Cachexia is a fatal disease that is associated with
several conditions, including acquired immunodeficiency syndrome,
multiple sclerosis, chronic obstructive pulmonary disease (COPD),
tuberculosis, congestive heart failure, chronic kidney disease and
cancer (1,2). Cancer-associated cachexia is a serious
wasting syndrome characterized by a continuous reduction in
skeletal muscle mass, with or without the loss of fat mass.
Distinct from hunger and nutritional deficiencies, cancer cachexia
(CC) cannot be reversed by food supplements, and leads to
progressive functional impairment (3,4). It is
prevalent in 50–80% of patients with advanced cancer (5). Unfortunately, cancer treatments such
as chemotherapy and radiotherapy aggravate cachexia (6). CC can have a negative impact on
physical function, tolerance to anticancer treatment, overall
survival and well-being in patients with cancer (7). Moreover, it can also increase
psychological stress and the financial burden on patients and their
families (2,8).
In 2011, an international panel of experts
identified a weight loss of >5% over 6 months, any degree of
weight loss >2% in an individual with a body mass index <20
kg/m2, or sarcopenia, defined as a skeletal muscle index
<7.26 kg/m2 in men and <5.45 kg/m2 in
women, as diagnostic criteria for CC (3). CC can be divided into three clinical
stages according to the degree of weight loss and metabolic
changes, namely pre-cachexia, cachexia and refractory cachexia
(3). The main clinical symptoms of
CC include anorexia, asthenia, fever, anemia, edema and wasting
(7,9). The occurrence of CC has been
attributed to systemic inflammation generated by tumor-host
interactions and tumor-derived catabolic factors such as
proteolysis-inducing factor, zinc-α2-glycoprotein (ZAG),
parathyroid hormone-related protein and microRNAs (miRNAs)
(6,7). Systemic inflammation is characterized
by increased circulating levels of cytokines, including tumor
necrosis factor (TNF)-α, TNF-like weak inducer of apoptosis,
interleukin (IL)-1, IL-6, IL-8, IL-20, interferon-γ, leukemia
inhibitory factor, myostatin, activin and growth differentiation
factor 15 (GDF15) (10–13). These factors drive metabolic
disorders in multiple tissues and organs during CC, including the
muscles (10,11,13,14),
adipose tissue (10–12,15,16),
heart (17), brain (10,11),
liver (10,18–21),
gallbladder (19), bone (22), pancreas (21), spleen (18), intestines (23), gonads (24) and blood (18,22,25).
Table I summarizes the cytokines
involved in the damage of various organs or tissues associated with
CC. In addition, other factors such as cancer type, stage, tumor
size, inter-individual genetics and sex can also influence the
development and progression of CC (3,26,27).
The current treatment protocol advocated for CC is a comprehensive
treatment system based on drug therapy, including anti-inflammatory
drugs, and measures to increase metabolism, inhibit catabolism and
stimulate appetite, supplemented by nutrition, exercise and
psychological support (28).
However, the effectiveness of these treatment options for CC is
unclear. Thus, an in-depth understanding of the key factors
associated with CC is crucial for the early identification and
development of novel therapeutic options.
GDF15 is a stress-induced cytokine that regulates
food intake, energy metabolism and body weight (29). Its levels have been shown to be
upregulated in patients with CC, as well as in animal models of
pancreatic, colon, head and neck, breast and prostate cancers
(30–33). Elevated circulating levels of GDF15
may lead to anorexia, weight loss and decreased survival in cases
of CC (32). Notably, GDF15 plasma
levels have been reported to be significantly higher in patients
with pre-cachexia than in those with cachexia and refractory
cachexia (30). These studies imply
that GDF15 is closely associated with CC. The present review aims
to clarify the role and molecular mechanisms of GDF15 in CC.
GDF15 is a novel transforming growth factor (TGF)-β
superfamily member that was first identified in activated
macrophages (34). It is also known
as macrophage inhibitory cytokine-1, nonsteroidal anti-inflammatory
drug activated gene-1, prostate-derived factor, placental TGF-β and
placental bone morphogenetic protein (29,35).
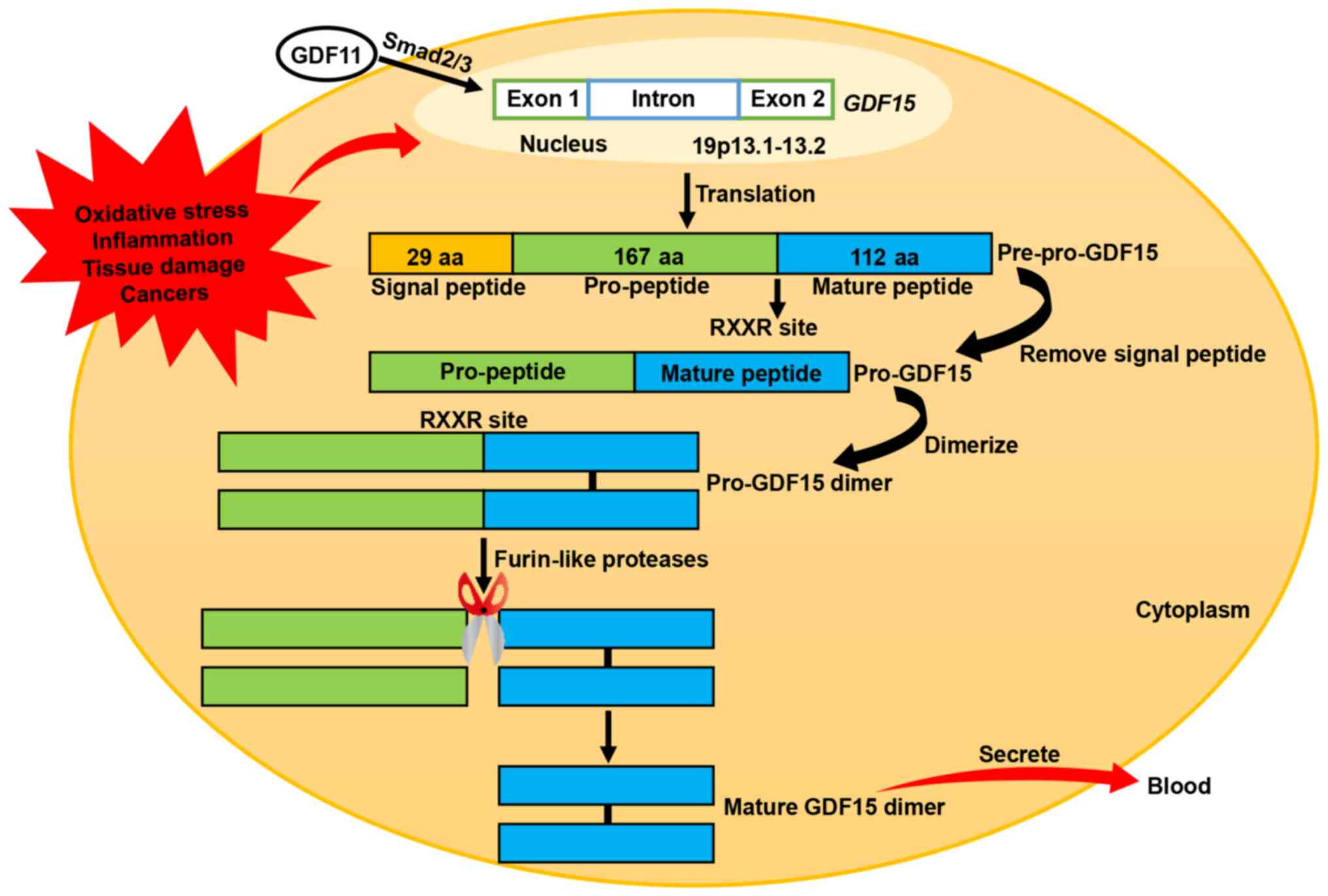
The human GDF15 gene is located on chromosome 19p13.1–13.2
and consists of two exons separated by an intron (36). GDF15 is synthesized as an inactive
precursor protein consisting of a chain of 308 amino acids,
including a signal peptide comprising 29 amino acids, a pro-peptide
comprising 167 amino acids and a mature peptide comprising 112
amino acids (29,35). Following removal of the signal
peptide, the remaining GDF15 pre-peptide dimerizes in the
endoplasmic reticulum through specific disulfide bonding to form a
pro-GDF15 dimer precursor. This precursor is subsequently cleaved
by furin-like proteases at the RXXR site (amino acid 196), thereby
releasing the C-terminal dimeric mature homodimer GDF15. Mature
GDF15 eventually diffuses into the circulation as a 25-kD dimer
(Fig. 1) (29,35).
GDF15 is widely expressed in body tissues at
different levels under normal conditions, with high expression in
the placenta, medium expression in the prostate and bladder, and
low expression in the kidney, liver, colon, pancreas, stomach,
gallbladder, breast, lung and endometrium (37–39).
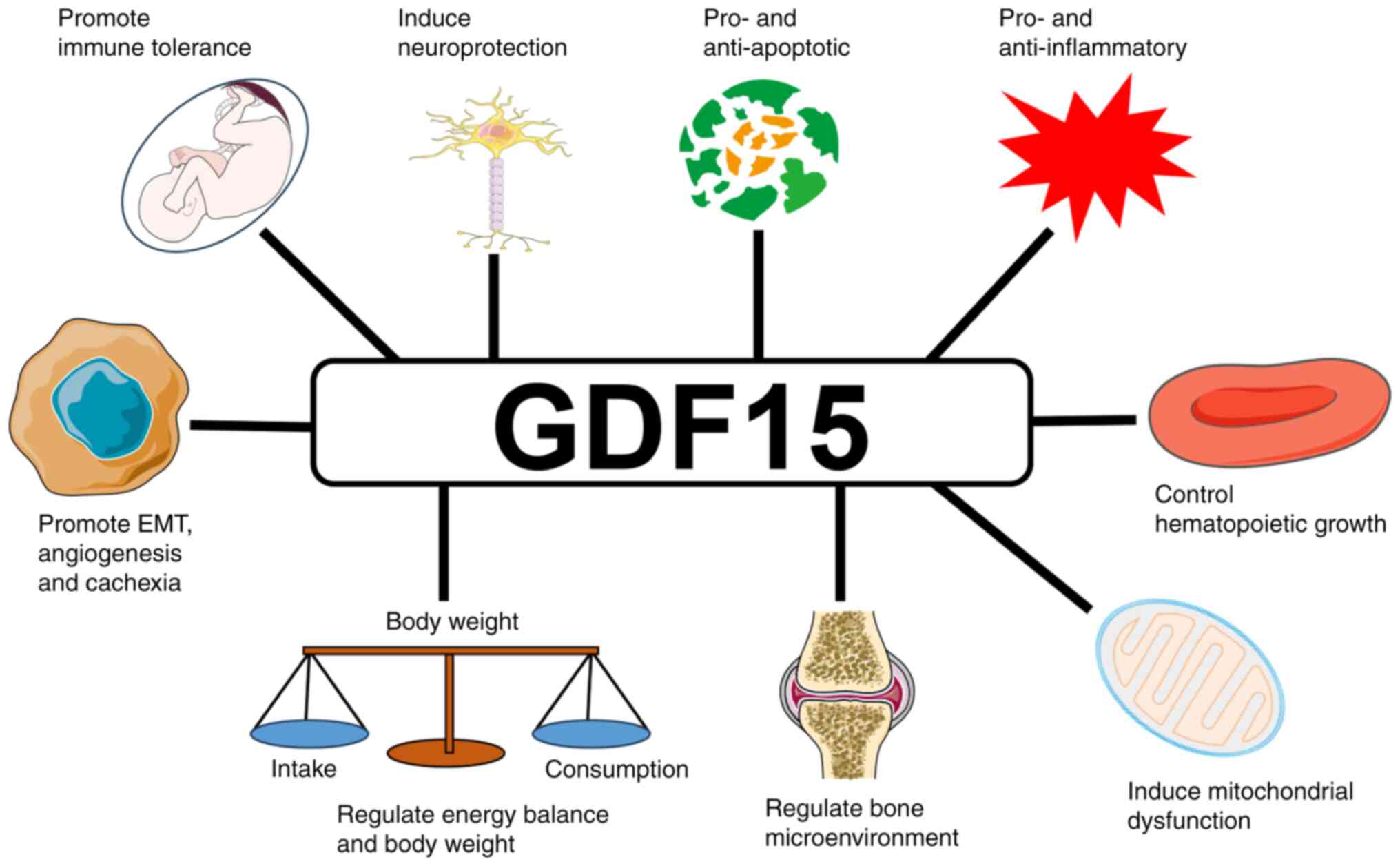
It has multiple biological functions (Fig. 2). The circulating concentrations of
GDF15 range between 0.2 and 1.2 ng/ml in healthy individuals
(38). These levels increase with
age, pregnancy, exercise, smoking and obesity, and are also
influenced by genetic and environmental factors (29,40).
GDF15 is highly expressed in vascular smooth muscle cells,
cardiomyocytes, endothelial cells, macrophages and adipocytes
during oxidative stress, inflammation, tissue damage and cancer
(41,42). GDF15 is elevated in a variety of
cancers, including those of the prostate, colon, pancreas and
breast (31,39). In one study, the mean value of serum
GDF15 was almost two-fold higher in cancer patients compared with
that in healthy controls (32).
GDF15 plays a number of roles in tumorigenesis. In the initial
stages of cancer, it induces tumor cell apoptosis and inhibits
cancer progression (43). In later
stages of cancer, it promotes tumor cell proliferation and
metastasis (44,45). GDF15 has been recognized as a tumor
biomarker that is closely implicated in tumor progression, cachexia
and reduced survival (31,46).
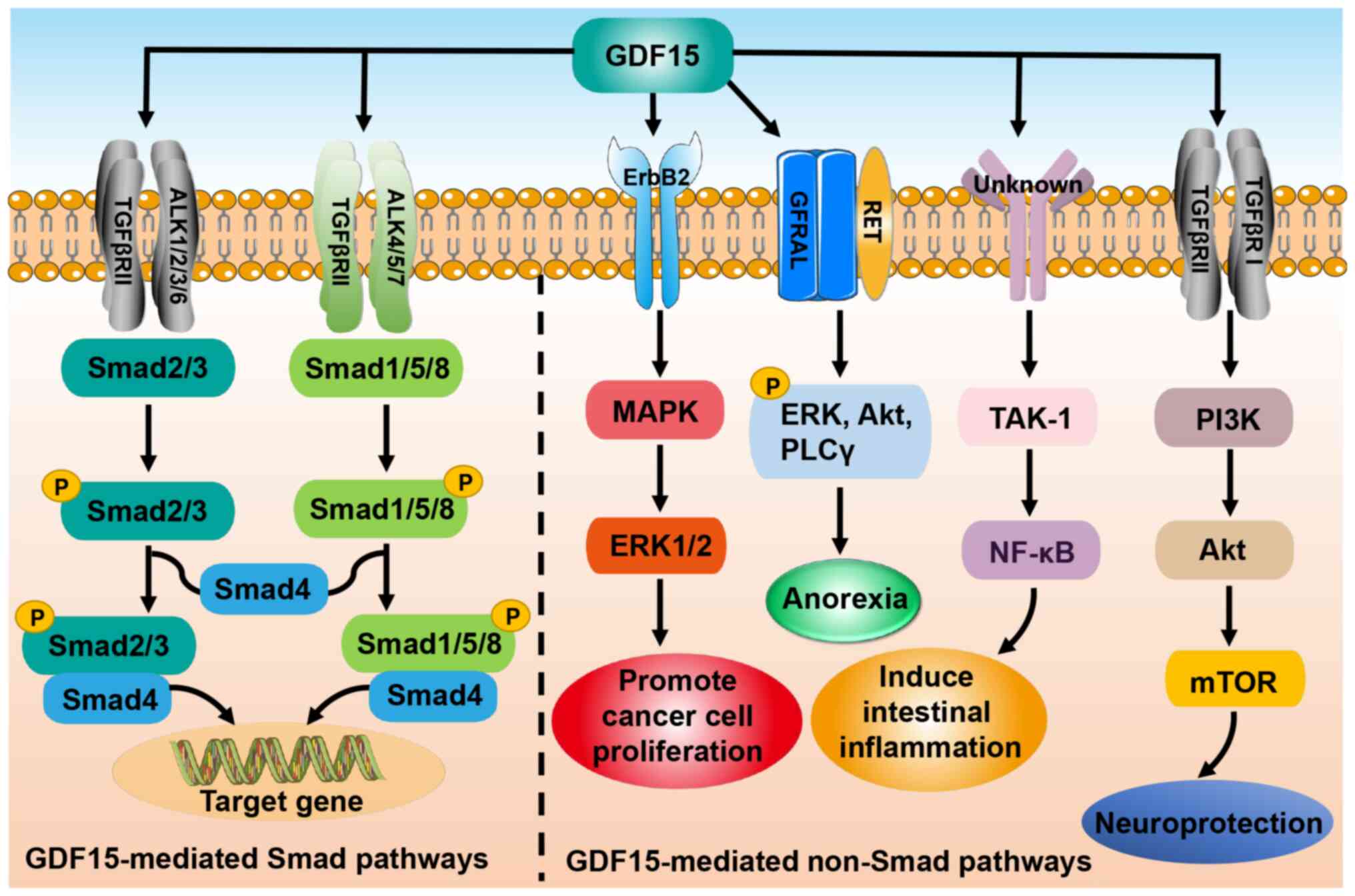
As a member of the TGF-β superfamily, GDF15 signals
through both Smad and non-Smad pathways. In the former pathway,
GDF15 binds to the type II TGF-β receptor (TGFβRII) and activates
the type I TGF-β receptor (TGFβRI), also known as activin
receptor-like kinase (47,48). Subsequently, TGFβRII and TGFβRI form
a heteromeric complex that induces the phosphorylation of Smad2/3
and Smad1/5/8. The phosphorylated Smad2/3 and Smad1/5/8 are then
able to bind to co-Smad (Smad4) and enter the nucleus to regulate
gene expression (47,48). In addition, GDF15 exerts its
biological functions through non-Smad-dependent pathways such as
phosphoinositide 3-kinase/Akt/mammalian target of rapamycin and
TGF-β-activated kinase-1 (TAK-1)/nuclear factor-κB (NF-κB), or
through other receptors such as glial cell-derived neurotrophic
factor receptor α-like (GFRAL) and epidermal growth factor receptor
2 (Fig. 3) (16,45,49,50).
A clinical study evaluated the association between
serum levels of GDF15 and anorexia in patients with cancer and
reported that GDF15 levels were significantly higher in anorexic
patients than in non-anorexic ones (51). This implies that high levels of
GDF15 in patients with cancer are associated with anorexia. There
is a body of evidence suggesting that GDF15 induces anorexia in
patients with CC (16,33,51).
Johnen et al (33)
originally described the role of GDF15 in CC and anorexia. They
observed that food intake was reduced in tumor-bearing mice that
were transgenically modified to overexpress GDF15. Lower food
intake indirectly resulted in fat loss, tibial and gastrocnemius
muscle atrophy and 28% weight loss in tumor-bearing mice (33). These effects were blocked by the
administration of a GDF15 monoclonal antibody and reproduced by the
injection of recombinant GDF15 (33). Johnen et al (33) further demonstrated that GDF15
promoted anorexia by interacting with TGFβRII to induce the
phosphorylation of extracellular signal-regulated kinase (ERK) 1/2
and transducer and activator of transcription 3 (STAT3) in the
hypothalamus. This process ultimately inhibited orexigenic
neuropeptide Y (NPY) neurons and stimulated anorexigenic
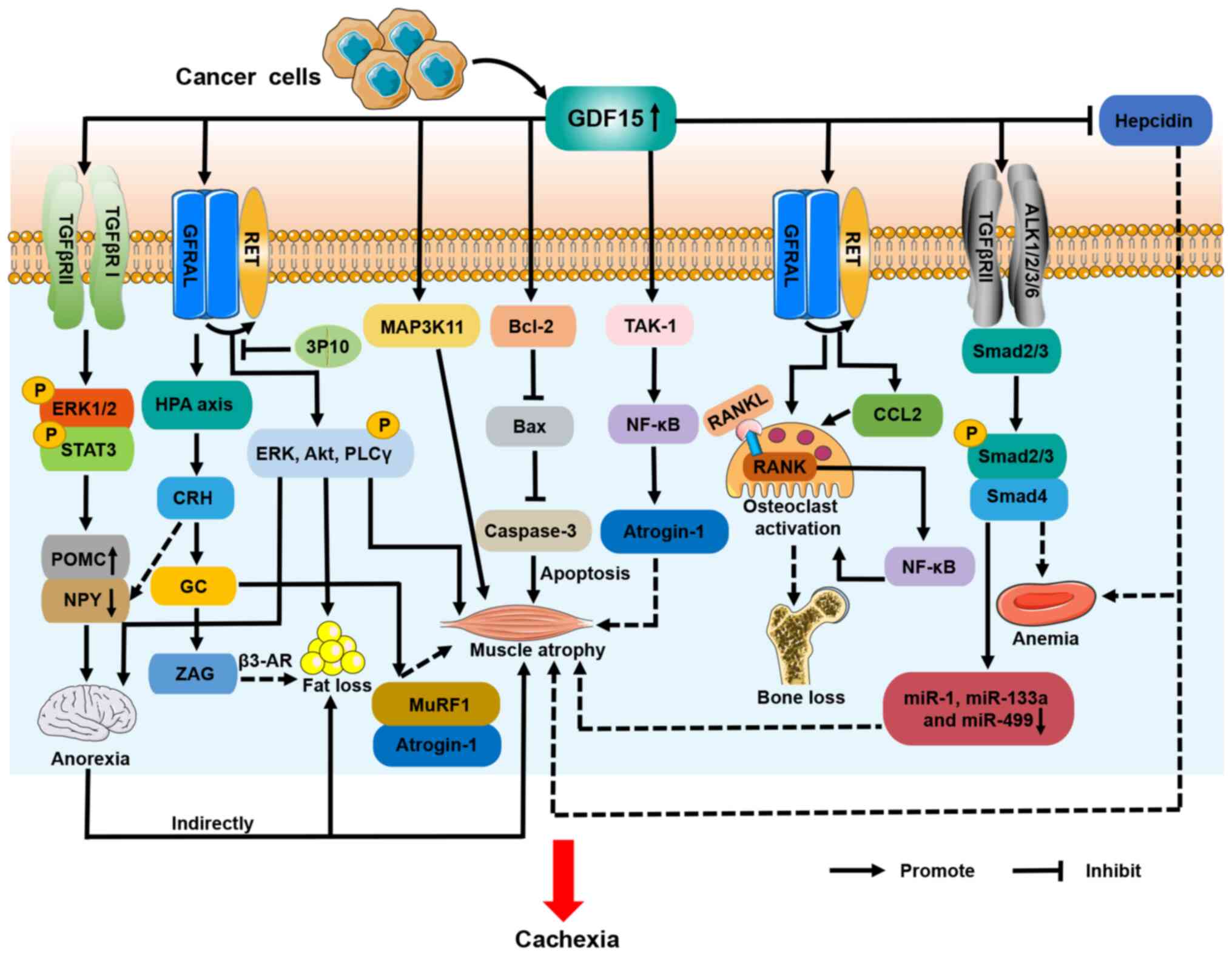
pro-opiomelanocortin (POMC) neurons (Fig. 4). GFRAL is a specific receptor for
GDF15 that is uniquely expressed in the hindbrain area postrema and
nucleus tractus solitarius (52).
The binding of GDF15 to GFRAL induces the activation of its
co-receptor Ret proto-oncogene (RET), which further promotes the
phosphorylation of ERK, Akt and phospholipase C γ, resulting in
decreased appetite in cachectic mice (Fig. 4) (16). The GDF15/GFRAL/RET pathway therefore
is considered a novel therapeutic target for anorexia and weight
loss in CC (16).
GDF15 may also influence CC anorexia through other
mechanisms. It has been shown that GDF15 activates the
hypothalamic-pituitary-adrenal (HPA) axis in a GFRAL-dependent
manner, leading to the secretion of corticotropin-releasing hormone
(CRH) and glucocorticoids (53).
CRH has been found to promote anorexia in CC, due to the inhibition
of NPY neurons (Fig. 4) (54).
As GDF15 is a myokine, its circulating levels are
negatively correlated with muscle mass in numerous diseases,
including COPD, intensive care unit-acquired weakness (ICUAW),
Crohn's disease, pulmonary hypertension (PH) and CC (30,55–58).
Muscle atrophy, including that of skeletal, chest, diaphragm and
cardiac muscle, is a hallmark of CC and a major cause of mortality
in patients with cancer (4,59).
One study indicated that GDF15 indirectly induced
muscle atrophy in CC via the inhibition of feeding centers in the
hypothalamus (33). However, in
other in vitro studies, GDF15 increased the expression of
muscle ring finger 1 (MuRF1) and muscle atrophy F-box
(MAFbx)/atrogin-1 and decreased myotube diameter (55,57).
Researchers also found that the expression of GDF15 and
MAFbx/atrogin-1 was elevated in the atrophied rectus abdominis
muscle of patients with ICUAW (55). The muscle-specific E3 ubiquitin
ligases MuRF1and MAFbx/atrogin-1 are primary factors that drive
muscle protein degradation in CC (2). These studies indicate that GDF15 may
contribute to muscle atrophy in CC, independently of food intake.
It has been reported that GDF15/GFRAL/RET directly induces muscle
atrophy in CC (16). In addition,
GDF15 has been shown to directly regulate muscle mass in CC through
other mechanisms. Lerner et al (30) demonstrated that the activation of
mitogen-activated protein kinase 11 (MAP3K11) by GDF15 promoted
gastrocnemius and flounder muscle reduction in a genetically
engineered mouse model of cachexia. These cachectic mice exhibited
weight gain and the retention of skeletal muscle when treated with
anti-GDF15 antibody (30).
Subsequently, Zhang et al (60) showed that elevated GDF15 levels in
the serum exosomes of mice with colon cancer caused the loss of
gastrocnemius muscle mass via the B cell lymphoma-2/caspase-3
pathway (Fig. 4).
Despite these studies, the mechanism by which GDF15
facilitates the loss of skeletal muscle mass in CC remains unclear.
It has been reported that GDF15 reduces muscle mass in patients
with PH and ICUAW through TAK-1/NF-κB/atrogin-1 signaling, and
downregulates the expression of various miRNAs, including miR-1,
miR-133a and miR-499, in muscle (55,57).
In one study, the upregulated phosphorylation of Smad2/3 was
observed in the muscles of patients with ICUAW, suggesting that
GDF15 may mediate muscle atrophy via the classical Smad pathway;
however, this was not observed in C2C12 murine cell-based myotubes
exposed to GDF15 in vitro (55). In addition, high levels of GDF15 are
known to inhibit the expression of hepcidin and lead to iron
overload (61). A recent study
revealed that iron overload induced muscle atrophy in patients with
gastric cancer and cachexia (62).
Moreover, GDF15/GFRAL signaling has been shown to activate the HPA
axis and trigger the release of glucocorticoids (53). Animal models of colon, Lewis lung
carcinoma (LLC) and pancreatic CC have shown that dysregulated
glucocorticoid levels increase the expression of MuRF1 and
MAFbx/atrogin-1 in skeletal muscle, driving the breakdown of muscle
protein (63). Additional studies
are warranted to identify whether GDF15 regulates muscle atrophy in
CC through these mechanisms (Fig.
4).
The molecular mechanisms of GDF15-induced fat loss
in CC have not been well studied, and pre-clinical models have
mainly been used. In one study, for example, mice with prostate CC
and high GDF15 expression lost all retroperitoneal fat and
exhibited a reduction of fat in the groin and epididymis of 54 and
89%, respectively, due to decreased food intake (33). In addition, in another study GDF15
increased the expression of differentiation and thermogenic genes
in brown adipocytes; the upregulated expression of iodothyronine
deiodinase 2, β3-adrenergic receptor (β3-AR), and very long chain
fatty acid-3 through GFRAL/RET signaling in cachectic mouse adipose
tissue led to a loss of fat and body weight (Fig. 4) (16). The therapeutic monoclonal antibody
3P10, which is a GFRAL antagonist and RET signaling inhibitor, has
been reported to reverse lipid hyperoxidation and prevent CC in
mice (16). Furthermore, ZAG is
known to be a lipid-mobilizing factor and has been demonstrated to
stimulate lipolysis via β3-AR during CC (66). Glucocorticoids have been shown to
increase ZAG expression and thereby promote lipolysis (67), suggesting that GDF15 may increase
adipose tissue depletion in CC via the HPA axis (Fig. 4).
Bone loss is caused by an imbalance between
bone-resorbing osteoclasts and bone-forming osteoblasts, which can
result in decreased bone mineral density (BMD), bone mass and bone
strength (68). Abnormal activation
of osteoclasts is the cause of various bone diseases, including
osteoporosis, rheumatoid arthritis, multiple myeloma and metastatic
cancers (69). A link between CC
and bone loss has been established in patients with CC and in
animal models (5,70–72).
Elevated levels of C-telopeptide of type I collagen (CTX-1) in
serum have been demonstrated to be indicative of increased bone
resorption and accelerated bone loss (73). Also, studies in humans have shown
that serum CTX-1 is significantly elevated in patients with
ovarian, lung and gastrointestinal CC compared with non-cachectic
controls (70–72). In a retrospective study, patients
with pancreatic CC were found to have significantly lower BMD than
control patients who underwent benign gallbladder surgery, and
those patients with pancreatic CC with osteopenia exhibited lower
median and 2-year postoperative survival times than those without
osteopenia (74). In an animal
model of LLC-induced CC, Yu et al (5) observed a reduction in bone trabecular
volume and BMD, and an increase in osteoclast activation. This
result was consistent with the findings of a previous study
regarding cachectic mice with colon cancer (71). The researchers further established
that the bone loss was induced via the Janus kinase/STAT3 pathway,
with the involvement of glucocorticoids (5). Surprisingly, bone loss preceded the
onset of muscle and fat loss in the LLC-induced model of cachexia
(5). These studies all demonstrate
that bone loss is closely associated with CC.
It has been demonstrated that GDF15 increases
osteoclast differentiation and inhibits osteoblast differentiation
in vivo and in vitro, leading to disturbances in bone
metabolism and bone loss (75,76).
Wakchoure et al (77)
injected a Du-145 human prostate cancer cell line overexpressing
GDF15 into the tibias of C57/B6 mice, and found that increased
GDF15 was associated with osteoclast activation and cachexia. This
result implies that GDF15 is involved in bone loss in CC, but the
biological mechanism underlying this has not yet been identified.
In a hypoxic mouse model induced by right femoral artery ligation,
the upregulation of GDF15 expression in osteoblasts was observed,
which stimulated the receptor activator of NF-κB ligand
(RANKL)-induced NF-κB signaling pathway and promoted osteoclast
activation in the mice, resulting in decreased bone mass (76). An anti-GDF15 antibody inhibited this
process and the associated bone loss (76). Moreover, Siddiqui et al
(78) demonstrated that elevated
GDF15 in prostate cancer upregulated the expression of C-C motif
chemokine ligand 2 and RANKL through the GFRAL/RET pathway, thereby
activating osteoclasts and leading to decreased bone mass. Other
studies have reported that the GDF15/GFRAL/RET receptor signaling
complex in the brain mediates anorexia in CC (16,79).
GFRAL and RET receptors have also been shown to be expressed in the
tibiae (Fig. 4) (78). These findings indicate that GDF15
may activate GFRAL and RET receptors expressed on osteocytes,
thereby inducing bone loss in CC.
A recent retrospective cohort study conducted across
multiple centers indicated that patients with CC had lower
hemoglobin levels than those without cachexia (80). The study demonstrated that patients
with CC developed anemia. However, the molecular mechanisms
underlying the CC-induced anemia remain unclear. A study in mice
with lung CC found that TGF-β activated the Smad2/3 signaling
pathway and inhibited hematopoietic stem cell and erythropoietic
cell production. The mice exhibited a significant reduction in
hemoglobin and erythrocyte levels in the peripheral blood (81). Additionally, GDF15 is produced by
erythroid precursor cells, and high levels of GDF15 are known to
inhibit effective erythropoiesis and the expression of hepcidin,
leading to anemia and iron overload (61). Hepcidin is a hepatic peptide hormone
that coordinates the systemic homeostasis of iron. Jiang et
al (82) showed that increased
serum levels of GDF15 in patients with cancer are associated with
downregulated hepcidin levels and cancer-related anemia. According
to these findings, we hypothesize that GDF15 may play a role in
CC-associated anemia (Fig. 4).
The TGF-β superfamily is a class of secreted peptide
cytokines with multiple members that are involved in the
development of CC (83). They are
categorized into four main subfamilies based on sequence
similarity, namely bone morphogenetic proteins/GDFs,
activins/inhibins/nodal, TGF-βs and others (84). GDF8, which is also known as
myostatin, and activin A have been reported to bind to activin
receptor type 2B on skeletal muscle to promote the phosphorylation
of Smad2/3 and inhibit Akt phosphorylation, leading to activation
of the ubiquitin-proteasome system that eventually leads to muscle
atrophy (54). Greco et al
(85) demonstrated that blocking
TGF-β reduces skeletal muscle catabolism and weight loss in mouse
models of pancreatic CC, and decreases phosphorylated Smad2/3
signaling in muscle tissues. This suggests that TGF-β is also
involved in skeletal muscle atrophy in CC, via activation of the
Smad2/3 signaling pathway. Another member of the TGF-β superfamily,
GDF11, is highly homologous to GDF8 (86). GDF11 directly increases the
expression of MuRF1 and MAFbx/atrogin-1 in skeletal muscle, leading
to muscle atrophy and cachexia (87). In addition, Zimmers et al
(88) demonstrated that elevated
circulating levels of GDF11 are associated with cardiac
atrophy.
Several clinical trials have been conducted to
investigate the role of GDF15 antibodies in CC. Ponsegromab is a
human monoclonal antibody that targets GDF15 (91). It binds to GDF15 and prevents it
from binding to GFRAL, thereby blocking GDF15/GFRAL-mediated
signaling. A recent phase-1b clinical trial (NCT04299048) conducted
by Pfizer evaluated the safety and efficacy of ponsegromab in
patients with non-small cell lung, colorectal and pancreatic
cancers accompanied by cachexia. In addition to standard anticancer
treatments, patients with cachexia received ponsegromab
subcutaneously every 3 weeks for a total of 12 weeks (92). The results showed that the median
circulating GDF15 levels in patients treated with ponsegromab were
lower than the those in healthy controls (92). Moreover, ponsegromab treatment
significantly increased weight, physical activity and appetite in
patients with CC (92). Clinical
studies of ponsegromab in CC remain ongoing and are currently in
phase II (91). Table II summarizes clinical trials
regarding GDF15 that have been conducted in patients with cancer
and CC (https://clinicaltrials.gov/). The
information obtained from these studies may ultimately enable
patients with CC to benefit from these treatments, and also guide
future studies.
Animal experiments further illustrate that the
inhibition of GDF15 is an effective strategy for the treatment of
CC (30,33,93).
In several animal models, including prostate, ovarian, colon and
breast cancer, leukemia and fibrosarcoma models, cachectic mice
treated with GDF15 antibodies exhibited increased food intake,
muscle mass and adipose tissue, ultimately leading to weight gain
(30,33,93).
These studies illustrate that targeting GDF15 alleviates CC via the
prevention of anorexia, and the loss of muscle and fat. In
addition, Hinoi et al (76)
demonstrated that an anti-GDF15 antibody inhibited bone loss and
osteoclast activation in the tibias of hypoxic mice. The inhibition
of GDF15 has also been observed to inhibit ineffective
erythropoiesis and improve anemia in patients with cancer (94). Thus, GDF15 may be a potential target
for the treatment of bone loss and anemia in CC. Moreover, one
study revealed that the combination of an anti-GDF15 antibody with
the angiogenesis inhibitor tivozanib significantly increased body
weight and survival in mice with CC, when compared with tivozanib
alone (30). This implies that the
inhibition of tumor growth and amelioration of CC may prolong the
lifespans of patients with this condition. The strategy represents
a new therapeutic prospect, but requires translation into clinical
studies.
In the present review, the roles and mechanisms of
GDF15 in CC are summarized. Studies have indicated that the effects
of GDF15 on CC are associated with inhibition of the feeding
center. However, since GFRAL was identified as an exclusive
receptor for GDF15, further studies have demonstrated that GDF15
also induces metabolic effects that are independent of food-intake
behavior. It has been established that GDF15 interacts with GFRAL
in the brainstem to suppress appetite, promote fat loss and reduce
muscle mass in CC. GDF15 also directly promotes muscle atrophy in
CC via the apoptotic pathway and MAP3K11, but the receptors
involved in these processes are currently unknown. It also appears
that GDF15 may have a role in bone loss and anemia in CC. However,
it remains uncertain whether other GDF15 receptors or pathways,
such as the GDF15/GFRAL/HPA axis, promote the development of CC.
Therefore, the roles and potential molecular mechanisms of GDF15 in
CC, particularly its receptors and downstream signaling pathways,
require further investigation. In addition, GDF15 is a potential
therapeutic target for CC, and clinical trials are being conducted
to study the safety and therapeutic value of GDF15 antibodies in
patients with CC. Notably, since CC is attributed to complex
interactions between tumor cells and the host, it may be necessary
to combine GDF15-targeting antibodies with anticancer therapies
such as immunotherapy or targeted therapy to improve the outcomes
of patients with CC.
Not applicable.
Funding: No funding was received.
Not applicable.
TL and JZ were responsible for drafting and revising
the manuscript, and creating the figures. FD and LM contributed to
manuscript conception. Data authentication is not applicable. All
authors have read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Evans WJ, Morley JE, Argilés J, Bales C,
Baracos V, Guttridge D, Jatoi A, Kalantar-Zadeh K, Lochs H,
Mantovani G, et al: Cachexia: A new definition. Clin Nutr.
27:793–799. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Baazim H, Antonio-Herrera L and Bergthaler
A: The interplay of immunology and cachexia in infection and
cancer. Nat Rev Immunol. 22:309–321. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fearon K, Strasser F, Anker SD, Bosaeus I,
Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N,
Mantovani G, et al: Definition and classification of cancer
cachexia: An international consensus. Lancet Oncol. 12:489–495.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Porporato PE: Understanding cachexia as a
cancer metabolism syndrome. Oncogenesis. 5:e2002016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yu J, Choi S, Park A, Do J, Nam D, Kim Y,
Noh J, Lee KY, Maeng CH and Park KS: Bone marrow homeostasis is
impaired via JAK/STAT and glucocorticoid signaling in cancer
cachexia model. Cancers (Basel). 13:10592021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Argilés JM, Stemmler B, López-Soriano FJ
and Busquets S: Inter-tissue communication in cancer cachexia. Nat
Rev Endocrinol. 15:9–20. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Baracos VE, Martin L, Korc M, Guttridge DC
and Fearon KCH: Cancer-associated cachexia. Nat Rev Dis Primers.
4:171052018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gupta A, Nshuti L, Grewal US, Sedhom R,
Check DK, Parsons HM, Blaes AH, Virnig BA, Lustberg MB, Subbiah IM,
et al: Financial burden of drugs prescribed for cancer-associated
symptoms. JCO Oncol Pract. 18:140–147. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu CA, Zhang Q, Ruan GT, Shen LY, Xie HL,
Liu T, Tang M, Zhang X, Yang M, Hu CL, et al: Novel diagnostic and
prognostic tools for lung cancer cachexia: Based on nutritional and
inflammatory status. Front Oncol. 12:8907452022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Malla J, Zahra A, Venugopal S, Selvamani
TY, Shoukrie SI, Selvaraj R, Dhanoa RK, Hamouda RK and Mostafa J:
What role do inflammatory cytokines play in cancer cachexia?
Cureus. 14:e267982022.PubMed/NCBI
|
|
11
|
Zeng R, Tong C and Xiong X: The molecular
basis and therapeutic potential of leukemia inhibitory factor in
cancer cachexia. Cancers (Basel). 14:29552022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lu SW, Pan HC, Hsu YH, Chang KC, Wu LW,
Chen WY and Chang MS: IL-20 antagonist suppresses PD-L1 expression
and prolongs survival in pancreatic cancer models. Nat Commun.
11:46112020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Di Girolamo D and Tajbakhsh S:
Pathological features of tissues and cell populations during cancer
cachexia. Cell Regen. 11:152022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Callaway CS, Delitto AE, Patel R, Nosacka
RL, D'Lugos AC, Delitto D, Deyhle MR, Trevino JG, Judge SM and
Judge AR: IL-8 released from human pancreatic cancer and
tumor-associated stromal cells signals through a CXCR2-ERK1/2 axis
to induce muscle atrophy. Cancers (Basel). 11:18632019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xiong H, Ye J, Xie K, Hu W, Xu N and Yang
H: Exosomal IL-8 derived from Lung Cancer and Colon Cancer cells
induced adipocyte atrophy via NF-κB signaling pathway. Lipids
Health Dis. 21:1472022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Suriben R, Chen M, Higbee J, Oeffinger J,
Ventura R, Li B, Mondal K, Gao Z, Ayupova D, Taskar P, et al:
Antibody-mediated inhibition of GDF15-GFRAL activity reverses
cancer cachexia in mice. Nat Med. 26:1264–1270. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Belloum Y, Rannou-Bekono F and Favier FB:
Cancer-induced cardiac cachexia: Pathogenesis and impact of
physical activity (Review). Oncol Rep. 37:2543–2552. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nissinen TA, Hentilä J, Penna F, Lampinen
A, Lautaoja JH, Fachada V, Holopainen T, Ritvos O, Kivelä R and
Hulmi JJ: Treating cachexia using soluble ACVR2B improves survival,
alters mTOR localization, and attenuates liver and spleen
responses. J Cachexia Sarcopenia Muscle. 9:514–529. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Thibaut MM, Sboarina M, Roumain M, Pötgens
SA, Neyrinck AM, Destrée F, Gillard J, Leclercq IA, Dachy G,
Demoulin JB, et al: Inflammation-induced cholestasis in cancer
cachexia. J Cachexia Sarcopenia Muscle. 12:70–90. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Peyta L, Jarnouen K, Pinault M, Coulouarn
C, Guimaraes C, Goupille C, de Barros JP, Chevalier S, Dumas JF,
Maillot F, et al: Regulation of hepatic cardiolipin metabolism by
TNFα: Implication in cancer cachexia. Biochim Biophys Acta.
1851:1490–1500. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Patel HJ and Patel BM: TNF-α and cancer
cachexia: Molecular insights and clinical implications. Life Sci.
170:56–63. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Black K, Garrett IR and Mundy GR: Chinese
hamster ovarian cells transfected with the murine interleukin-6
gene cause hypercalcemia as well as cachexia, leukocytosis and
thrombocytosis in tumor-bearing nude mice. Endocrinology.
128:2657–2659. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bindels LB, Neyrinck AM, Loumaye A, Catry
E, Walgrave H, Cherbuy C, Leclercq S, Van Hul M, Plovier H,
Pachikian B, et al: Increased gut permeability in cancer cachexia:
mechanisms and clinical relevance. Oncotarget. 9:18224–18238. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
White JP, Puppa MJ, Narsale A and Carson
JA: Characterization of the male ApcMin/+ mouse as a hypogonadism
model related to cancer cachexia. Biol Open. 2:1346–1353. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tracey KJ, Wei H, Manogue KR, Fong Y,
Hesse DG, Nguyen HT, Kuo GC, Beutler B, Cotran RS, Cerami A, et al:
Cachectin/tumor necrosis factor induces cachexia, anemia, and
inflammation. J Exp Med. 167:1211–1227. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Johns N, Stretch C, Tan BH, Solheim TS,
Sørhaug S, Stephens NA, Gioulbasanis I, Skipworth RJ, Deans DA,
Vigano A, et al: New genetic signatures associated with cancer
cachexia as defined by low skeletal muscle index and weight loss. J
Cachexia Sarcopenia Muscle. 8:122–130. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhong X, Narasimhan A, Silverman LM, Young
AR, Shahda S, Liu S, Wan J, Liu Y, Koniaris LG and Zimmers TA: Sex
specificity of pancreatic cancer cachexia phenotypes, mechanisms,
and treatment in mice and humans: Role of Activin. J Cachexia
Sarcopenia Muscle. 13:2146–2161. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Avancini A, Trestini I, Tregnago D, Lanza
M, Menis J, Belluomini L, Milella M and Pilotto S: A multimodal
approach to cancer-related cachexia: from theory to practice.
Expert Rev Anticancer Ther. 21:819–826. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lockhart SM, Saudek V and O'Rahilly S:
GDF15: A hormone conveying somatic distress to the brain. Endocr
Rev. 41:bnaa0072020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lerner L, Tao J, Liu Q, Nicoletti R, Feng
B, Krieger B, Mazsa E, Siddiquee Z, Wang R, Huang L, et al:
MAP3K11/GDF15 axis is a critical driver of cancer cachexia. J
Cachexia Sarcopenia Muscle. 7:467–482. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Suzuki H, Mitsunaga S, Ikeda M, Aoyama T,
Yoshizawa K, Yoshimatsu H, Kawai N, Masuda M, Miura T and Ochiai A:
Clinical and tumor characteristics of patients with high serum
levels of growth differentiation factor 15 in advanced pancreatic
cancer. Cancers (Basel). 13:48422021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lerner L, Hayes TG, Tao N, Krieger B, Feng
B, Wu Z, Nicoletti R, Chiu MI, Gyuris J and Garcia JM: Plasma
growth differentiation factor 15 is associated with weight loss and
mortality in cancer patients. J Cachexia Sarcopenia Muscle.
6:317–324. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Johnen H, Lin S, Kuffner T, Brown DA, Tsai
VW, Bauskin AR, Wu L, Pankhurst G, Jiang L, Junankar S, et al:
Tumor-induced anorexia and weight loss are mediated by the TGF-beta
superfamily cytokine MIC-1. Nat Med. 13:1333–1340. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li P, Lv H, Zhang B, Duan R, Zhang X, Lin
P, Song C and Liu Y: Growth differentiation factor 15 protects
SH-SY5Y cells from rotenone-induced toxicity by suppressing
mitochondrial apoptosis. Front Aging Neurosci. 14:8695582022.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Asrih M, Wei S, Nguyen TT, Yi HS, Ryu D
and Gariani K: Overview of growth differentiation factor 15 in
metabolic syndrome. J Cell Mol Med. 27:1157–1167. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wan Y and Fu J: GDF15 as a key disease
target and biomarker: Linking chronic lung diseases and ageing. Mol
Cell Biochem. Apr 24–2023.(Epub ahead of print). View Article : Google Scholar
|
|
37
|
Assadi A, Zahabi A and Hart RA: GDF15, an
update of the physiological and pathological roles it plays: A
review. Pflugers Arch. 472:1535–1546. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Johann K, Kleinert M and Klaus S: The Role
of GDF15 as a Myomitokine. Cells. 10:29902021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wischhusen J, Melero I and Fridman WH:
Growth/Differentiation Factor-15 (GDF-15): From biomarker to novel
targetable immune checkpoint. Front Immunol. 11:9512020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tsai VW, Brown DA and Breit SN: Targeting
the divergent TGFβ superfamily cytokine MIC-1/GDF15 for therapy of
anorexia/cachexia syndromes. Curr Opin Support Palliat Care.
12:404–409. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Siddiqui JA, Pothuraju R, Khan P, Sharma
G, Muniyan S, Seshacharyulu P, Jain M, Nasser MW and Batra SK:
Pathophysiological role of growth differentiation factor 15 (GDF15)
in obesity, cancer, and cachexia. Cytokine Growth Factor Rev.
64:71–83. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Niu Y, Zhang W, Shi J, Liu Y, Zhang H, Lin
N, Li X, Qin L, Yang Z and Su Q: The relationship between
circulating growth differentiation factor 15 levels and diabetic
retinopathy in patients with type 2 diabetes. Front Endocrinol
(Lausanne). 12:6273952021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tsui KH, Hsu SY, Chung LC, Lin YH, Feng
TH, Lee TY, Chang PL and Juang HH: Growth differentiation
factor-15: A p53- and demethylation-upregulating gene represses
cell proliferation, invasion, and tumorigenesis in bladder
carcinoma cells. Sci Rep. 5:128702015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Joo M, Kim D, Lee MW, Lee HJ and Kim JM:
GDF15 promotes cell growth, migration, and invasion in gastric
cancer by inducing STAT3 activation. Int J Mol Sci. 24:29252023.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li S, Ma YM, Zheng PS and Zhang P: GDF15
promotes the proliferation of cervical cancer cells by
phosphorylating AKT1 and Erk1/2 through the receptor ErbB2. J Exp
Clin Cancer Res. 37:802018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Spanopoulou A and Gkretsi V: Growth
differentiation factor 15 (GDF15) in cancer cell metastasis: From
the cells to the patients. Clin Exp Metastasis. 37:451–464. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Rochette L, Dogon G, Zeller M, Cottin Y
and Vergely C: GDF15 and cardiac cells: Current concepts and new
insights. Int J Mol Sci. 22:88892021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li L, Zhang R, Yang H, Zhang D, Liu J, Li
J and Guo B: GDF15 knockdown suppresses cervical cancer cell
migration in vitro through the TGF-β/Smad2/3/Snail1 pathway. FEBS
Open Bio. 10:2750–2760. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wang CY, Huang AQ, Zhou MH and Mei YA:
GDF15 regulates Kv2.1-mediated outward K+ current through the
Akt/mTOR signalling pathway in rat cerebellar granule cells.
Biochem J. 460:35–47. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Park SH, Yu M, Kim J and Moon Y: C/EBP
homologous protein promotes NSAID-activated gene 1-linked
pro-inflammatory signals and enterocyte invasion by
enteropathogenic Escherichia coli. Microbes Infect. 19:110–121.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Molfino A, Amabile MI, Imbimbo G, Rizzo V,
Pediconi F, Catalano C, Emiliani A, Belli R, Ramaccini C, Parisi C,
et al: Association between growth differentiation factor-15
(GDF-15) serum levels, anorexia and low muscle mass among cancer
patients. Cancers (Basel). 13:992020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sabatini PV, Frikke-Schmidt H, Arthurs J,
Gordian D, Patel A, Rupp AC, Adams JM, Wang J, Beck Jørgensen S,
Olson DP, et al: GFRAL-expressing neurons suppress food intake via
aversive pathways. Proc Natl Acad Sci USA. 118:e20213571182021.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cimino I, Kim H, Tung YCL, Pedersen K,
Rimmington D, Tadross JA, Kohnke SN, Neves-Costa A, Barros A,
Joaquim S, et al: Activation of the hypothalamic-pituitary-adrenal
axis by exogenous and endogenous GDF15. Proc Natl Acad Sci USA.
118:e21068681182021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Setiawan T, Sari IN, Wijaya YT, Julianto
NM, Muhammad JA, Lee H, Chae JH and Kwon HY: Cancer cachexia:
Molecular mechanisms and treatment strategies. J Hematol Oncol.
16:542023. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Bloch SA, Lee JY, Syburra T, Rosendahl U,
Griffiths MJ, Kemp PR and Polkey MI: Increased expression of GDF-15
may mediate ICU-acquired weakness by down-regulating muscle
microRNAs. Thorax. 70:219–228. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yamamoto H, Takeshima F, Haraguchi M,
Akazawa Y, Matsushima K, Kitayama M, Ogihara K, Tabuchi M,
Hashiguchi K, Yamaguchi N, et al: High serum concentrations of
growth differentiation factor-15 and their association with Crohn's
disease and a low skeletal muscle index. Sci Rep. 12:65912022.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Garfield BE, Crosby A, Shao D, Yang P,
Read C, Sawiak S, Moore S, Parfitt L, Harries C, Rice M, et al:
Growth/differentiation factor 15 causes TGFβ-activated kinase
1-dependent muscle atrophy in pulmonary arterial hypertension.
Thorax. 74:164–176. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Deng M, Bian Y, Zhang Q, Zhou X and Hou G:
Growth differentiation factor-15 as a biomarker for sarcopenia in
patients with chronic obstructive pulmonary disease. Front Nutr.
9:8970972022. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Song M, Zhang Q, Tang M, Zhang X, Ruan G,
Zhang X, Zhang K, Ge Y, Yang M, Li Q, et al: Associations of low
hand grip strength with 1 year mortality of cancer cachexia: A
multicentre observational study. J Cachexia Sarcopenia Muscle.
12:1489–1500. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhang W, Sun W, Gu X, Miao C, Feng L, Shen
Q, Liu X and Zhang X: GDF-15 in tumor-derived exosomes promotes
muscle atrophy via Bcl-2/caspase-3 pathway. Cell Death Discov.
8:1622022. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Tanno T, Bhanu NV, Oneal PA, Goh SH,
Staker P, Lee YT, Moroney JW, Reed CH, Luban NL, Wang RH, et al:
High levels of GDF15 in thalassemia suppress expression of the iron
regulatory protein hepcidin. Nat Med. 13:1096–1101. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhou D, Zhang Y, Mamtawla G, Wan S, Gao X,
Zhang L, Li G and Wang X: Iron overload is related to muscle
wasting in patients with cachexia of gastric cancer: using
quantitative proteome analysis. Med Oncol. 37:1132020. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Martin A, Castells J, Allibert V, Emerit
A, Zolotoff C, Cardot-Ruffino V, Gallot YS, Vernus B, Chauvet V,
Bartholin L, et al: Hypothalamic-pituitary-adrenal axis activation
and glucocorticoid-responsive gene expression in skeletal muscle
and liver of Apc mice. J Cachexia Sarcopenia Muscle. 13:1686–1703.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Laurens C, Parmar A, Murphy E, Carper D,
Lair B, Maes P, Vion J, Boulet N, Fontaine C, Marquès M, et al:
Growth and differentiation factor 15 is secreted by skeletal muscle
during exercise and promotes lipolysis in humans. JCI Insight.
5:e1318702020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Fouladiun M, Körner U, Bosaeus I, Daneryd
P, Hyltander A and Lundholm KG: Body composition and time course
changes in regional distribution of fat and lean tissue in
unselected cancer patients on palliative care-correlations with
food intake, metabolism, exercise capacity, and hormones. Cancer.
103:2189–2198. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Elattar S, Dimri M and Satyanarayana A:
The tumor secretory factor ZAG promotes white adipose tissue
browning and energy wasting. FASEB J. 32:4727–4743. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Weber BZC, Arabaci DH and Kir S: Metabolic
reprogramming in adipose tissue during cancer cachexia. Front
Oncol. 12:8483942022. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Plotkin LI, Sanz N and Brun LR: Messages
from the Mineral: How bone cells communicate with other tissues.
Calcif Tissue Int. 113:39–47. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhang Y, Wang H, Zhu G, Qian A and Chen W:
F2r negatively regulates osteoclastogenesis through inhibiting the
Akt and NFκB signaling pathways. Int J Biol Sci. 16:1629–1639.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zwickl H, Zwickl-Traxler E, Haushofer A,
Seier J, Podar K, Weber M, Hackner K, Jacobi N, Pecherstorfer M and
Vallet S: Effect of cachexia on bone turnover in cancer patients: A
case-control study. BMC Cancer. 21:7442021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Bonetto A, Kays JK, Parker VA, Matthews
RR, Barreto R, Puppa MJ, Kang KS, Carson JA, Guise TA, Mohammad KS,
et al: Differential bone loss in mouse models of colon cancer
cachexia. Front Physiol. 7:6792016.PubMed/NCBI
|
|
72
|
Pin F, Jones AJ, Huot JR, Narasimhan A,
Zimmers TA, Bonewald LF and Bonetto A: RANKL blockade reduces
cachexia and bone loss induced by non-metastatic ovarian cancer in
mice. J Bone Miner Res. 37:381–396. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Adesina OO, Jenkins IC, Wu QV, Fung EB,
Narla RR, Lipkin EW, Mahajan K, Konkle BA and Kruse-Jarres R:
Urinary cross-linked carboxyterminal telopeptide, a bone resorption
marker, decreases after vaso-occlusive crises in adults with sickle
cell disease. Blood Cells Mol Dis. 80:1023692020. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Cameron ME, Underwood PW, Williams IE,
George TJ, Judge SM, Yarrow JF, Trevino JG and Judge AR: Osteopenia
is associated with wasting in pancreatic adenocarcinoma and
predicts survival after surgery. Cancer Med. 11:50–60. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Westhrin M, Moen SH, Holien T, Mylin AK,
Heickendorff L, Olsen OE, Sundan A, Turesson I, Gimsing P, Waage A
and Standal T: Growth differentiation factor 15 (GDF15) promotes
osteoclast differentiation and inhibits osteoblast differentiation
and high serum GDF15 levels are associated with multiple myeloma
bone disease. Haematologica. 100:e511–e514. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Hinoi E, Ochi H, Takarada T, Nakatani E,
Iezaki T, Nakajima H, Fujita H, Takahata Y, Hidano S, Kobayashi T,
et al: Positive regulation of osteoclastic differentiation by
growth differentiation factor 15 upregulated in osteocytic cells
under hypoxia. J Bone Miner Res. 27:938–949. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Wakchoure S, Swain TM, Hentunen TA,
Bauskin AR, Brown DA, Breit SN, Vuopala KS, Harris KW and Selander
KS: Expression of macrophage inhibitory cytokine-1 in prostate
cancer bone metastases induces osteoclast activation and weight
loss. Prostate. 69:652–661. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Siddiqui JA, Seshacharyulu P, Muniyan S,
Pothuraju R, Khan P, Vengoji R, Chaudhary S, Maurya SK, Lele SM,
Jain M, et al: GDF15 promotes prostate cancer bone metastasis and
colonization through osteoblastic CCL2 and RANKL activation. Bone
Res. 10:62022. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Ahmed DS, Isnard S, Lin J, Routy B and
Routy JP: GDF15/GFRAL pathway as a metabolic signature for cachexia
in patients with cancer. J Cancer. 12:1125–1132. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zhang XW, Zhang Q, Song MM, Zhang KP,
Zhang X, Ruan GT, Yang M, Ge YZ, Tang M, Li XR, et al: The
prognostic effect of hemoglobin on patients with cancer cachexia: A
multicenter retrospective cohort study. Support Care Cancer.
30:875–885. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Wang B, Wang Y, Chen H, Yao S, Lai X, Qiu
Y, Cai J, Huang Y, Wei X, Guan Y, et al: Inhibition of TGFβ
improves hematopoietic stem cell niche and ameliorates
cancer-related anemia. Stem Cell Res Ther. 12:652021. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Jiang F, Yu WJ, Wang XH, Tang YT, Guo L
and Jiao XY: Regulation of hepcidin through GDF-15 in
cancer-related anemia. Clin Chim Acta. 428:14–19. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Balsano R, Kruize Z, Lunardi M,
Comandatore A, Barone M, Cavazzoni A, Re Cecconi AD, Morelli L,
Wilmink H, Tiseo M, et al: Transforming growth factor-beta
signaling in cancer-induced cachexia: From molecular pathways to
the clinics. Cells. 11:26712022. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Liu S, Ren J and Ten Dijke P: Targeting
TGFβ signal transduction for cancer therapy. Signal Transduct
Target Ther. 6:82021. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Greco SH, Tomkötter L, Vahle AK, Rokosh R,
Avanzi A, Mahmood SK, Deutsch M, Alothman S, Alqunaibit D, Ochi A,
et al: TGF-β blockade reduces mortality and metabolic changes in a
validated murine model of pancreatic cancer cachexia. PLoS One.
10:e01327862015. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wang Z, Jiang P, Liu F, Du X, Ma L, Ye S,
Cao H, Sun P, Su N, Lin F, et al: GDF11 Regulates PC12 neural stem
cells via ALK5-Dependent PI3K-Akt signaling pathway. Int J Mol Sci.
23:122792022. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Jones JE, Cadena SM, Gong C, Wang X, Chen
Z, Wang SX, Vickers C, Chen H, Lach-Trifilieff E, Hadcock JR and
Glass DJ: Supraphysiologic administration of GDF11 induces cachexia
in part by upregulating GDF15. Cell Rep. 22:1522–1530. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Zimmers TA, Jiang Y, Wang M, Liang TW,
Rupert JE, Au ED, Marino FE, Couch ME and Koniaris LG: Exogenous
GDF11 induces cardiac and skeletal muscle dysfunction and wasting.
Basic Res Cardiol. 112:482017. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Ebner N, Anker SD and von Haehling S:
Recent developments in the field of cachexia, sarcopenia, and
muscle wasting: highlights from the 11th Cachexia Conference. J
Cachexia Sarcopenia Muscle. 10:218–225. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Ebner N, Anker SD and von Haehling S:
Recent developments in the field of cachexia, sarcopenia, and
muscle wasting: Highlights from the 12th Cachexia Conference. J
Cachexia Sarcopenia Muscle. 11:274–285. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
da Fonseca GWP, Sato R, de Nazaré Nunes
Alves MJ and von Haehling S: Current advancements in
pharmacotherapy for cancer cachexia. Expert Opin Pharmacother.
24:629–639. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Crawford J, Calle RA, Collins SM, Weng Y,
Lubaczewski SL, Buckeridge C, Wang EQ, Harrington MA, Tarachandani
A, Rossulek MI, et al: CT108/16-First-in-patient study of the
GDF-15 inhibitor ponsegromab in patients with cancer and cachexia:
Safety, tolerability, and exploratory measures of efficacy.
Proceedings of the 114th Annual Meeting of the American Association
for Cancer Research. 14–19. 2023.https://www.abstractsonline.com/pp8/#!/10828/presentation/10304
|
|
93
|
Kim-Muller JY, Song L, LaCarubba Paulhus
B, Pashos E, Li X, Rinaldi A, Joaquim S, Stansfield JC, Zhang J,
Robertson A, et al: GDF15 neutralization restores muscle function
and physical performance in a mouse model of cancer cachexia. Cell
Rep. 42:1119472023. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Vignjević Petrinović S, Jauković A,
Milošević M, Bugarski D and Budeč M: Targeting stress
erythropoiesis pathways in cancer. Front Physiol. 13:8440422022.
View Article : Google Scholar : PubMed/NCBI
|