray value ratio of the target protein to GAPDH was calculated
to be the relative expression of the target protein. The primary
antibodies used included anti-RANK, anti-RANKL, anti-OPG,
anti-E-cadherin, anti-vimentin, anti-N-cadherin and anti-GAPDH, all
from Abcam (Table SII).
Statistical analysis
Statistical analyzes were performed using GraphPad
Prism 6.0 Software (GraphPad Software, Inc.) from ≥ three
independent experiments. The results were expressed as the mean ±
SD and data between normal and cancer tissues were compared using
paired t test, data among three or more groups were compared using
One-way ANOVA, time-based measurement data were compared using
ANOVA for repeated measurements. The Bonferroni method was
performed for the post hoc test. Statistical differences between
groups were indicated to be significant at P<0.05.
Results
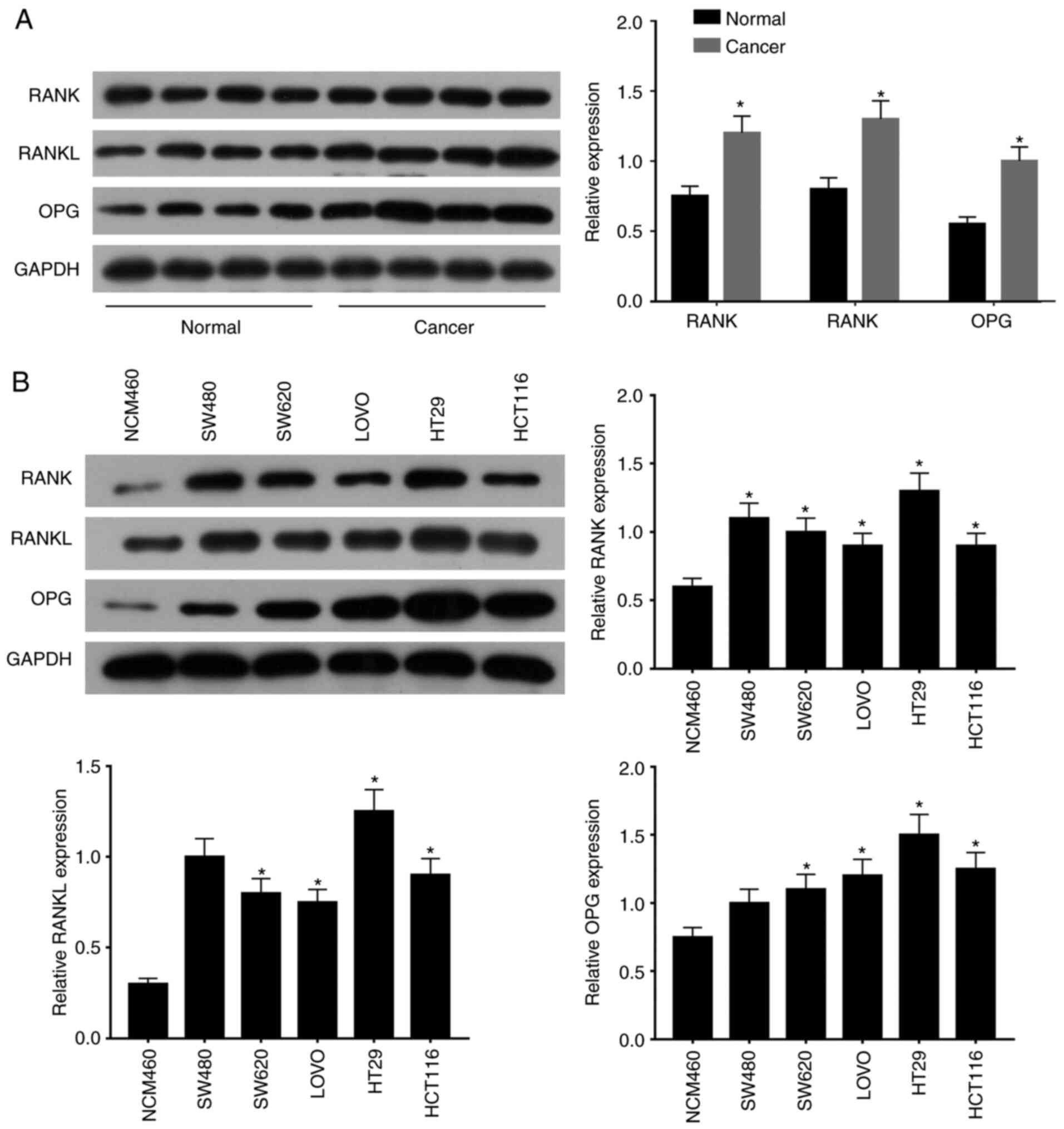
RANK/RANKL pathway is upregulated in
CRC
To explore the possible expression pattern of
components of the RANK/RANKL pathway in CRC, the expression of
proteins in the RANK/RANKL pathway was measured in CRC tissues and
cells. Compared with that in normal tissues, the expression levels
of RANK, RANKL and OPG were all significantly elevated in cancerous
tissues compared with those in normal tissues as assessed by
western blotting (Fig. 1A).
Furthermore, compared with those in the normal control cell line
(colonic epithelial cell NCM460), RANK, RANKL and OPG all exhibited
significantly higher expression levels in the five CRC cell lines
tested (Fig. 1B). HT29 cells
exhibited the highest expression of RANK, RANKL and OPG (Fig. 1B). These data suggest that the
expression of RANK/RANKL in CRC tissues and cells was elevated
compared with their non-cancerous counterparts.
Capecitabine reduces the levels of
proteins in the RANK/RANKL pathway in HT29 cells
To determine the effects of capecitabine on the
RANK/RANKL pathway and the optimal concentration, the expression
levels of proteins in the RANK/RANKL pathway were measured in HT29
cells after treatment with ascending concentrations of capecitabine
(Fig. 2). It was found that as the
concentration of capecitabine increased, the expression of RANK,
RANKL and OPG also decreased, where the difference became
statistically significant compared with that in untreated cells
(Fig. 2). These findings suggest
that capecitabine may function by inhibiting the RANK/RANKL
pathway. The dose range of capecitabine between 50 and 400 µM could
significantly reduce the expression of RANK, RANKL and OPG
(Fig. 2). Furthermore, the cell
viability of other CRC cells (HT29, SW480, SW620, LOVO and HCT116)
decreased when the concentration of capecitabine increased.
Additionally, the dose range of capecitabine between 50 and 400 µM
could significantly decrease cell viability (Fig. S1). Although protein expression
levels and cell viability were lower after treatment with 400 µM,
50 µM was selected for subsequent experiments, as this
concentration had a certain inhibitory effect on CRC cells and was
non-toxic in the control cell (NCM460) group.
Capecitabine treatment and RANK
knockdown can both inhibit HT29 cell proliferation
Cells were first transfected with si-RANK. To verify
if RANK protein expression was knocked down after si-RANK
transfection, the expression of RANK in the DMSO, si-NC and si-RANK
groups was measured by western blotting. Compared with those in the
si-NC group, the protein levels of RANK in the si-RANK group were
lower, the difference of which was statistically significant
(Fig. 3A). This finding suggests
that RANK was successfully knocked down by si-RANK transfection.
Functional experiments were subsequently performed to study the
role of capecitabine in the RANK/RANKL pathway further in HT29
cells. Capecitabine at the dose of 50 µM was applied to treat HT29
cells in parallel to si-RANK transfection. Results from MTT assay
revealed that the cell viability of HT29 cells was significantly
reduced after the cells were either treated with capecitabine or
transfected with si-RANK compared with their respective negative
controls (Fig. 3B). According to
data from EdU assay, the proportion of EdU-positive cells was
decreased after the HT29 cells were subjected to either
capecitabine treatment or transfection with si-RANK compared with
that in their corresponding negative controls (Fig. 3C). Taken together, these findings
suggest that HT29 cell proliferation was inhibited by capecitabine
treatment and RANK silencing.
Capecitabine treatment and RANK
knocked down inhibit EMT in HT29 cells
To measure cell migration, Transwell and wound
healing assays were performed whereas western blotting was
performed to measure the expression levels of EMT-associated
proteins E-cadherin, vimentin and N-cadherin. According to results
from Transwell assays, the number of migratory HT29 cells was
significantly reduced after the cells were treated with
capecitabine or transfected with si-RANK compared with that in
their corresponding negative controls (Fig. 4A). From wound healing assays, the
migration rate was found to be significantly reduced in both
capecitabine-treated and si-RANK-transfected HT29 cells compared
with that in their corresponding negative controls (Fig. 4B). These two observations
implicates the inhibitory effects on cell migration exerted by both
capecitabine administration and RANK silencing. Compared with those
in their corresponding negative controls, the protein expression
levels for E-cadherin were significantly increased by the either
the addition of capecitabine or silencing of RANK expression
(Fig. 4C). By contrast, the
protein expression levels of vimentin and N-cadherin were
significantly decreased by either the addition of capecitabine or
silencing of RANK compared with those in their corresponding
negative controls (Fig. 4C). These
results suggest that both capecitabine and RANK knockdown can both
inhibit EMT in HT29 cells.
RANK overexpression reverses the
inhibitory effects of capecitabine on HT29 cell proliferation
To verify the effects of capecitabine on the
RANK/RANKL pathway and transfection efficiency, western blotting
and RT-qPCR. RANK, RANKL and OPG protein expression were all found
to be inhibited by treatment with capecitabine in the Cap +
pcDNA3.1 vector group and the Cap + pcDNA3.1-RANK group, but
increased by transfection with pcDNA3.1-RANK in the DMSO +
pcDNA3.1-RANK group and the Cap + pcDNA3.1-RANK group (Fig. 5A). After the combined intervention
of capecitabine + pcDNA3.1-RANK, RANK, RANKL and OPG protein
expression recovered back to their original levels in the DMSO +
pcDNA3.1 vector group (Fig. 5A).
Subsequently, the expression of RANK mRNA and protein in the
pcDNA3.1-RANK group was significantly increased compared with that
in the control group (Fig. 5B).
Additionally, the expression levels of RANK mRNA in the other CRC
cells (SW480, SW620, LOVO and HCT116) exhibited the same trend
(Fig. S2). These results suggest
that RANK overexpression was successful. Inhibition of cell
viability induced by capecitabine was revealed to be significantly
reversed by RANK overexpression as a result of transfection with
the pcDNA3.1-RANK plasmid (Fig.
5C). By contrast, cell viability was significantly reduced in
HT29 cells overexpressing RANK were treated with capecitabine
compared with that in cells treated with DMSO and transfected with
pcDNA3.1-RANK (Fig. 5C). EdU assay
data revealed that the number of EdU-positive cells was
significantly reduced after capecitabine treatment, which was
significantly reversed after RANK overexpression (Fig. 5D). However, the number of
EdU-positive cells was significantly reduced after the HT29 cells
were treated with capecitabine and transfected with pcDNA3.1-RANK
compared with that in cells treated with DMSO and transfected with
pcDNA3.1-RANK (Fig. 5D). Taken
together, these findings suggest that RANK overexpression
counteracted the inhibitory effects of capecitabine on HT29 cell
proliferation.
RANK overexpression reverses the
inhibitory effects of capecitabine on EMT in HT29 cells
Expression of EMT markers were also measured in CRC
cell lines (HT29, SW480, SW620, LOVO and HCT116) after they were
treated with capecitabine and transfected with pcDNA3.1-RANK.
According to the Transwell assays, capecitabine treatment
significantly inhibited HT29 cell migration, which was
significantly reversed by RANK overexpression upregulation of RANK
(Fig. 6A). Simultaneous treatment
with capecitabine and RANK overexpression reduced HT29 cell
migration compared with that in cells treated with DMSO and
transfected with pcDNA3.1-RANK (Fig.
6A). Results from wound healing assays revealed that the cell
migration was also significantly inhibited by capecitabin treatment
but was significantly reversed by RANK overexpression (Fig. 6B). Compared with that in cells
treated with DMSO and transfected with pcDNA3.1-RANK, cell
migration was significantly reduced by the simultaneous RANK
overexpression and capecitabin addition (Fig. 6B). Finally, western blotting
demonstrated that the capecitabin-induced increase in the protein
expression of E-cadherin and capecitabin-induced decrease in the
protein expression of vimentin and N-cadherin were significantly
reversed by RANK overexpression (Figs.
6C and S3). Furthermore,
reductions in E-cadherin expression and elevations in vimentin and
N-cadherin induced by RANK overexpression were both significantly
reversed by treatment with capecitabin (Figs. 6C and S3). In conclusion, these observations
suggest that RANK overexpression reversed the suppressive effects
of capecitabine on HT29 cells.
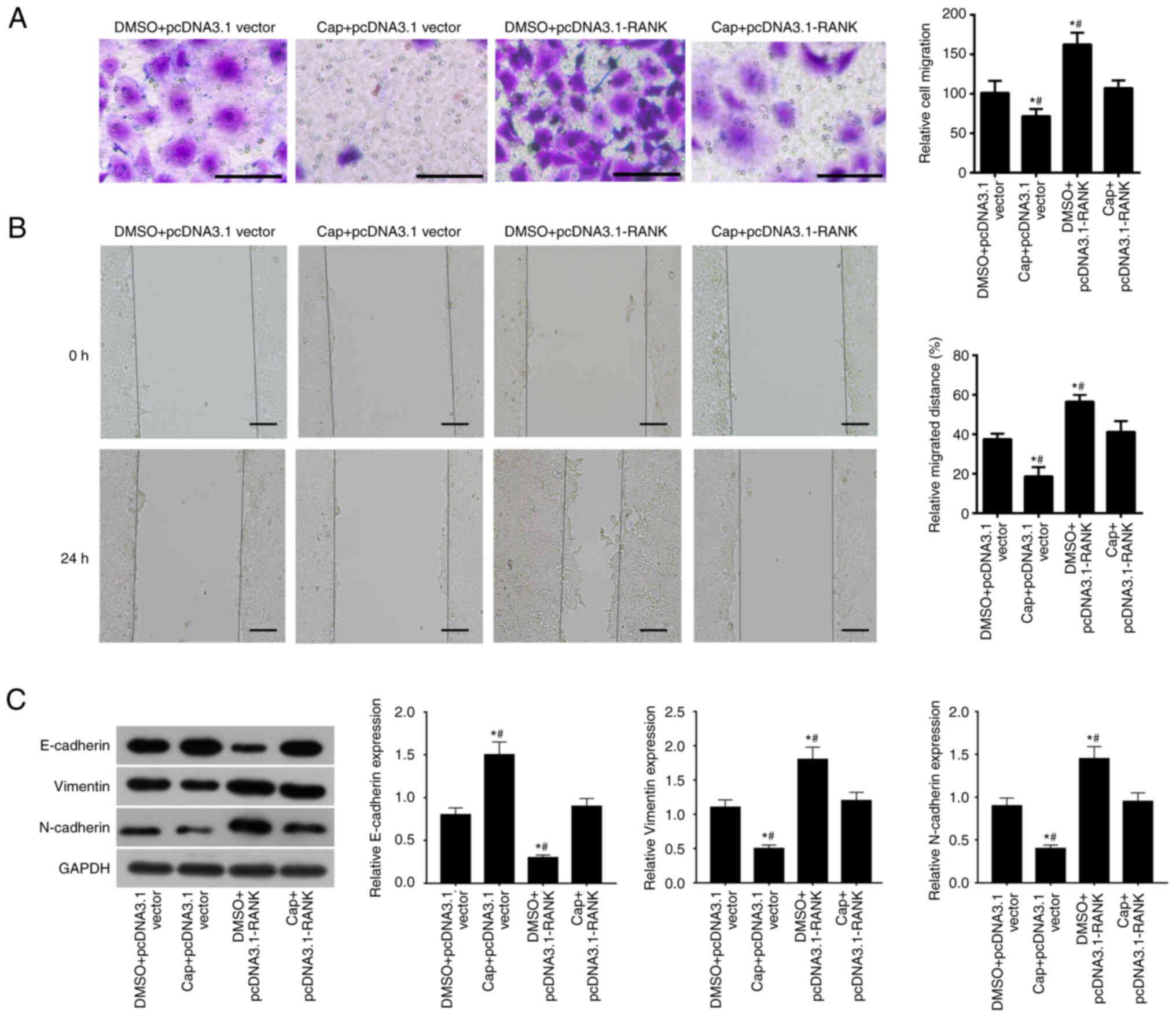 | Figure 6.Overexpression of RANK reverses the
inhibitory effects of capecitabine on EMT and migration in HT29
cells. The migration ability of HT29 cells in the DMSO + pcDNA3.1
vector, capecitabine (50 µM) + pcDNA3.1 vector, DMSO +
pcDNA3.1-RANK and Cap + pcDNA3.1-RANK groups were separately
evaluated by (A) Transwell and (B) wound healing assays. The
concentration of capecitabine was 50 µM. Scale bar, 50 µm, 200 µm.
(C) Western blotting was performed to assess EMT-associated
proteins in HT29 cells in the DMSO + pcDNA3.1 vector, capecitabine
(50 µM) + pcDNA3.1 vector, DMSO + pcDNA3.1-RANK and Cap +
pcDNA3.1-RANK groups. *P<0.05 vs. DMSO + pcDNA3.1 vector and
#P<0.05 vs. Cap + pcDNA3.1-RANK. Cap, capecitabine;
RANK, receptor activator of nuclear factor-κB; RANKL, RANK ligand;
OPG, osteoprotegerin. |
Discussion
Over the past number of decades, the rates of
morbidity and mortality as a result of malignancies have been
increasing worldwide (34). At
present, CRC is the most prevalent malignancy in the digestive
tract, where morbidity and mortality caused by this disease are
increasing annually (1,2). There is accumulating evidence
highlighting the dysregulation of specific signaling pathways in
CRC. Bone morphogenic protein 3 has been found to suppress the
initiation of CRC through the activin receptor type IIB/SMAD2 and
TGF-β-activated kinase 1/JNK pathways (35). In addition, depletion of kinesin
family member 22 can attenuates the colon carcinoma proliferation
and tumor growth (36). Proline
rich 14 upregulation can facilitate the growth, EMT progression and
metastasis of CRC through the AKT pathway (37). Among the signaling pathways that
have been shown to be aberrantly dysregulated in CRC, the
RANK/RANKL pathway was focused upon in the present study. The
RANK/RANKL pathway has been implicated in the development of
several types of cancer, including breast cancer (38), myeloma (39) and endometrial cancer (40). The present study demonstrated that
the RANK/RANKL pathway was upregulated in CRC, suggesting that this
pathway was involved in its pathogenesis.
CRC typically becomes refractory to chemotherapy
treatment whilst retaining its malignancy (41). This results in poor
chemotherapeutic efficacy and survival rates in patients with CRC
(42). Therefore, discovery of
novel treatments that are effective is vital for improving patient
outcome. Capecitabine is a fluorouracil derivative chemotherapeutic
drug that can prolong the period of high 5-FU sensitivity in breast
cancer and gastric cancer cells (43). In addition, previous studies have
confirmed that 5-FU is involved in regulating RANKL signaling and
hence osteoclast differentiation (44). This suggests that the effects
exerted by capecitabine may be associated with the RANK/RANKL
pathway. At present, capecitabine is known as a ‘failure agent’ for
treating patients with CRC (13,14).
Therefore, the present study investigated the potential effects of
capecitabine on CRC with specific focus on the RANK/RANKL pathway.
Through the measurement of the expression of proteins in the
RANK/RANKL signaling pathway in multiple CRC cell lines and
tissues, it was found that RANK, RANKL and OPG expression were all
increased compared with that in their corresponding controls,
suggesting that the occurrence of CRC was closely associated with
activation of the RANK/RAKL signaling pathway. Furthermore,
following treatment of HT29 cells with different concentrations of
capecitabine, it was found that capecitabine mediated an inhibitory
effect on the expression of RANK/RANKL pathway proteins. It was
also found that the silencing of RANK expression could inhibit HT29
cell proliferation. By contrast, after RANK was overexpressed, the
proliferation of HT29 cells was increased significantly, which can
in turn be reversed by capecitabine. These findings implicate a
suppressive function for capecitabine in CRC and to the best of our
knowledge, the present study was the first to reveal a promotional
role for the RANK/RANKL pathway in CRC. The present study
demonstrated that capecitabine could inactivate the RANK/RANKL
pathway in CRC cells. The present study found that capecitabine and
the RANK/RANKL pathway mediated opposite functions in CRC.
Therefore, the hypothesis that resistance to capecitabine typically
observed in CRC was due to an activated RANK/RANKL pathway was
tested. Rescue experiments revealed that stimulation of the
RANK/RANKL pathway reversed the inhibitory effects of capecitabine
on CRC proliferation and EMT.
In the present study, it was found that the failure
of capecitabine chemotherapy in CRC was at least in part attributed
to the upregulated activation of the RANK/RANKL pathway. In the
present study, high expression levels of RANK were detected in CRC
tumor tissues. In conclusion, capecitabine inhibited EMT and
proliferation in CRC through modulation of the RANK/RANKL pathway.
Therefore, to treat patients more effectively, capecitabine
chemotherapy combined with a RANK-targeted therapy would be
desirable.
There remain a number of limitations to the present
study. Only in vitro studies were performed. Ideally,
further studies in animal models of CRC should be performed.
Furthermore, the precise modulatory mechanism between capecitabine
and the RANK/RANKL pathway in CRC is required clinically. Future
investigations should focus on deepening understanding into the
detailed mechanism between capecitabine and the RANK/RANKL pathway
in CRC both in vitro and in vivo, in addition to
identifying novel diagnostic and prognostic markers.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Science and Technology
Agency of Taizhou City of Zhejiang Province (grant no.
131KY13).
Availability of data and materials
The data used to support the findings of this study
are available from the corresponding author upon request.
Authors' contributions
MS and XM were responsible for the concept and study
design. CJ and CY were responsible for performing the experiments,
data collection and raw data authentication. HJ and YW were
responsible for data analysis and interpretation. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
The Ethics Committee of Taizhou Hospital of Zhejiang
Province affiliated to Wenzhou Medical University reviewed and
approved the present study. Each patient had provided appropriately
signed consent forms.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Arnold M, Sierra MS, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global patterns and trends in
colorectal cancer incidence and mortality. Gut. 66:683–691. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carethers JM and Doubeni CA: Causes of
socioeconomic disparities in colorectal cancer and intervention
framework and strategies. Gastroenterology. 158:354–367. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sivamaruthi BS, Kesika P and Chaiyasut C:
The role of probiotics in colorectal cancer management. Evid Based
Complement Alternat Med. Feb 14–2020.(Epub ahead of print). doi:
10.1155/2020/3535982. View Article : Google Scholar
|
|
5
|
Müller MF, Ibrahim AE and Arends MJ:
Molecular pathological classification of colorectal cancer.
Virchows Arch. 469:125–134. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Quan Y, Xu M, Cui P, Ye M, Zhuang B and
Min Z: Grainyhead-like 2 promotes tumor growth and is associated
with poor prognosis in colorectal cancer. J Cancer. 6:342–350.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang Y, Wang L, Wang W and Guo X:
Overexpression of circular RNA hsa_circ_0001038 promotes cervical
cancer cell progression by acting as a ceRNA for miR-337-3p to
regulate cyclin-M3 and metastasis-associated in colon cancer 1
expression. Gene. 733:1442732020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
De Falco V, Napolitano S, Roselló S,
Huerta M, Cervantes A, Ciardiello F and Troiani T: How we treat
metastatic colorectal cancer. ESMO Open. 4 (Suppl 2):e0008132020.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thorat SG, Chikhale RV and Tajne MR:
Development and validation of HPLC and HPTLC methods for
therapeutic drug monitoring of capecitabine in colorectal cancer
patients. J Chromatogr Sci. 57:892–900. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim JH: Chemotherapy for colorectal cancer
in the elderly. World J Gastroenterol. 21:5158–5166. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Aglago EK, Huybrechts I, Murphy N,
Casagrande C, Nicolas G, Pischon T, Fedirko V, Severi G,
Boutron-Ruault MC, Fournier A, et al: Consumption of fish and
long-chain n-3 polyunsaturated fatty acids is associated with
reduced risk of colorectal cancer in a large european cohort. Clin
Gastroenterol Hepatol. 18:654–66.e6. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Van Cutsem E, Verslype C and Tejpar S:
Oral capecitabine: Bridging the Atlantic divide in colon cancer
treatment. Semin Oncol. 32:43–51. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen Q, Mao Y, Meng F, Wang L, Zhang H,
Wang W and Hua D: Rs7911488 modified the efficacy of
capecitabine-based therapy in colon cancer through altering
miR-1307-3p and TYMS expression. Oncotarget. 8:74312–74319. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hirsch BR and Zafar SY: Capecitabine in
the management of colorectal cancer. Cancer Manag Res. 3:79–89.
2011.PubMed/NCBI
|
|
15
|
Silva I and Branco JC: Rank/Rankl/opg:
Literature review. Acta Reumatol Port. 36:209–218. 2011.PubMed/NCBI
|
|
16
|
Khosla S: Minireview: The OPG/RANKL/RANK
system. Endocrinology. 142:5050–5055. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sisay M, Mengistu G and Edessa D: The
RANK/RANKL/OPG system in tumorigenesis and metastasis of cancer
stem cell: Potential targets for anticancer therapy. Onco Targets
Ther. 10:3801–3810. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Luo G, Li F, Li X, Wang ZG and Zhang B:
TNFα and RANKL promote osteoclastogenesis by upregulating RANK via
the NFκB pathway. Mol Med Rep. 17:6605–6611. 2018.PubMed/NCBI
|
|
19
|
Wood MB, Rios D and Williams IR: TNF-α
augments RANKL-dependent intestinal M cell differentiation in
enteroid cultures. Am J Physiol Cell Physiol. 311:C498–C507. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chu GC, Zhau HE, Wang R, Rogatko A, Feng
X, Zayzafoon M, Liu Y, Farach-Carson MC, You S, Kim J, et al: RANK-
and c-Met-mediated signal network promotes prostate cancer
metastatic colonization. Endocr Relat Cancer. 21:311–326. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Casimiro S, Mohammad KS, Pires R,
Tato-Costa J, Alho I, Teixeira R, Carvalho A, Ribeiro S, Lipton A,
Guise TA and Costa L: RANKL/RANK/MMP-1 molecular triad contributes
to the metastatic phenotype of breast and prostate cancer cells in
vitro. PLoS ONE. 8:e631532013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Buckle CH, Neville-Webbe HL, Croucher PI
and Lawson MA: Targeting RANK/RANKL in the treatment of solid
tumours and myeloma. Curr Pharm Des. 16:1272–1283. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Okimoto G, Zeinalzadeh A, Wenska T, Loomis
M, Nation JB, Fabre T, Tiirikainen M, Hernandez B, Chan O, Wong L
and Kwee S: Joint analysis of multiple high-dimensional data types
using sparse matrix approximations of rank-1 with applications to
ovarian and liver cancer. BioData Min. 9:242016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Khan MA, Sharif M, Akram T, Yasmin M and
Nayak RS: Stomach deformities recognition using rank-based deep
features selection. J Med Syst. 43:3292019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Infante M, Fabi A, Cognetti F, Gorini S,
Caprio M and Fabbri A: RANKL/RANK/OPG system beyond bone
remodeling: Involvement in breast cancer and clinical perspectives.
J Exp Clin Cancer Res. 38:122019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sood SK, Balasubramanian S, Higham S,
Fernando M and Harrison B: Osteoprotegerin (OPG) and related
proteins (RANK, RANKL and TRAIL) in thyroid disease. World J Surg.
35:1984–1992. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Retraction, Antitumor and antimetastatic
activities of docetaxel are enhanced by genistein through
regulation of osteoprotegerin/receptor activator of nuclear
factor-κB (RANK)/RANK Ligand/MMP-9 signaling in prostate cancer.
Cancer Res. 78:54752018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pritchard L, Cardle L, Quinn S and Dufton
M: Simple intrasequence difference (SID) analysis: An original
method to highlight and rank sub-structural interfaces in protein
folds. Application to the folds of bovine pancreatic trypsin
inhibitor, phospholipase A2, chymotrypsin and carboxypeptidase A.
Protein Eng. 16:87–101. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liang Q, Wang Y, Lu Y, Zhu Q, Xie W, Tang
N, Huang L, An T, Zhang D, Yan A, et al: RANK promotes colorectal
cancer migration and invasion by activating the
Ca(2+)-calcineurin/NFATC1-ACP5 axis. Cell Death Dis. 12:3362021.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhao W, Chen B, Guo X, Wang R, Chang Z,
Dong Y, Song K, Wang W, Qi L, Gu Y, et al: A rank-based
transcriptional signature for predicting relapse risk of stage II
colorectal cancer identified with proper data sources. Oncotarget.
7:19060–19071. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
National Health Commission of the People's
Republic of China, . [Chinese Protocol of Diagnosis and Treatment
of Colorectal Cancer. ((2020 edition)]). Zhonghua Wai Ke Za Zhi.
58:561–585. 2020.(In Chinese). PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Park DW, Ham YM, Lee YG, So R, Seo YJ and
Kang SC: Multioside, an active ingredient from adonis amurensis,
displays anti-cancer activity through autophagosome formation.
Phytomedicine. 65:1531142019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Choi MY, Flood K, Bernatsky S,
Ramsey-Goldman R and Clarke AE: A review on SLE and malignancy.
Best Pract Res Clin Rheumatol. 31:373–396. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wen J, Liu X, Qi Y, Niu F, Niu Z, Geng W,
Zou Z, Huang R, Wang J and Zou H: BMP3 suppresses colon
tumorigenesis via ActRIIB/SMAD2-dependent and TAK1/JNK signaling
pathways. J Exp Clin Cancer Res. 38:4282019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li B, Zhu FC, Yu SX, Liu SJ and Li BY:
Suppression of KIF22 inhibits cell proliferation and xenograft
tumor growth in colon cancer. Cancer Biother Radiopharm. 35:50–57.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li F, Zhang C and Fu L: PRR14
overexpression promotes cell growth, epithelial to mesenchymal
transition and metastasis of colon cancer via the AKT pathway. PLoS
ONE. 14:e02188392019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kiesel L and Kohl A: Role of the
RANK/RANKL pathway in breast cancer. Maturitas. 86:10–16. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tsubaki M, Takeda T, Yoshizumi M, Ueda E,
Itoh T, Imano M, Satou T and Nishida S: RANK-RANKL interactions are
involved in cell adhesion-mediated drug resistance in multiple
myeloma cell lines. Tumour Biol. 37:9099–9110. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang J, Sun X, Zhang H, Wang Y and Li Y:
MPA influences tumor cell proliferation, migration, and invasion
induced by RANKL through PRB involving the MAPK pathway in
endometrial cancer. Oncol Rep. 33:799–809. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yamaue H, Tanimura H, Nakamori M, Noguchi
K, Iwahashi M, Tani M, Hotta T, Murakami K and Ishimoto K: Clinical
evaluation of chemosensitivity testing for patients with colorectal
cancer using MTT assay. Dis Colon Rectum. 39:416–422. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Singh G, Graffner HO, Milsom JW and
Chaudry IH: Tauromustine is more effective than conventional
chemotherapy in the treatment of colonic tumors. Dis Colon Rectum.
36:394–399. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lam SW, Guchelaar HJ and Boven E: The role
of pharmacogenetics in capecitabine efficacy and toxicity. Cancer
Treat Rev. 50:9–22. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Song D, Meng T, Xu W, Hou T, Lin Z, Yin H,
Li B, Zhou L, Wang T, Han S, et al: 5-Fluoruracil blocked giant
cell tumor progression by suppressing osteoclastogenesis through
NF-kappaB signals and blocking angiogenesis. Bone. 78:46–54. 2015.
View Article : Google Scholar : PubMed/NCBI
|