Introduction
The lung cancer incidence rate (14%) and mortality
rate (18%) remain high worldwide (1), posing an extreme threat to human
health. Chemotherapy plays an irreplaceable role in the treatment
of lung cancer. Most chemotherapeutic agents kill cells by
interfering with DNA replication or inducing DNA damage, leading to
apoptosis (2). Gemcitabine, a
deoxycytidine analog, has been used as the first-line chemotherapy
drug for lung cancer (3,4). The most important mechanism of
gemcitabine is inhibition of DNA synthesis. Gemcitabine is not only
a cell cycle-specific antimetabolic drug that acts mainly during
the DNA synthesis (S) phase of the cell cycle but also blocks the
G1/S transition. These effects are detrimental to cell
cycle progression. Therefore, gemcitabine can prevent DNA
replication (5–7). Unfortunately, acquired resistance
limits the efficacy of gemcitabine. Factors contributing to
resistance include alterations in gemcitabine uptake and
metabolism, gene regulation and apoptotic pathways (8,9).
Therefore, studying the mechanisms of lung cancer drug resistance
and reversing this resistance are urgent issues to be solved in the
treatment of lung cancer.
p21, also called p21Waf1/Cip1 or
cyclin-dependent kinase inhibitor 1A (CDKN1A), plays an important
role in regulating cell proliferation and maintaining genomic
stability (10). It can induce
G1 arrest and cell senescence in response to various
stimuli, including oncogene-induced proliferation (11,12). A
number of studies have reported that p21 plays dual roles in
maintaining the stability of the genome and regulating cell
proliferation and drug resistance (11,13).
Previous studies have suggested that upregulation of
p21 expression could increase the sensitivity of lung cancer cells
to cisplatin. For instance, Wang et al showed that
overexpression of p21 in drug-resistant cells blocked the
G1/S transition. In addition, it reduced the expression
of Bcl-2 and Bcl-XL while increasing the expression of Bax and Bak,
which may promote apoptosis and reverse cisplatin resistance
(14). Liu et al also
revealed that upregulation of p21 expression induced cell cycle
arrest in lung cancer cells, inhibited cell proliferation and
increased the sensitivity of lung adenocarcinoma cells to cisplatin
(15). However, some studies
arrived at the opposite conclusions. In these studies, high
expression of p21 was considered to promote drug resistance. For
example, Zhao et al demonstrated that overexpression of p21
induced G1 arrest and inhibited DNA synthesis. However,
at the same time, DNA repair in these cells was enhanced, and the
DNA damage induced by cisplatin was mitigated. These factors
resulted in resistance to cisplatin (16). In addition, Guo et al
revealed that the expression of p21 was increased in A549 cells
under hypoxic conditions, resulting in cell cycle arrest at the
G1/G0 phase boundary; however, cisplatin
resistance in these A549 cells was simultaneously increased. One
potential reason for this phenomenon is that cell cycle arrest led
to an increase in the number of cells in a non-proliferative state
and reduced the effect of cisplatin on these cells (17).
Given the mechanism by which gemcitabine inhibits
cell proliferation and the role of p21 in the cell cycle, in the
present study, gemcitabine-resistant A549 (A549/G+)
cells were established to confirm that p21 plays a critical role in
the drug resistance of lung cancer cells by altering cell cycle
progression and apoptosis. In addition, p21 knockdown was
demonstrated to increase DNA damage and apoptosis induced by
gemcitabine, providing a therapeutic strategy to overcome drug
resistance.
Materials and methods
Cell culture
A549 is a non-small cell lung cancer cell line and
was purchased from the research facilities of Peking Union Medical
College (Beijing, China). The A549/G+ cell line was
induced and established by the authors of the present study and
continues to be maintained (18).
Gemcitabine is produced by Eli Lilly and Company. All cells were
cultured in DMEM (HyClone; Cytiva) supplemented with 10% fetal
bovine serum (Sangon Biotech Co., Ltd.) and 1%
penicillin/streptomycin in humidified air at 37°C with 5%
CO2.
Antibodies and reagents
Anti-γH2AX (1:1,000; cat. no. 9718; Cell Signaling
Technology, Inc.), anti-GAPDH (1:1,000; cat. no. AF0006; Beyotime
Institute of Biotechnology), anti-P21 (1:1,000; cat. no. 2947; Cell
Signaling Technology, Inc.), anti-cyclin D1 (1:1,000; cat. no.
55506), anti-cyclin A2 (1:1,000; cat. no. 4656), anti-cyclin E1
(1:1,000; cat. no. 4129), and anti-cleaved caspase-3 (1:1,000; cat.
no. 9661; all from Cell Signaling Technology, Inc.) antibodies were
used.
Cell survival and cytotoxicity
assays
Cell proliferation in vitro was monitored by
a Cell Counting Kit-8 (CCK-8) assay in 96-well plates. A total of
2×103 cells in complete medium were seeded into each
well, and 10 µl of CCK-8 reagent (cat. no. CK04-13; Dojindo
Molecular Technologies, Inc.) was added for an additional 1 h of
incubation at 37°C. The plates were read and the absorbance was
measured at 450 nm using a microplate reader (BioTek Synergy HTX;
BioTek Instruments, Inc.).
Reverse transcription-quantitative PCR
(qPCR) assay
Total RNA was isolated from cells with RNAiso Plus
(Takara Bio, Inc.) and the quantity was measured by using NanoDrop
equipment (Thermo Fisher Scientific, Inc.). Total RNA was reverse
transcribed using the Prime Script RT Master Mix (Takara Bio, Inc.)
according to the manufacturer's instructions. The TB Green Premix
Ex Taq™ II Kit (Takara Bio, Inc.) was used to detect relative mRNA
expression using the PIKOREAL 96 Real-Time PCR System (Thermo
Fisher Scientific, Inc.) with 40 cycles of PCR thermocycling at
95°C for 15 sec and 60°C for 60 sec, followed by 95°C for 10 min.
The primer sequences were as follows: GAPDH forward,
5′-CAGGAGGCATTGCTGATGAT-3′ and reverse, 5′-GAAGGCTGGGGCTCATTT-3′;
p21 forward, 5′-GCCCGTGAGCGATGGAACTTC-3′ and reverse,
5′-CCTGCCTCCTCCCAACTCATCC-3′. All the target genes were calculated
using the 2−ΔΔCq method (19) and normalized against the expression
level of GAPDH.
Western blot analysis
Cells were lysed with RIPA buffer (cat. no. P0013B;
Beyotime Institute of Biotechnology) containing a protease and
phosphatase inhibitor cocktail. Protein concentration was detected
by the BCA method (Thermo Fisher Scientific, Inc.). SDS-PAGE gels
(10%) (Beyotime Institute of Biotechnology) were used for protein
electrophoresis with equal amounts of protein (30 µg). The
separated proteins were transferred to PVDF membranes
(MilliporeSigma) and blocked with 5% skimmed milk in TBS with 0.2%
Tween-20 at room temperature for 2 h. The membranes were then
incubated with the aforementioned antibodies at 4°C overnight. On
the second day, the membranes were washed and incubated with
HRP-conjugated anti-rabbit or anti-mouse secondary antibodies at
the dilution of 1:1,000 (cat. nos. A0208 and A0216; Beyotime
Institute of Biotechnology) at room temperature for 2 h. Labeled
proteins were detected by an enhanced chemiluminescence detection
system (Azure Biosystems), and the protein band grayscale analysis
was performed using ImageJ 1.51k software (National Institute of
Health).
Cell cycle analyses
Harvested cells were washed with PBS, fixed with 70%
ethanol at 4°C for 2 h and then treated with 450 µl of RNase and 50
µl of propidium iodide (PI; cat. no. KGA512; Nanjing KeyGen Biotech
Co., Ltd.) at room temperature for 15 min. The samples were
analyzed by flow cytometry (FACS Canto™ II; BD Biosciences) and the
data were analyzed using Flowjo.7.6 software (BD Biosciences).
Apoptosis assay
Cells were washed once in PBS and incubated with 5
µl of 7-AAD in 50 µl of binding buffer (cat. no. KGA1017; Nanjing
KeyGen Biotech Co., Ltd.) for 15 min at 37°C in a humidified
atmosphere in the dark. Then, 450 µl of binding buffer and 1 µl of
Annexin V-PE (cat. no. KGA1017; Nanjing KeyGen Biotech Co., Ltd.)
were added at room temperature. The apoptosis rate was determined
by flow cytometry (FACS Canto II; BD Biosciences) and the data were
analyzed using Flowjo.7.6. software within 1 h of staining.
siRNA transfection
Cells were seeded at a density of 2×105
cells per well in a six-well plate. On the next day, transfection
of siRNA (50 nM) into cells was performed with Lipofectamine™ 3000
Transfection Reagent (cat. no. L3000015; Invitrogen; Thermo Fisher
Scientific, Inc.) at room temperature for 24 or 48 h according to
the manufacturer's instructions. siRNAs and the control siRNAs were
purchased from Guangzhou Ribobio Co., Ltd. The sequence of the
siRNA targeting p21 was 5′-GAATGAGAGGTTCCTAAGA-3′; and the
sequence of the control is not disclosed by Ribobio company;
however, the product number was provided by the company
(siN0000001-1-5).
Plasmid transfection
Cells were seeded 1 day before transfection. Empty
vector GV658 was used as the control group and p21 expression
recombinant plasmid GV658-p21 was purchased from Shanghai GeneChem
Co., Ltd. Plasmid (2 µg) was transfected into cells using
Lipofectamine 3000 Transfection Reagent at room temperature (cat.
no. L3000015; Invitrogen; Thermo Fisher Scientific, Inc.) at room
temperature for 24 h or 48 h according to the manufacturer's
instructions, and then 24 h or 48 h later, the transfected cells
were used for the following experiments.
Comet assay
A neutral comet assay was performed to evaluate DNA
double-strand break (DSB) damage. Collected cells were washed with
PBS. The first layer of agarose was prepared, 75 µl of 1% normal
melting point agarose (Biofroxx; neoFroxx) was added to the slides,
which were quickly covered with a coverslip and placed at 4°C for
10 min in the dark. Subsequently, 75 µl of 0.7% low melting point
(LMP)-agarose (Beijing Solarbio Science & Technology Co., Ltd.)
and 10 µl of the cell suspension were mixed, quickly added to the
first layer of gel and covered with a coverslip. After the second
layer of agarose had solidified, 75 µl of 0.7% LMP-agarose was
added to make the third agarose layer. After this agarose layer had
solidified, the slides were immersed in ice-cold lysis solution for
2 h at 4°C in the dark, and electrophoresis was then performed in a
horizontal electrophoresis apparatus [BG-subMIDI(V); Beijing
Baygene Biotech Co., Ltd.]. Following electrophoresis, the slides
were immersed in neutralization buffer for 10 min at room
temperature. PI was pipetted onto each sample, and the slides were
incubated for 10 min at room temperature in the dark. Images were
acquired with an Olympus microscope connected to a charge-coupled
device (CCD) camera (final magnification, ×200). The tail moment
was calculated using Comet Assay Software Project 4 (CASP 4;
Perspective Instruments, Ltd.) as an indicator of DNA damage.
Statistical analysis
Data are presented as the mean ± standard deviation
from three independent assays. One-way analysis of variance (ANOVA)
was used to compare the sample mean and pairwise comparisons
between multiple groups (followed by Dunnett's if all comparisons
were vs. a single control or Tukey/Bonferroni if the groups were
compared with each other). Statistical analysis was performed using
GraphPad Prism 8.0 software (GraphPad Software, Inc.; Dotmatics).
P-values of <0.05 were considered to indicate statistically
significant differences.
Results
p21 is markedly upregulated in
A549/G+ cells and p21 upregulation potentially induces
gemcitabine resistance in A549 cells
In a previous study conducted by the authors
(20), it was observed that
miR-17-5p and let-7i-5p overexpression in A549/G+ cells
could increase their sensitivity to gemcitabine, while the
expression of p21 protein in these cells was decreased. To
investigate whether the expression level of p21 is related to the
gemcitabine tolerance of A549 cells, the mRNA and protein
expression levels of p21 in A549 and A549/G+ cells were
verified by reverse transcription-quantitative PCR and western
blotting, respectively. Compared with that in A549 cells (Fig. 1A and B), the p21 expression level in
A549/G+ cells were increased (P<0.001). p21 plays a
dual role in tumors (13,21,22).
Therefore, it was hypothesized that the high level of p21
expression in A549/G+ cells contributed to the
acquisition of drug resistance, and this hypothesis was later
verified. p21 was overexpressed in A549 cells, and
A549/G+ cells were transfected with siRNA-p21 (Fig. 1C and D). Subsequently, a CCK-8 assay
was performed to measure the IC50 value of gemcitabine.
The results (Fig. 1E and F)
revealed that knockdown of p21 expression in A549/G+
cells resulted in a decrease in the IC50 value of
gemcitabine from 84.2 to 26.7 µM (P<0.05); by contrast,
overexpression of p21 expression in A549 cells increased the
IC50 value from 15.4 to 40.1 µM (P<0.001). Thus, p21
knockdown sensitized A549 cells to gemcitabine. These results
indicated that p21 upregulation in A549/G+ cells may
contribute to gemcitabine resistance.
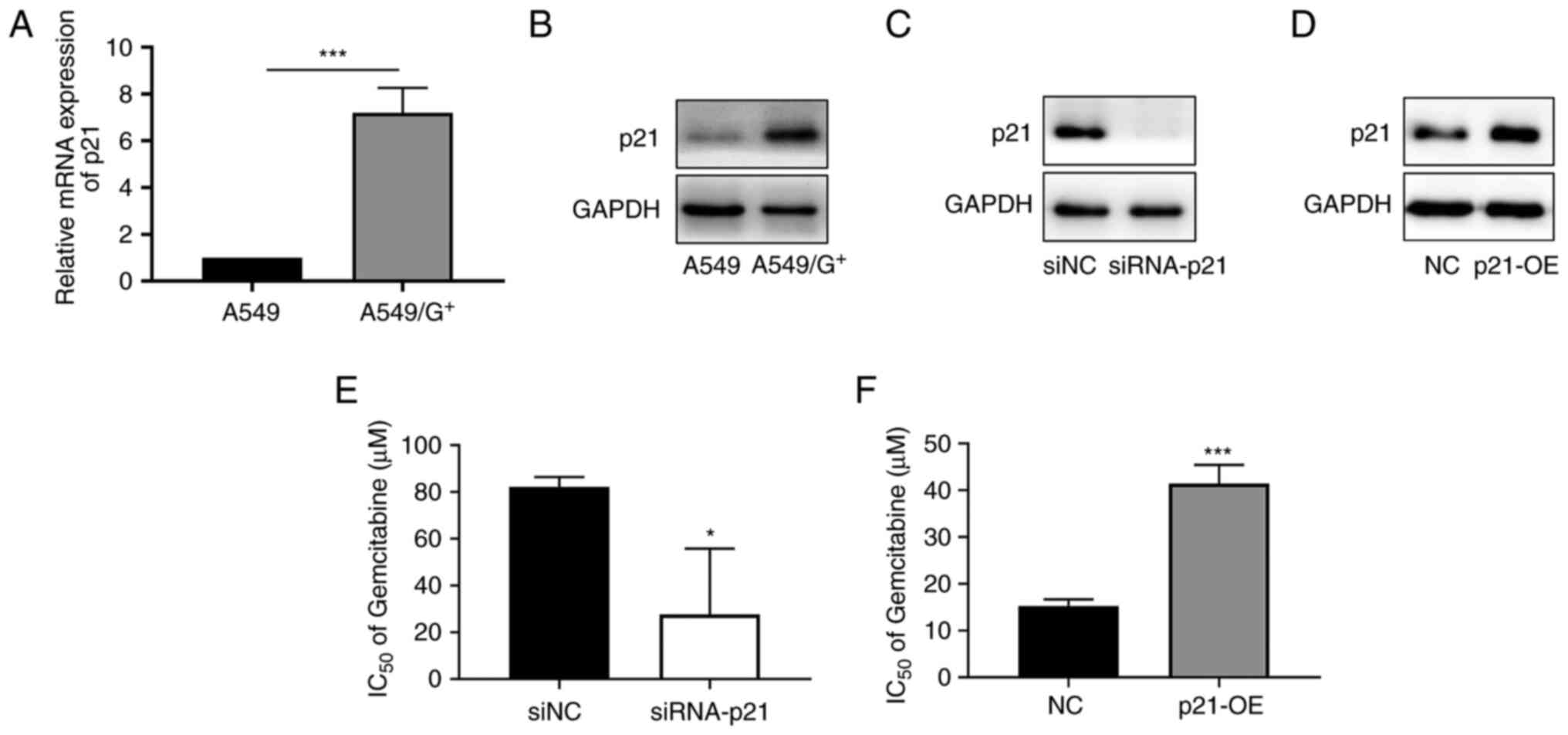 | Figure 1.p21 is markedly upregulated in
gemcitabine-resistant A549 cells. (A) p21 mRNA and (B) protein
expression levels in A549/G+ and parental A549 cells.
(C) A549 cells were transfected with siRNA, and western blotting
was used to verify the interference efficiency of p21. (D)
A549/G+ cells were transfected with plasmids, and
western blotting was used to verify the efficiency of p21
overexpression. (E) After p21-siRNA or siNC was successfully
transfected into A549/G+ cells, the cells were treated
with different concentrations of gemcitabine (1, 5, 10, 25, 50,
100, 500 and 1,000 µM), and a CCK8 proliferation assay was
performed to detect the IC50. (F) Overexpression of p21
in A549 cells was assessed after treatment with different
concentrations of gemcitabine (1, 5, 10, 25, 50, 100, 500 and 1,000
µM), and then a CCK8 proliferation assay was performed to detect
the IC50. siRNA, small interfering RNA; NC, negative
control; OE, overexpressed; A549/G+,
gemcitabine-resistant A549; CCK-8, Cell Counting Kit 8. *P<0.05
and ***P<0.001 vs. respective controls. |
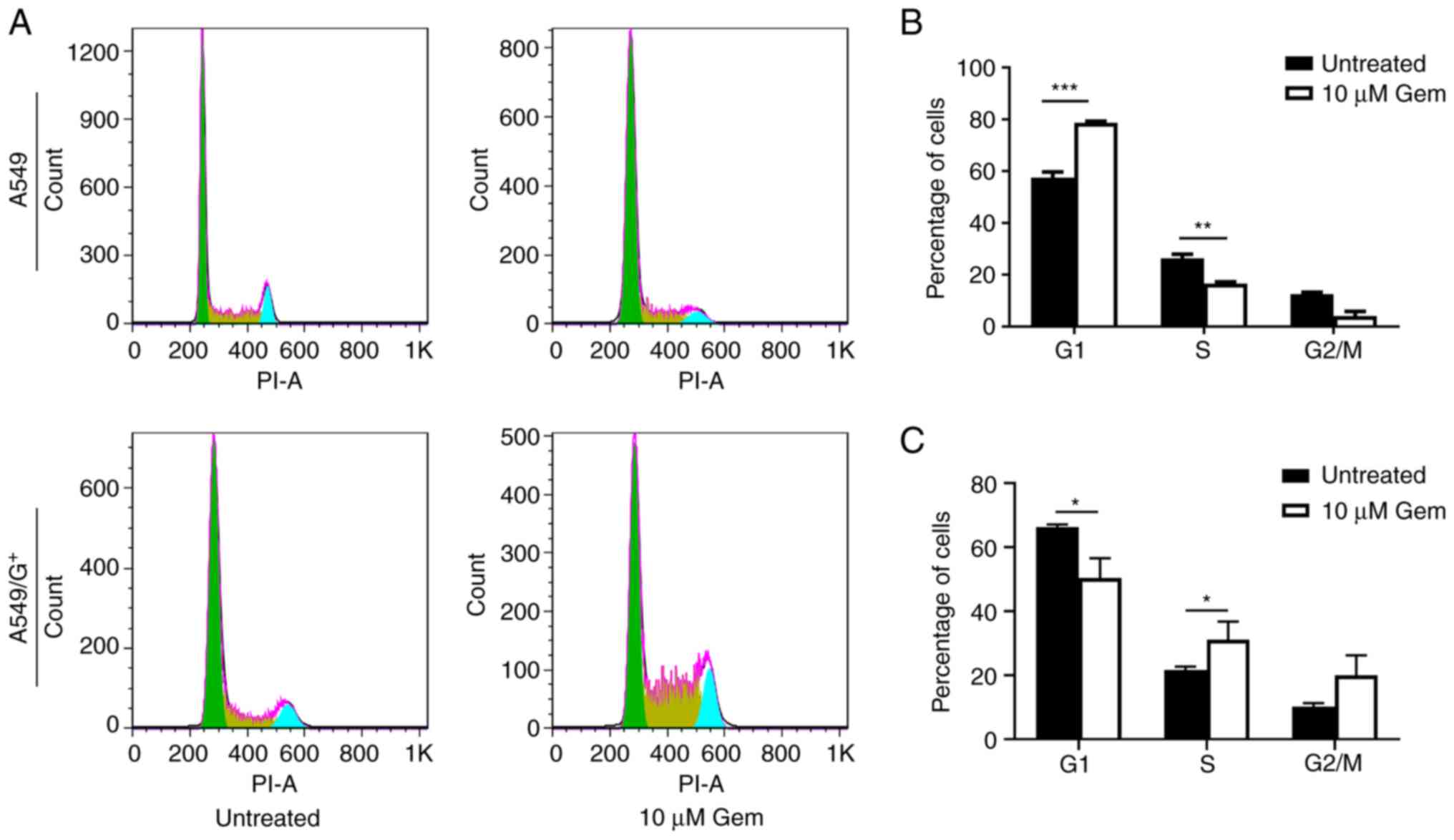
Treatment with 10 µM gemcitabine
increases the percentage of A549 cells in the
G0/G1 phase and reduces the percentage of
A549/G+ cells in the G1 and S phase
Gemcitabine is a potent replication inhibitor, but
whether A549 cell tolerance to gemcitabine is associated with the
cell cycle remains unclear. Subsequently, the two aforementioned
cell lines were treated with the same concentration of gemcitabine,
and flow cytometry was performed to assess the cell cycle
distribution. As illustrated in Fig.
2A, in A549 cells, treatment with 10 µM gemcitabine resulted in
a slight decrease in the percentage of cells in the S phase
(Fig. 2B; 16.62% vs. untreated
26.38%) and an increase in the percentage of cells in the
G0/G1 phase (Fig.
2B; 78.70% vs. untreated 57.55%). However, when
A549/G+ cells were treated with 10 µM gemcitabine, there
was a decrease in the percentage of cells in the G1
phase (Fig. 2C; 50.35% vs.
untreated 66.29%), accompanied by an increase in the percentage of
cells in the S phase (Fig. 2C;
31.08% vs. untreated 21.61%) and a lesser increase in the
percentage of cells in the G2/M phase (Fig. 2C; 19.96% vs. untreated 10.24%).
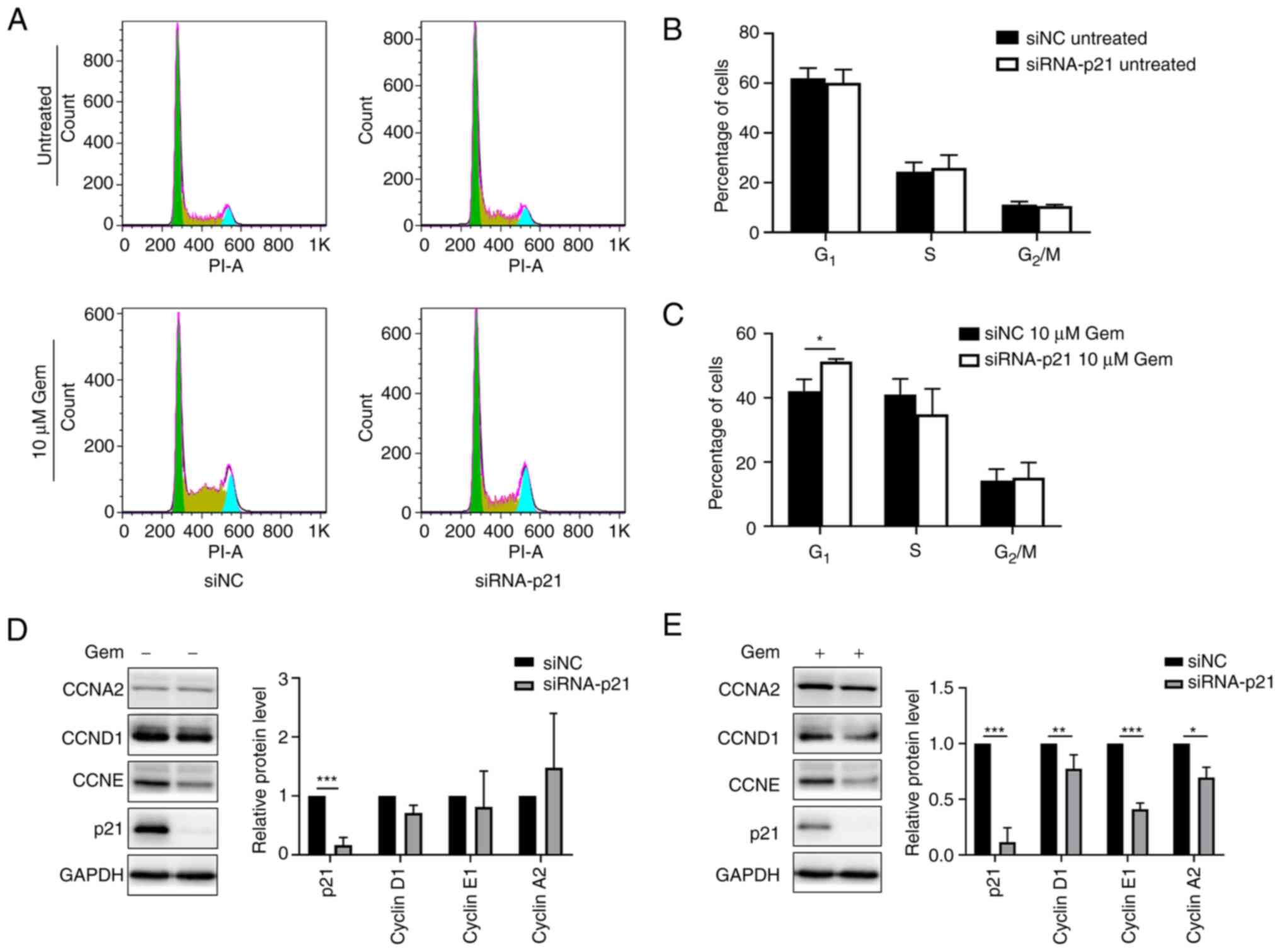
Knockdown of p21 promotes
gemcitabine-induced G1 arrest
Considering the role of p21 in the cell cycle, it
was speculated that the different changes in the cell cycle in
gemcitabine-resistant cells are involved in the difference in p21
expression. To test this hypothesis, A549/G+ cells were
transfected with siRNA-p21 to knockdown p21 expression prior to
gemcitabine treatment for 24 h and cell cycle analysis was
performed using flow cytometry. The reason why sensitive cells were
not selected is that the expression level of p21 in the sensitive
cells is already extremely low. If siRNA is used to interfere with
its expression, the knockdown effect is not significant. Therefore,
drug-resistant cells were selected for interference experiments.
The results revealed that the expression of p21 in
A549/G+ cells was decreased, but there was no
significant change in the cell cycle distribution in the absence of
gemcitabine (Fig. 3A and B).
However, the percentage of G1-phase cells was
significantly increased after gemcitabine treatment (Fig. 3C; 51.14% vs. untreated 42.14%).
Furthermore, as revealed in Fig.
3D, the expression levels of cyclin E1, cyclin A2 and cyclin
D1, which are related to the G1 and S phases, did not
change significantly when p21 expression was knocked down in
A549/G+ cells in the absence of gemcitabine. However, as
revealed in Fig. 3E, following
treatment with gemcitabine, cyclin E1, cyclin A2, and cyclin D1
expression was reduced in the siRNA-p21 group. The aforementioned
results indicated that p21 expression in A549/G+ cells
is involved in modulating the expression of cell cycle regulators
and that a reduction in p21 expression could promote
gemcitabine-induced G1 arrest. Moreover, similar results
demonstrated that an increase in the number of G1-phase
cells is related to apoptosis (23). Considering these observations, it
was assumed that the failure of gemcitabine to induce G1
arrest may be an important cause of gemcitabine resistance and that
the reduction in p21 expression could inhibit progression through
the G1 phase.
Knocked down p21 expression results in
accumulation of unrepaired DSBs and induction of apoptosis by
gemcitabine, which enhances sensitivity to gemcitabine
Gemcitabine, a nucleoside analog, can be
incorporated into replicating DNA, resulting in partial chain
termination and stalling of replication forks, which can cause DNA
DSB damage and apoptosis to kill tumor cells (7,24). To
assess the difference in gemcitabine-induced DNA DSB damage between
A549 cells and A549/G+ cells, the two cell lines were
treated with the same concentration of gemcitabine. As depicted in
Fig. 4A, the level of the DSB
indicator γ-H2AX was increased by gemcitabine treatment, but its
change trend was opposite that of p21 expression. The γ-H2AX
expression level in A549 cells was higher than that in
A549/G+ cells (P<0.05), suggesting that
gemcitabine-induced DNA DSB damage was more severe in A549 cells
than in A549/G+ cells. In addition, it was hypothesized
that upregulation of p21 in drug-resistant cells reduced
gemcitabine-induced DSB formation and apoptosis. Therefore, p21 was
overexpressed in A549 cells and siRNA-p21 was transfected into
A549/G+ cells, and these cells were then treated with
gemcitabine to evaluate the level of γ-H2AX. As revealed in
Fig. 4B, the apoptosis activity, as
indicated by caspase-3 cleavage, was upregulated, as was the level
of γ-H2AX, in the siRNA-p21 group compared with the siNC group.
However, overexpression of p21 reduced gemcitabine-induced DNA
damage (Fig. 4C). Furthermore, to
confirm the role of p21 in gemcitabine-induced DSB formation and
apoptosis, a comet assay was performed to detect DSBs and flow
cytometry was conducted to determine the apoptosis rate after
A549/G+ cells were treated with siRNA-p21 and
gemcitabine. Consistent with the western blot results, the decrease
in p21 expression in A549/G+ cells promoted
gemcitabine-induced DNA damage and apoptosis (Fig. 4D and E).
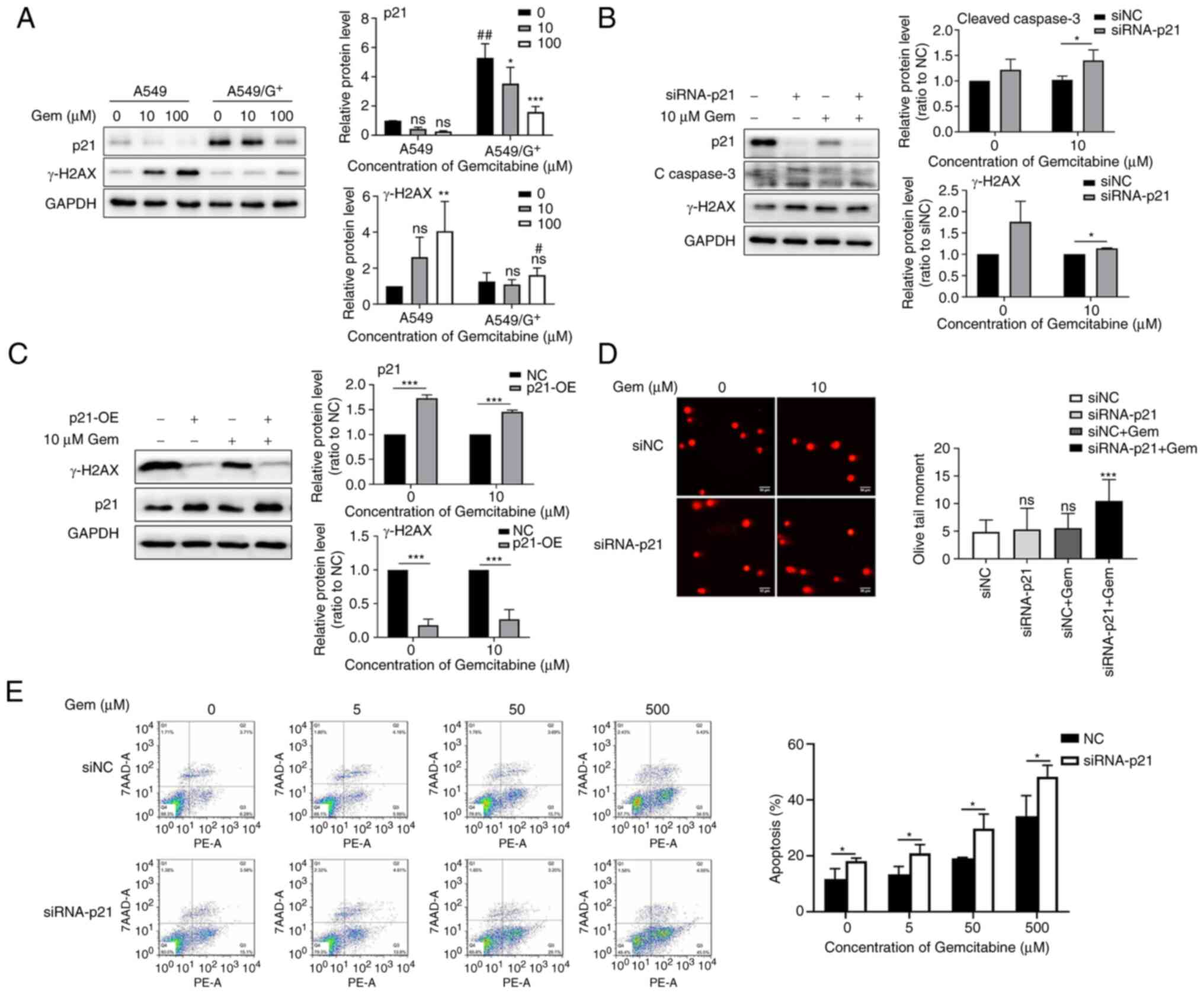 | Figure 4.Knockdown of p21 expression results
in accumulation of unrepaired DSBs and induction of apoptosis by
gemcitabine. (A) Western blotting detection of DNA double-strand
replication-related protein (γ-H2AX) in A549 cells and
A549/G+ cells coupled with various concentrations of
gemcitabine (0, 10 and 100 µM). (B) Western blotting detection of
apoptosis and DNA double-strand replication-related protein (γ-H2AX
and cleaved caspase-3) in siNC- or siRNA-p21-transfected
A549/G+ cells treated with different concentrations of
gemcitabine (0 and 10 µM). (C) Western blotting detection of DNA
double-strand damage was observed with p21 overexpression. (D)
Representative images of the comet assay in A549/G+
cells treated with siRNA-p21 (or siNC) combined with gemcitabine.
(E) Flow cytometric analysis of apoptosis in siNC- or
siRNA-p21-transfected A549/G+ cells treated with various
concentrations of gemcitabine (0.0, 5.0, 50 and 500 µM). Gem,
gemcitabine; A549/G+, gemcitabine-resistant A549; siRNA,
small interfering RNA; NC, negative control; OE, overexpressed.
*P<0.05, **P<0.01 and ***P<0.001 vs. respective controls,
#P<0.05 and ##P<0.01 vs. the same drug
concentration in A549 cells. |
Based on the aforementioned results, it was deduced
that the reduction in p21 expression in A549/G+ cells
promoted gemcitabine-induced accumulation of DNA damage and
apoptosis, which may sensitize cells to gemcitabine. Consistent
with the findings of the present study, it was reported that p21
contributed to the ability of Langerhans cells to resist the
detrimental effects of ionizing radiation by the rapid repair of
DSBs and inhibition of apoptosis (25).
Discussion
p21 is downstream of the tumor suppressor p53 and
mediates cell cycle arrest upon DNA damage (26). p21 has always been considered a
tumor suppressor gene. However, an increasing number of studies
have revealed that p21 plays two roles in the progression of cancer
and in radio- and chemoresistance. Numerous studies have shown that
high expression of p21 plays a primary role in promoting drug
resistance in lung cancer or increasing A549 cell survival by
protecting cells against cytotoxic anticancer agents (23,27).
However, a small number of studies have indicated that high
expression of p21 can increase the resistance of lung cancer cells
to gemcitabine. Similarly, previous research conducted by the
authors revealed that downregulation of miR-17-5p in
A549/G+ cells can directly or through RRM2 increase p21
mRNA and protein expression (20).
It is generally considered that the role of p21 in
tumors depends mainly on its localization and expression status in
cells. Nuclear p21 can inhibit cell proliferation and promote
apoptosis, while cytoplasmic p21 has carcinogenic and antiapoptotic
effects. For example, a number of studies have reported that
cytoplasmic p21 is involved in the chemoresistance of ovarian
cancer cells (28), testicular
embryonal tumors (29). It plays an
important role in protecting cells against apoptosis and promoting
tumor invasion, migration, and drug resistance (30,31).
Consistent with the findings of the present study,
Ikeda et al revealed that the p21 expression level in
A549/G+ cells were increased. Their findings suggested
that overexpression of p21 may be a molecular marker associated
with gemcitabine resistance in the A549 cell line but did not
elucidate the underlying mechanism (32). In the present study, upon treatment
with the same concentration of gemcitabine, the cell cycle in the
gemcitabine-sensitive and A549/G+ cell lines was
differentially altered. Gemcitabine successfully induced
G1 arrest in A549 but not in A549/G+ cells.
However, p21 knockout and treatment with gemcitabine increased the
proportion of G1-phase cells among A549/G+ cells. This
finding was consistent with the study conducted by Gaben et
al, which showed that inhibition of the expression of p21 could
inhibit G1 phase progression and was related to the
antiproliferative effects of rapamycin in BP-A31 cells (33). Similarly, a recent study
demonstrated that in RAL cells treated with the
cisplatin-pemetrexed combination, knockdown of p21 could increase
the number of apoptotic cells and induce G1 arrest.
These results suggested that p21 is involved in regulating the
response of RAL cells under treatment with the combination of
cisplatin and pemetrexed (23).
In addition to alterations in the cell cycle
distribution, significant increases in the number of apoptotic
cells, apparent G1 arrest and accumulation of DNA DSB
damage in CDKN1A-knockdown A549/G+ cells were observed.
These findings indicated that p21 is involved in modulating the
response of A549 cells to gemcitabine and demonstrated that p21 may
be a target for reversing drug resistance in lung cancer.
Of note, the reason why the authors of the present
study used the A549 cell line for their present research is that it
focuses on chemoresistance, and the chemical sensitivity and drug
resistance characteristics of the A549 cell line are very obvious.
In particular, A549 cells are more sensitive to doxycycline,
gemcitabine, and other chemotherapy drugs but relatively resistant
to paclitaxel, vincristine, and platinum compounds. Therefore, the
A549 cell line is widely used in the screening and evaluation of
chemotherapy drugs for lung cancer, in research on cellular signal
transduction in lung cancer, and in antitumor immunotherapy
research (34).
In several tumors, increased p21 expression mediates
drug resistance and promotes cell survival (35,36).
Reducing the expression of p21 may be a solution to reverse drug
resistance. Recently, a novel p21 inhibitor, UC2288, was
demonstrated to decrease p21 mRNA expression independent of p53 and
attenuate p21 protein expression with a minimal effect on p21
protein stability (37). Yan et
al discovered that inhibiting p21 in QGP-1 and NCI-H727 cells
by treatment with UC2288, further enhanced the cytotoxicity of
artesunate, implying that p21 may confer resistance to artesunate
on these two cell lines (38). In
addition, p21 inhibitor-induced death in cells with high p21
expression suggests that p21 suppression could be a therapeutic
strategy for patients with clear cell carcinoma (39). Although the present research has
certain limitations, such as only conducting experiments on a few
cell lines and lacking evidence from in vivo experiments, it
still suggests that p21 may be a new marker of drug resistance, and
that p21 suppression could be a therapeutic strategy for patients
with cancer.
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural Science
Foundation of China (grant no. 81071853).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Author contributions
ALL and YJL contributed to the conceptualization and
supervision of the study. TF and XM performed the experiments and
confirm the authenticity of all the raw data. TF, XM, SLD, ZYK,
XCW, HHY and WXW contributed to acquiring and analyzing the data.
TF and XM contributed to the writing of the original draft. TF, XM,
ALL and YJL contributed to the manuscript review and editing. YJL
contributed to the funding acquisition. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
He L, Luo L, Zhu H, Yang H, Zhang Y, Wu H,
Sun H, Jiang F, Kathera CS, Liu L, et al: FEN1 promotes tumor
progression and confers cisplatin resistance in non-small-cell lung
cancer. Mol Oncol. 11:640–654. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang X, Wang D, Li Z, Jiao D, Jin L, Cong
J, Zheng X and Xu L: Low-Dose gemcitabine treatment enhances
immunogenicity and natural killer cell-driven tumor immunity in
lung cancer. Front Immunol. 11:3312020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Toschi L, Finocchiaro G, Bartolini S,
Gioia V and Cappuzzo F: Role of gemcitabine in cancer therapy.
Future Oncol. 1:7–17. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mini E, Nobili S, Caciagli B, Landini I
and Mazzei T: Cellular pharmacology of gemcitabine. Ann Oncol. 17
(Suppl 5):v7–v12. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li Y, Wang LR, Chen J, Lou Y and Zhang GB:
First-line gemcitabine plus cisplatin in nonsmall cell lung cancer
patients. Dis Markers. 2014:9604582014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jones RM, Kotsantis P, Stewart GS, Groth P
and Petermann E: BRCA2 and RAD51 promote double-strand break
formation and cell death in response to gemcitabine. Mol Cancer
Ther. 13:2412–2421. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
de Sousa Cavalcante L and Monteiro G:
Gemcitabine: Metabolism and molecular mechanisms of action,
sensitivity and chemoresistance in pancreatic cancer. Eur J
Pharmacol. 741:8–16. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu JS, Yeh CA, Huang IC, Huang GY, Chiu
CH, Mahalakshmi B, Wen SY, Huang CY and Kuo WW: Signal transducer
and activator of transcription 3 mediates apoptosis inhibition
through reducing mitochondrial ROS and activating Bcl-2 in
gemcitabine-resistant lung cancer A549 cells. J Cell Physiol.
236:3896–3905. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Deng T, Yan G, Song X, Xie L, Zhou Y, Li
J, Hu X, Li Z, Hu J, Zhang Y, et al: Deubiquitylation and
stabilization of p21 by USP11 is critical for cell-cycle
progression and DNA damage responses. Proc Natl Acad Sci USA.
115:4678–4683. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zlotorynski E: Cancer biology: The dark
side of p21. Nat Rev Mol Cell Biol. 17:4612016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Warfel NA and El-Deiry WS: p21WAF1 and
tumourigenesis: 20 years after. Curr Opin Oncol. 25:52–58. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Georgakilas AG, Martin OA and Bonner WM:
p21: A two-faced genome guardian. Trends Mol Med. 23:310–319. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang H, Zhu LJ, Yang YC, Wang ZX and Wang
R: MiR-224 promotes the chemoresistance of human lung
adenocarcinoma cells to cisplatin via regulating G1/S
transition and apoptosis by targeting p21WAF1/CIP1. Br J
Cancer. 111:339–354. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu Z, Sun M, Lu K, Liu J, Zhang M, Wu W,
De W, Wang Z and Wang R: The long noncoding RNA HOTAIR contributes
to cisplatin resistance of human lung adenocarcinoma cells via
downregualtion of p21(WAF1/CIP1) expression. PLoS One.
8:e772932013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao J, Fu W, Liao H, Dai L, Jiang Z, Pan
Y, Huang H, Mo Y, Li S, Yang G and Yin J: The regulatory and
predictive functions of miR-17 and miR-92 families on cisplatin
resistance of non-small cell lung cancer. BMC Cancer. 15:7312015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guo Q, Lan F, Yan X, Xiao Z, Wu Y and
Zhang Q: Hypoxia exposure induced cisplatin resistance partially
via activating p53 and hypoxia inducible factor-1α in non-small
cell lung cancer A549 cells. Oncol Lett. 16:801–808.
2018.PubMed/NCBI
|
|
18
|
Liang AL, Du SL, Zhang B, Zhang J, Ma X,
Wu CY and Liu YJ: Screening miRNAs associated with resistance
gemcitabine from exosomes in A549 lung cancer cells. Cancer Manag
Res. 11:6311–6321. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ma X, Fu T, Ke ZY, Du SL, Wang XC, Zhou N,
Zhong MY, Liu YJ and Liang AL: MiR-17-5p/RRM2 regulated gemcitabine
resistance in lung cancer A549 cells. Cell Cycle. 22:1367–1379.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Galanos P, Vougas K, Walter D, Polyzos A,
Maya-Mendoza A, Haagensen EJ, Kokkalis A, Roumelioti FM, Gagos S,
Tzetis M, et al: Chronic p53-independent p21 expression causes
genomic instability by deregulating replication licensing. Nat Cell
Biol. 18:777–789. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zohny SF, Al-Malki AL, Zamzami MA and
Choudhry H: p21Waf1/Cip1: Its paradoxical effect in the
regulation of breast cancer. Breast Cancer. 26:131–137. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zamagni A, Pasini A, Pirini F, Ravaioli S,
Giordano E, Tesei A, Calistri D, Ulivi P, Fabbri F, Foca F, et al:
CDKN1A upregulation and cisplatin-pemetrexed resistance in
non-small cell lung cancer cells. Int J Oncol. 56:1574–1584.
2020.PubMed/NCBI
|
|
24
|
Ewald B, Sampath D and Plunkett W: H2AX
phosphorylation marks gemcitabine-induced stalled replication forks
and their collapse upon S-phase checkpoint abrogation. Mol Cancer
Ther. 6:1239–1248. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Price JG, Idoyaga J, Salmon H, Hogstad B,
Bigarella CL, Ghaffari S, Leboeuf M and Merad M: CDKN1A regulates
Langerhans cell survival and promotes Treg cell generation upon
exposure to ionizing irradiation. Nat Immunol. 16:1060–1068. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Karimian A, Ahmadi Y and Yousefi B:
Multiple functions of p21 in cell cycle, apoptosis and
transcriptional regulation after DNA damage. DNA Repair (Amst).
42:63–71. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jana S, Patra K, Jana J, Mandal DP and
Bhattacharjee S: Nrf-2 transcriptionally activates
P21Cip/WAF1 and promotes A549cell survival against
oxidative stress induced by H2O2. Chem Biol
Interact. 285:59–68. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xia X, Ma Q, Li X, Ji T, Chen P, Xu H, Li
K, Fang Y, Weng D, Weng Y, et al: Cytoplasmic p21 is a potential
predictor for cisplatin sensitivity in ovarian cancer. BMC Cancer.
11:3992011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Koster R, di Pietro A, Timmer-Bosscha H,
Gibcus JH, van den Berg A, Suurmeijer AJ, Bischoff R, Gietema JA
and de Jong S: Cytoplasmic p21 expression levels determine
cisplatin resistance in human testicular cancer. J Clin Invest.
120:3594–3605. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Maiuthed A, Ninsontia C, Erlenbach-Wuensch
K, Ndreshkjana B, Muenzner JK, Caliskan A, Husayn AP, Chaotham C,
Hartmann A, Vial Roehe A, et al: Cytoplasmic p21 mediates
5-fluorouracil resistance by inhibiting pro-apoptotic Chk2. Cancers
(Basel). 10:3732018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhao WS, Yan WP, Chen DB, Dai L, Yang YB,
Kang XZ, Fu H, Chen P, Deng KJ, Wang XY, et al: Genome-scale CRISPR
activation screening identifies a role of ELAVL2-CDKN1A axis in
paclitaxel resistance in esophageal squamous cell carcinoma. Am J
Cancer Res. 9:1183–1200. 2019.PubMed/NCBI
|
|
32
|
Ikeda R, Vermeulen LC, Lau E, Jiang Z,
Sachidanandam K, Yamada K and Kolesar JM: Isolation and
characterization of gemcitabine-resistant human non-small cell lung
cancer A549 cells. Int J Oncol. 38:513–519. 2011.PubMed/NCBI
|
|
33
|
Gaben AM, Saucier C, Bedin M, Barbu V and
Mester J: Rapamycin inhibits cdk4 activation, p 21(WAF1/CIP1)
expression and G1-phase progression in transformed mouse
fibroblasts. Int J Cancer. 108:200–206. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dijk SN, Protasoni M, Elpidorou M, Kroon
AM and Taanman JW: Mitochondria as target to inhibit proliferation
and induce apoptosis of cancer cells: The effects of doxycycline
and gemcitabine. Sci Rep. 10:43632020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Santarelli R, Pompili C, Gilardini Montani
MS, Romeo MA, Gonnella R, D'Orazi G and Cirone M: Lovastatin
reduces PEL cell survival by phosphorylating ERK1/2 that blocks the
autophagic flux and engages a cross-talk with p53 to activate p21.
IUBMB Life. 73:968–977. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tay VSY, Devaraj S, Koh T, Ke G, Crasta KC
and Ali Y: Increased double strand breaks in diabetic β-cells with
a p21 response that limits apoptosis. Sci Rep. 9:193412019.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wettersten HI, Hee Hwang S, Li C, Li C,
Shiu EY, Wecksler AT, Hammock BD and Weiss RH: A novel p21
attenuator which is structurally related to sorafenib. Cancer Biol
Ther. 14:278–285. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yan G, Dawood M, Böckers M, Klauck SM,
Fottner C, Weber MM and Efferth T: Multiple modes of cell death in
neuroendocrine tumors induced by artesunate. Phytomedicine.
79:1533322020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Minagawa Y, Ishino K, Wada R, Kudo M,
Naito Z, Takeshita T and Ohashi R: High Expression of p21 as a
potential therapeutic target in ovarian clear-cell carcinoma.
Anticancer Res. 40:5631–5639. 2020. View Article : Google Scholar : PubMed/NCBI
|