Introduction
Hepatocellular carcinoma (HCC) is the most common
form of liver cancer, which accounts for ~80% of liver cancer
cases; the number of cases in China exceeds 400,000 per year
(1). Liver cancer is difficult to
diagnose in its early stages and, thus, has a poor prognosis in the
later stages; patients with liver cancer have a poor prognosis and
a low five-year survival rate (1,2).
Therefore, it is important to identify effective therapeutic drugs
(2). Curcumin is extracted from the
rhizome of turmeric, zedoary and other ginger plants; it has a
variety of pharmacological activities, including anti-inflammatory,
antioxidant, lipid-lowering and anti-tumor effects (3). However, its low stability and poor
bioavailability limit its clinical application (3). To improve its anti-tumor effects and
bioavailability, structural modification and optimization of
curcumin are performed. For example, transformation of a central
double α,β-unsaturated ketone into a piperidone-related
α,β-unsaturated ketone to yield
(3E,5E)-3,5-bis(arylidene)-4-piperidone (BAP) derivatives (4,5).
Symmetrical and unsymmetrical BAPs with a variety of substituents
on both sides, such as pyridine-, methoxy-, halogen- and nitro-,
have been proven to have both antitumor and anti-inflammatory
activity by inhibiting the activation of NF-κB and MAPK signal
pathways (6). However, BAPs still
have the disadvantages of having low water solubility and high
toxicity (6). For optimization of
physicochemical properties of BAPs, novel
1,4,5,6,7,8-hexahydropyrido[4,3-d]pyrimidine (PPM) derivatives
based on scaffold hopping have been generated through Michael
condensation reaction between BAPs and guanidine hydrochloride;
PPMs are obtained from the structural optimization of curcumin and
BAPs (7,8). Recently, certain fluoro- or
trifluoromethyl-substituted PPMs have been reported to exhibit
increased water solubility and decreased toxicity of human
wild-type (wt) hepatocytes, but increased cytotoxicity to the liver
cancer cells compared with curcumin and BAPs (7,8). More
importantly, PPM serves a potential anti-hepatoma role by
increasing Bax and Caspase-3 expression and decreasing Bcl-2
expression, as well as inducing HepG2 cell apoptosis by inhibiting
the activation of NF-κB signaling (8). The present study aimed to investigate
the underlying mechanisms to understand the role of PPM in
preventing HCC.
MicroRNAs (miRNAs) regulate biological functions,
serve as a diagnostic tool for cancer and are involved in
tumorigenesis and cancer progression by pairing with the
3′-untranslated region (UTR) of mRNA to increase or decrease mRNA
translation (9–11). miRNA (miR)-26-5p increased the
sensitivity of breast cancer cells toward chemotherapeutic drugs
and inhibited the migration and invasion of tumor cells (12). In bladder cancer cells, miR-26a/b-5p
regulates migration and invasion (13). miR-26b-5p inhibits HCC cells
migration and invasion (14). Both
miR-26b-5p and heat shock 70 kDa protein 8 are molecular targets
for selectively eliminating epithelial cancer stem cells in human
HCC (14,15). miR-26b-5p is involved in the
induction of apoptosis in ovarian granulosa cells of rats, whereas
downregulation of miR-26b-5p inhibits Bax and promotes the
expression of Bcl-2, thereby inhibiting apoptosis (16). In A549 lung adenocarcinoma cells,
miR-26b-5p mimics inhibit expression of Bcl-2 and induce apoptosis
(17). miR-26b-5p can promote the
apoptosis of SKOV3 ovarian cancer cells by regulating the
expression of Bax and Bcl-2 (18).
miR-26b can promote the expression of Bax and Caspase 3, and
increase apoptosis of liver cancer cells (19). In addition, miR-26b-5p may regulate
NF-κB activation; miR-26b inhibits the expression of IκB and
phosphorylated (p-)p65 by targeting the proteins TGF-β-activated
kinase 1 and TGF-β-activated kinase 1/MAP3K7 binding protein 3,
thus inhibiting NF-κB signaling and increasing sensitivity of liver
cancer cells to apoptosis (20).
miR-26b was also reported to inhibit PI3K/AKT/NF-κB signaling
(19).
The present study aimed to investigate the
biological roles of PPM in HepG2 cells, its effects on miR-26b-5p
expression and whether PPM has an anti-liver cancer role by
regulating miR-26b-5p. Proliferation, invasion and apoptosis
assays, as well as western blotting and reverse
transcription-quantitative PCR (RT-qPCR), were performed. Results
from the present study identified some potential treatment methods
and elucidated the mechanism by which PPM induces apoptosis in HCC
cells. In addition, these results identified potential effective
treatment mechanisms for HCC through miRNA-targeting
treatments.
Materials and methods
Cell culture
The human hepatoma cancer cell line HepG2 was
purchased from Nanjing KeyGen Biotech Co., Ltd. (cat. no. KG020).
Short tandem repeat analysis showed that there was no cross
contamination of human cells was found in the cell line. DNA typing
of this cell line had 100% match with HepG2 cells obtained from
American Type Culture Collection cell bank. Cells were cultured in
HyClone DMEM with high glucose medium (Cytiva) containing 1%
penicillin/streptomycin and 10% FBS (Newzerum, Ltd.) in humidified
conditions with 5% CO2 at 37°C.
Cell transfection and PPM
treatment
miR-26b-5p inhibitor, inhibitor negative control
(NC), miR-26b-5p mimics and NC mimics were purchased from Shanghai
GenePharma Co., Ltd. To investigate the effect of PPM on the
expression of miR-26b-5p in HepG2 cells, cells were seeded
(1.5×105 cells/well) and cultured at 37°C for 24 h.
Then, 2 µM PPM was added and the cells were incubated at 37°C for
an additional 24 h. miR-26b-5p inhibitor concentrations of 20, 50
and 80 nM were used to screen for the miR inhibitor concentration
with the highest transfection efficiency; miR-26b-5p mimics and NC
mimics were used at 50 nM. In the Inhibitor NC + PPM and Inhibitor
+ PPM group, HepG2 (1.5×105 cells/well) cells were
seeded in 6-well plates, and transfection reagent Lipofectamine™
2000 (Invitrogen; Thermo Fisher Scientific, Inc.) was added when
cell density reached 30%, according to the manufacturer's
instructions. Cells were transfected in serum-free HyClone DMEM
with high glucose medium at 37°C for 6 h. Following transfection,
the medium was replaced with complete medium containing 10% FBS
(Newzerum, Ltd.) and the cells were incubated for a further 24 h. A
total of 2 µM PPM was added and the cells were incubated at 37°C
for 24 h. The sequences were as follows: miR-26b-5p inhibitor,
5′-ACCUAUCCUGAAUUACUUGAA-3′; miR-26b-5p mimics forward,
5′-UUCAAGUAAUUCAGGAUAGGU-3′ and reverse,
5′-CUAUCCUGAAUUACUUGAAUU-3′; miR inhibitor NC
5′-CAGUACUUUUGUGUAGUACAA-3′; and miR mimics NC forward,
5′-UUCUCCGAACGUGUCACGUTT-3′ and reverse,
5′-ACGUGACACGUUCGGAGAATT-3′. Transfection efficiency was determined
using RT-qPCR.
RNA extraction and RT-qPCR
analysis
To detect the effect of PPM on miR-26b-5p
expression, the experiment was divided into two groups: Untreated
Control and PPM treatment. To screen the concentration of
inhibitors, the experiment was divided into five groups: Control,
Inhibitor NC, Inhibitor (20 nM), Inhibitor (50 nM), Inhibitor (80
nM). To detect the effect of inhibitors on miR-26b-5p expression
after PPM addition, the experiment was divided into two groups:
Inhibitor NC + PPM and Inhibitor + PPM. RNA was isolated from the
different groups using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's instructions.
RT-qPCR was used to measure the expression of miR-26b-5p. Total RNA
was extracted from HepG2 cells using TRIzol reagent (Invitrogen;
Thermo Fisher Scientific, Inc.). The RNA was reverse-transcribed
into cDNA using the SPARKscript II RT Plus kit with gDNA Eraser
(cat. no. AG0304; Shandong Sparkjade Scientific Instruments Co.,
Ltd.), according to the manufacturer's protocol, and amplification
was performed using a 2X SYBR Green qPCR Mix (with ROX) kit (cat.
no. AH0104; Shandong Sparkjade Scientific Instruments Co., Ltd.).
Using 0.2 µl cDNA as the template, thermocycling was performed as
follows: 94°C for 3 min; followed by 40 cycles of 94°C for 15 sec,
60°C for 15 sec and 72°C for 25 sec. β-actin was used as a
housekeeping gene. β-actin and miR-26b-5p primers were designed and
synthesized by Shanghai Shenggong Biology Engineering Technology
Service, Ltd. Changes in miR-26b-5p expression levels were
calculated using the 2−ΔΔCq method (18). The primer sequences were as follows:
miR-26b-5p forward, 5′-CCGGGACCCAGTTCAAGTAA-3′ and reverse,
5′-CCCCGAGCCAAGTAATGGAG-3′ and β-actin forward,
5′-TGGCACCCAGCACAATGAA-3′ and reverse,
5′-CTAAGTCATAGTCCGCCTAGAAGCA-3′.
Wound healing assay
HepG2 cells were transfected with miR-26b-5p or
inhibitor NC for 24 h at 37°C when the cell density reached 30%,
followed by treatment with PPM for 24 h at 37°C. When the cells
reached 90% confluence, a scratch was made with a 200 µl pipette
tip and debris was washed with PBS; the cells were then cultured in
serum-free medium at 37°C. At 0 and 48 h, the wound was observed
with an inverted light microscope (Olympus Corporation) at ×100
magnification, and images were captured for analysis.
Transwell migration and invasion
assay
The migratory and invasive abilities of HepG2 cells
were examined using Transwell chambers with 8 µm pore membranes
(Guangzhou Jet Bio-Filtration Co., Ltd.). Following miR-26b-5p
inhibitor transfection and PPM treatment, HepG2 cells were seeded
(2.5×104 cells/well) into the upper chamber containing
HyClone DMEM with high glucose medium (Cytiva). The lower chamber
was filled with complete medium containing 10% FBS (Newzerum,
Ltd.), After 24 h of incubation at 37°C, cells in the upper chamber
were removed and those migrating into the lower chamber were fixed
with 4% paraformaldehyde for 15 min and stained with 0.1% crystal
violet (Beyotime Institute of Biotechnology) for 10 min, both at
room temperature. A total of five randomly selected fields of view
were captured under a light microscope (Zeiss AG) at ×200
magnification. ImageJ v1.53a (National Institutes of Health,) was
used to count the migrated cells.
For the cell invasion assay, the upper chamber was
precoated with Matrigel (dilution, 1:9; Corning Inc.) at 4°C for 12
h. HepG2 cells were seeded (2.5×104 cells/well) in
tissue culture plate inserts coated with Matrigel (Corning Inc.) at
37°C for 6 h. The subsequent procedures were the same as for the
cell migration assay.
EdU proliferation assay
For the cell proliferation assay, HepG2 cells were
seeded (2.5×104 cells/well) into 24-well plates at 37°C
for 24 h. Subsequently, cells were transfected with miR-26b-5p
inhibitor for 24 h, then treated with 2 µM PPM for 37°C for 24 h.
Cell proliferation was detected using BeyoClick™ EdU-488 Cell
Proliferation Assay kit (Beyotime Institute of Biotechnology)
according to the manufacturer's protocol. Briefly, the cells were
incubated with EdU solution for 2 h at 37°C, fixed with 4%
paraformaldehyde for 15 min and then washed with PBS three times,
both at room temperature. To perforate the cells, PBS containing
0.3% Triton X-100 was added for 10 min at 37°C, and the Click
Additive solution (provided in the kit) was added and the cells
were incubated at room temperature for 30 min in the dark. Nuclei
were stained with 100 µl Hoechst (1,000X diluted to 1:1,000) in
dark for 10 min at 37°C. After washing with PBS, cells were
observed under a fluorescence microscope (Echo) at ×100
magnification, and images were analyzed using ImageJ v1.53a
(National Institutes of Health).
Western blotting
HepG2 cells were lysed using RIPA lysis buffer
(Beyotime Institute of Biotechnology) and collected by
centrifugation (16,904 × g for 15 min at 4°C) to extract the total
protein. BCA protein concentration kit (Beyotime Institute of
Biotechnology) was used to quantify protein concentration. A total
of 20 µg protein/lane was separated by 10% SDS-PAGE and transferred
onto PVDF membranes for 1.5 h at 200 mA. Following blocking with 5%
skimmed milk for 1 h at room temperature, the membranes were
incubated overnight at 4°C with the following rabbit primary
antibodies: Rabbit polyclonal anti-Bax (1:2,000; cat. no.
50599-2-Ig) and anti-GAPDH (1:10,000; cat. no. 10494-1-AP) from
Proteintech Group, Inc.; monoclonal anti-Bcl-2 (1:2,000; cat. no.
A19693) from ABclonal Technology; anti-p65 (cat. no. 4767) and
anti-p-p65 (both 1:2,000; cat. no. 3033S) from Cell Signaling
Technology, Inc.; and anti-CDK8 (1:2,000; cat. no. AF2467) from
Beyotime Institute of Biotechnology. The PVDF membranes were then
incubated with an HRP-conjugated goat anti-rabbit IgG H&L
antibody (1:5,000; cat. no. SA00001-2; Proteintech Group, Inc.) for
1 h at room temperature. The membranes were washed with TBST
containing 1% Tween 20 (BioFroxx; NeoFroxx GmbH) and Super
Excellent Chemiluminescent Substrate (ECL) Detection kit reagent
(cat. no. E-IR-R308; Elabscience, Co., Ltd.) was added dropwise
onto the PVDF membrane. The immunoreactive bands were detected
using a Tanon 4600SF chemiluminescence imaging system (Tanon
Science and Technology Co., Ltd.), and the relative levels of each
protein were semi-quantified using ImageJ v1.53a (National
Institutes of Health).
Apoptosis assay
The Annexin V-FITC Cell Apoptosis kit (cat. no.
G003-1; Nanjing Jiancheng Bioengineering Institute) was used to
determine apoptotic rates according to the manufacturer's
instructions. HepG2 cells were seeded (1.5×105
cells/well) in 6-well plates and transfected with inhibitor NC,
miR-26b-5p inhibitor, inhibitor NC + PPM or inhibitor + PPM for 24
h, then 2 µM PPM was added and the cells were incubated at 37°C for
24 h. Cells were washed with cold PBS and pelleted by
centrifugation at 1,000 × g for 5 min at room temperature. Then,
the cells were resuspended in 1X Annexin-binding buffer (500 µl)
and 5 µl each Annexin V-FITC and PI was added to the binding
solution and mixed according to the manufacturer's instructions.
After 10 min at room temperature, cell apoptosis was analyzed using
FACSCanto™ II flow cytometer (BD Biosciences). Total apoptosis
included early and late apoptotic cells, and the results were
analyzed using FACSDiva Version 6.1.3 (BD Biosciences).
Cell cycle assay
For the cell cycle assay, a PI Cell Cycle and
Apoptosis kit (cat. no. abs50005; Absin Bioscience, Inc.) was used.
HepG2 cells were seeded (1.5×105 cells/well) in 6-well
plates at 37°C for 24 h. When the cells reached 30% confluence,
they were transfected with miR-26b inhibitor or inhibitor NC for 24
h, with or without subsequent treatment with 2 µM PPM for 37°C for
24 h. Cells were collected by centrifugation at 1,000 × g for 5 min
at room temperature. A total of 1 ml 70% ethanol was added to each
well and incubated at 4°C for 18 h for fixation. Cells were then
washed with PBS and centrifuged at 1,000 × g for 5 min at room
temperature. The cell pellets were resuspended in 100 µl propidium
iodide staining buffer and stained with PI working solution for 30
min at room temperature in the dark. The cells were analyzed by
FACSCanto™ II flow cytometry (BD Biosciences), and ModFit LT V4.1.7
software (Verity Software House) was used to analyze the DNA
content.
Bioinformatics
TargetScan (www.targetscan.org/vert_72), miRDB (www.mirdb.org) and Starbase (https://starbase.sysu.edu.cn/starbase2) were used to
predict downstream targets of miR-26b-5p, as well as the binding
site between miR-26b-5p and the target mRNAs.
NCBI (http://www.ncbi.nlm.nih.gov) was used to find the
3′-UTR of the target mRNA as well as to design the plasmid vector
according to the binding site between miR-26b-5p and the target
mRNA, which was experimentally validated by dual-luciferase
reporter gene assay.
Dual-luciferase reporter gene
assay
miR-26b-5p mimics, NC mimics and the Dual Luciferase
Reporter Assay kit were purchased from Shanghai GenePharma Co.,
Ltd. wt and mutant (mut) plasmids of CDK8 (pmirGLO-CDK8-wt and
pmirGLO-CDK8-mut) were designed by Shandong Scientific Cloud
Biotechnology Co., Ltd. Both wt and mut 3′-UTRs of CDK8 mRNAs were
cloned and inserted downstream of firefly luciferase in the pmiRGLO
vector. HepG2 cells were seeded (1.5×105 cells/well) in
6-well plates. When the cell density reached 30%, they were
co-transfected, using 8 µl Lipofectamine 2000, with either 1 µg wt
or mut plasmid and 50 nM of either miR-26b-5p mimics or mimics NC,
transfected; after 6 h, the cells were incubated in complete medium
containing 10% FBS (Newzerum, Ltd.) for 24 h at 37°C. Cells were
collected and luciferase activities were detected using the Dual
Luciferase Reporter Assay, according to the manufacturer's
instructions. Firefly luciferase activity was normalized to
Renilla luciferase activity.
Construction of CDK8 overexpression
vector
The human CDK8 coding sequences (NM_001346501) were
obtained from NCBI (http://www.ncbi.nlm.nih.gov). CDK8 overexpression
plasmid of HepG2 cells were purchased from KeyBio Scientific, Inc.
The HepG2 cells were seeded, at a density of 1.5×105
cells/well in six-well plates and incubated for at 37°C 24 h. HepG2
cells grown to 30% confluence were transfected with 8 µl
Lipofectamine 2000 and 1 µg plasmid, Cells were incubated at 37°C
for 2 days, followed by extraction of cellular proteins.
Statistical analysis
Data are expressed as the mean ± SD; experiments
were repeated in triplicate. Comparisons between groups were
performed using one-way analysis of variance using GraphPad Prism
v8.0.1 (Dotmatics) followed by Bonferroni's correction. P<0.05
was considered to indicate a statistically significant
difference.
Results
PPM upregulates expression of
miR-26b-5p in HepG2 cells
RT-qPCR was performed to determine the effects of
PPM treatment on the expression of miR-26b-5p. PPM significantly
increased the expression of miR-26b-5p compared with the Control
group (untreated cells) (Fig. 1A).
To investigate the effects of miR-26b-5p inhibition, cells were
transfected with varying concentrations of miR-26b-5p. miR-26b-5p
inhibitor transfection at 50 nM induced the greatest inhibition of
miR-26b-5p expression compared with the Control group. The
knockdown efficiency of the inhibitor at 80 nM was lower compared
with that of 50 nM, likely because high concentration of Inhibitor
may lead to the increased of off-target effects and cytotoxicity to
cells, thereby reducing the transfection efficiency. Therefore, 50
nM was selected for subsequent experiments (Fig. 1B). RT-qPCR was used to determine the
relative expression levels of miR-26b-5p in liver cancer cells. The
results demonstrated that miR-26b-5p expression levels were
downregulated in Inhibitor + PPM group compared with Inhibitor NC +
PPM group (Fig. 1C).
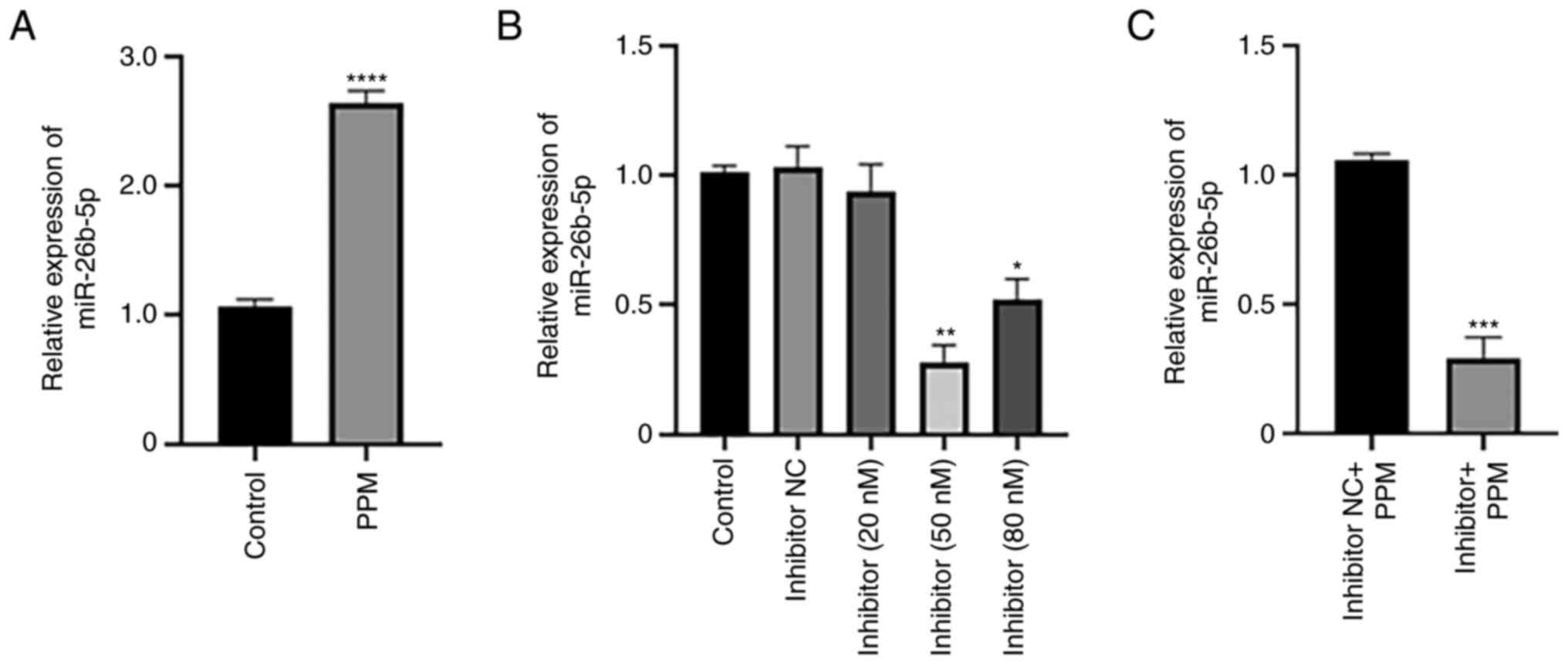 | Figure 1.PPM upregulates expression of
miR-26b-5p. Reverse transcription-quantitative PCR analysis of
miR-26b-5p expression in HepG2 cells following treatment with (A)
PPM and (B) miR-26b-5p inhibitor at various concentrations. (C) The
effect of miR-26b-5p inhibitor and PPM on miR-26b-5p expression.
Data are presented as the mean ± SD. *P<0.05, **P<0.01,
***P<0.001 and *****P<0.0001 vs. Control. miR, microRNA; NC,
negative control; PPM,
1,4,5,6,7,8-hexa-hydropyrido[4,3-d]pyrimidine. |
PPM inhibits migration of HepG2 cells
by upregulating miR-26b-5p
Wound healing and Transwell assays were performed to
examine cell migration. Wound closure increased in the Inhibitor
group and decreased in the PPM group compared with the untreated
Control group (Fig. 2A and B).
Cells transfected with miR-26b-5p inhibitor alleviated the
PPM-induced inhibition of HepG2 cell migration. The number of
migrating cells increased in the miR-26b-5p Inhibitor group, but
decreased in the PPM-treated group compared with Inhibitor + PPM
group (Fig. 2C and D). These
results suggested that PPM may inhibit HepG2 cell migration by
upregulating miR-26b-5p.
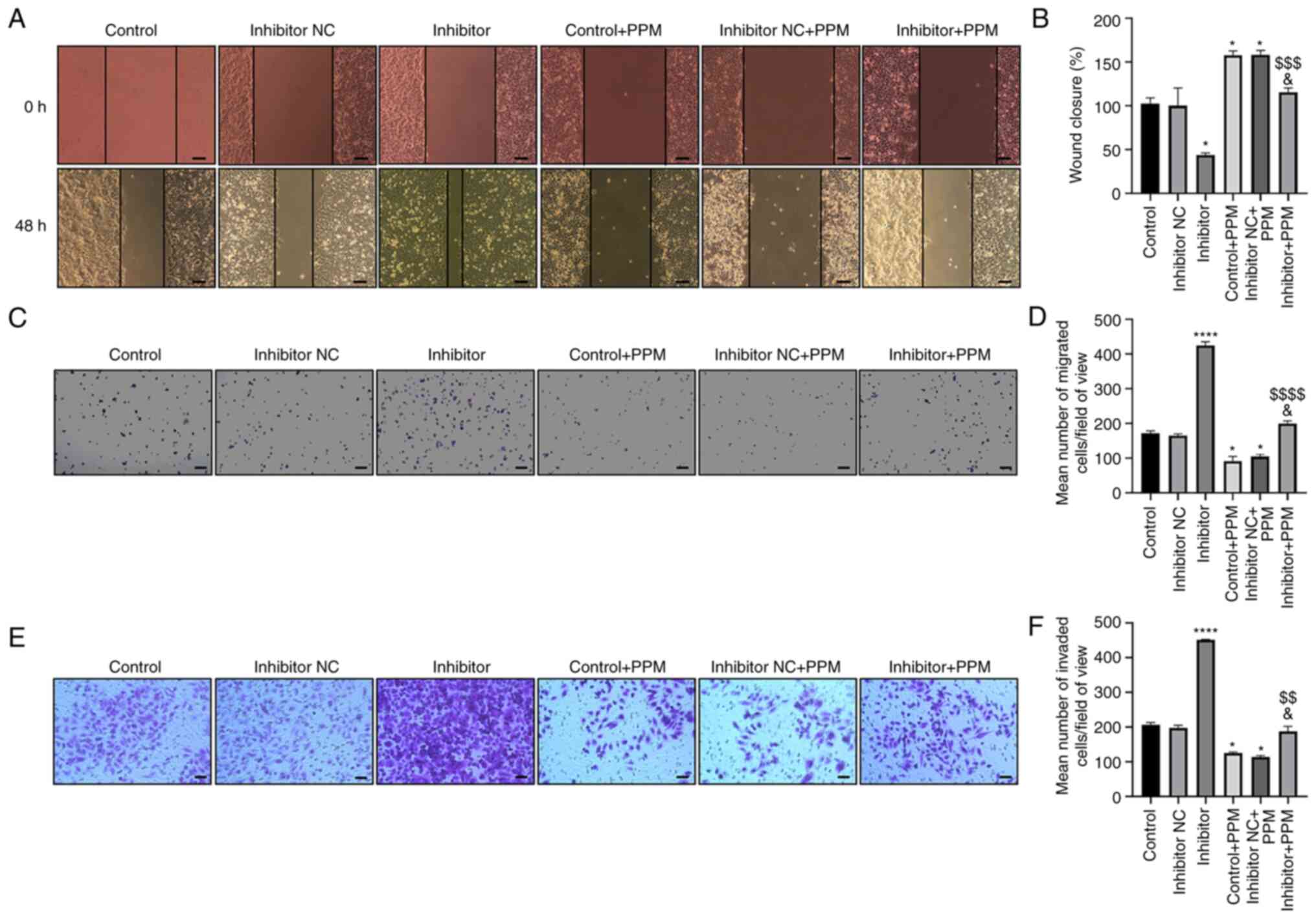 | Figure 2.PPM inhibits migration and invasion
of HepG2 cells through upregulation of miR-26b-5p. (A) Wound
healing assay was used to detect the effect of PPM upregulation of
miR-26b-5p on migration of HepG2 cells. Scale bar, 100 µm. (B)
Quantitated wound healing area from part (A). (C) Transwell assays
were used to detect the effect of PPM on cell migration. Scale bar,
200 µm. (D) Average number of migrated cells from part (C). (E)
Transwell Matrigel assays were used to detect the effect of PPM on
cell invasion. Scale bar, 50 µm. (F) Average number of invaded
cells from part (E). *P<0.05, ****P<0.0001 vs. Control;
&P<0.05 vs. Control + PPM;
$$P<0.01, $$$P<0.001,
$$$$P<0.0001 vs. Inhibitor. miR, microRNA; NC,
negative control; PPM,
1,4,5,6,7,8-hexahydropyrido[4,3-d]pyrimidine. |
PPM inhibits the invasion of HepG2
cells by upregulating miR-26b-5p
Transwell invasion assay results showed that PPM
significantly decreased the cell invasion rate, whereas miR-26b-5p
inhibitor transfection significantly increased cell invasion, as
compared with the Control group (Fig.
2E and F). PPM significantly inhibited the invasion of HepG2
cells, as compared with Inhibitor + PPM group. These results
indicated that PPM inhibited the invasion of HepG2 cells by
upregulating miR-26b-5p.
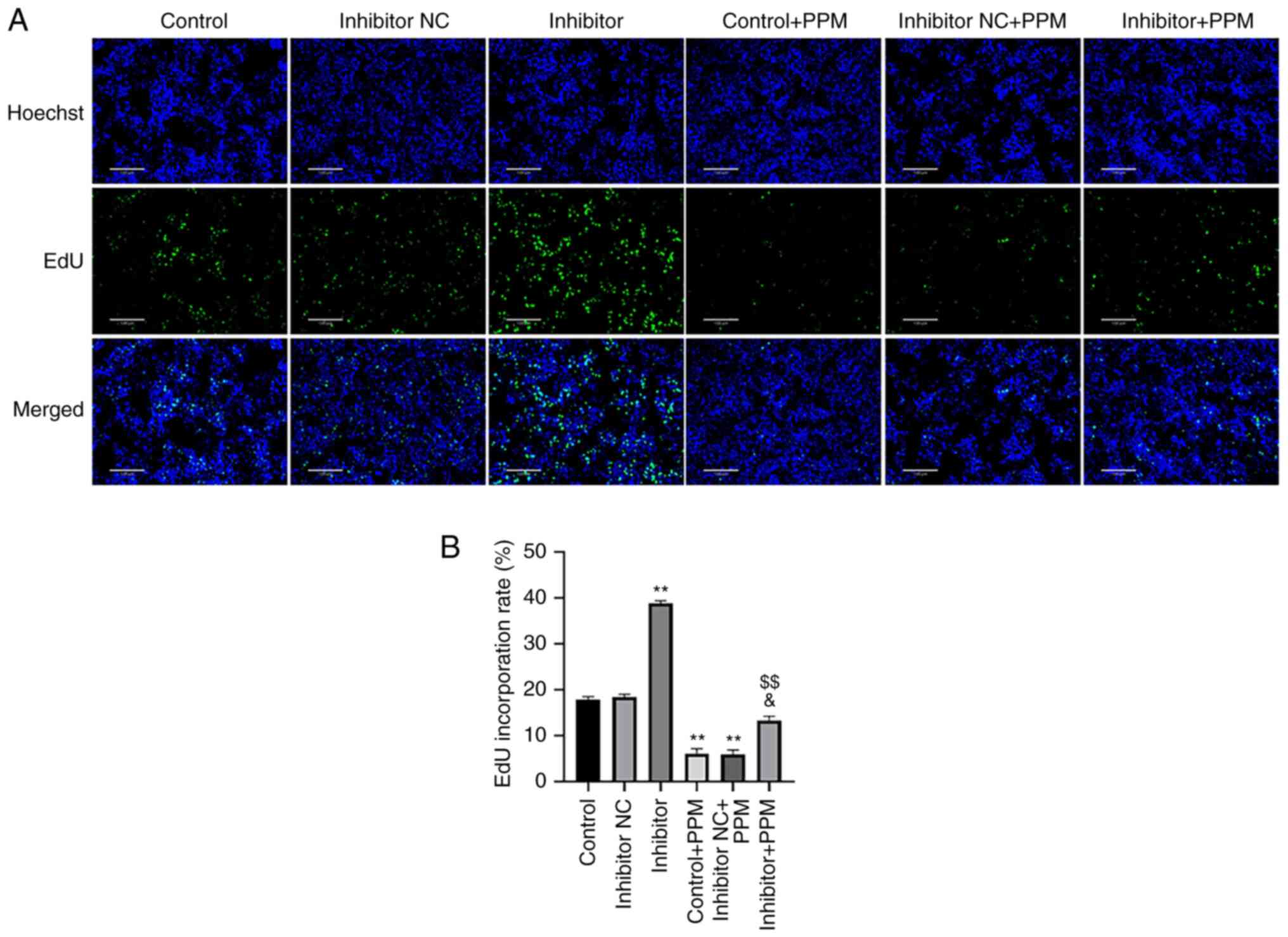
PPM inhibits HepG2 cell proliferation
by upregulating miR-26b-5p
EdU staining was performed to determine whether PPM
inhibited the proliferation of HepG2 cells through miR-26b-5p. PPM
treatment significantly decreased the number of EdU-positive cells
compared with the Control group (Fig.
3), whereas miR-26b-5p inhibitor alone significantly increased
the EdU-positive cells. PPM significantly decreased proliferation
of HepG2 cells compared with Inhibitor + PPM group. These results
suggested that PPM may inhibit the proliferation of HepG2 cells by
upregulating miR-26b-5p.
PPM promotes HepG2 cell apoptosis by
upregulating miR-26b-5p
An Annexin V-FITC/PI staining kit was used to
investigate the effects of PPM treatment on apoptosis by
upregulating miR-26b-5p. In the Control, Inhibitor NC and Inhibitor
group, the mean apoptotic rate of HepG2 cells was 5.5, 7.1 and
2.5%, respectively, demonstrating that miR-26b-5p inhibition
decreased apoptosis compared with the Control group (Fig. 4A and B). When treated with PPM, the
mean apoptotic rates in these groups increased to 11.3, 11.5 and
5.4%, respectively, indicating that PPM significantly increased
apoptosis, and that miR-26b-5p Inhibitor group significantly
decreased the apoptosis compared with Inhibitor + PPM group. These
results suggested that PPM may promote apoptosis by upregulating
miR-26b-5p.
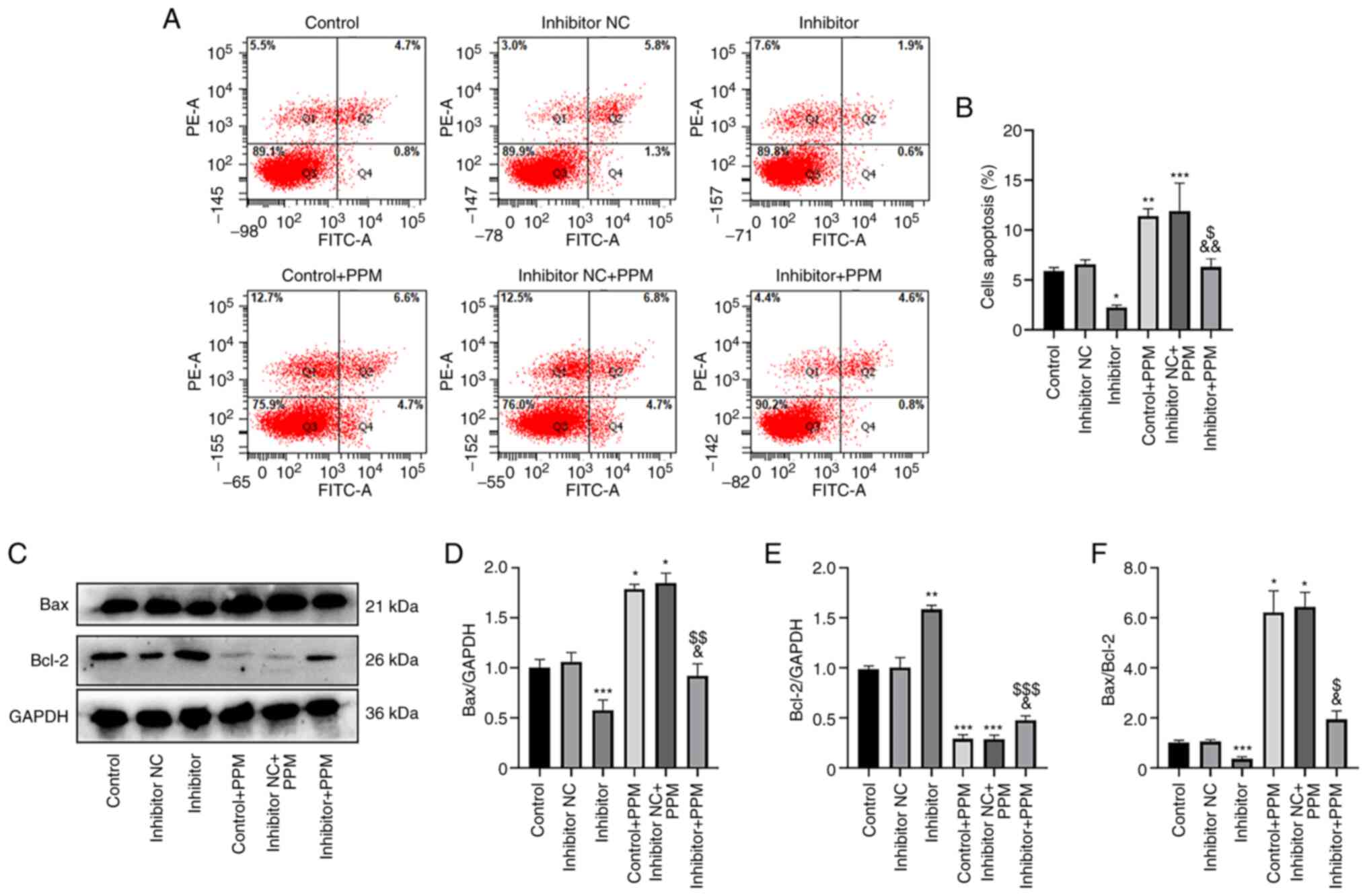 | Figure 4.PPM promotes apoptosis and Bax/Bcl2
ratio through upregulation of miR-26b-5p in HepG2 cells. (A) Flow
cytometry was used to analyze apoptosis in HepG2 cells. (B)
Histograms presenting the percentage of apoptotic cells in the
various groups. (C) The protein expression levels of Bax and Bcl-2
in HepG2 cells were detected by western blotting. Relative protein
expression levels of (D) Bax and (E) Bcl-2; GAPDH was used as the
internal control. (F) The Bax/Bcl-2 ratio in the different groups.
*P<0.05, **P<0.01, ***P<0.001 vs. Control;
&P<0.05, &&P<0.01 vs.
Control + PPM; $P<0.05, $$P<0.01,
$$$P<0.001 vs. Inhibitor. miR, microRNA; NC, negative
control; PPM, 1,4,5,6,7,8-hexahydropyrido[4,3-d]pyrimidine. |
Results from western blotting demonstrated that PPM
treatment significantly increased Bax and decreased Bcl-2 compared
with the Control group. The miR-26b-5p Inhibitor significantly
decreased Bax but increased Bcl-2 compared with the Control group.
However, PPM co-treatment significantly inhibited the miR-26b-5p
inhibitor-induced increased expression of Bax and decreased
expression of Bcl-2 (Fig. 4C-E).
miR-26b-5p inhibitor significantly reduced the expression ratio of
Bax/Bcl-2 compared with the Control, whereas PPM significantly
increased expression ratio of Bax/Bcl-2, PPM co-treatment
significantly reduced the expression ratio of Bax/Bcl-2 compared
with the PPM group (Fig. 4F). These
results suggested that PPM may promote the expression of Bax and
reduced the expression of Bcl-2 by upregulating miR-26b-5p in HepG2
cells.
PPM arrests HepG2 cells in the
G0/G1 phase without the involvement of
miR-26b-5p
The percentage of cells in the
G0/G1, S and G2/M phases was
analyzed using flow cytometry. The percentage of
G0/G1 cells in the Control, miR-26b-5p
Inhibitor NC and miR-26b-5p Inhibitor group was 22.44, 21.10 and
22.48%, respectively (Fig. 5A and
B), indicating that miR-26b-5p inhibitor did not regulate the
cell cycle in HepG2 cells. When treated with PPM, the percentage of
G0/G1 cells was 40.92, 37.56 and 37.54%, in
the Control, miR-26b-5p Inhibitor NC and miR-26b-5p Inhibitor
groups, respectively (Fig. 5A and
B), indicating that PPM significantly increased cell count in
the G0/G1 phase. Collectively, these data
indicated that PPM arrested HepG2 cell cycle in
G0/G1 phase independently of miR-26b-5p.
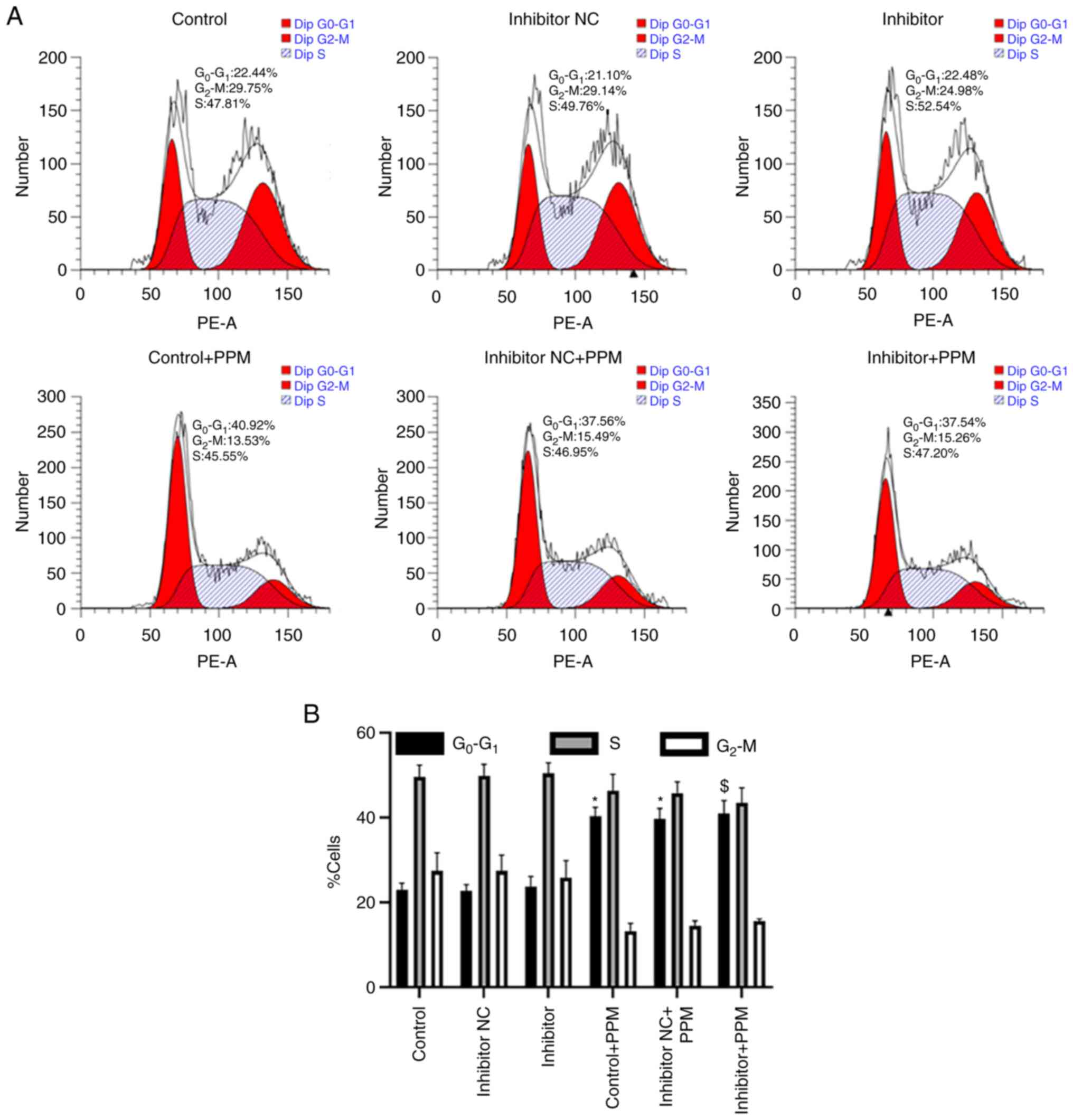 | Figure 5.PPM regulates HepG2 cell cycle
progression independently of miR-26b-5p. (A) Cell cycle phase
distribution was assessed in HepG2 cells, including
G0/G1, S and G2/M. (B)
Quantitative data of HepG2 cells cycle phase distribution.
*P<0.05 vs. Control; $P<0.05 vs. Inhibitor. miR,
microRNA; NC, negative control; PPM,
1,4,5,6,7,8-hexahydropyrido[4,3-d]pyrimidine. |
CDK8 is a target gene of
miR-26b-5p
To study the mechanism by which miR-26b-5p controls
HepG2 proliferation, invasion and apoptosis, the target genes of
miR-26b-5p were predicted using TargetScan, miRDB and Starbase,
which identified 1,045, 1,134 and 2,018 miR-26b-5p target genes,
respectively, with 494 overlapping genes (Fig. 6A). CDK8 was predicted to be the
target gene of miR-26b-5p; a recent study reported the involvement
of CDK8 in the progression of HepG2, and binding sites between
miR-26b-5p and CDK8 were identified (14). The 3′-UTR of CDK8 mRNA has three
predicted miR-26b-5p target sites; in the present study, the first
target site was mutated (192–198 sequence) as inserted into a
dual-luciferase reporter assay plasmid containing a part of the
CDK8 3′-UTR was used (Fig. 6B).
miR-26b-5p mimics transfection upregulated the expression of
miR-26b-5p (Fig. 6C). The results
of the dual-luciferase reporter assay demonstrated that miR-26b-5p
significantly suppressed the relative luciferase activity of the
pmirGLO-CDK8-wt compared with pmirGLO-CDK8-mut (Fig. 6D); however, no suppression in
luciferase activity was observed with the mut 3′-UTR of CDK8. These
results indicated the presence of binding sites between CDK8 and
miR-26b-5p.
 | Figure 6.CDK8 is a potential target of
miR-26b-5p. (A) Venn diagram of the overlapping predicted target
genes of miR-26b-5p from TargetScan, miRDB and Starbase. (B) Three
predicted target sites of miR-26b-5p in CDK8 were identified from
TargetScan, miRDB and Starbase. (C) Reverse
transcription-quantitative PCR showed that miR-26b-5p was
upregulated in HepG2 cells after adding miR-26b-5p mimics.
**P<0.01 vs. NC mimics. (D) Dual-luciferase reporter assay
results demonstrated an interaction between miR-26b-5p and CDK8.
%P<0.05 vs. NC. (E) The protein expression levels of
CDK8 in the HepG2 cells were detected by western blot. (F)
Semi-quantification of the western blot data in (E), from there
independent experiments. (G) Western blot analysis was performed to
detect the protein expression levels of p65 and p-p65 in HepG2
cells. (H) p-p65 to total p65 ratio of each group. *P<0.05,
**P<0.01 vs. Control; &&&P<0.001 vs.
Control + PPM; $$P<0.01 vs. Inhibitor. (I)
Representative western blots and (J) semi-quantitative expression
of CDK8 protein expression levels following transfection of the
CDK8-OE vector. **P<0.01 vs. CDK8-NC. (K) Representative western
blots and (L) semi-quantitative expression of p65 and p-p65
following CDK8-OE vector transfection and treated with PPM. GAPDH
expression was used as a loading control. *P<0.05,
****P<0.0001 vs. Control; &P<0.05 vs. Control
+ PPM; $P<0.05, vs. Inhibitor. CDS, coding sequence;
hsa, Homo sapiens; miR, microRNA; mut, mutant; NC, negative
control; OE, overexpression; p-, phosphorylated; PGK,
phosphoglycerate kinase; PPM,
1,4,5,6,7,8-hexahydropyrido[4,3-d]pyrimidine; UTR, untranslated
region; wt, wild-type. |
Western blot assay results demonstrated that
miR-26b-5p negatively regulated CDK8 levels in HepG2 cells; that
is, miR-26b-5p inhibitor transfection significantly increased the
expression of CDK8 compared with the Control, whereas PPM
significantly decreased CDK8 expression. PPM co-treatment
significantly decreased the miR-26b-5p inhibitor-induced expression
of CDK8 of HepG2 cells (Fig. 6E and
F). Taken together, these results indicated that CDK8 is a
target of miR-26b-5p.
PPM upregulation of miR-26b-5p
suppresses NF-κB/p65 signaling pathway in HepG2 cells by targeting
of CDK8
NF-κB activation is associated with apoptosis and
tumorigenesis (12). PPM treatment
inhibited phosphorylation of NF-κB/p65 compared with the Control
group (Fig. 6G and H). PPM
co-treatment significantly inhibited NF-κB/p65 activation induced
by miR-26b-5p inhibitor (Fig. 6G and
H). CDK8-OE vector transfection significantly increased the
CDK8 expression compared with the CDK8-NC group (Fig. 6I and J). CDK8-OE promoted activation
of NF-κB/p65, whereas PPM significantly inhibited NF-κB/p65
activation induced by CDK8-OE group (Fig. 6K and L). These results suggested
that PPM upregulation of miR-26b-5p suppresses NF-κB/p65 signaling
pathway in HepG2 cells by targeting of CDK8.
Discussion
PPM induces HepG2 cell apoptosis by inhibiting
NF-κB/p65 activation (8); however,
it is still unknown whether microRNAs are involved in this process.
The present study demonstrated that PPM treatment increased
expression of miR-26b-5p and inhibited cell migration,
proliferation and NF-κB/p65 activation. PPM also promoted cell
apoptosis by upregulating miR-26b-5p.
miR-26b-5p serves an important role in many
diseases, such as promoting angiogenesis and endothelial cell
proliferation after acute ischemia (21). In addition, it disrupts cancer
progression and EMT, and promotes apoptosis of tumor cells. For
example, miR-26b-5p was reported to inhibit non-small cell lung
cancer progression, promote apoptosis and enhance radiosensitivity
in lung adenocarcinoma cells (22,23).
miRNA-26b-5p also targets transcriptional repressor GATA binding 1
mRNA to promote apoptosis of breast cancer cells and inhibit their
proliferation (24). In addition,
miR-26b-5p inhibits EMT, migration and invasion of HCC cells. It
also promotes expression of apoptotic proteins Bax and Caspase 3,
and increases the sensitivity of liver cancer cells to apoptosis
(19,20). NF-κB activation is associated with
apoptosis and tumorigenesis (12);
miR-26b inhibits NF-κB signaling to promote apoptosis of liver
cancer cells (19,20). In the present study, miR-26b-5p
inhibitor transfection increased p-p65 expression, whereas PPM
decreased this induced expression of p-p65, demonstrating that PPM
may upregulate miR-26b-5p to promote cell apoptosis, which is
associated with activation of NF-κB/p65 (20).
Dual-luciferase reporter assay, western blotting and
bioinformatics were used to verified that miR-26b-5p targeted and
downregulated the expression of CDK8. Bcl-2 is an oncogene
responsible for promoting liver cancer occurrence and metastasis
(24); it also inhibits
stemness-related protein c-Myc (25) and reduces breast cancer cell
proliferation (26). The present
study results indicated that PPM upregulation of miR-26b-5p
suppresses NF-κB/p65 signaling pathway in HepG2 cells by targeting
of CDK8. The present study verified the association between CDK8
and NF-κB/p65 by OE experiments and western blotting. CDK8-OE
transfection upregulated the expression of NF-κB/p65. Therefore PPM
was hypothesized to induce HepG2 cell apoptosis through
upregulation of miR-26b-5p and subsequent targeting of CDK8, which
leads to the inhibition of NF-κB activation.
Furthermore, PPM induced HepG2 cell arrest in the
G0/G1 phase without the involvement of
miR-26b-5p. Previous studies have demonstrated that miR-26b-5p
mimics affect the cell cycle; for example, CAL27 tongue squamous
cell carcinoma cells are arrested at the S/G2 transition
(27) and EC9706 human esophageal
cancer cells are arrested in G1 phase (28). However, miR-26b-5p mimics
transfection does not induce apoptosis or cell cycle arrest in
mouse spermatocyte-derived GC-2 cells (29). Taken together, the aforementioned
results suggested that regulation of the cell cycle by miR-26b-5p
may be dependent on cell type; further studies are required to
verify this.
In the present study, PPM promoted HepG2 cell
apoptosis, decreased cell migration and proliferation and inhibited
NF-κB/p65 activation by upregulating miR-26b-5p expression. CDK8
was identified as a target gene of miR-26b-5p. The present study
provided novel insights into the anti-tumor effects of PPM.
However, the lack of miRNA knockout experiments and PPM + miR
mimics transfection experiments are limitation of the present
study. In vivo studies are required to validate the
potential use of miR-26b-5p as a therapeutic target for liver
cancer and may further prove that PPM is an effective drug in the
treatment of liver cancer. The present study data may provide a
basis for further investigation of PPM as a promising drug for the
treatment of liver cancer.
Acknowledgements
Not applicable
Funding
The present study was supported by Technology Project of Yantai
(grant no. 2022XDRH003).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HL and YH confirm the authenticity of all the raw
data. HL performed the experiments and statistical analysis and
wrote the manuscript. YY performed western blotting. YZ performed
the cell culture. XB designed the experiments. YH designed the
experiments. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yang X, Qu S, Wang L, Zhang H, Yang Z,
Wang J, Dai B, Tao K, Shang R, Liu Z, et al: PTBP3 splicing factor
promotes hepatocellular carcinoma by destroying the splicing
balance of NEAT1 and pre-miR-612. Oncogene. 37:6399–6413. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang Y, Yang L, Chen T, Liu X, Guo Y, Zhu
Q, Tong X, Yang W, Xu Q, Huang D and Tu K: A novel lncRNA
MCM3AP-AS1 promotes the growth of hepatocellular carcinoma by
targeting miR-194-5p/FOXA1 axis. Mol Cancer. 18:282019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nelson KM, Dahlin JL, Bisson J, Graham J,
Pauli GF and Walters MA: The essential medicinal chemistry of
curcumin. J Med Chem. 60:1620–1637. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yin DL, Liang YJ, Zheng TS, Song RP, Wang
JB, Sun BS, Pan SH, Qu LD, Liu JR, Jiang HC and Liu LX: EF24
inhibits tumor growth and metastasis via suppressing NF-kappaB
dependent pathways in human cholangiocarcinoma. Sci Rep.
6:321672016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li N, Xin WY, Yao BR, Cong W, Wang CH and
Hou GG: N-phenylsulfonyl-3,5-bis(arylidene)-4-piperidone
derivatives as activation NF-κB inhibitors in hepatic carcinoma
cell lines. Eur J Med Chem. 155:531–544. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yao BR, Sun Y, Chen SL, Suo HD, Zhang YL,
Wei H, Wang CH, Zhao F, Cong W, Xin WY and Hou GG: Dissymmetric
pyridyl-substituted 3,5-bis(arylidene)-4-piperidones as
anti-hepatoma agents by inhibiting NF-κB pathway activation. Eur J
Med Chem. 167:187–199. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sun Y, Gao ZF, Yan WB, Yao BR, Xin WY,
Wang CH, Meng QG and Hou GG: Discovery of novel NF-кB inhibitor
based on scaffold hopping:
1,4,5,6,7,8-hexahydropyrido[4,3-d]pyrimidine. Eur J Med Chem.
198:1123662020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou YQ, Sun Y, Luo HL, Gao ZF, Zhang HQ,
Meng QG, Bai XY, Hou GG and Hou Y: Discovery of anti-hepatoma
agents from 1,4,5,6,7,8-hexahydropyrido[4,3-d]pyrimidine by
inhibiting PI3K/AKT/NF-κB pathway activation. Eur J Med Chem.
225:1137962021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Iacona JR and Lutz CS: miR-146a-5p:
Expression, regulation, and functions in cancer. Wiley Interdiscip
Rev RNA. 10:e15332019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gao X, Han D and Fan W: Down-regulation of
RBP-J mediated by microRNA-133a suppresses dendritic cells and
functions as a potential tumor suppressor in osteosarcoma. Exp Cell
Res. 349:264–272. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nussbacher JK and Yeo GW: Systematic
discovery of RNA binding proteins that regulate MicroRNA levels.
Mol Cell. 69:1005–1016.e7. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Du Q, Yuan Z, Huang X, Huang Y, Zhang J
and Li R: miR-26b-5p suppresses chemoresistance in breast cancer by
targeting serglycin. Anticancer Drugs. 33:308–319. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Miyamoto K, Seki N, Matsushita R, Yonemori
M, Yoshino H, Nakagawa M and Enokida H: Tumour-suppressive
miRNA-26a-5p and miR-26b-5p inhibit cell aggressiveness by
regulating PLOD2 in bladder cancer. Br J Cancer. 115:354–363. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin Y, Jian Z, Jin H, Wei X, Zou X, Guan R
and Huang J: Long non-coding RNA DLGAP1-AS1 facilitates
tumorigenesis and epithelial-mesenchymal transition in
hepatocellular carcinoma via the feedback loop of
miR-26a/b-5p/IL-6/JAK2/STAT3 and Wnt/β-catenin pathway. Cell Death
Dis. 11:342020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Khosla R, Hemati H, Rastogi A, Ramakrishna
G, Sarin SK and Trehanpati N: miR-26b-5p helps in EpCAM+cancer stem
cells maintenance via HSC71/HSPA8 and augments malignant features
in HCC. Liver Int. 39:1692–1703. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu S, Li L, Li M and Zhang J: Effect of
miR-26b-5p on cis-diamine dichloroplatinum-induced ovarian
granulosa cell injury by targeting MAP3K9. In Vitro Cell Dev Biol
Anim. 56:213–221. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu T, Chen W, Liu S, Lu H, Wang H, Kong D,
Huang X, Kong Q, Ning Y and Lu Z: Huaier suppresses proliferation
and induces apoptosis in human pulmonary cancer cells via
upregulation of miR-26b-5p. FEBS Lett. 588:2107–2114. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhu L and Mei M: Interference of long
non-coding RNA HAGLROS inhibits the proliferation and promotes the
apoptosis of ovarian cancer cells by targeting miR-26b-5p. Exp Ther
Med. 22:8792021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Feng Y, Zu LL and Zhang L: MicroRNA-26b
inhibits the tumor growth of human liver cancer through the
PI3K/Akt and NF-κB/MMP-9/VEGF pathways. Oncol Rep. 39:2288–2296.
2018.PubMed/NCBI
|
|
20
|
Zhao N, Wang R, Zhou L, Zhu Y, Gong J and
Zhuang SM: MicroRNA-26b suppresses the NF-κB signaling and enhances
the chemosensitivity of hepatocellular carcinoma cells by targeting
TAK1 and TAB3. Mol Cancer. 13:352014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen Y, Li S, Zhang Y, Wang M, Li X, Liu
S, Xu D, Bao Y, Jia P, Wu N, et al: The lncRNA Malat1 regulates
microvascular function after myocardial infarction in mice via
miR-26b-5p/Mfn1 axis-mediated mitochondrial dynamics. Redox Biol.
41:1019102021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang R, Niu Z, Pei H and Peng Z: Long
noncoding RNA LINC00657 induced by SP1 contributes to the non-small
cell lung cancer progression through targeting miR-26b-5p/COMMD8
axis. J Cell Physiol. 235:3340–3349. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Han F, Huang D, Huang X, Wang W, Yang S
and Chen S: Exosomal microRNA-26b-5p down-regulates ATF2 to enhance
radiosensitivity of lung adenocarcinoma cells. J Cell Mol Med.
24:7730–7742. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wilke CM, Hess J, Klymenko SV, Chumak VV,
Zakhartseva LM, Bakhanova EV, Feuchtinger A, Walch AK,
Selmansberger M, Braselmann H, et al: Expression of miRNA-26b-5p
and its target TRPS1 is associated with radiation exposure in
post-Chernobyl breast cancer. Int J Cancer. 142:573–583. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang X, Zhang X, Wang T, Wang L, Tan Z,
Wei W, Yan B, Zhao J, Wu K, Yang A, et al: MicroRNA-26a is a key
regulon that inhibits progression and metastasis of c-Myc/EZH2
double high advanced hepatocellular carcinoma. Cancer Lett.
426:98–108. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li J, Li X, Kong X, Luo Q, Zhang J and
Fang L: MiRNA-26b inhibits cellular proliferation by targeting CDK8
in breast cancer. Int J Clin Exp Med. 7:558–565. 2014.PubMed/NCBI
|
|
27
|
Yi L, Liu Y, Xu A, Li S, Zhang H, Peng M,
Li Z, Ren H, Dai J, Luo C, et al: MicroRNA-26b-5p suppresses the
proliferation of tongue squamous cell carcinoma via targeting
proline rich 11 (PRR11). Bioengineered. 12:5830–5838. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang K, Zhang B, Bai Y and Dai L: E2F1
promotes cancer cell sensitivity to cisplatin by regulating the
cellular DNA damage response through miR-26b in esophageal squamous
cell carcinoma. J Cancer. 11:301–310. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu Y, Liu WB, Liu KJ, Ao L, Cao J, Zhong
JL and Liu JY: Overexpression of miR-26b-5p regulates the cell
cycle by targeting CCND2 in GC-2 cells under exposure to extremely
low frequency electromagnetic fields. Cell Cycle. 15:357–367. 2016.
View Article : Google Scholar : PubMed/NCBI
|