Introduction
Breast cancer is one of the common malignant tumors
in women and the incidence rate ranks first among female malignant
tumors (1). Triple-negative breast
cancer (TNBC), a subtype of breast cancer with negative expression
of estrogen receptor (ER), progesterone receptor (PR) and
proto-oncogene human epidermal growth factor receptor 2 receptor
(HER2), is resistant to endocrine and molecular targeted therapy
and prone to local recurrence and distant metastasis (2). Chemotherapy is therefore the main
treatment option for triple-negative breast cancer (3). However, patients with breast cancer
are usually resistant to chemical therapeutics, which is the main
reason of poor prognosis (4,5).
According to the theory of tumor stem cells, tumor stem cells serve
an important role in the survival, proliferation, metastasis and
recurrence of tumors. Thus, targeted killing of tumor stem cells
may be the key to treat tumors (6,7).
Therefore, it is urgent to develop new chemotherapeutic drugs that
can kill triple negative breast cancer rand study its specific
mechanism, so as to find effective therapeutic targets.
Salinomycin (SAL), an ionophore antibiotic isolated
from the fermentation broth of Streptomyces albicularis
(strain no. 80614), kills pathogenic microorganisms by interfering
with the cation (Na+ and K+) balance inside
or outside the cell and altering the osmotic pressure (8). Gupta et al (9) showed that SAL selectively
exterminates breast cancer stem cells (CSCs), with an efficiency
100 times higher than that of paclitaxel. Since then, a number of
in vivo and in vitro studies (10,11)
have demonstrated that SAL inhibits CSCs in various types of tumor
(12). SAL helps tumor cells or
CSCs overcome the apoptosis resistance caused by P53 gene mutation,
or Bcl-2, 26S proteasome and P-glycoprotein overexpression
(13), increasing the DNA damage
caused by oxidative stress (14)
and inducing autophagy and subsequent apoptosis (15). In addition, SAL reverses drug
resistance and increases the sensitivity of chemical drugs by
inhibiting the activity of ATP binding box transporter superfamily
and blocking the signaling pathway of Wnt/β-catenin, Akt/NF-κB and
others (16–18). Therefore, SAL might be a potential
clinically effective and highly selective anti-tumor drug. However,
its severe neurotoxic and muscular toxicity cannot be ignored. To
reduce the side effects of SAL, the development of combined
treatment strategy or targeting delivery system is required.
17-allylamino-17-demethoxygeldanamycin (17-AAG) is
an inhibitor of chaperone heat shock protein 90 (Hsp90), which
binds to ATP-binding region of Hsp90, inhibits the formation of
multiprotein complex comprising co-chaperone proteins and induces
the degradation of client proteins through the ubiquitin-proteasome
pathway (19). During the process
of malignant transformation, Hsp90 stabilizes and protects mutated
proteins from proteasomal degradation and enables the sustained
survival and cell growth of cancer cells. By inhibiting Hsp90,
17-AAG has been widely investigated in preclinical and clinical
research as a single agent or in combination with other anticancer
agents for a wide range of types of human cancer. For example,
17-AAG inducing cell apoptosis by blocking the customer protein
Hsp90 hypoxia-inducible factor 1α transcription function (20) and influencing the colony formation
ability of CSCs and the growth of tumor cells (21). 17-AAG can directly interact with
voltage-dependent anion channel through hydrophobic interaction,
independent of Hsp90, to increase intracellular calcium ion
concentration, prompting increased intracellular calcium ion
concentration (22). In addition,
17-AAG can kill cancer cells through the synergistic action of the
upstream molecules of CD95 death receptor with MAPK/ERK 1/2
inhibitors (23). In addition,
17-AAG can affect the stability of Akt, leading to the deletion and
expression of Akt and enhancing the oxidative stress mediated by
sulfhydryl in cancer cells, thus effectively increasing the
sensitivity of cancer cells to chemotherapy (24).
In our previous study, SAL significantly inhibited
the proliferation of breast cancer cells via reducing the
expression of breast CSCs marker ALDH (25). To further enhance efficacy and
reduce toxicity, the present study investigated the combined effect
of SAL with 17-AAG on apoptosis and autophagy in breast cancer
cells and the corresponding molecular mechanism. It may provide a
theoretical basis for the potential combined treatment strategy
involving SAL.
Materials and methods
Cell lines and cell culture
Human breast cancer cell line MDA-MB-231 was
obtained from the American Type Culture Collection. The cells were
incubated with RPMI-1640 medium (HyClone; Cytiva) containing 10%
fetal bovine serum (FBS; HyClone; Cytiva), 100 U/ml penicillin and
100 U/ml streptomycin and passaged at a ratio of 1:2 or 1:3. All
cell lines were tested for mycoplasma and characterized by short
tandem repeat profiling analysis (Cenvino).
Drugs
SAL was purchased from China Institute of Veterinary
Drug Control, and 17-AAG was purchased from MedChemExpress. SAL and
17-AAG were dissolved in DMSO and administrated at the
concentration of 1–32 µM or 1.25 nM, respectively.
MTT assay
Single cell suspension with concentration of
5×104 cells/ml was prepared from logarithmic growth
stage cells. Briefly, 5×103 cells were seeded to each
well of 96-well plates and were cultured overnight. Then, cells
were treated with SAL alone (1, 2, 4, 8, 16 and 32 µM) or in
combination with 17-AAG (1.25 nM) for 24, 48 and 72 h. Next, cells
were incubated with 20 µl MTT (5 mg/ml; Sigma; cat. no. M2003) for
4 h, followed by 150 µl DMSO for dissolving the crystals. The
optical density values were detected at 490 nm and the relative
cell growth was calculated and expressed as IC50 values
using GraphPad Prism 6.0 (GraphPad Software, Inc.). Untreated cells
were chosen as a control, which were cells incubated with DMSO
(solvent for SAL) and DMSO (solvent for 17-AAG). Combination index
(CI) was calculated by the CompuSyn software (version 1.0; Biosoft)
using the Chou-Talalay method: CI=1, additive effect; CI<1,
synergistic effect; CI>1, antagonistic effect (26,27).
Apoptosis assay
Cells were treated with SAL and/or 17-AAG for 48 h.
Then, 5×105 cells were labeled with the Annexin V and
propidium iodide (PI) using FITC Annexin V Apoptosis Detection kit
with PI (BioLegend, Inc, cat. no. 641904) according to the
manufacturer's protocol. The labeled cells were immediately
measured with a BD FACSCalibur Flow Cytometer (BD Biosciences) and
quantified with CellQuest software (version 5.1; BD Biosciences)
for early and late apoptotic cells.
RT-qPCR
Total RNA was extracted from 5×105 cells
treated with SAL and/or 17-AAG treated cells for 48 h using
TRIzol® (Thermo Fisher Scientific, Inc.), and the
quantity and purity of RNA were detected using NanoDrop 1000
spectrophotometer (Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Total RNAs (1 µg) was reversely
transcribed into cDNA by Super Script first-strand synthesis system
(Invitrogen; Thermo Fisher Scientific, Inc.) as described
previously (9). Prepared cDNA was
then subjected to quantitative PCR analysis using the Strata gene
Mx3005P Multiplex quantitative PCR system (Agilent Technologies,
Inc.) with 2X SYBR Green qPCR Master Mix (Bimake; cat. no. B21203).
A total of 20 µl real-time fluorescence PCR reaction mixture was
used and the reaction conditions were as follows: Pre-denaturation
at 95°C for 5 min, reaction at 95°C for 20 sec, 58°C for 20 sec,
72°C for 20 sec and 72°C for 10 min for a total of 40 cycles. The
relative expression of genes was analyzed by the comparative Ct
method. The data are presented as the fold change, which was
calculated as 2−ΔΔCq
(ΔΔCq=ΔCqtreated-ΔCqcontrol) (28). Cq is the cycle number at which
fluorescence first exceeds the threshold. The ΔCq values from each
target gene were obtained by subtracting the GAPDH Cq from the
sample Cq. The experiment was repeated three times. The primer
sequences are shown in Table
I.
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Gene | Orientation | Primer sequence
(5′-3′) |
|---|
| Bcl-2 | Forward |
GGTGGGGTCATGTGTGTGG |
|
| Reverse |
CGGTTCAGGTACTCAGTCATCC |
| Bax | Forward |
CCCGAGAGGTCTTTTTCCGAG |
|
| Reverse |
CCAGCCCATGATGGTTCTGAT |
| Caspase
3 | Forward |
CATGGAAGCGAATCAATGGACT |
|
| Reverse |
CTGTACCAGACCGAGATGTCA |
| LC3 | Forward |
AACATGAGCGAGTTGGTCAAG |
|
| Reverse |
GCTCGTAGATGTCCGCGAT |
| Beclin1 | Forward |
CCATGCAGGTGAGCTTCGT |
|
| Reverse |
GAATCTGCGAGAGACACCATC |
| P62 | Forward |
GCACCCCAATGTGATCTGC |
|
| Reverse |
CGCTACACAAGTCGTAGTCTGG |
| GAPDH | Forward |
GGAGCGAGATCCCTCCAAAAT |
|
| Reverse |
GGCTGTTGTCATACTTCTCATGG |
Western blotting
SAL and/or 17-AAG treated cells were collected and
the total protein was extracted using the phospho-RIPA buffer (1 M
Tris-HCl at pH 7.5, 5 M NaCl, 0.01% NP-40, 0.5 M EGTA and 10% SDS)
supplemented with a complete EDTA-free protease inhibitor cocktail
(Roche Diagnostics). Protein was quantitated by a bicinchoninic
acid kit (Abcam), separated in SDS-PAGE gels (8 or 10%) with
protein loaded of 10 µl in per lane and electrically transferred
onto polyvinylidene fluoride membranes (MilliporeSigma) membrane.
Following blocking in PBS + 5% bovine serum albumin (Abcam) at room
temperature, the membranes were then incubated with the following
primary antibodies: Cleaved-caspase 3 (1:500; ProteinTech Group,
Inc.; cat. no. 25546-1-AP), Bcl-2 (1:1,000; Cell Signaling
Technology, Inc.; cat. no. 15071S), Bax (1:1,000; Cell Signaling
Technology, Inc.; cat. no. 89477), microtubule-associated protein 1
light chain 3 (LC3)B (1:1,000; Abcam; cat. no. ab51520), Beclin1
(1:1,000; Abcam; cat. no. ab210498), P62 (1:1,000; Abcam; cat. no.
ab56416), JNK (1:1,000; Abcam; cat. no. ab76125) and phosphorylated
(p-) JNK (1:1,000; Cell Signaling Technology, Inc.; cat. no. 9255S)
at 4°C overnight; and subsequently with the Rabbit Anti-Mouse IgG
H&L (HRP) (1:5,000; Abcam; cat. no. ab6728) or Goat Anti-Rabbit
IgG H&L (HRP) (1:5,000; Abcam; cat. no. ab6721) at room
temperature for 1 h. The protein signals were developed using the
Enlight Western blot ECL reagents (Engreen Biosystem Co., Ltd.).
GAPDH (1:2,500; Abcam; cat. no. ab9485) or α-tubulin (1:5,000;
Abcam; cat. no. ab7292) was used as a loading control.
Transmission electron microscopy
Cells were collected 48 h after treatment with SAL
alone and in combination with 17-AAG. The cell concentration was
adjusted to 4×106−1×107 cells/ml Cells were
fixed with 2.5% glutaraldehyde buffered in 0.1 M sodium cacodylate
(pH 7.4) at 5°C for ~4 h on ice, then rinsed in sodium cacodylate
buffer and post-fixed in 1% aqueous osmium tetroxide (buffered in
0.1 M sodium cacodylate) at 5°C for ~2 h and then rinsed and stored
in the buffer at 4°C. Cells were later dehydrated in an
acetone/ethanol series and transferred to propyleneoxide and then
subsequently embedded in Glycidether 100 (formerly Epon) (Abcam;
cat. no. c5318). Following polymerization, semi-thin sections (1
µm) and ultrathin sections (60–90 nm) were cut and stained with
toluidine blue for 20–30 min at room temperature. Digital
micrographs were captured with a JEOL JEM1010 electron microscope
(JEOL, Ltd. Tokyo:6951).
Tandem Mass Tag (TMT) quantification
for proteomics
Cells were collected 48 h after treatment with SAL
alone and in combination with 17-AAG. The cells were lysed by
adding five cell-pellet volumes of lysis buffer (100 µl of Lysis
Buffer for a 20 µl cell pellet) according to the TMT labelling kit
instructions (Thermo Fisher Scientific, Inc). Peptides were
labelled with the TMT Iso baric Mass Tags (Thermo Fisher
Scientific, Inc.); the lysate was centrifuged at 16,000 × g for 10
min at 4°C, and adjusted to a final volume of 1,000 µl with 100 nM
TEAB. After adding 5 µl TCEP (200 nM) and incubating at 55°C for 1
h, TEAB was diluted away from light in 132 µl iodoacetamide to a
concentration 375 nM. Next, 6 volumes of pre-chilled acetone
(−20°C) and 20 µl trypsin storage solution (room temperature) were
added and incubated for 5 min. Lastly, 2.5 µl trypsin per 100 µg of
protein was added to digest the sample overnight at 37°C. Finally,
Proteome Discoverer (version 2.1; Thermo Fisher Scientific, Inc.)
was used to analyze data, screen differential proteins.
Mito-ROS
Cells were treated with SAL and/or 17-AAG for 48 h
at 37°C. The concentration of cell suspension was adjusted to
5×105 cells/ml. The cell suspension was transferred to
an Eppendorf test tube, centrifuged at 1,000 × g for 5 min at room
temperature and the supernatant discarded. The blank control group
and experimental group were set up. The Mito Tracker Red CMX Ros
using Mito Tracker Red CMX Ros-Special Packaging (Thermo Fisher
Scientific, Inc.; cat. no. M7512) was diluted in serum-free medium
to a final concentration of 1 mM according to the manufacturer's
protocol and incubated at 37°C for 15–35 min away from light and
mixed every 5 min. Following incubation, the centrifuged at 1,000 ×
g for 5 min and the supernatant discarded. Following washing with
PBS three times, the cells were suspended with 500 µl PBS and
placed on ice for flow cytometry detection. Mito-tracker Red CMXRos
fluoresce in red with a maximum excitation wavelength of 579 nm and
a maximum emission wavelength of 599 nm. A FACSCalibur flow
cytometer (BD Biosciences) was used to detect the fluorescence
intensity of different groups of cells at specific wavelengths to
compare the ROS content in different groups of mitochondria using
FlowJo (version 10; BD Biosciences).
Statistical analysis
All data are obtained from three replicate
experiments and expressed as mean ± standard deviation of three
independent experiments. GraphPad Prism 6.0 software (GraphPad
Software, Inc.) was used for statistical analysis and graph
rendering. One-way ANOVA followed by Tukey's test was used for
comparing continuous variables multiple groups. P<0.05 was
considered to indicate a statistically significant difference.
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was
carried out on the proteomics data. In the KEGG database
(https://www.kegg.jp/), KO (KEGG Orthology) is a
classification system for genes and their products. Lineal
homologous genes with similar functions on the same pathway and
their products are grouped together and assigned the same KO label
(29). To annotate KEGG pathways
in the target protein set, KEGG Orthology and Links Annotation
(KOALA) (30) was used to classify
the target proteins by KO the KEGG database (http://www.kegg.jp/kegg-bin/show_pathway?ko04010+K04440).
The pathway information associated with the target protein was
obtained automatically according to KO classification. For KEGG
pathway enrichment analysis, Fisher's Exact Test was used to screen
the proteins associated with specific KEGG pathway and analyse
their relative enrichment. Pathways with a false discovery rate
<0.01 were considered significantly enriched.
Results
Combination of SAL and 17-AAG
synergistically inhibits cell growth in human breast cancer
cells
To explore the possible Synergistic inhibitory
effect of SAL and 17-AAG, the MTT assay was used to detect the
relative cell growth in human breast cancer cells treated with SAL
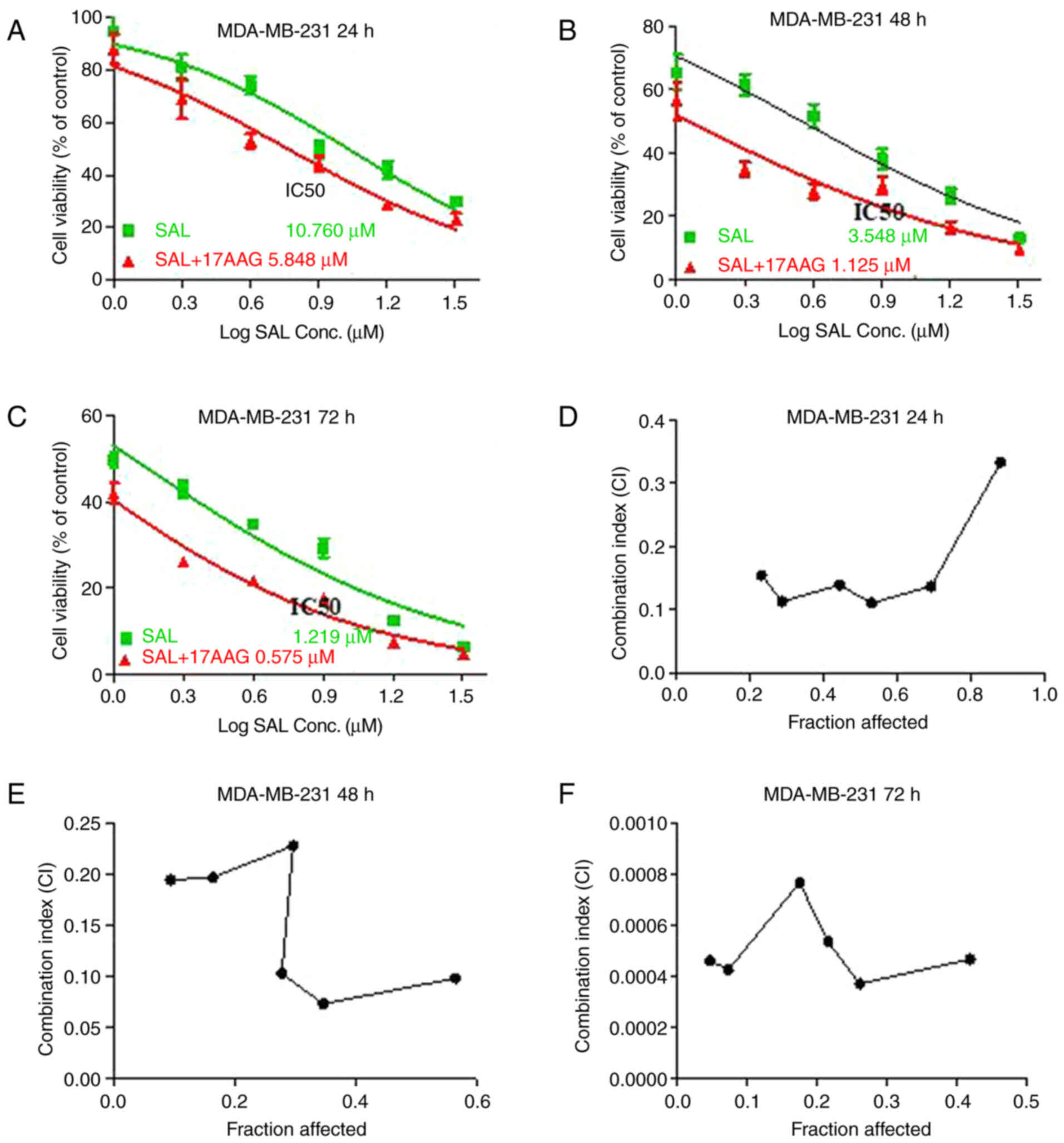
alone or in combination with 17-AAG. The results showed that SAL
alone significantly reduced cell growth in a dose- and
time-dependent manner, with IC50 of SAL being 10.760,
3.548 and 1.219 µM in MDA-MB-231 cells for 24, 48 and 72 h,
respectively (Fig. 1A-C). In
addition, the combination of 17-AAG with SAL clearly decreased
IC50 of SAL by 50% compared with that in cells treated
with SAL alone, with the highest reduction occurring at 48 h. These
results indicated that the combination treatment of SAL and 17-AAG
was more effective in inhibiting cell growth when compared with the
single treatment of SAL, which implying an interaction between SAL
and 17-AAG.
To further clarify the combined growth inhibitory
effect of SAL and 17-AAG on MDA-MB-231 cells, the Chou-Talalay
combined index method was used. The results showed that the growth
inhibition ratio of SAL combined with 17-AAG in MDA-MB-231 cells
for 24 h was 0.8807773, 0.6917017, 0.5309874, 0.4438025, 0.2883403
and 0.2326681 and the corresponding combination index (CI) was
0.33311, 0.13707, 0.11066, 0.13916, 0.11332 and 0.15435. As CI
values <1 indicate synergism in drug combinations, the data
suggested the combination of SAL and 17-AAG had a synergistic
effect on the cell growth of breast cancer cells (Fig. 1D-F). With time, CI was
significantly increased, indicating that the synergistic inhibition
between SAL and 17-AAG occurred in time-dependent manner.
Combination of SAL and 17-AAG
synergistically induces apoptosis in human breast cancer cells
Synergism of anticancer drug can be occurred from
the combination with different mechanisms and/or modes of actions.
SAL and 17-AAG was reported to have a role in inducing apoptosis,
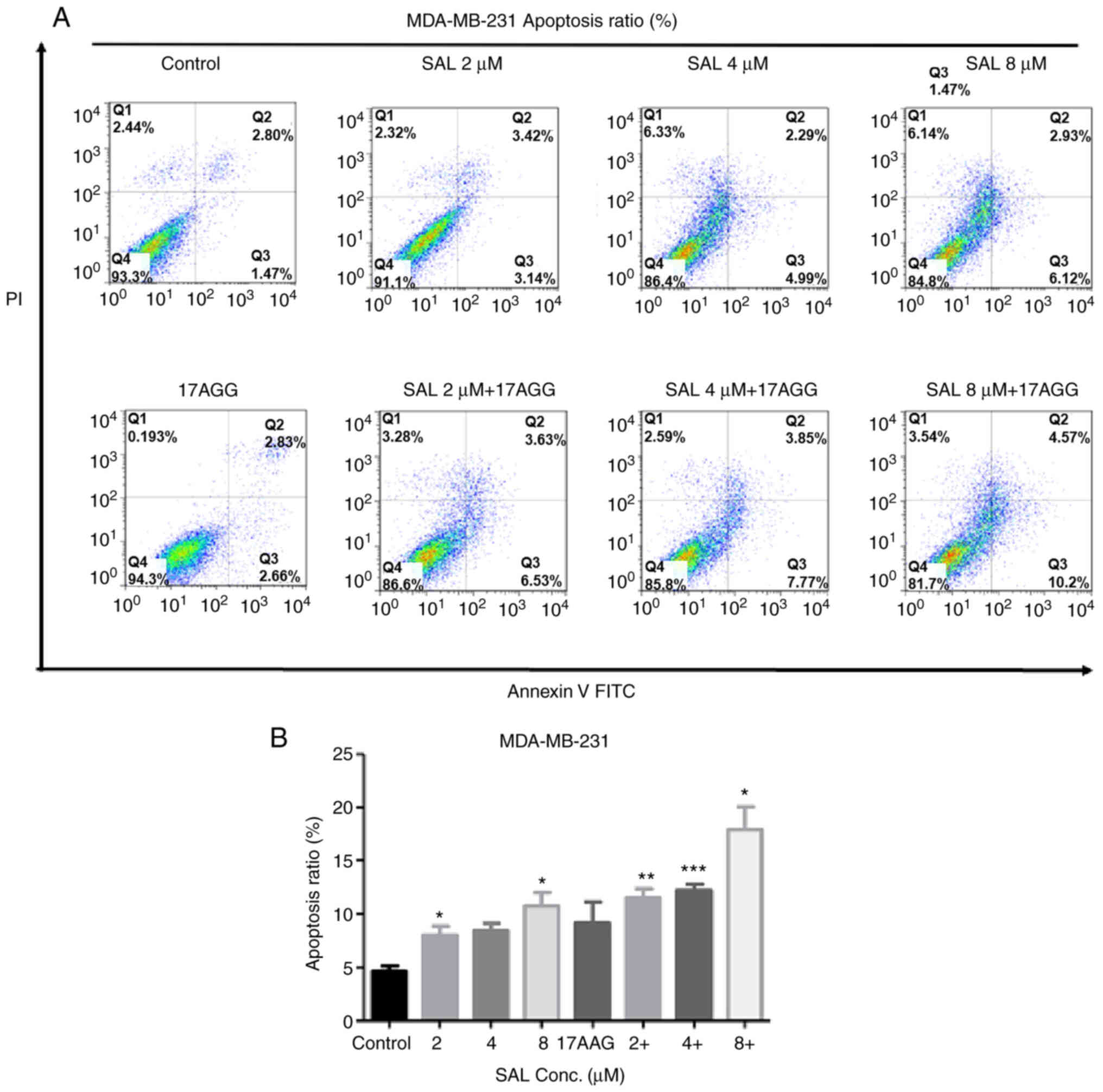
thus the apoptotic ratios in human breast cancer cells treated with
SAL and/or 17-AAG was first detected. As shown in Fig. 2, the apoptotic ratio increased in a
dose-dependently manner in SAL treated cells. The SAL-induced
apoptotic ratios were further significantly enhanced when SAL and
17-AAG were combined. Correspondingly, the mRNA and protein
expression of genes involving in apoptosis signaling pathway, such
as Bcl-2 and caspase-3, in SAL treated cells were decreased, while
the mRNA and protein expression of Bax and cleaved caspase-3 was
increased, as compared with the cells treated with combined SAL and
17-AAG (Fig. 3). The results
showed that SAL in combination with 17-AAG greatly activated the
apoptosis-related pathways and then promoted apoptosis in breast
cancer cells.
Combination of SAL and 17-AAG
synergistically inhibits autophagy in human breast cancer
cells
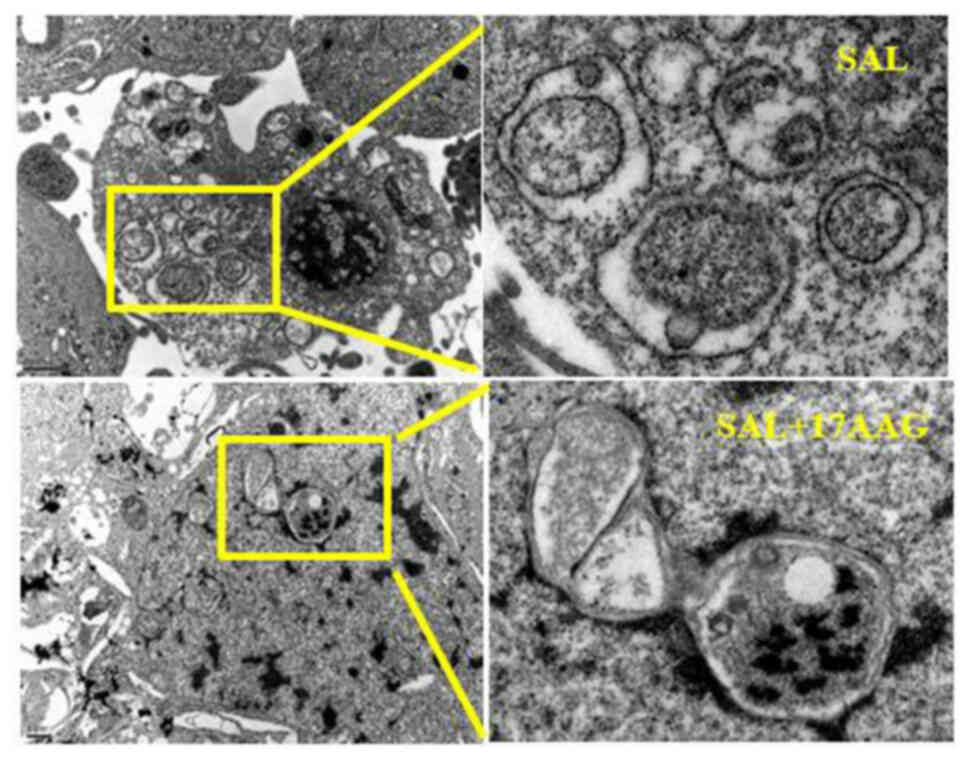
Transmission electron microscopy was used to observe
the effect of SAL alone and in combination with 17-AAG on
MDA-MB-231 cell apoptosis and autophagy. The results showed that
SAL alone and in combination with 17-AAG could induce apoptosis and
autophagy in human breast cancer MDA-MB-231 cells. Under the
microscope, the formation of autophagic bodies, swelling and
deformation of mitochondria, as well as obvious nuclear
fragmentation and vacuoles in the cytoplasm, could be observed,
suggesting the occurrence of apoptosis and autophagy (Fig. 4). Compared with the single drug
group, the autophagosomes in the combined drug group were reduced
and the cells in the combined drug group were mainly apoptotic,
with obvious nuclear fragmentation observed under the microscope.
As compared with the single drug group, apoptosis increased and
autophagy decreased in the combination group. It was suggested that
the combination of SAL and 17-AAG may induce apoptosis and inhibit
autophagy, thereby affecting the growth of MDA-MB-231 cells in
triple-negative breast cancer.
SAL was used alone (2, 4 and 8 µM) or in combination
with 17-AAG (1.25 nM) on breast cancer MDA-MB-231 cells for 48 h.
The protein and mRNA expression levels of LC3, Beclin1 and P62 were
all lower than those of the control group and the protein bands
became lighter and narrower. Compared with the single drug group,
the histone and mRNA expression levels of the combined drug group
were more significantly reduced (Fig.
5).
Combination of SAL and 17-AAG
synergistically induces apoptosis and inhibits autophagy through
the reactive oxygen species (ROS)-JNK signaling pathway in human
breast cancer cells
TMT results showed that SAL alone and in combination
with 17-AAG can cause changes in apoptosis and autophagy pathways
(Fig. 6A). Furthermore, it was
found that SAL combined with 17-AAG caused significant changes in
the MAPK signaling pathway proteins, with JNK and p-JNK proteins
significantly upregulated. Therefore, the combination of SAL and
17-AAG may induce apoptosis and autophagy through the JNK signaling
pathway and eventually affect the growth of breast cancer
MDA-MB-231 cells. The proteomic results were further verified to
investigate whether the mechanism of SAL and 17-AAG inhibiting the
growth of breast cancer cells through apoptosis and autophagy was
associated with the JNK signaling pathway. The protein expression
of JNK and p-JNK in the MAPK pathway were detected by western
blotting (Fig. 6B and C). The
results showed that after breast cancer cells were treated with SAL
alone and in combination with 17-AAG, the upregulation of JNK and
p-JNK proteins was observed. The expression of proteins in the
combination group was higher than that in the single drug group. It
was also found that SAL alone and combined with 17-AAG can
synergistically induce the production of ROS in MDA-MB-231 cells
and the ROS content in the combined group was higher than that in
the single group (Fig. 6D and E),
suggesting that SAL and 17-AAG may further activate the JNK pathway
through ROS to further induce apoptosis and autophagy and influence
the growth of breast cancer MDA-MB-231 cells.
 | Figure 6.Combination of SAL and 17-AAG
synergistically induced apoptosis and inhibited autophagy through
the ROS-JNK signaling pathway in human breast cancer cells. (A)
Kyoto Encyclopedia of Genes and Genomes pathway statistics with
significant enrichment (top 10). (B) Effects of JNK and p-JNK
protein expression levels. (C) Effect of ROS content in cells.
condition for 48 h. (D) Quantitative statistics of protein. (E)
Peak statistics of Mito-Ros. *P<0.05, **P<0.01 vs. control
group. SAL, salinomycin; 17-AAG,
17-allylamino-17-demethoxygeldanamycin; ROS, reactive oxygen
species; p-, phosphorylated; SLE, systemic lupus erythematosus; MP,
metabolic pathways; HTLV1, human T-cell lymphotropic virus 1; TMIC,
transcriptional misregulation in cancer; HSV, herpes simplex virus;
EB, Epstein-Barr virus; VC, viral carcinogenesis, HIV1, human
immunodeficiency virus 1; PEC, pathogenic Escherichia coli
infection; APP, antigen processing and presentation; SA,
Staphylococcus aureus; EP, estrogen pathway; KSAH, Kaposi
sarcoma-associated herpesvirus; Conc., concentration. |
Discussion
In 2012, Verdoodt et al (31) first discovered that SAL can
activate autophagy after acting on colon and breast cancer cell
lines. Subsequent studies showed that SAL could affect breast
cancer cell proliferation by inducing apoptosis and autophagy
(18,31). However, the cytotoxicity of SAL is
also a problem that cannot be ignored, which is the key factor
affecting its curative effect. Therefore, how to enhance the
selective killing effect of SAL between cancer or CSCs cells and
normal cells and improve its therapeutic effect has become an
important issue that requires urgent attention.
Combined drug use refers to the simultaneous or
continuous use of two or more drugs to improve the efficacy and
reduce the possibility of drug resistance without increasing
toxicity, so as to achieve therapeutic effects. Through a
literature review, it was found that SAL and 17-AAG overlap in
certain anti-tumor mechanisms. The two drugs can affect the growth
of cancer cells by inducing apoptosis, targeting CSCs and
increasing oxidative stress, as well as affecting drug resistance
in cancer cells. Therefore, the combined application of SAL and
17-AAG may reduce the dose of SAL, thus reducing its cytotoxicity
and effectively killing cancer cells or CSCs while reducing the
occurrence of drug tolerance. The present study also showed that
the use of SAL and 17-AAG alone and in combination can induce
apoptosis, leading to an increase in the apoptotic ratio and
upregulation of apoptosis-related protein expression, as well as
the downregulation of autophagy-related protein expression in
breast cancer cells, inhibition of protein expression and reduction
of autophagosomes. The results also indicated that the combination
of SAL and 17-AAG can affect breast cancer cell growth by inducing
apoptosis and autophagy. The proteomic results showed that both SAL
and 17-AAG alone and in combination could induce apoptosis and the
autophagy signaling pathway, as well as inflict significant changes
in the MAPK signaling pathway.
In addition, studies (32,33)
have shown that there is an association between autophagy and ROS.
The inhibition of autophagy through autophagy inhibitors can
significantly increase the level of ROS, and ROS elimination can
significantly induce cell death. Studies have also shown that
autophagy can promote cell apoptosis and ROS are considered to be
the main molecules associated with cell apoptosis and autophagy
(34,35). ROS can regulate cell growth and
survival, as well as inhibit the PI3K/Akt signaling pathway
(36) and activate the MAPK
signaling pathway (37). The
present study found that SAL alone and in combination with 17-AAG
could cause an increase in the ROS content. In cancer treatment,
ROS are not only associated with autophagy, but are also a key
factor affecting cell apoptosis and proliferation (38).
ROS can activate JNK through bispecific kinase JNKK
and the activated JNK can promote the expression of pro-apoptotic
proteins such as P53, Bax, Fas-ligand (FasL) and TNF, through
transcription factor AP-1 (39).
High expression of pro-apoptotic proteins, such as Bax and Bak, can
promote the release of cytochrome c into the cytoplasm. The binding
of cytochrome c and caspase-9 can activate caspase-3 (40). The activated caspase-3 serves a
very important role in that it can lyse autophagy-related proteins,
which can enter the mitochondria, promote the release of cytochrome
c and further promote the occurrence of cell apoptosis (41). Studies have shown that Beclin 1 can
be cleaved by caspase-3 to produce C-terminal fragment of Beclin 1,
which can enter the mitochondria to promote the release of
cytochrome c, inhibit autophagy and induce apoptosis (42,43).
In addition, expressed ligands, such as FasL and TNF, bind to the
death receptor on the cell membrane to form a death-inducing
signaling complex, which promotes the cleavage of the precursor
caspase-8 to generate the activated caspase-8 (44). On the one hand, caspase-8 can
activate the downstream caspase-8 of cell apoptosis to initiate the
apoptosis signal and on the other hand, it can induce the
production of cellular FLICE-inhibitory protein, viral
FLICE-inhibitory protein and other substances that bind to Atg3.
Thus, the binding of Atg3 and LC3 is inhibited, as is the
occurrence of autophagy (45).
However, there are some limitations of the present
study. First, it only focused on the synergistic inhibition effects
on the MDA-MB-231 cell line, which is not enough to judge the
efficacy of combined drugs and further application in human breast
cancer. Other triple-negative breast cancer cell line should be
used to verify the effect of this combination therapy in a future
study. Second, the present study only explored the JNK signaling
pathway and stemness-associated biomarkers was not performed.
Moreover, SAL has been shown to inhibit CSCs via blocking
Wnt/beta-catenin signaling (46).
So the Wnt/beta-catenin signaling or other more CSC associated
mechanisms and stemness-associated biomarkers should be performed
in the future.
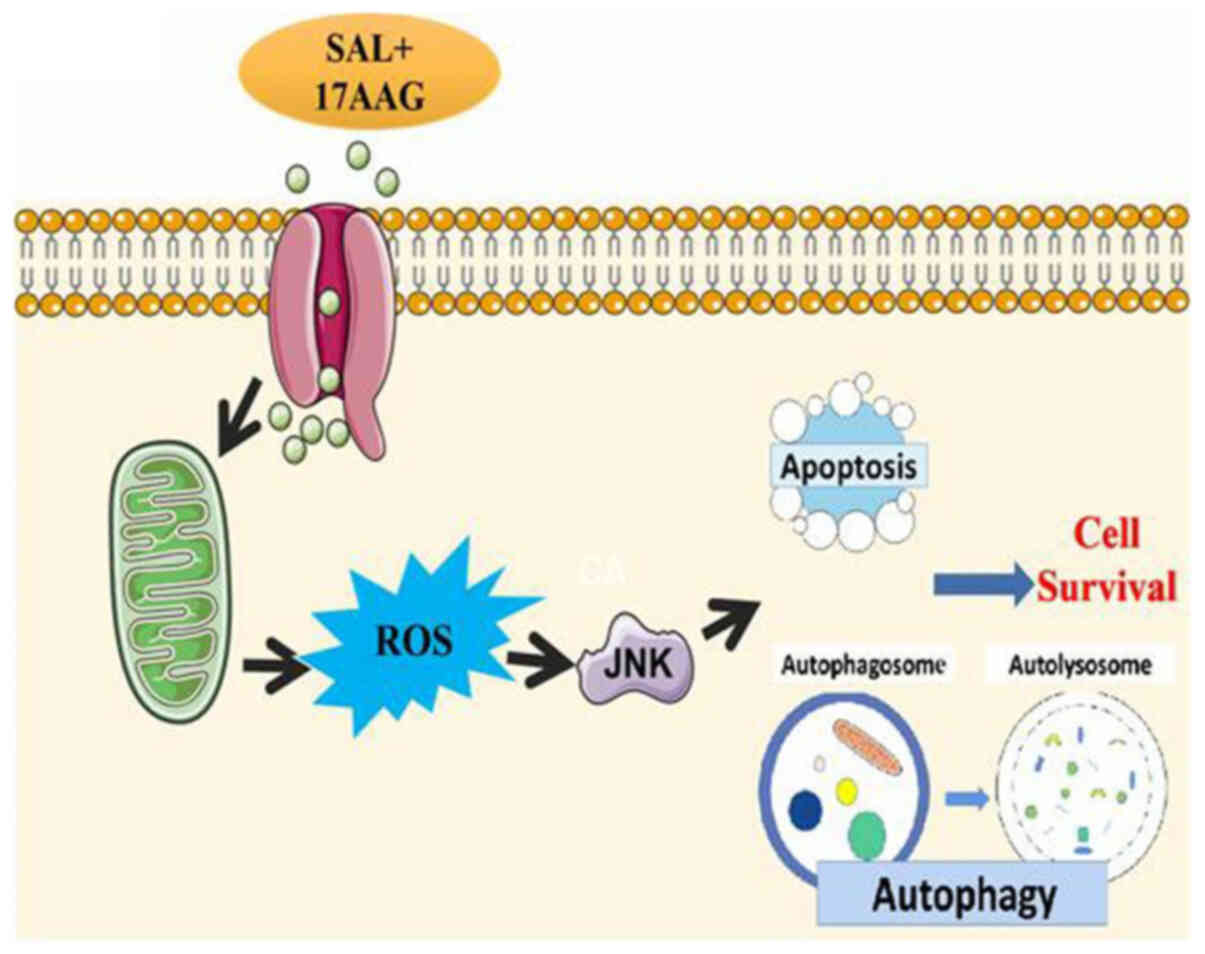
In conclusion, the combination of SAL and 17-AAG is
likely to activate the JNK signaling pathway through the production
of ROS, to induce apoptosis and inhibit autophagy, thus affecting
the proliferation of breast cancer cells or CSCs (Fig. 7).
Acknowledgements
Not applicable.
Funding
The present study was supported in part by grants from the
National Science and Technology Major Project (grant no.
2015ZX09501-009), National Natural Science Foundation (grant nos.
81760484, 31571469 and 81872349), Scientific Research Plan Projects
of Shaanxi Education Department (grant no. 19JK0970), Scientific
research project of Yan'an University (grant no. YDQ2019-38) and
Free Exploring Projects of State Key Laboratory of Cancer Biology
(grant no. CBSKL2014Z08).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BW conceived the idea for the study and performed
the preliminary experiments. DH performed the experiment and
prepared the original draft. JD analyzed and interpreted the data.
LL and JZ contributed to the conception and design of the study and
revised the final manuscript. All authors read and approved the
final version of the manuscript submitted for publication. LL and
JZ confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2021. CA Cancer J Clin. 71:7–33. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chaudhary LN, Wilkinson KH and Kong A:
Triple-negative breast cancer: Who should receive neoadjuvant
chemotherapy? Surg Oncol Clin N Am. 27:141–153. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lee A and Djamgoz MBA: Triple negative
breast cancer: Emerging therapeutic modalities and novel
combination therapies. Cancer Treat Rev. 62:110–122. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Al-Mahmood S, Sapiezynski J, Garbuzenko OB
and Minko T: Metastatic and triple-negative breast cancer:
Challenges and treatment options. Drug Deliv Transl Res.
8:1483–1507. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tang Y, Wang Y, Kiani MF and Wang B:
Classification, treatment strategy, and associated drug resistance
in breast cancer. Clin Breast Cancer. 16:335–343. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Turdo A, Veschi V, Gaggianesi M, Chinnici
A, Bianca P, Todaro M and Stassi G: Meeting the challenge of
targeting cancer stem cells. Front Cell Dev Biol. 7:162019.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mummery C, Wilmut SI, van de Stolpe A and
Roelen BAJ: Stem cells in cancer and cancer stem cells. Stem Cells.
27. Academic Press; Cambridge, MA: pp. 237–256. 2011, View Article : Google Scholar
|
|
8
|
Dewangan J, Srivastava S and Rath SK:
Salinomycin: A new paradigm in cancer therapy. Tumour Biol.
39:10104283176950352017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gupta PB, Onder TT, Jiang G, Tao K,
Kuperwasser C, Weinberg RA and Lander ES: Identification of
selective inhibitors of cancer stem cells by high-throughput
screening. Cell. 138:645–659. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou J, Liu S, Wang Y, Dai W, Zou H, Wang
S, Zhang J and Pan J: Salinomycin effectively eliminates cancer
stem-like cells and obviates hepatic metastasis in uveal melanoma.
Mol Cancer. 18:1592019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao Y, Zhao W, Lim YC and Liu T:
Salinomycin-loaded gold nanoparticles for treating cancer stem
cells by ferroptosis-induced cell death. Mol Pharm. 16:2532–2539.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Miyazaki Y, Shibuya M, Sugawara H,
Kawaguchi O and Hirsoe C: Salinomycin, a new polyether antibiotic.
J Antibiot (Tokyo). 27:814–821. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Qin LS, Jia PF, Zhang ZQ and Zhang SM:
ROS-p53-cyclophilin-D signaling mediates salinomycin-induced glioma
cell necrosis. J Exp Clin Cancer Res. 34:572015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim KY, Lee SG, Baek SY, Lee EH, Jang EJ,
Lee JH, Ahn SC, Chang JH, Oh TW, Kim SH, et al: Salinomycin
ameliorates oxidative hepatic damage through AMP-activated protein
kinase, facilitating autophagy. Toxicol Appl Pharmacol.
360:141–149. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiang J, Li H, Qaed E, Zhang J, Song Y, Wu
R, Bu X, Wang Q and Tang Z: Salinomycin, as an autophagy
modulator-a new avenue to anticancer: A review. J Exp Clin Cancer
Res. 37:262018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fuchs D, Daniel V, Sadeghi M, Opelz G and
Naujokat C: Salinomycin overcomes ABC transporter-mediated
multidrug and apoptosis resistance in human leukemia stem cell-like
KG-1a cells. Biochem Biophys Res Commun. 394:1098–1104. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Z, Zhou L, Xiong Y, Yu S, Li H, Fan
J, Li F, Su Z, Song J, Sun Q, et al: Salinomycin exerts
anti-colorectal cancer activity by targeting the β-catenin/T-cell
factor complex. Br J Pharmacol. 176:3390–3406. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim KY, Park KI, Kim SH, Yu SN, Park SG,
Kim YW, Seo YK, Ma JY and Ahn SC: Inhibition of autophagy promotes
salinomycin-induced apoptosis via reactive oxygen species-mediated
PI3K/AKT/mTOR and ERK/p38 MAPK-dependent signaling in human
prostate cancer cells. Int J Mol Sci. 18:10882017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Talaei S, Mellatyar H, Asadi A, Akbarzadeh
A, Sheervalilou R and Zarghami N: Spotlight on 17-AAG as an Hsp90
inhibitor for molecular targeted cancer treatment. Chem Biol Drug
Des. 93:760–786. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hirakawa H, Fujisawa H, Masaoka A, Noguchi
M, Hirayama R, Takahashi M, Fujimori A and Okayasu R: The
combination of Hsp90 inhibitor 17AAG and heavy-ion irradiation
provides effective tumor control in human lung cancer cells. Cancer
Med. 4:426–436. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Moon HJ, Park SY, Lee SH, Kang CD and Kim
SH: Nonsteroidal anti-inflammatory drugs sensitize
CD44-overexpressing cancer cells to Hsp90 inhibitor through
autophagy activation. Oncol Res. 27:835–847. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xie Q, Wondergem R, Shen Y, Cavey G, Ke J,
Thompson R, Bradley R, Daugherty-Holtrop J, Xu Y, Chen E, et al:
Benzoquinone ansamycin 17AAG binds to mitochondrial
voltage-dependent anion channel and inhibits cell invasion. Proc
Natl Acad Sci USA. 108:4105–4110. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Walker T, Mitchell C, Park MA, Yacoub A,
Rahmani M, Häussinger D, Reinehr R, Voelkel-Johnson C, Fisher PB,
Grant S and Dent P: 17-allylamino-17-demethoxygeldanamycin and
MEK1/2 inhibitors kill GI tumor cells via Ca2+-dependent
suppression of GRP78/BiP and induction of ceramide and reactive
oxygen species. Mol Cancer Ther. 9:1378–1395. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Piredda ML, Gaur G, Catalano G, Divona M,
Banella C, Travaglini S, Puzzangara MC, Voso MT, Lo-Coco F and
Noguera NI: PML/RARA inhibits expression of HSP90 and its target
AKT. Br J Haematol. 184:937–948. 2019.PubMed/NCBI
|
|
25
|
He D, Wo B and Zhao JM: Effect of
salinomycin on the proliferation and apoptosis of triple negative
breast cancer cell line MDA-MB-231 by target-regulating ALDH.
Chinese J Oncol Prevention Treatment. 12:72020.
|
|
26
|
Jafari-Gharabaghlou D,
Pilehvar-Soltanahmadi Y, Dadashpour M, Mota A, Vafajouy-Jamshidi S,
Faramarzi L, Rasouli S and Zarghami N: Combination of metformin and
phenformin synergistically inhibits proliferation and hTERT
expression in human breast cancer cells. Iran J Basic Med Sci.
21:1167–1173. 2018.PubMed/NCBI
|
|
27
|
Chatran M, Pilehvar-Soltanahmadi Y,
Dadashpour M, Faramarzi L, Rasouli S, Jafari-Gharabaghlou D,
Asbaghi N and Zarghami N: Synergistic anti-proliferative effects of
metformin and silibinin combination on T47D breast cancer cells via
hTERT and cyclin D1 inhibition. Drug Res (Stuttg). 68:710–716.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kanehisa M, Furumichi M, Tanabe M, Sato Y
and Morishima K: KEGG: New perspectives on genomes, pathways,
diseases and drugs. Nucleic Acids Res. 45:D353–D361. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kanehisa M, Sato Y and Morishima K:
BlastKOALA and GhostKOALA: KEGG tools for functional
characterization of genome and metagenome sequences. J Mol Biol.
428:726–731. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Verdoodt B, Vogt M, Schmitz I, Liffers ST,
Tannapfel A and Mirmohammadsadegh A: Salinomycin induces autophagy
in colon and breast cancer cells with concomitant generation of
reactive oxygen species. PLoS One. 7:e441322012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Park E and Chung SW: ROS-mediated
autophagy increases intracellular iron levels and ferroptosis by
ferritin and transferrin receptor regulation. Cell Death Dis.
10:8222019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zheng Q, Li Q, Zhao G, Zhang J, Yuan H,
Gong D, Guo Y, Liu X, Li K and Lin P: Alkannin induces cytotoxic
autophagy and apoptosis by promoting ROS-mediated mitochondrial
dysfunction and activation of JNK pathway. Biochem Pharmacol.
180:1141672020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Su LJ, Zhang JH, Gomez H, Murugan R, Hong
X, Xu D, Jiang F and Peng ZY: Reactive oxygen species-induced lipid
peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med
Cell Longev. 2019:50808432019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang S, Li Z, Liu W, Wei G, Yu N and Ji G:
Neohesperidin induces cell cycle arrest, apoptosis, and autophagy
via the ROS/JNK signaling pathway in human osteosarcoma cells. Am J
Chin Med. 49:1251–1274. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu
Y and Dong W: ROS and ROS-mediated cellular signaling. Oxid Med
Cell Longev. 2016:43509652016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang ZD, Yang YJ, Liu XW, Qin Z, Li SH
and Li JY: Aspirin eugenol ester ameliorates paraquat-induced
oxidative damage through ROS/p38-MAPK-mediated mitochondrial
apoptosis pathway. Toxicology. 453:1527212021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Diwanji N and Bergmann A: An unexpected
friend-ROS in apoptosis-induced compensatory proliferation:
Implications for regeneration and cancer. Semin Cell Dev Biol.
80:74–82. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu J, Chang F, Li F, Fu H, Wang J, Zhang
S, Zhao J and Yin D: Palmitate promotes autophagy and apoptosis
through ROS-dependent JNK and p38 MAPK. Biochem Biophys Res Commun.
463:262–267. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Peña-Blanco A and García-Sáez AJ: Bax, Bak
and beyond-mitochondrial performance in apoptosis. FEBS J.
285:416–431. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gu J, Zhan AJ, Jiang JL, Chen Y, Xu J, Ye
L and Mao MG: Conserved function of pacific cod caspase-3 in
apoptosis. Gene. 732:1443702020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tsapras P and Nezis IP: Caspase
involvement in autophagy. Cell Death Differ. 24:1369–1379. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu X, Li Q, Sun L, Chen L, Li Y, Huang B,
Liu Y and Jiang C: miR-30e-5p regulates autophagy and apoptosis by
targeting Beclin1 involved in contrast-induced acute kidney injury.
Curr Med Chem. 28:7974–7984. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Uriarte SM, Joshi-Barve S, Song Z, Sahoo
R, Gobejishvili L, Jala VR, Haribabu B, McClain C and Barve S: Akt
inhibition upregulates FasL, downregulates c-FLIPs and induces
caspase-8-dependent cell death in Jurkat T lymphocytes. Cell Death
Differ. 12:233–242. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Oral O, Oz-Arslan D, Itah Z, Naghavi A,
Deveci R, Karacali S and Gozuacik D: Cleavage of Atg3 protein by
caspase-8 regulates autophagy during receptor-activated cell death.
Apoptosis. 17:810–820. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mao J, Fan S, Ma W, Fan P, Wang B, Zhang
J, Wang H, Tang B, Zhang Q, Yu X, et al: Roles of Wnt/β-catenin
signaling in the gastric cancer stem cells proliferation and
salinomycin treatment. Cell Death Dis. 5:e10392014. View Article : Google Scholar : PubMed/NCBI
|