Introduction
To date, the relationship between breast-related
cancer antigen (BRCA1/2) and poly (ADP ribose) polymerase (PARP)
enzymes has been well studied (1).
Based on the genetic concept of synthetic lethality, several PARP
inhibitors have been developed and approved for various clinical
indications (1,2). PARP inhibitors are small-molecule
targeted drugs that trap the PARP enzymes in DNA damage sites and
prevent DNA repair, resulting in the accumulation of double-strand
DNA breaks (DSBs) during the S phase of the cell cycle (3). Homologous recombination
(HR)-proficient tumor cells are able to repair DSBs and restart
(3), whereas HR-deficient tumor
cells (i.e., those with BRCA mutation) that lost a functional HR
pathway rely primarily on the nonhomologous end joining pathway to
repair DSBs, resulting in the accumulation of genome instability
and cell death (3). Olaparib,
niraparib, rucaparib and talazoparib are currently approved for
clinical use and veliparib is still under clinical investigation
(2,4). PARP inhibitors are widely used in the
treatment of numerous types of solid tumor, particularly in
patients with BRCA1/2 mutations (2–4). PARP
inhibitors are administered orally, which has the advantages of
improved flexibility and convenience for the patients, but it may
be affected by numerous factors, such as transporters (5).
Transporters are able to transport a wide range of
endogenous and exogenous substrates and have an important role in
their disposition (5). Transporters
are generally divided into the solute carrier (SLC) family and the
ATP-binding cassette (ABC) family. The SLC transporters are mainly
involved in the uptake of small molecules into cells, whereas the
ABC transporters harness energy from ATP hydrolysis and primarily
function as efflux transporters. The SLC transporters mainly
include organic anion transporters (OATs), organic
anion-transporting polypeptides (OATPs), organic cation transporter
(OCTs), organic cation and carnitine transporters (OCTNs) and
peptide transporters (PEPTs) (6).
Most SLC transporters are influx transporters and mediate the
uptake of substrates into cells. The ABC transporters are
classified into seven subfamilies designated ABCA to ABCG based on
their gene structure, amino acid sequence, domain organization and
phylogenetic analysis (7). Among
the ABC transporters, P-glycoprotein (P-gp), multi-drug resistance
proteins (MRPs) and breast cancer resistance protein (BCRP) are the
most extensively studied (7). Most
ABC transporters are efflux transporters and export substrates out
of cells using ATP as driving energy. Transporters are located
throughout the body and they are involved in drug absorption,
distribution, metabolism and excretion (8). An alteration in the activity and
expression of a transporter may significantly change the PK profile
of a drug and cause clinically relevant drug-drug interactions
(DDIs) (9).
The five PARP inhibitors are the substrate of
transporters and some of them are also transporter inhibitors
(2–4). Therefore, it is essential to know the
relationship between PARP inhibitors and transporter inhibitors,
inducers or substrates. The purpose of the present review was to
characterize and summarize the transporter-mediated DDIs for each
PARP inhibitor. In addition, practical recommendations for managing
DDIs involving PARP inhibitors were provided.
Expression and function of SLC
transporters
The SLC transporters mainly include OATs, OATPs,
OCTs, OCTNs, PEPTs and multi-drug and toxin extrusion proteins
(MATEs). These transporters mediate the influx and efflux of
various substrates across cellular membranes (6). The OATPs consist of 11 members grouped
into 6 subfamilies. The OATPs transport large and fairly
hydrophobic organic anions (10).
Among the 11 OATPs, OATP1A2, OATP1B1, OATP1B3 and OATP2B1 have been
identified as being critical for drug disposition (11,12).
OATP1A2 and OATP2B1 are highly expressed in the intestinal
epithelium, renal epithelium, retina, brain capillary endothelial
cells, hepatocytes and red blood cells, where they have critical
roles in the intestinal absorption, renal reabsorption and
secretion, brain distribution and hepatic absorption (13). OATP1B1 and OATP1B3 are highly
expressed in hepatocytes, where they are responsible for the
hepatic uptake of substrates, such as conjugated bilirubin
(13). In addition to their
expression in normal tissues, several studies have indicated that
certain OATPs are highly expressed in certain cancer cells.
The OCTs mainly include OCT1, OCT2 and OCT3, and
they transport organic cations into cells (14). OCT1 is mainly expressed in the liver
and at lower levels in certain other tissues; it is considered to
be a liver-specific transporter, along with OATP1B1 and OATP1B3,
having important roles in the uptake of substrates by hepatocytes
(15). OCT2 is primarily expressed
in the proximal kidney tubule cells and is generally considered to
be a renal uptake transporter; it mediates the uptake of substrates
into the kidneys and the excretion of substrates into urine
(16). OCT3 is widely expressed in
tissues, with moderate to high expression in the intestines,
kidneys and liver; it is associated with intestinal absorption and
hepatic and renal uptake (16).
There are two OCTN isomers in humans, namely OCTN1
and OCTN2 (17). OCTN1 is highly
expressed in the kidneys and, to a lesser extent, in other tissues
(17). OCTN2 is expressed in
numerous tissues, such as the liver, kidneys, intestines, skeletal
muscles, heart and placenta (17).
OCTN1 and OCTN2 are involved in the intestinal absorption of
carnitine and organic cations and their distribution to tissues
(17).
The PEPTs mainly include PEPT1 and PEPT2; they are
responsible for the uptake of peptides and peptide-like compounds
(18). PEPT1 is primarily expressed
in the small intestine; it mediates the absorption of substrates
into the enterocyte (6,18). However, PEPT2 is primarily expressed
in the kidneys and mediates the renal reabsorption of small
peptides and peptide-like compounds (6,18).
The OATs mainly include OAT1, OAT2, OAT3 and OAT4;
they transport organic anions (19). OAT1, OAT3 and OAT4 are highly
expressed in the kidneys, where they are responsible for the uptake
of substrates from the blood into the proximal tubule cells and the
reabsorption of substrates from the ultrafiltrate (20). OAT2 is highly expressed in the liver
and, to a lesser extent, in the kidneys and other tissues; it has a
critical role in hepatic organic anion transport (21).
In contrast to other SLC transporters, the MATEs,
including MATE1 and MATE2-K, are responsible for the efflux of
organic cations (22). MATE1 is
highly expressed in the kidneys and bile canaliculi, whereas
MATE2-K exhibits a kidney-specific expression. These transporters
mediate the export of substrates taken up by OCT1 and OCT2
(23).
Expression and function of ABC
transporters
The ABC transporters are primary transporters that
utilize energy derived from ATP hydrolysis to transport substrates
across membranes (24). P-gp is the
first described and identified ABC transporter; it is expressed in
various tissues, including the intestine, kidneys, liver, brain and
placenta (25). P-gp transports
substrates from the intracellular to the extracellular space
(25). In the intestine, P-gp
inhibits the entrance of substrates from the intestinal lumen into
the bloodstream, leading to reduced bioavailability of several
orally administered drugs (26). In
the liver and kidneys, P-gp mediates the transport of agents into
bile and urine, respectively (25).
In the brain, P-gp is crucial for the blood-brain barrier to limit
the entrance of toxins and drugs into the central nervous system,
protecting the brain from the toxic effects of exogenous compounds
(27).
Similar to P-gp, BCRP is an efflux pump located in
numerous tissue types, such as the intestines, liver, kidneys,
brain, testis and placenta (28).
The bile salt export pump is predominantly expressed in the liver
and functions to mediate the efflux of conjugated and unconjugated
bile salts into bile (28).
The MRP family consists of nine MRP proteins. MRP2
is highly expressed in the liver, kidneys, small intestine, gall
bladder and placenta (29). MRP1 is
highly expressed in tumor cells, the lungs, brain, testis, kidneys,
skeletal muscles and peripheral blood mononuclear cells, and to a
lesser extent in the liver (29).
Both MRP1 and MRP2 are associated with the excretion of numerous
phase II metabolites and endogenous agents into bile, urine and the
intestinal lumen (29,30). MRP3 is mainly expressed in
hepatocytes and enterocytes. MRP4 is widely located in most
tissues; both MRP3 and MRP4 are involved in the efflux of organic
anions (31).
Transporter-mediated DDIs involving PARP
inhibitors
Transporters have different tissue expression
patterns; if they are expressed in the small intestine, liver,
kidneys and blood-tissue barriers, they significantly affect the
drug disposition, leading to clinically significant DDIs (32). The location and function of
transporters are illustrated in Fig.
1. Most drugs are the substrates, inducers or inhibitors of
transporters; thus, inhibition or induction of transporters may
lead to alterations in the intestinal absorption, hepatic uptake,
renal reabsorption, and biliary and renal excretion of drugs,
causing clinically relevant DDIs, undesired toxicities or reduced
therapeutic effects (9,32). The potential DDIs between PARP
inhibitors and transporter inhibitors/inducers are listed in
Table I. The potential DDIs between
PARP inhibitors and transporter substrates are listed in Table II.
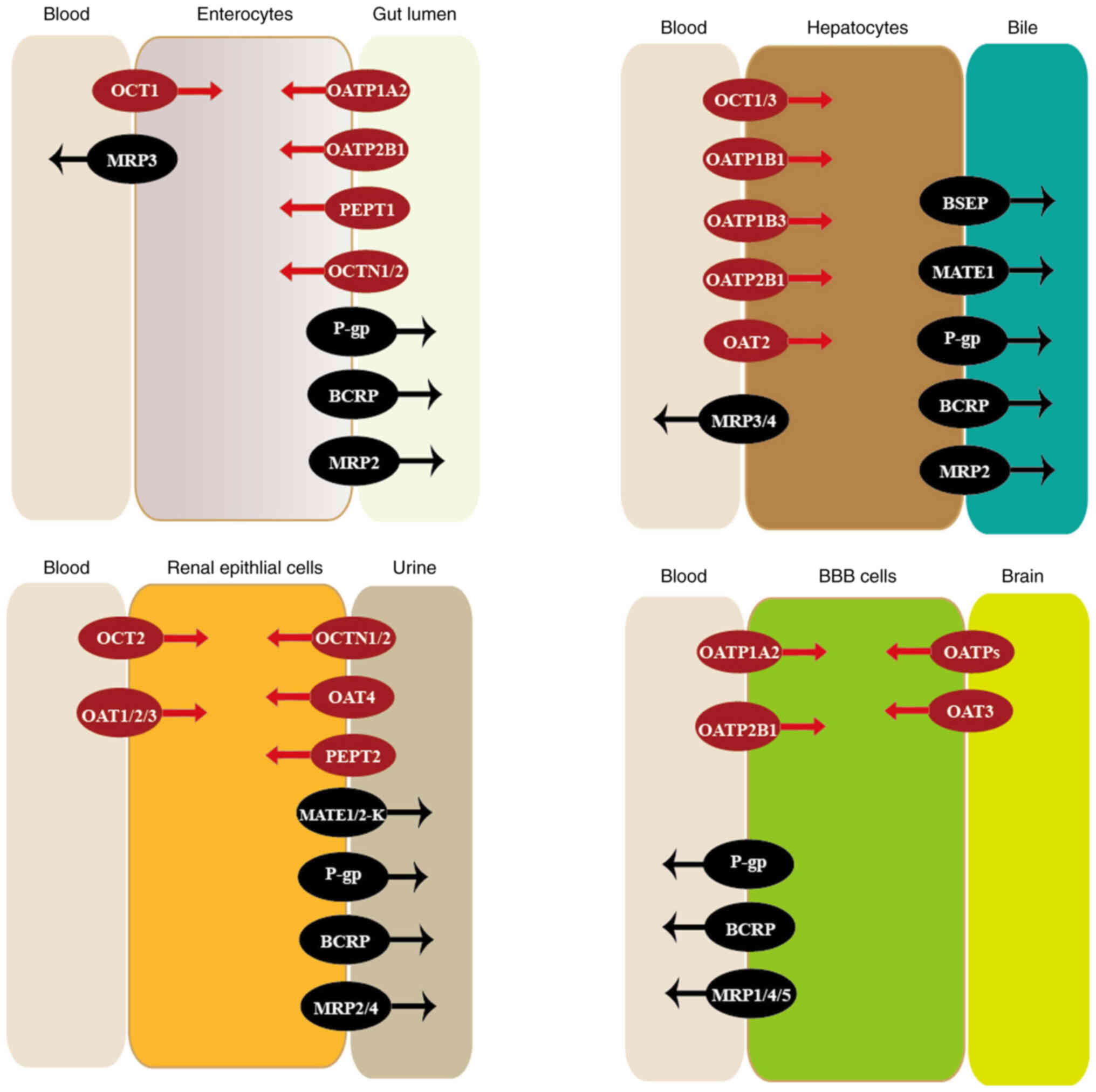 | Figure 1.Location and function of
transporters. Transporters are widely expressed in numerous
tissues, where they have important roles in the membrane transport
of various drugs. Influx transporters are colored in red, while
efflux transporters are colored in black. BBB, blood-brain barrier;
OCTs, organic cation transporters; MRPs, multi-drug resistance
proteins; OATPs, organic anion-transporting polypeptides; PEPTs,
peptide transporters; OCTNs, organic cation and carnitine
transporters; P-gp, P-glycoprotein; BCRP, breast cancer resistance
protein; OATs, organic anion transporters; BSEP, bile salt export
pump; MATEs, multi-drug and toxin extrusion proteins. |
 | Table I.Potential drug-drug interactions
between poly (ADP ribose) polymerase inhibitors and transporter
inhibitors/inducers. |
Table I.
Potential drug-drug interactions
between poly (ADP ribose) polymerase inhibitors and transporter
inhibitors/inducers.
| First author,
year | Substrates | Transporters |
Recommendations | (Refs.) |
|---|
| Song, 2022;
Vaidyanathan, 2016 | Olaparib | P-gp, BRCP | Adverse reactions
monitoring is required | (34,35) |
| Martins, 2021; Sun,
2018; Morosi, 2020 | Niraparib | P-gp, BCRP | Close monitoring of
adverse reactions is needed when niraparib administered
concomitantly with P-gp and BCRP inhibitors | (37–39) |
| Durmus, 2015; Liao,
2020; Chen, 2020 | Rucaparib | P-gp, BCRP | Caution must be
exercised when rucaparib is co-administered with P-gp and BCRP
inhibitors | (42–44) |
| Elmeliegy, 2020;
Yu, 2020; US Food and Drug Administration, 2020; European Medicines
Agency, 2021 | Talazoparib | P-gp, BCRP | Co-administration
with strong P-gp inhibitors should be avoided. If co-administration
is unavoidable, the talazoparib dose should be reduced to 0.75 mg
once daily. Dose adjustment is not required for talazoparib when
co-administered with rifampin. Concomitant use of strong BCRP
inhibitors during treatment with talazoparib must be avoided; if
co-administration cannot be avoided, the potential increased
adverse reactions must be monitored | (51–54) |
| Lin, 2017 | Veliparib | P-gp, BCRP, OCT2,
MATE1, MATE2-K | Close clinical
surveillance is required when veliparib is combined with P-gp and
BCRP inhibitors | (57) |
| Table II.Potential drug-drug interactions
between poly (ADP ribose) polymerase inhibitors and transporter
substrates. |
Table II.
Potential drug-drug interactions
between poly (ADP ribose) polymerase inhibitors and transporter
substrates.
| First author,
year | Inhibitor
drugs | Transporters |
Recommendations | (Refs.) |
|---|
| McCormick,
2017 | Olaparib | P-gp, BCRP,
OATP1B1, OCT1, OCT2, OAT3, MATE1, MATE2-K | Caution must be
exercised when olaparib is combined with any statins | (36) |
| US Food and Drug
Administration, 2021; European Medicines Agency, 2022 | Niraparib | MATE1, MATE2-K,
BCRP, OCT1 | Caution is
recommended when niraparib is combined with active substrates
transported by MATE1, MATE2-K, BCRP and OCT1 | (40,41) |
| Liao, 2020; US Food
and Drug Administration, 2020; European Medicines Agency, 2022 | Rucaparib | P-gp, BCRP,
OATP1B1, OATP1B3, OAT1, OAT3, MATE1, MATE2-K, OCT1, OCT2, MRP4 | Dose adjustment is
not required for P-gp and BRCP substrates when they are
co-administered with rucaparib. Caution is advised when metformin
is co-administered with rucaparib | (43,45,46) |
| Chang, 2020 | Veliparib | OAT1, OAT3, OCT2,
MATE1, MATE2-K | Appropriate
clinical monitoring is required when veliparib is co-administered
with P-gp substrates | (58) |
Olaparib is a substrate for P-gp and BRCP (33). The effects of P-gp and BRCP
inhibitors or inducers on olaparib have not been evaluated in
humans. In an in vivo study, the brain concentration and
brain-to-plasma unbound concentration ratios of olaparib increased
10.7- and 5.3-fold, respectively, in mice treated with a
combination of elacridar (a dual inhibitor of P-gp and BRCP) and
olaparib relative to those that were not treated with elacridar
(34). However, no significant
differences in the plasma concentration of olaparib were observed
between the two groups. A study revealed that olaparib resistance
correlates with increased P-gp expression and the resistance is
reversible following combination treatment with verapamil or
elacridar (35). Therefore,
olaparib resistance mayn be overcome by inhibiting P-gp and BRCP
(35). However, the combination of
olaparib with P-gp and BRCP inhibitors may lead to increased
adverse reactions (e.g., anemia, gastrointestinal toxicities,
fatigue, nasopharyngitis, arthralgia, myalgia, dysgeusia, headache
and stomatitis); thus, adverse reaction monitoring is required.
Olaparib is an inhibitor of P-gp, BCRP, OATP1B1,
OCT1, OCT2, OAT3, MATE1 and MATE2-K (36). Although it is unknown whether
olaparib exhibits any clinically significant DDI when
co-administered with substrates of these transporters, it cannot be
ruled out (36). In particular,
caution should be exercised when olaparib is combined with statins
(36).
Niraparib is a substrate for P-gp and BCRP; its
major primary metabolite M1 is a substrate for MATE1 and MATE2-K
(37). In vitro studies have
indicated that P-gp and BCRP do not have any significant impact on
the bioavailability and liver disposition of niraparib (37). However, P-gp and BCRP may
significantly increase the brain distribution of niraparib. In a
study conducted in mice, co-administration of elacridar
significantly increased the brain concentration of niraparib
without significantly altering the plasma concentration (38). Therefore, P-gp and BCRP inhibitors
may increase the brain concentration of niraparib, improving the
treatment outcome in patients with brain tumors (37–39).
However, close monitoring of adverse reactions [e.g.,
hematotoxicity, palpitations, gastrointestinal toxicities,
mucositis, aspartate aminotransferase (AST) alanine
aminotransferase (ALT) elevation, urinary tract infection and rash]
is required when niraparib is administered concomitantly with P-gp
and BCRP inhibitors.
Niraparib is an inhibitor of MATE1 and MATE2-K, as
well as a weak inhibitor of BCRP and OCT1 (40,41).
Clinically significant DDIs between niraparib and substrates of
these transporters are unlikely to occur but cannot be ruled out
(40,41). Thus, caution is needed when
niraparib is combined with active substrates that are transported
by these transporters, including metformin, irinotecan,
rosuvastatin, simvastatin, atorvastatin and methotrexate.
Rucaparib is a substrate for P-gp and BCRP (42). An in vitro study revealed
that P-gp and BCRP markedly reduce the oral bioavailability and
brain accumulation of rucaparib (43). Similarly, an in vitro study
revealed that verapamil reverses rucaparib resistance by inhibiting
P-gp (44). Therefore, the effect
of P-gp and BCRP inhibitors on the PK profile of rucaparib cannot
be ruled out and there must be strict monitoring for toxicities
(e.g., gastrointestinal toxicities, fatigue, hematotoxicities,
dysgeusia, AST/ALT elevation, stomatitis and rash) when rucaparib
is co-administered with P-gp and BCRP inhibitors.
Rucaparib was found to be an inhibitor of P-gp,
BCRP, OATP1B1, OATP1B3, OAT1, OAT3, MATE1, MATE2-K, OCT1, OCT2 and
MRP4 (45,46). A phase I study revealed that
co-administration of rucaparib and digoxin increased the digoxin
area under the curve (AUC) by 20% (47). Another phase I study conducted in
patients with advanced solid tumors demonstrated that rucaparib
slightly increased the plasma concentration of rosuvastatin (a BRCP
substrate) (48). Therefore, dose
adjustment is not recommended for P-gp and BRCP substrates when
they are co-administered with rucaparib (45,46).
As the inhibition of MATE1, MATE2-K, OCT1 and OCT2 may decrease the
renal elimination and hepatic uptake of metformin, caution is
advised when metformin is co-administered with rucaparib (43,45,46).
Talazoparib is a substrate for P-gp and BCRP
(49). An in vivo study
revealed that overexpression of P-gp decreases the brain
accumulation of talazoparib (50).
In patients with advanced solid tumors, concomitant administration
of itraconazole (a P-gp inhibitor) with talazoparib increased the
AUC and maximum concentration (Cmax) of talazoparib by
56 and 40%, respectively (51). In
addition, PK analysis revealed that strong P-gp inhibitors,
including amiodarone, carvedilol, clarithromycin, itraconazole and
verapamil, increased talazoparib exposure by 45% (52). Therefore, the concomitant use of
strong P-gp inhibitors must be avoided. If co-administration is
unavoidable, the talazoparib dose must be reduced to 0.75 mg once
daily (52–54). Co-administration of P-gp inhibitors,
including azithromycin, atorvastatin, diltiazem, felodipine,
fluvoxamine and quercetin, increased talazoparib exposure by 8%
(52–54). It is recommended to monitor for
potential adverse reactions when these P-gp inhibitors are
co-administered with talazoparib and dose adjustment is based on
tolerability (54).
Co-administration of rifampin (a P-gp inducer) with talazoparib
increased the talazoparib Cmax by 37% with no effect on
the AUC. These results suggest that dose adjustment for talazoparib
is not required when co-administered with rifampin (51,53,54).
However, the effect of other P-gp inducers on talazoparib exposure
remains elusive (53,54). Thus, caution is advised when
rucaparib is co-administered with other P-gp inducers.
Co-administration with BCRP inhibitors may increase talazoparib
exposure. Therefore, the concomitant use of strong BCRP inhibitors
during treatment with talazoparib must be avoided. If
co-administration cannot be avoided, the potential adverse
reactions must be monitored (54).
In vitro studies have revealed that talazoparib is not a
transporter inhibitor or inducer (53,54).
Veliparib is a substrate for P-gp, BCRP, OCT2, MATE1
and MATE2-K (55). Population PK
analysis has revealed that P-gp, MATE1, MATE2-K and OCT2 did not
significantly impact the plasma AUC of veliparib (56). A study conducted in mice and cells
revealed that P-gp and BCRP did not have any critical role in the
systemic clearance of veliparib but an essential role in the brain
accumulation of veliparib (57).
The results indicated that elacridar slightly increased the plasma
AUC of veliparib but significantly increased the brain accumulation
of veliparib (57). Therefore,
inhibition of P-gp and BCRP may improve the efficacy of veliparib
in patients with brain tumors, but close clinical surveillance is
required when veliparib is combined with P-gp and BCRP inhibitors
(57).
In a DDI study, veliparib inhibited OAT1, OAT3,
OCT2, MATE1 and MATE2-K with half-maximal inhibitory concentration
(IC50) values of 1,371, 505, 3,913, 69.9 and 69.5 µM,
respectively (55). The maximum
unbound plasma concentration of veliparib after a single oral dose
of 50 mg was lower than the IC50 values for these
transporters (55). These results
indicated that veliparib has a minimal potential for DDIs with
these transporters (55). However,
in an in vitro study, veliparib significantly increased the
accumulation of doxorubicin in liver cancer cells with P-gp
overexpression by inhibiting the expression of P-gp (58). Therefore, veliparib may reverse
P-gp-mediated multidrug resistance (MDR) in liver cancer cells and
this may benefit patients with liver cancer, particular those who
are not sensitive to chemotherapy due to the overexpression of P-gp
(58). However, appropriate
clinical monitoring is required when veliparib is co-administered
with P-gp substrates.
Discussion
Transporters are membrane-bound proteins that
mediate the movement of substrates across biological membranes. In
addition, these proteins mediate the transport of drug molecules
(5). Transporters are located
throughout the body, suggesting their crucial roles in drug
disposition (5–7). By altering the expression and activity
of transporters, a perpetrator agent may change the PK parameters
for an affected drug, leading to clinically significant DDIs
(9,32). In addition, when two agents are able
to be carried by the same transporter, there may be competition for
the same transporter site, producing a competitive inhibition and
leading to clinically significant DDIs (32).
Transporters have a broad substrate spectrum and
facilitate the transport of several drugs. The five PARP inhibitors
are substrates for P-gp and BRCP. In a DDI study, P-gp and BRCP
inhibitors did not have any significant impact on the systemic
exposure of olaparib, niraparib and veliparib, but significantly
increased their brain accumulation, indicating that P-gp and BRCP
inhibitors may improve treatment outcomes when co-administered with
olaparib, niraparib or veliparib in patients with brain tumors
(34,37,57).
For rucaparib and talazoparib, P-gp and BRCP have critical roles in
their systemic exposure and brain accumulation, indicating that
dose adjustment or caution is required when rucaparib and
talazoparib are co-administered with strong P-gp and BRCP
inhibitors or inducers (43,51,52).
PARP inhibitors are also transporter inhibitors. In vitro
and in vivo studies have demonstrated that, apart from
talazoparib, the other four PARP inhibitors inhibit several
transporters (36,40,41,47,48,55).
As it cannot be ruled out that the four PARP inhibitors may cause
clinically relevant DDIs with certain transporter substrates, it is
recommended that close monitoring of adverse reactions is ensured
when olaparib, niraparib, rucaparib and veliparib are administered
concomitantly with these substrates.
Metabolism- and transporter-mediated DDIs are the
most common DDIs that affect the PK profile of a drug (59,60).
Metabolic enzymes are primarily localized in the liver and small
intestine, whereas transporters are widely distributed throughout
the body (5,59). As a result, the effects of
transporters on the PK profile of drugs are more complex (5,32).
Metabolic enzymes significantly contribute to drug absorption and
metabolism, leading to alterations in the systemic exposure of
drugs (61). Similarly,
transporters have crucial roles in drug absorption, distribution
and elimination, leading to alterations in the distribution and
systemic exposure of drugs (5).
Furthermore, because numerous transporters and metabolic enzymes
share common locations, substrates, inhibitors and inducers, they
may exert coordinated effects on drug absorption and metabolism
(59,62).
Increased expression of ABC drug transporters in
cancer cells is one of the mechanisms leading to cancer MDR; these
transporters may export drugs from cells, leading to reduced
intracellular drug concentrations (63,64).
P-gp and BRCP are two of the most extensively studied ABC multidrug
transporters; these transporters are highly expressed in numerous
solid tumors and have broad substrate specificity. In vitro
studies have indicated that P-gp and BRCP inhibitors, such as
elacridar, partially reverse the resistance to olaparib and
rucaparib (35,65,66).
Based on the available studies, the present review
comprehensively summarized the expression and function of
transporters and the DDIs between transporters and PARP inhibitors.
In addition, specific recommendations for managing the potential
DDIs were also provided. However, studies on the DDIs between
transporters and PARP inhibitors are limited and further clinical
studies are needed to evaluate the efficacy and safety of PARP
inhibitors when they are combined with transporter inhibitors,
inducers or substrates.
Conclusion
Transporters have critical roles in drug disposition
and DDIs. The five PARP inhibitors are substrates for transporters.
Alterations in the activity and expression of these transporters
may influence the oral bioavailability, tissue distribution and
elimination of PARP inhibitors and consequently cause clinically
relevant DDIs. Furthermore, apart from talazoparib, the other four
PARP inhibitors may inhibit certain transporters; thus, they may
have a significant impact on the PK profile of substrates for
transporters. In summary, PARP inhibitors may exhibit
transporter-mediated DDIs and various DDIs may lead to reduced
efficacy and increased toxicity. However, certain DDIs may
contribute to increasing the tissue distribution and intracellular
concentrations of certain drugs, which may enhance the drug
efficacy and overcome MDR.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
DZ and XL conceived the study and performed the
literature search. DZ drafted the manuscript. DZ and JW revised the
manuscript. All authors have read and approved the final
manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mittica G, Ghisoni E, Giannone G, Genta S,
Aglietta M, Sapino A and Valabrega G: PARP inhibitors in ovarian
cancer. Recent Pat Anticancer Drug Discov. 13:392–410. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tew WP, Lacchetti C, Ellis A, Maxian K,
Banerjee S, Bookman M, Jones MB, Lee JM, Lheureux S, Liu JF, et al:
PARP inhibitors in the management of ovarian cancer: ASCO
Guideline. J Clin Oncol. 38:3468–3493. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rose M, Burgess JT, O'Byrne K, Richard DJ
and Bolderson E: PARP inhibitors: Clinical relevance, mechanisms of
action and tumor resistance. Front Cell Dev Biol. 8:5646012020.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hennes ER, Dow-Hillgartner EN, Bergsbaken
JJ and Piccolo JK: PARP-inhibitor potpourri: A comparative review
of class safety, efficacy, and cost. J Oncol Pharm Pract.
26:718–729. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nigam SK: What do drug transporters really
do? Nat Rev Drug Discov. 14:29–44. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rives ML, Javitch JA and Wickenden AD:
Potentiating SLC transporter activity: Emerging drug discovery
opportunities. Biochem Pharmacol. 135:1–11. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Beis K: Structural basis for the mechanism
of ABC transporters. Biochem Soc Trans. 43:889–893. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zamek-Gliszczynski MJ, Sangha V, Shen H,
Feng B, Wittwer MB, Varma MVS, Liang X, Sugiyama Y, Zhang L and
Bendayan R; International Transporter Consortium, : Transporters in
drug development: International transporter consortium update on
emerging transporters of clinical importance. Clin Pharmacol Ther.
112:485–500. 2022. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
König J, Müller F and Fromm MF:
Transporters and drug-drug interactions: Important determinants of
drug disposition and effects. Pharmacol Rev. 65:944–966. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thakkar N, Lockhart AC and Lee W: Role of
organic anion-transporting polypeptides (OATPs) in cancer therapy.
AAPS J. 17:535–545. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen M, Hu S, Li Y, Gibson AA, Fu Q, Baker
SD and Sparreboom A: Role of Oatp2b1 in drug absorption and
drug-drug interactions. Drug Metab Dispos. 48:419–425. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou Y, Yuan J, Li Z, Wang Z, Cheng D, Du
Y, Li W, Kan Q and Zhang W: Genetic polymorphisms and function of
the organic anion-transporting polypeptide 1A2 and its clinical
relevance in drug disposition. Pharmacology. 95:201–218. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Alam K, Crowe A, Wang X, Zhang P, Ding K,
Li L and Yue W: Regulation of organic anion transporting
polypeptides (OATP) 1B1- and OATP1B3-Mediated Transport: An updated
review in the context of OATP-Mediated drug-drug interactions. Int
J Mol Sci. 19:8552018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Koepsell H: Role of organic cation
transporters in drug-drug interaction. Expert Opin Drug Metab
Toxicol. 11:1619–1633. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brosseau N and Ramotar D: The human
organic cation transporter OCT1 and its role as a target for drug
responses. Drug Metab Rev. 51:389–407. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Samodelov SL, Kullak-Ublick GA, Gai Z and
Visentin M: Organic cation transporters in human physiology,
pharmacology, and toxicology. Int J Mol Sci. 21:78902020.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pochini L, Galluccio M, Scalise M, Console
L and Indiveri C: OCTN: A small transporter subfamily with great
relevance to human pathophysiology, drug discovery, and
diagnostics. SLAS Discov. 24:89–110. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Brecht K, Schäfer AM and Meyer Zu
Schwabedissen HE: Uptake transporters of the SLC21, SLC22A, and
SLC15A families in anticancer therapy-modulators of cellular entry
or pharmacokinetics? Cancers (Basel). 12:22632020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nigam SK, Bush KT, Martovetsky G, Ahn SY,
Liu HC, Richard E, Bhatnagar V and Wu W: The organic anion
transporter (OAT) family: A systems biology perspective. Physiol
Rev. 95:83–123. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ivanyuk A, Livio F, Biollaz J and Buclin
T: Renal drug transporters and drug interactions. Clin
Pharmacokinet. 56:825–892. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Izat N and Sahin S: Hepatic
transporter-mediated pharmacokinetic drug-drug interactions: Recent
studies and regulatory recommendations. Biopharm Drug Dispos.
42:45–77. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nies AT, Damme K, Kruck S, Schaeffeler E
and Schwab M: Structure and function of multidrug and toxin
extrusion proteins (MATEs) and their relevance to drug therapy and
personalized medicine. Arch Toxicol. 90:1555–1584. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Motohashi H and Inui K: Organic cation
transporter OCTs (SLC22) and MATEs (SLC47) in the human kidney.
AAPS J. 15:581–588. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Locher KP: Mechanistic diversity in
ATP-binding cassette (ABC) transporters. Nat Struct Mol Biol.
23:487–493. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Elmeliegy M, Vourvahis M, Guo C and Wang
DD: Effect of P-glycoprotein (P-gp) inducers on exposure of P-gp
substrates: Review of clinical drug-drug interaction studies. Clin
Pharmacokinet. 59:699–714. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Suzuki K, Taniyama K, Aoyama T and
Watanabe Y: Evaluation of the role of P-glycoprotein
(P-gp)-Mediated efflux in the intestinal absorption of common
substrates with elacridar, a P-gp inhibitor, in rats. Eur J Drug
Metab Pharmacokinet. 45:385–392. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Storelli F, Anoshchenko O and Unadkat JD:
Successful prediction of human steady-state unbound brain-to-plasma
concentration ratio of P-gp substrates using the
proteomics-informed relative expression factor approach. Clin
Pharmacol Ther. 110:432–442. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mao Q and Unadkat JD: Role of the breast
cancer resistance protein (BCRP/ABCG2) in drug transport-an update.
AAPS J. 17:65–82. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Amawi H, Sim HM, Tiwari AK, Ambudkar SV
and Shukla S: ABC Transporter-mediated multidrug-resistant cancer.
Adv Exp Med Biol. 1141:549–580. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cole SP: Targeting multidrug resistance
protein 1 (MRP1, ABCC1): Past, present, and future. Annu Rev
Pharmacol Toxicol. 54:95–117. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Järvinen E, Deng F, Kidron H and Finel M:
Efflux transport of estrogen glucuronides by human MRP2, MRP3, MRP4
and BCRP. J Steroid Biochem Mol Biol. 178:99–107. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu X: Transporter-Mediated drug-drug
interactions and their significance. Adv Exp Med Biol.
1141:241–291. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dufour R, Daumar P, Mounetou E, Aubel C,
Kwiatkowski F, Abrial C, Vatoux C, Penault-Llorca F and Bamdad M:
BCRP and P-gp relay overexpression in triple negative basal-like
breast cancer cell line: A prospective role in resistance to
Olaparib. Sci Rep. 5:126702015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Song YK, Kim MJ, Kim MS, Lee JH, Chung SJ,
Song JS, Chae YJ and Lee KR: Role of the efflux transporters Abcb1
and Abcg2 in the brain distribution of olaparib in mice. Eur J
Pharm Sci. 173:1061772022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vaidyanathan A, Sawers L, Gannon AL,
Chakravarty P, Scott AL, Bray SE, Ferguson MJ and Smith G: ABCB1
(MDR1) induction defines a common resistance mechanism in
paclitaxel- and olaparib-resistant ovarian cancer cells. Br J
Cancer. 115:431–441. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
McCormick A and Swaisland H: In vitro
assessment of the roles of drug transporters in the disposition and
drug-drug interaction potential of olaparib. Xenobiotica.
47:903–915. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Martins MLF, Loos NHC, Mucuk S, de Jong D,
Lebre MC, Rosing H, Tibben M, Beijnen JH and Schinkel AH:
P-Glycoprotein (ABCB1/MDR1) controls brain penetration and
intestinal disposition of the PARP1/2 inhibitor niraparib. Mol
Pharm. 18:4371–4384. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sun K, Mikule K, Wang Z, Poon G,
Vaidyanathan A, Smith G, Zhang ZY, Hanke J, Ramaswamy S and Wang J:
A comparative pharmacokinetic study of PARP inhibitors demonstrates
favorable properties for niraparib efficacy in preclinical tumor
models. Oncotarget. 9:37080–37096. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Morosi L, Matteo C, Ceruti T, Giordano S,
Ponzo M, Frapolli R, Zucchetti M, Davoli E, D'Incalci M and Ubezio
P: Quantitative determination of niraparib and olaparib tumor
distribution by mass spectrometry imaging. Int J Biol Sci.
16:1363–1375. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
US Food and Drug Administration, . Label.
Available from. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/208447s022s024lbl.pdfJune
20–2022
|
|
41
|
European Medicines Agency, . Product
information. Available from. https://www.ema.europa.eu/en/documents/product-information/zejula-epar-product-information_en.pdfJune
20–2022
|
|
42
|
Durmus S, Sparidans RW, van Esch A,
Wagenaar E, Beijnen JH and Schinkel AH: Breast cancer resistance
protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1) restrict oral
availability and brain accumulation of the PARP inhibitor rucaparib
(AG-014699). Pharm Res. 32:37–46. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liao M, Jaw-Tsai S, Beltman J, Simmons AD,
Harding TC and Xiao JJ: Evaluation of in vitro absorption,
distribution, metabolism, and excretion and assessment of drug-drug
interaction of rucaparib, an orally potent poly(ADP-ribose)
polymerase inhibitor. Xenobiotica. 50:1032–1042. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen Z, Ling K, Zhu Y, Deng L, Li Y and
Liang Z: Rucaparib antagonize multidrug resistance in cervical
cancer cells through blocking the function of ABC transporters.
Gene. 759:1450002020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
US Food and Drug Administration, . Label.
Available from. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/209115s008lbl.pdfJune
20–2022
|
|
46
|
European Medicines Agency, . Product
information. Available from. https://www.ema.europa.eu/en/documents/product-information/rubraca-epar-product-information_en.pdfJune
20–2022
|
|
47
|
Xiao JJ, Nowak D, Ramlau R,
Tomaszewska-Kiecana M, Wysocki PJ, Isaacson J, Beltman J, Nash E,
Kaczanowski R, Arold G and Watkins S: Evaluation of drug-drug
interactions of rucaparib and CYP1A2, CYP2C9, CYP2C19, CYP3A, and
P-gp substrates in patients with an advanced solid tumor. Clin
Transl Sci. 12:58–65. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liao M, Jeziorski KG, Tomaszewska-Kiecana
M, Láng I, Jasiówka M, Skarbová V, Centkowski P, Ramlau R, Górnaś
M, Lee J, et al: A phase 1, open-label, drug-drug interaction study
of rucaparib with rosuvastatin and oral contraceptives in patients
with advanced solid tumors. Cancer Chemother Pharmacol. 88:887–897.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Guney Eskiler G, Cecener G, Egeli U and
Tunca B: Talazoparib nanoparticles for overcoming multidrug
resistance in triple-negative breast cancer. J Cell Physiol.
235:6230–6245. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kizilbash SH, Gupta SK, Chang K, Kawashima
R, Parrish KE, Carlson BL, Bakken KK, Mladek AC, Schroeder MA,
Decker PA, et al: Restricted delivery of talazoparib across the
blood-brain barrier limits the sensitizing effects of PARP
inhibition on temozolomide therapy in glioblastoma. Mol Cancer
Ther. 16:2735–2746. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Elmeliegy M, Láng I, Smolyarchuk EA, Chung
CH, Plotka A, Shi H and Wang D: Evaluation of the effect of
P-glycoprotein inhibition and induction on talazoparib disposition
in patients with advanced solid tumours. Br J Clin Pharmacol.
86:771–778. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yu Y, Durairaj C, Shi H and Wang DD:
Population pharmacokinetics of talazoparib in patients with
advanced cancer. J Clin Pharmacol. 60:218–228. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
US Food and Drug Administration, . Label.
Available from. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/211651s006lbl.pdfJune
20–2022
|
|
54
|
European Medicines Agency, . Product
information. Available from. https://www.ema.europa.eu/en/documents/product-information/talzenna-epar-product-information_en.pdfJune
20–2022
|
|
55
|
Kikuchi R, Lao Y, Bow DA, Chiou WJ,
Andracki ME, Carr RA, Voorman RL and De Morais SM: Prediction of
clinical drug-drug interactions of veliparib (ABT-888) with human
renal transporters (OAT1, OAT3, OCT2, MATE1, and MATE2K). J Pharm
Sci. 102:4426–4432. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Stodtmann S, Nuthalapati S, Eckert D,
Kasichayanula S, Joshi R, Bach BA, Mensing S, Menon R and Xiong H:
A population pharmacokinetic meta-analysis of veliparib, a PARP
Inhibitor, Across Phase 1/2/3 trials in cancer patients. J Clin
Pharmacol. 61:1195–1205. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lin F, de Gooijer MC, Roig EM, Buil LC,
Christner SM, Beumer JH, Würdinger T, Beijnen JH and van Tellingen
O: ABCB1, ABCG2, and PTEN determine the response of glioblastoma to
temozolomide and ABT-888 therapy. Clin Cancer Res. 20:2703–2713.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chang L, Hou Y, Zhu L, Wang Z, Chen G, Shu
C and Liu Y: Veliparib overcomes multidrug resistance in liver
cancer cells. Biochem Biophys Res Commun. 521:596–602. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
van Leeuwen RW, van Gelder T, Mathijssen
RH and Jansman FG: Drug-drug interactions with tyrosine-kinase
inhibitors: A clinical perspective. Lancet Oncol. 15:e315–e326.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhao D, Long X and Wang J: Dose adjustment
of poly (ADP-Ribose) Polymerase inhibitors in patients with hepatic
or renal impairment. Drug Des Devel Ther. 16:3947–3955. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Koinuma K, Tsuchitani T, Imaoka A,
Akiyoshi T and Ohtani H: Relative contributions of metabolic
enzymes to systemic elimination can be estimated from clinical DDI
studies: Validation using an in silico approach. Int J Clin
Pharmacol Ther. 59:231–238. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yu J, Petrie ID, Levy RH and
Ragueneau-Majlessi I: Mechanisms and clinical significance of
pharmacokinetic-based drug-drug interactions with drugs approved by
the U.S. Food and Drug Administration in 2017. Drug Metab Dispos.
47:135–144. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Choi YH and Yu AM: ABC transporters in
multidrug resistance and pharmacokinetics, and strategies for drug
development. Curr Pharm Des. 20:793–807. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Chen Z, Shi T, Zhang L, Zhu P, Deng M,
Huang C, Hu T, Jiang L and Li J: Mammalian drug efflux transporters
of the ATP binding cassette (ABC) family in multidrug resistance: A
review of the past decade. Cancer Lett. 370:153–164. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Lombard AP, Liu C, Armstrong CM, D'Abronzo
LS, Lou W, Chen H, Dall'Era M, Ghosh PM, Evans CP and Gao AC:
Overexpressed ABCB1 induces olaparib-taxane cross-resistance in
advanced prostate cancer. Transl Oncol. 12:871–878. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Lawlor D, Martin P, Busschots S, Thery J,
O'Leary JJ, Hennessy BT and Stordal B: PARP Inhibitors as
P-glyoprotein Substrates. J Pharm Sci. 103:1913–1920. 2014.
View Article : Google Scholar : PubMed/NCBI
|














