Introduction
Non-small cell lung cancer (NSCLC) is one of the
most common malignant tumors, and the 5-year survival rate
following diagnosis is <15% (1).
The epidermal growth factor receptor (EGFR) is associated with the
growth and progression of cancer and an activation mutation in the
tyrosine kinase domain (exon 21 L858R point mutation or exon-19
deletion) has been identified as an oncogenic driver of NSCLC
(2). A study has shown that 30–40%
of Asian patients with NSCLC harbor EGFR mutations at the time of
diagnosis (3). Although it was
demonstrated that the overall response and disease control rates in
gefitinib- and erlotinib-treated groups (via targeting of EGFR)
were 76.9 vs. 74.4% and 90.1 vs. 86.8%, respectively (4), drug resistance is inevitable, and ~36%
of patients with NSCLC have EGFR T790M/L858R resistance mutations
(5). Although a second-generation
EGFR-tyrosine kinase inhibitor (TKI), afatinib, showed high
activity in an EGFR T790M/L858R driven xenotransplantation model,
it did not increase the objective response rate in patients with
NSCLC with drug-resistant mutations (6). Third-generation EGFR-TKIs, such as
CO1686 and AZD9291, are the only effective drugs for treating NSCLC
with the EGFR L858R/T790M mutation (7,8).
However, because the drug resistance molecular subtypes emerged
(such as acquisition of the EGFR C797S mutation or loss of the
T790M mutation), drug resistance is still inevitable after a period
of treatment (9). Additionally,
EGFR-TKIs may not be superior to chemotherapy in patients with
NSCLC with wild-type EGFR (10). A
study demonstrated that chemotherapy showed a superiority in terms
of PFS (HR, 1.84; 95% CI, 1.35–2.52) and ORR (16.8 vs. 7.2%;
relative risk, 1.11; 95% CI, 1.02–1.21) compared with EGFR-TKIs in
patients with NSCLC with wild-type EGFR (11). Chemotherapy remains the primary
treatment strategy for patients with wild-type EGFR NSCLC (10). Thus, identifying additional
treatment strategies for EGFR-resistant mutant and EGFR wild-type
NSCLC is important.
Ferroptosis is a newly discovered type of programmed
cell death that differs from apoptosis, necrosis and autophagy.
Ferroptosis is characterized by iron-dependent cell death, which
progresses with the accumulation of lipid peroxide and the
generation of reactive oxygen species (ROS) (12). During lipid peroxidation, the
activity of glutathione peroxidase 4 (GPX4) decreases, and
therefore lipid oxides cannot be metabolized through the
glutathione reductase reaction catalyzed by GPX4 (13,14).
The redox-active iron form (Fe2+) oxidizes lipids in a
manner similar to that of the Fenton reaction to produce large
amounts of ROS, causing an increase in malondialdehyde (MDA), which
promotes ferroptosis (15).
Ferritin is a major intracellular iron storage protein complex that
includes ferritin light-chain polypeptide 1 and ferritin
heavy-chain polypeptide 1 (FTH1) (16), and increased ferritin expression
limits ferroptosis (17). In
addition, iron levels are actively regulated in cells through
transferrin, which transports iron into the cells, and ferroportin,
which exports iron from the cells (18). The degradation of iron storage
proteins and changes in iron transporters can increase
iron-mediated ROS production, eventually leading to ferroptosis
(19). A study has shown that
inducing ferroptosis can improve therapeutic effects in
gefitinib-resistant lung cancer (20). In addition, another report has shown
that inducing ferroptosis enhances the cytotoxicity of erlotinib in
NSCLC cells, thereby overcoming erlotinib resistance (21), suggesting that inducing ferroptosis
may be an effective treatment for EGFR-TKI-resistant NSCLC.
The phosphatidylinositol 3-kinase (PI3K)/AKT
signaling pathway is also associated with tumor development
(22). Mammalian target of
rapamycin (mTOR), an important serine/threonine protein kinase
downstream of PI3K/AKT, regulates the proliferation, survival,
invasion and metastasis of tumor cells by activating ribosomal
kinases (23). mTOR inhibitors
(notably RAD001, an oral rapamycin derivative) have been approved
by the US Food and Drug Administration (FDA) for wider use in
antitumor clinical treatment, providing more treatment options for
patients with pancreatic neuroendocrine tumors (PNETs), estrogen
receptor+ + HER2− breast cancer and other
solid tumors (24). A study has
shown that RAD001 promotes the death of T790M+ NSCLC
cells (25), suggesting that RAD001
is a potential treatment strategy for EGFR-TKI resistant tumors. In
addition, inhibition of mTOR overcomes lapatinib resistance by
inducing ferroptosis in NSCLC (26), suggesting that the regulation of
mTOR may promote ferroptosis and overcome EGFR-TKI resistance.
Other studies have shown that the induction of ferroptosis in
wild-type EGFR NSCLC enhances the therapeutic effect of cisplatin
(27) and overcomes cell resistance
to gefitinib (28). A previous
report also demonstrated that dual inhibition of the EGFR/mTOR
pathway had a significant antitumor effect on wild-type EGFR NSCLC
(29). However, whether mTOR
inhibition enhances the antitumor effect in EGFR-TKI-resistant
mutants and wild-type EGFR NSCLC by inducing ferroptosis and the
related mechanisms remain unknown.
The present study investigated the effect and
mechanism of targeting the mTOR pathway to regulate ferroptosis in
NSCLC with EGFR wild-type, EGFR sensitive mutant cells and
EGFR-resistant mutant cells through MTT assay, western blotting,
reverse transcription-quantitative PCR (RT-qPCR), ROS assay and MDA
assay.
Materials and methods
Cell culture
Human NSCLC (H1975, A549 and H3255) cell lines were
purchased from The Cell Bank of Type Culture Collection of The
Chinese Academy of Sciences and cultured in RPMI 1640 medium
supplemented with 10% fetal bovine serum and 1%
penicillin/streptomycin (all from Gibco; Thermo Fisher Scientific,
Inc.). Cells were incubated at 37°C in a 5% CO2
incubator and routinely passaged every 3 days.
MTT assay
H1975, A549 and H3255 cells were seeded into 96-well
plates with 4×103 cells/well and treated with various
concentrations of RAD001 (0, 0.25, 0.5, 1, 2, 4, 8 and 16 µM) or
ferroptosis regulators, erastin (0, 0.25, 0.5, 1, 2, 4, 8 and 16
µM), ferrostatin-1 (0, 0.125, 0.25, 0.5, 1, 2, 4 and 8 µM) and RSL3
(0, 0.25, 0.5, 1, 2, 4, 8 and 16 µM). After 24 h, these cells were
incubated with MTT reagent (20 µl/well) at 37°C for 4 h. Then the
supernatant was replaced with 150 µl/well DMSO. This mixture was
shaken for 10 min and the absorbance at 490 nm was measured using a
CLARIOstarPlus microplate reader (BMG Labtech GmbH).
Western blotting
The H1975, A549 and H3255 cells with different doses
of RAD001 or RSL3 treatment were lysed using RIPA lysis buffer
containing the protease and phosphatase inhibitors (Beyotime
Institute of Biotechnology). The supernatant was centrifuged at
15,000 × g at 4°C for 15 min and a BCA protein assay kit (Takara
Bio, Inc.) was used to determine the concentration of proteins in
the samples. The extracted protein (50 µg/lane) was mixed with 5X
sodium dodecyl sulfate (SDS) loading buffer (BioTeke Corporation),
which was loaded in a 10% gel (Beyotime Institute of Biotechnology)
and the proteins were separated using SDS-polyacrylamide gel
electrophoresis, transferred to a polyvinylidene fluoride membrane
and the membrane was blocked in 5% non-fat milk at 22–24°C for 1.5
h. The membranes were then incubated overnight at 4°C with
different primary antibodies (listed below). After washing with
TBS-1% Tween 20 for three times, the membranes were incubated with
the corresponding secondary antibodies for 1 h at 22–24°C (listed
below). The proteins were visualized using an enhanced
chemiluminescence kit (cat. no. P0018AS; Beyotime Institute of
Biotechnology) and analyzed with Quantity One software (version
4.6.9; Bio-Rad Laboratories, Inc.). Primary antibodies against
Caspase 3/Cleaved-Caspase 3 (1:1,000; cat. no. 9662; Cell Signaling
Technology, Inc.), FTH1 (1:1,000; cat. no. 3998; Cell Signaling
Technology, Inc.), transferrin (1:1,000; cat. no. ab109503; Abcam),
transferrin receptor, CD71 (1:1,000; cat. no. ab84036; Abcam),
ferroportin (1:1,000; cat. no. ab235166; Abcam), GPX4 (1:1,000;
cat. no. ab125066; Abcam), phosphorylated (p-)mTOR (1:500; cat. no.
2971; Cell Signaling Technology, Inc.), mTOR (1:500; cat. no. 2972;
Cell Signaling Technology, Inc.), AKT (1:1,000; cat. no. 9272; Cell
Signaling Technology, Inc.) and anti-glyceraldehyde-3-phosphate
dehydrogenase (GAPDH; 1:1,000; cat. no. ab9485; Abcam), and
secondary antibodies against goat anti-rabbit IgG (1:1,000; cat.
no. WLA023; Wanleibio Co., Ltd.) and goat anti-mouse IgG (1:1,000;
cat. no. WLA024; Wanleibio Co., Ltd.) were used.
Reverse transcription-quantitative PCR
(RT-qPCR)
H1975, A549 and H3255 cells were seeded into a
6-well microplate at a density of 5×105 cells/well.
Total RNA from the cells in each group was extracted using TRIzol
reagent (Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. After RNA quality was tested, cDNA was
synthesized according to the manufacturer's protocols using a
PrimeScript II Reverse Transcriptase kit (Takara Bio, Inc.). qPCR
was then performed using a LightCycler 480 system (Roche
Diagnostics) and a SYBR Green Master Kit (Takara Bio, Inc.). The
reaction conditions were as follows: 95°C for 3 min, followed by 40
cycles of 95°C for 30 sec, 57°C for 30 sec and 72°C for 30 sec.
GAPDH mRNA was used to normalize relative mRNA levels. The relative
quantification of the PCR product was calculated, using the
2−ΔΔCq method (30).
Primer sequences are listed in Table
I.
 | Table I.Primer sequences used in reverse
transcription-quantitative PCR. |
Table I.
Primer sequences used in reverse
transcription-quantitative PCR.
| Gene name | Sense primer | Antisense
primer |
|---|
| xCT |
5′-TGGAACGAGGAGGTGGAGAA-3′ |
5′-TGGTGGACACAACAGGCTTT-3′ |
| FTH1 |
5′-CCAGAACTACCACCAGGACTC-3′ |
5′-GAAGATTCGGCCACCTCGTT-3′ |
| GPX4 |
5′-GCTGGACGAGGGGAGGAG-3′ |
5′-GGAAAACTCGTGCATGGAGC-3′ |
| Ferroportin |
5′-GAAAATCCCTGGGCCCCTTT-3′ |
5′-CTCTCGCTGAGGTGCTTGTT-3′ |
| Transferrin |
5′-CAGAAGCGAGTCCGACTGTG-3′ |
5′-CGCTTTTCATATGGTCGCGG-3′ |
| Transferrin
receptor |
5′-GGACGCGCTAGTGTTCTTCT-3′ |
5′-CATCTACTTGCCGAGCCAGG-3′ |
| GAPDH |
5′-ACCACAGTCCATGCCATCAC-3′ |
5′-TCCACCACCCTGTTGCTGTA-3′ |
ROS assay
H1975, A549 and H3255 cells with different doses of
RAD001 treatment were seeded into a 6-well microplate at a density
of 3×105 cells/well. After 24 h of treatment with RAD001
(0, 0.5, 1, 8 µM), the cells were collected, 10 µmol/l DCFH-DA was
added and the reaction mixture and was incubated at 37°C for 20
min. The cells were then washed three times with serum-free cell
culture medium of RPMI 1640 and the rate of ROS was detected using
a flow cytometer (FACSCanto; BD Biosciences). The results were
analysed using BD FACSDiva software (BD Biosciences).
MDA assay
H1975, A549 and H3255 cells with different doses of
RAD001 treatment were seeded into a 6-well microplate at a density
of 3×105 cells/well. After 24 h of treatment with RAD001
(0, 0.5, 1, 8 µM), the cells were collected and the levels of
intracellular MDA were measured using a Lipid Peroxidation MDA
Assay Kit (cat. no. S0131S; Beyotime Institute of Biotechnology)
according to the manufacturer's instructions.
Statistical analysis
All data were analyzed using SPSS 17.0 (SPSS, Inc.)
and GraphPad Prism (version 6.01; Dotmatics) software. Statistical
data are presented as the mean ± standard deviation. One-way
analysis of variance was used to compare the means among three or
more groups. Dunnett's test was used for all comparisons against a
single control. P-values were based on two-tailed statistical
analyses. P<0.05 was considered to indicate a statistically
significant difference.
Results
Different EGFR genotypes of NSCLC have
a different sensitivity to ferroptosis inducers
To explore the sensitivity of wild-type EGFR and
EGFR-mutated NSCLC cells to ferroptosis, the expression levels of
ferroptosis markers in H1975, A549 and H3255 cells were first
detected. When compared with H3255 (EGFR L858R mutation) cells, the
expression levels of FTH1, GPX4 and the transferrin receptor were
higher and the level of transferrin was lower in H1975 (EGFR
T790M/L858R) and A549 (EGFR wild type) cells (Fig. 1A). However, compared with H3255, the
expression levels of xCT and ferroportin were notably higher in
A549, but not H1975. When the A549, H1975 and H3255 cells were
treated with the ferroptosis inducer, erastin, cell death was
induced in a dose-dependent manner. Compared with H3255/A549 cells,
H1975 cells were more sensitive to erastin (IC50: 15.66
µM vs. 6.989 µM vs. 13.83 µM, Fig.
1B). These results suggest that the NSCLC cell lines with
wild-type EGFR and EGFR T790M/L858R mutations were more prone to
erastin-induced ferroptosis.
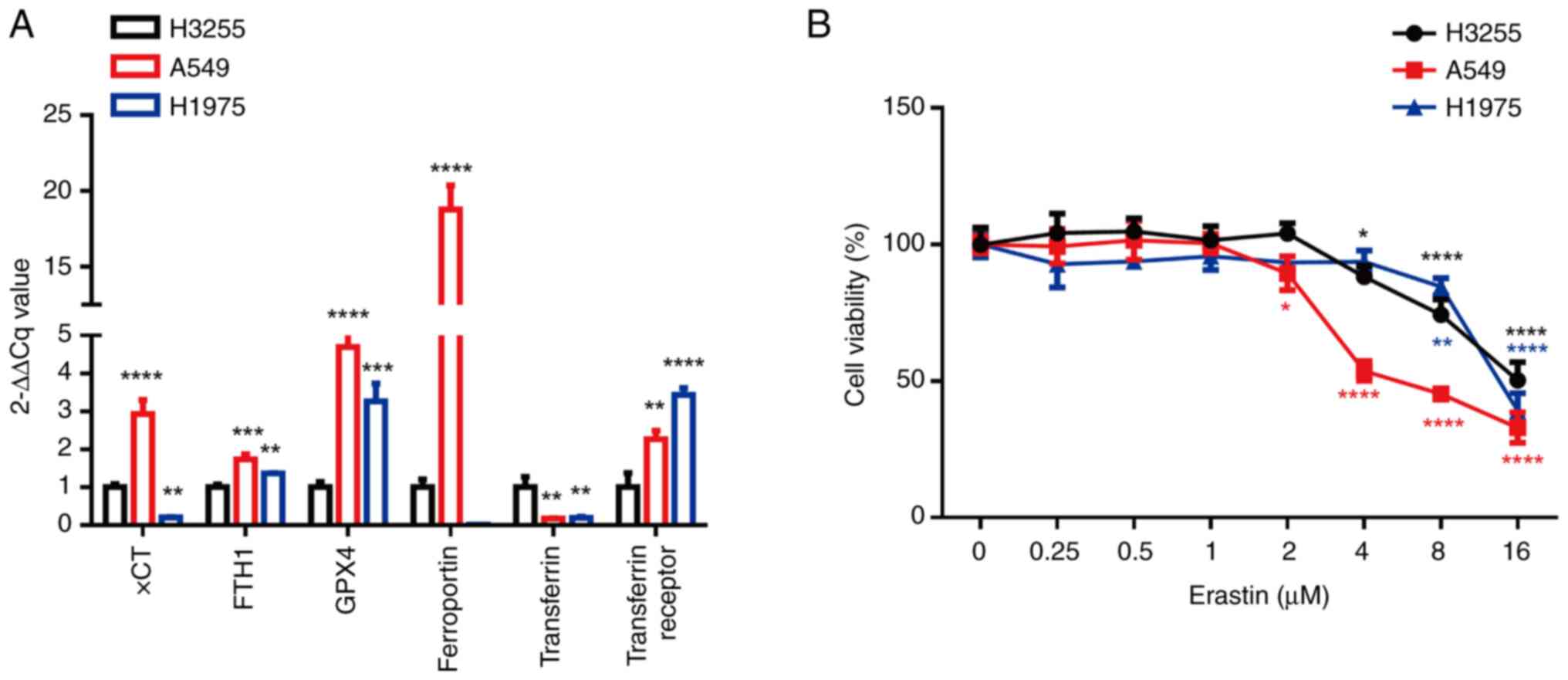 | Figure 1.Different epidermal growth factor
receptor mutant non-small cell lung cancer cell lines have a
different sensitivity to a ferroptosis inducer. (A) mRNA expression
levels of ferroptosis markers (xCT, FTH1, GPX4, ferroportin,
transferrin and the transferrin receptor) in A549, H1975 and H3255
cells were analyzed using the 2−ΔΔCq method.
**P<0.01, ***P<0.001 and ****P<0.0001 vs. H3255 cells
using Dunnett's test. (B) A549, H1975 and H3255 cells were treated
with different concentrations of erastin (0, 0.25, 0.5, 1, 2, 4 and
8 µM) and the MTT assay was used to detect the cell viability after
24 h. *P<0.05, **P<0.01, and ****P<0.0001 vs. the control
group (0 µM of Erastin) using Dunnett's test. xCT,
cysteine-glutamate antiporter; FTH1, ferritin heavy-chain
polypeptide 1; GPX4, glutathione peroxidase 4. |
Inhibition of mTOR may induce
ferroptosis in EGFR-TKI resistant mutant and EGFR wild-type
NSCLC
To explore whether RAD001 regulates ferroptosis in
NSCLC with different EGFR mutations, H3255 (EGFR-TKI-sensitive),
H1975 (EGFR-TKI-resistant) and A549 (EGFR wild-type) cells were
treated with RAD001 at different concentrations. It was found that
as the drug concentration increased, cell viability gradually
decreased in a dose-dependent manner. Compared with H3255 cells,
H1975 and A549 cells were more sensitive to RAD001
(IC50: 4.474 mM vs. 8.869 µM vs. 50.79 µM; Fig. 2A). Since ferroptosis is a form of
apoptosis independent cell death, it was first tested whether
different concentrations of RAD001 induce cell apoptosis, and then
explored which concentration of RAD001 might induce ferroptosis.
The three cell types were treated with low- and high-dose RAD001.
Only 0.5 and 1 µΜ RAD001 treatment in A549 cells induced an
increase in cleaved-Caspase3/Caspase3 (Fig. 2B-D). The three cell lines were then
treated with RAD001, ferrostatin-1 (Fer-1) and RAD001 + Fer-1,
respectively. The results showed that compared with RAD001 (1 µM)
control, Fer-1 reduced the cell death induced by RAD001 (1 µM) in
H1975 and A549 cells obviously. The difference was statistically
significant. However, Fer-1 did not reverse cell death in H3255
cells (Fig. 2E-G).
 | Figure 2.RAD001 may regulate ferroptosis in
EGFR T790M/L858R and wild type EGFR non-small cell lung cancer. (A)
H3255, A549 and H1975 cells were treated with different
concentrations of RAD001 (0, 0.25, 0.5, 1, 2, 4, 8 and 16 µM).
After 24 h, cell viability was detected using MTT. **P<0.01,
***P<0.001 and ****P<0.0001 vs. the control group (0 µM of
RAD001) using Dunnett's test. (B) H3255, (C) A549 and (D) H1975
cells were treated with different concentrations of RAD001 (0, 0.5,
1 and 8 µM) for 24 h and cleaved-Caspase3/Caspase3 was detected
using western blotting. GAPDH was used as an internal standard. (E)
H3255, (F) A549 and (G) H1975 cells were treated with different
concentrations of RAD001 (0, 0.5 and 1 µM) combined with Fer-1 (0,
0.125, 0.25, 0.5, 1, 2, 4 and 8 µM). After 24 h, the cell viability
was detected using MTT. *P<0.05, **P<0.01, and
****P<0.0001 vs. the control group (0 µM of Fer-1) using
Dunnett's test. EGFR, epidermal growth factor receptor; GAPDH,
glyceraldehyde-3-phosphate dehydrogenase; Fer-1, ferrostatin-1. |
mTOR inhibitor combined with an
ferroptosis inducer promotes ferroptosis in EGFR resistant mutant
and wild type EGFR NSCLC cells
The H3255, A549 and H1975 cells were treated with
RAD001, erastin (ferroptosis inducer) and RAD001 + erastin,
respectively. After 24 h, the results showed that erastin induced
H1975, A549 and H3255 cell death in a dose-dependent manner. In
addition, compared with H3255/A549 cells, H1975 cells were more
sensitive to RAD001 combined with erastin. The IC50s of
RAD001 (0.5 µM) + erastin were 15.15, 15.35 and 4.106 µM in H3255,
A549 and H1975 cells, respectively. The IC50s of RAD001
(1 µM) + erastin were 15.87, 15.46 and 1.653 µM in H3255, A549 and
H1975 cells, respectively (Fig.
3A-C).
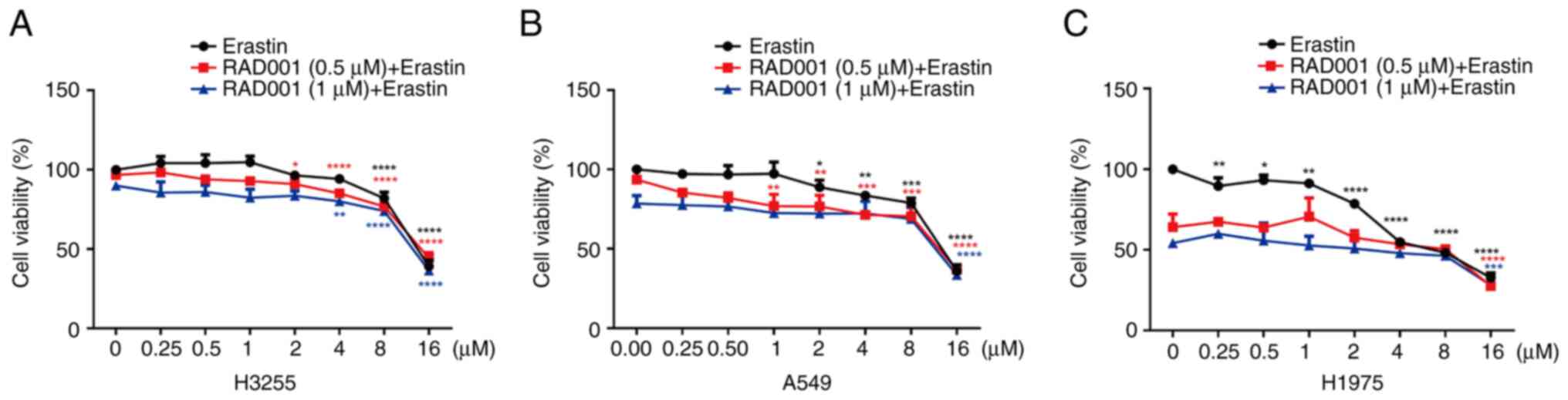 | Figure 3.RAD001 combined with erastin promotes
ferroptosis in EGFR T790M/L858R and wild-type EGFR non-small cell
lung cancer. (A) H3255, (B) A549 and (C) H1975 cells were treated
with different concentrations of RAD001 (0, 0.5 and 1 µM) combined
with erastin (0, 0.25, 0.5, 1, 2, 4, 8 and 16 µM). After 24 h, the
cell viability was detected using MTT. *P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001 vs. the control group (0 µM of
erastin) using Dunnett's test. EGFR, epidermal growth factor
receptor. |
Inhibiting mTOR promotes ferroptosis
in EGFR resistant mutant and wild-type EGFR NSCLC cells by inducing
lipid peroxidation
H3255, A549 and H1975 cells were treated with low or
high concentrations of RAD001 (Fig.
4A-C). The results showed that 0.5, M and 8 µM of RAD001
inhibited mTOR activity by 56, 43 and 40% in A549 cells,
respectively. In addition, 0.5, 1 and 8 µM of RAD001 inhibited mTOR
activity by 5, 14 and 22% in H1975 cells, respectively. However,
these concentrations of RAD001 did not inhibit mTOR activity in
H3255 cells. It was found that 8 µΜ RAD001 significantly decreased
the expression of GPX4, FTH1 and ferroportin by 43, 31 and 22%,
respectively, in A549 cells and by 41, 29 and 26% in H1975 cells,
respectively. However, RAD001 did not markedly alter GPX4, FTH1 or
ferroportin expression in H3255 cells. Following treatment with
different concentrations of RAD001 in A549, H3255 and H1975 cells
for 4 h, high concentrations (8 µM) of RAD001 significantly induced
the accumulation of ROS by 52, 33 and 55% in these three types of
cells, respectively (Fig. 4D).
Moreover, 8 µM RAD001 increased the level of MDA by 23% in A549
cells and by 31% in H1975 cells, respectively. However, 8 µM of
RAD001 did not significantly alter MDA in H3255 cells (Fig. 4E). These results suggested that
RAD001 induced lipid peroxidation in EGFR-resistant mutant and
wild-type EGFR NSCLC cells by inhibiting mTOR, leading to
ferroptosis.
 | Figure 4.Effect of RAD001 on ferroptosis
related proteins and ROS in EGFR T790M/L858R and wild-type EGFR
non-small cell lung cancer. (A) H3255, (B) A549 and (C) H1975 cells
were treated with different concentrations of RAD001 (0, 0.5, 1 and
8 µM). After 24 h, the expression levels of p-mTOR, mTOR, GPX4,
FTH1, ferroportin, transferrin and the transferrin receptor were
detected using western blotting. GAPDH was used as an internal
standard. *P<0.05, **P<0.01, ***P<0.001 and
****P<0.0001 vs. the control group (0 µM of RAD001) using
Dunnett's test. (D) H3255, A549 and H1975 cells were treated with
different concentrations of RAD001 (0, 0.5, 1 and 8 µM) for 4 h.
The ROS levels were detected using flow cytometry. *P<0.05,
**P<0.01 and ***P<0.001 vs. the control group (0 µM of
RAD001) using Dunnett's test. (E) H3255, A549, and H1975 cells were
treated with different concentrations of RAD001 (0, 0.5, 1, and 8
µM) for 4 h. The accumulation of lipid peroxidation was detected
using an MDA kit *P<0.05 and ****P<0.0001 vs. the control
group (0 µM of RAD001) using Dunnett's test. EGFR, epidermal growth
factor receptor; GAPDH, glyceraldehyde-3-phosphate dehydrogenase;
ROS, reactive oxygen species; MDA, malondialdehyde; p-,
phosphorylated; mTOR, mammalian target of rapamycin; FTH1, ferritin
heavy-chain polypeptide 1; GPX4, glutathione peroxidase 4. |
Inhibition of GPX4 inhibits AKT/mTOR
and promotes ferroptosis in EGFR resistant mutant NSCLC cells
To clarify the regulatory relationship between mTOR
and GPX4, H3255, A549 and H1975 cells were treated with the GPX4
inhibitor, RSL3. The results showed that RSL3 notably inhibited
cell viability in a dose-dependent manner. Furthermore, compared
with A549 and H1975 cells, H3255 cells were the most sensitive to
RSL3 (Fig. 5A). H1975, A549 and
H3255 cells were also treated with different concentrations of
RAD001 in combination with RSL3. Subsequent, western blotting
showed that high-dose RAD001 markedly inhibited the AKT/mTOR
pathway in H1975 and A549 cells compared with H3255 cells.
High-dose RSL3 inhibited AKT/mTOR pathway notably in H1975 cells
and the combination of RAD001 and RSL3 further enhanced the
inhibitory effect on the AKT/mTOR pathway in this type of cells;
however, this effect was not observed in A549 cells, possibly due
to the low mTOR expression in A549 cells (Fig. 5B-D).
 | Figure 5.Inhibition of GPX4 inhibits mTOR/AKT
pathway of EGFR T790M/L858R and wild-type EGFR non-small cell lung
cancer cells. (A) H3255, A549 and H1975 cells were treated with
different concentrations of RSL3 (0, 0.25, 0.5, 1, 2, 4, 8 and 16
µM) for 24 h. The cell viability was detected using MTT.
**P<0.01, ***P<0.001 and ****P<0.0001 vs. the control
group (0 µM of RSL3) using Dunnett's test. (B) H1975, (C) A549 and
(D) H3255 cells were treated with different concentrations of
RAD001 (0, 0.5, 1 and 8 µM) combined with RSL3 (0, 0.1 and 0.5 µM).
After 24 h, the expression of p-mTOR, mTOR and AKT were detected
using western blotting, and GAPDH expression was used as an
internal standard. EGFR, epidermal growth factor receptor; GAPDH,
glyceraldehyde-3-phosphate dehydrogenase; p-, phosphorylated; mTOR,
mammalian target of rapamycin. |
Discussion
TKIs have greatly changed the clinical prospects of
patients with NSCLC with EGFR activation mutations (31). Although disease control is prolonged
and the tumor response rate is high, all patients eventually
progress after EGFR-TKI treatment (32). Therefore, there is an urgent need to
develop novel treatment strategies for these patients. In addition,
if there are no co-mutations in driving genes such as anaplastic
lymphoma kinase and ROS1, it is difficult for patients with NSCLC
with wild-type EGFR to benefit from targeted therapy. In the
present study, compared with H3255 cells harboring the EGFR L858R
mutation, H1975 cells with the EGFR T790M/L858R mutation and A549
cells with the EGFR wild-type mutation were more sensitive to the
ferroptosis inducer, erastin, suggesting that targeted ferroptosis
may be a potential treatment against EGFR T790M/L858R resistant
mutant and wild-type NSCLC. In addition, A549 cells harbor the KRAS
G12S mutation. KRAS mutation is a common carcinogenic driver
mutation that accounts for ~35% of lung adenocarcinomas (33). Although sotorasib (AMG510), a drug
targeting the KRAS G12C mutation, has been approved by the FDA as a
second-line treatment for locally advanced or metastatic NSCLC with
the KRAS G12C mutation (34), there
are no approved targeted drugs for other KRAS mutation types
(35). A study has shown that KRAS
mutations are associated with EGFR-TKI resistance (36). In NSCLC, 15–30% of patients with
adenocarcinoma have functional mutations in the KRAS gene, which
means that the tumors in these patients do not respond to EGFR-TKIs
(37). The targeted regulation of
ferroptosis has a significant antitumor effect on EGFR-TKI
resistant NSCLC treated with EGFR-TKIs such as gefitinib (20). Therefore, based on the findings of
the present and aforementioned studies, the regulation of
ferroptosis may be an effective treatment strategy against tumors
harboring KRAS G12S and EGFR-TKI resistance.
Abnormal activation of mTOR is common in a variety
of cancer types such as lung adenocarcinoma, lymphoma and melanoma
(38–40), and is related to its role as a key
effector downstream of several carcinogenic pathways, such as
PI3K/AKT and RAS/RAF/MEK/ERK, as well as tumor inhibitory pathways,
such as p53 and LKB1 (38,41–44). A
study has shown that mTOR is an important target for the regulation
of ferroptosis in many types of tumor cells, like breast cancer and
prostate cancer (45). The results
of the present study showed that, compared with EGFR L858R mutant
NSCLC, EGFR T790M/L858R and EGFR wild-type NSCLC were more
sensitive to the mTOR inhibitor, RAD001. RAD001-induced cell death
in these two cell types was reduced by the ferroptosis inhibitor,
Fer-1. Data from a clinical study suggest that mTOR inhibitors, as
single drugs, typically have relatively moderate therapeutic
effects, possibly due to the lack of strong cytotoxic effects
induced by these inhibitors (40).
RAD001 has been approved for the treatment of advanced renal cell
carcinoma, PNET and advanced breast cancer (46). However, designing an appropriate
combination therapy to enhance the cytotoxicity induced by mTOR
inhibition remains an unmet demand in clinical research (40). Recent preclinical studies have
further proven that co-targeting mTOR and ferroptosis may be a
promising cancer treatment strategy (45,47,48).
The results of the present study showed that compared with EGFR
L858R mutant NSCLC cells/EGFR wild-type NSCLC cells, EGFR
T790M/L858R mutant NSCLC cells were more sensitive to RAD001
combined with the ferroptosis inducer, erastin, suggesting that the
combination of mTOR inhibitor and erastin may be a potential
treatment strategy for EGFR-resistant mutant NSCLC cells.
mTOR complex 1 mediates cystine-induced GPX4 protein
synthesis, inhibits lipid peroxidation and protects cells from
ferroptosis (40). The results of
the present study showed that RAD001 markedly reduced mTOR activity
in EGFR T790M/L858R mutant and wild-type NSCLC cells, resulting in
decreased GPX4 protein levels and increased ROS levels. Therefore,
the molecular mechanisms underlying the inhibition of mTOR-induced
ferroptosis may be related to the accumulation of lipid
peroxidation caused by the reduction in GPX4. In addition, the
results of the present study demonstrated that the inhibition of
mTOR not only changed the expression of GPX4 protein but also
reduced the expression of the iron storage protein, FTH1, and the
iron transporter, ferroportin. Since FTH1 and ferroportin have
important roles in the regulation of iron metabolism in cells,
their reduction can lead to iron metabolism disorders and induce
ferroptosis (49). The results of
the present study indicated that inhibition of mTOR induced
ferroptosis in EGFR T790M/L858R mutant and wild-type NSCLC cells by
inducing lipid peroxidation and iron metabolism disorder. In
addition, the effect of GPX4 regulation on the AKT/mTOR pathway was
observed and it was found that GPX4 inhibition effectively
inhibited the AKT/mTOR pathway in H1975 cells. This may be related
to the positive regulatory relationship between AKT/mTOR and GPX4
(50,51). Notably, compared with H1975 and A549
cells, H3255 cells were the most sensitive to the GPX4 inhibitor,
RSL3, and the least sensitive to erastin, which may be related to
the different mechanisms of these drugs. Erastin exerts a
significant antitumor effect on wild-type EGFR cells by inducing
ROS-mediated caspase-independent cell death (52). Erastin binds directly to
voltage-dependent anion channel 2 and produces ROS in an
NADH-dependent manner, leading to mitochondrial damage. In certain
tumor cells with activated mutations, erastin induces cell death
via the RAS/RAF/MEK pathway (53).
Another study has shown that RSL3 inactivated GPX4, induced ROS
production by lipid peroxidation and rapidly induced ferroptosis in
RAS-mutant cells (54). Therefore,
for NSCLC with different EGFR mutations, different drugs that
regulate ferroptosis may be selected to achieve the most effective
antitumor effects. There were certain limitations to the present
study. In this study, H1975 (EGFR T790M/L858R), H3255 (EGFR L858R)
and A549 (EGFR wild type) cells were selected to test our
hypothesis. Experiments performed on other EGFR-mutant NSCLC types
of cells would strengthen the significance of the present results
and conclusion. The experiments using in vivo animal models
could also provide more robust results. Moreover, small cell lung
cancer (SCLC), squamous cell lung cancer or large cell
neuroendocrine lung carcinoma cell lines were not screened. Bebber
et al (55) reported that
non-neuroendocrine SCLC is vulnerable to ferroptosis. In our
ongoing study, it was found that, compared with neuroendocrine
SCLC, non-neuroendocrine SCLC cells with upregulated GPX4
expression were not sensitive to mTOR inhibitors. Therefore,
targeting mTOR may not induce ferroptosis by inhibiting GPX4 in
SCLC. We will further explore the role and molecular mechanism of
targeting mTOR to regulate ferroptosis in NSCLC, SCLC and large
cell neuroendocrine lung carcinoma.
In conclusion, the sensitivity of different EGFR
mutation types to ferroptosis was described in the present study
and the induction of ferroptosis by targeting mTOR was confirmed to
be a potential treatment strategy for EGFR T790M/L858R and
wild-type EGFR NSCLC.
Acknowledgements
Not applicable.
Funding
The work was supported by the Health and Family Planning
Commission of Jilin Province (grant no. 2021JC095) and the
Department of Science and Technology of Jilin Province (grant no.
YDZJ202301ZYTS512).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YC, HL, CJW and RZ designed the study; RZ and TXW
performed the experiments; RZ and CYH analyzed the data; CJW and RZ
wrote the manuscript. YLJ performed flow cytometry. All authors
read and approved the final version of the manuscript. CJW and RZ
confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: The impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. CA Cancer J
Clin. 61:212–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ochi N, Takeyama M, Miyake N, Fuchigami M,
Yamane H, Fukazawa T, Nagasaki Y, Kawahara T, Nakanishi H and
Takigawa N: The complexity of EGFR exon 19 deletion and L858R
mutant cells as assessed by proteomics, transcriptomics, and
metabolomics. Exp Cell Res. 424:1135032023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hamaguchi R, Okamoto T, Sato M, Hasegawa M
and Wada H: Effects of an alkaline diet on EGFR-TKI therapy in EGFR
mutation-positive NSCLC. Anticancer Res. 37:5141–5145.
2017.PubMed/NCBI
|
|
4
|
Lim SH, Lee JY, Sun JM, Ahn JS, Park K and
Ahn MJ: Comparison of clinical outcomes following gefitinib and
erlotinib treatment in non-small-cell lung cancer patients
harboring an epidermal growth factor receptor mutation in either
exon 19 or 21. J Thorac Oncol. 9:506–511. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liang H, Pan Z, Wang W, Guo C, Chen D,
Zhang J, Zhang Y, Tang S, He J and Liang W; written on behalf of
AME Lung Cancer Cooperative Group, : The alteration of T790M
between 19 del and L858R in NSCLC in the course of EGFR-TKIs
therapy: A literature-based pooled analysis. J Thorac Dis.
10:2311–2320. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Miller VA, Hirsh V, Cadranel J, Chen YM,
Park K, Kim SW, Zhou C, Su WC, Wang M, Sun Y, et al: Afatinib
versus placebo for patients with advanced, metastatic
non-small-cell lung cancer after failure of erlotinib, gefitinib,
or both, and one or two lines of chemotherapy (LUX-Lung 1): A phase
2b/3 randomised trial. Lancet Oncol. 13:528–538. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Walter AO, Sjin RT, Haringsma HJ, Ohashi
K, Sun J, Lee K, Dubrovskiy A, Labenski M, Zhu Z, Wang Z, et al:
Discovery of a mutant-selective covalent inhibitor of EGFR that
overcomes T790M-mediated resistance in NSCLC. Cancer Discov.
3:1404–1415. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ward RA, Anderton MJ, Ashton S, Bethel PA,
Box M, Butterworth S, Colclough N, Chorley CG, Chuaqui C, Cross DA,
et al: Structure- and reactivity-based development of covalent
inhibitors of the activating and gatekeeper mutant forms of the
epidermal growth factor receptor (EGFR). J Med Chem. 56:7025–7048.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thress KS, Paweletz CP, Felip E, Cho BC,
Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, et
al: Acquired EGFR C797S mutation mediates resistance to AZD9291 in
non-small cell lung cancer harboring EGFR T790M. Nat Med.
21:560–562. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou F and Zhou CC: Targeted therapies for
patients with advanced NSCLC harboring wild-type EGFR: What's new
and what's enough. Chin J Cancer. 34:310–319. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee JK, Hahn S, Kim DW, Suh KJ, Keam B,
Kim TM, Lee SH and Heo DS: Epidermal growth factor receptor
tyrosine kinase inhibitors vs conventional chemotherapy in
non-small cell lung cancer harboring wild-type epidermal growth
factor receptor: A meta-analysis. JAMA. 311:1430–1437. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee JY, Kim WK, Bae KH, Lee SC and Lee EW:
Lipid metabolism and ferroptosis. Biology (Basel).
10:1842021.PubMed/NCBI
|
|
13
|
Seibt TM, Proneth B and Conrad M: Role of
GPX4 in ferroptosis and its pharmacological implication. Free Radic
Biol Med. 133:144–152. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK,
Kagan VE, et al: Ferroptosis: A regulated cell death nexus linking
metabolism, redox biology, and disease. Cell. 171:273–285. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Badgley MA, Kremer DM, Maurer HC,
DelGiorno KE, Lee HJ, Purohit V, Sagalovskiy IR, Ma A, Kapilian J,
Firl CEM, et al: Cysteine depletion induces pancreatic tumor
ferroptosis in mice. Science. 368:85–89. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wish JB: Assessing iron status: Beyond
serum ferritin and transferrin saturation. Clin J Am Soc Nephrol. 1
(Suppl 1):S4–S8. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh
HJ III, Kang R and Tang D: Autophagy promotes ferroptosis by
degradation of ferritin. Autophagy. 12:1425–1428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gkouvatsos K, Papanikolaou G and
Pantopoulos K: Regulation of iron transport and the role of
transferrin. Biochim Biophys Acta. 1820:188–202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fang X, Wang H, Han D, Xie E, Yang X, Wei
J, Gu S, Gao F, Zhu N, Yin X, et al: Ferroptosis as a target for
protection against cardiomyopathy. Proc Natl Acad Sci USA.
116:2672–2680. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang C, Lu X, Liu X, Xu J, Li J, Qu T,
Dai J and Guo R: Carbonic anhydrase IX controls vulnerability to
ferroptosis in gefitinib-resistant lung cancer. Oxid Med Cell
Longev. 2023:13679382023. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu C, Jiang ZB, Shao L, Zhao ZM, Fan XX,
Sui X, Yu LL, Wang XR, Zhang RN, Wang WJ, et al: β-Elemene enhances
erlotinib sensitivity through induction of ferroptosis by
upregulating lncRNA H19 in EGFR-mutant non-small cell lung cancer.
Pharmacol Res. 191:1067392023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vasan N, Razavi P, Johnson JL, Shao H,
Shah H, Antoine A, Ladewig E, Gorelick A, Lin TY, Toska E, et al:
Double PIK3CA mutations in cis increase oncogenicity and
sensitivity to PI3Kα inhibitors. Science. 366:714–723. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Aylett CH, Sauer E, Imseng S, Boehringer
D, Hall MN, Ban N and Maier T: Architecture of human mTOR complex
1. Science. 351:48–52. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hasskarl J: Everolimus. Recent Results
Cancer Res. 211:101–123. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fujiwara R, Taniguchi Y, Rai S, Iwata Y,
Fujii A, Fujimoto K, Kumode T, Serizawa K, Morita Y, Espinoza JL,
et al: Chlorpromazine cooperatively induces apoptosis with tyrosine
kinase inhibitors in EGFR-mutated lung cancer cell lines and
restores the sensitivity to gefitinib in T790M-harboring resistant
cells. Biochem Biophys Res Commun. 626:156–166. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ni J, Chen K, Zhang J and Zhang X:
Inhibition of GPX4 or mTOR overcomes resistance to Lapatinib via
promoting ferroptosis in NSCLC cells. Biochem Biophys Res Commun.
567:154–160. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lou JS, Zhao LP, Huang ZH, Chen XY, Xu JT,
Tai WC, Tsim KWK, Chen YT and Xie T: Ginkgetin derived from Ginkgo
biloba leaves enhances the therapeutic effect of cisplatin via
ferroptosis-mediated disruption of the Nrf2/HO-1 axis in EGFR
wild-type non-small-cell lung cancer. Phytomedicine. 80:1533702021.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yan WY, Cai J, Wang JN, Gong YS and Ding
XB: Co-treatment of betulin and gefitinib is effective against EGFR
wild-type/KRAS-mutant non-small cell lung cancer by inducing
ferroptosis. Neoplasma. 69:648–656. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Huang Y, Chen Y, Mei Q, Chen Y, Yu S and
Xia S: Combined inhibition of the EGFR and mTOR pathways in EGFR
wild-type non-small cell lung cancer cell lines with different
genetic backgrounds. Oncol Rep. 29:2486–2492. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Singh S, Sadhukhan S and Sonawane A: 20
Years since the approval of first EGFR-TKI, gefitinib: Insight and
foresight. Biochim Biophys Acta Rev Cancer. 1878:1889672023.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Morimoto K, Yamada T, Takeda T, Shiotsu S,
Date K, Tamiya N, Goto Y, Kanda H, Chihara Y, Kunimatsu Y, et al:
Clinical efficacy and safety of first- or second-generation
EGFR-TKIs after osimertinib resistance for EGFR mutated lung
cancer: A prospective exploratory study. Target Oncol. 18:657–665.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Reck M, Carbone DP, Garassino M and
Barlesi F: Targeting KRAS in non-small-cell lung cancer: Recent
progress and new approaches. Ann Oncol. 32:1101–1110. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nakajima EC, Drezner N, Li X,
Mishra-Kalyani PS, Liu Y, Zhao H, Bi Y, Liu J, Rahman A, Wearne E,
et al: FDA approval summary: Sotorasib for KRAS G12C-mutated
metastatic NSCLC. Clin Cancer Res. 28:1482–1486. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hofmann MH, Gerlach D, Misale S,
Petronczki M and Kraut N: Expanding the reach of precision oncology
by drugging all KRAS mutants. Cancer Discov. 12:924–937. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pao W, Wang TY, Riely GJ, Miller VA, Pan
Q, Ladanyi M, Zakowski MF, Heelan RT, Kris MG and Varmus HE: KRAS
mutations and primary resistance of lung adenocarcinomas to
gefitinib or erlotinib. PLoS Med. 2:e172005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rodenhuis S, Slebos RJ, Boot AJ, Evers SG,
Mooi WJ, Wagenaar SS, van Bodegom PC and Bos JL: Incidence and
possible clinical significance of K-ras oncogene activation in
adenocarcinoma of the human lung. Cancer Res. 48:5738–5741.
1988.PubMed/NCBI
|
|
38
|
Saxton RA and Sabatini DM: mTOR signaling
in growth, metabolism, and disease. Cell. 168:960–976. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu GY and Sabatini DM: mTOR at the nexus
of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol.
21:183–203. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lei G, Zhuang L and Gan B: mTORC1 and
ferroptosis: Regulatory mechanisms and therapeutic potential.
Bioessays. 43:e21000932021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Feng Z, Zhang H, Levine AJ and Jin S: The
coordinate regulation of the p53 and mTOR pathways in cells. Proc
Natl Acad Sci USA. 102:8204–8209. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shaw RJ, Bardeesy N, Manning BD, Lopez L,
Kosmatka M, DePinho RA and Cantley LC: The LKB1 tumor suppressor
negatively regulates mTOR signaling. Cancer Cell. 6:91–99. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Byun JK, Park M, Yun JW, Lee J, Kim JS,
Cho SJ, Lee YM, Lee IK, Choi YK and Park KG: Oncogenic KRAS
signaling activates mTORC1 through COUP-TFII-mediated lactate
production. EMBO Rep. 20:e474512019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Prabowo AS, Iyer AM, Veersema TJ, Anink
JJ, Schouten-van Meeteren AY, Spliet WG, van Rijen PC, Ferrier CH,
Capper D, Thom M and Aronica E: BRAF V600E mutation is associated
with mTOR signaling activation in glioneuronal tumors. Brain
Pathol. 24:52–66. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yi J, Zhu J, Wu J, Thompson CB and Jiang
X: Oncogenic activation of PI3K-AKT-mTOR signaling suppresses
ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci USA.
117:31189–31197. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hua H, Kong Q, Zhang H, Wang J, Luo T and
Jiang Y: Targeting mTOR for cancer therapy. J Hematol Oncol.
12:712019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang Y, Swanda RV, Nie L, Liu X, Wang C,
Lee H, Lei G, Mao C, Koppula P, Cheng W, et al: mTORC1 couples
cyst(e)ine availability with GPX4 protein synthesis and ferroptosis
regulation. Nat Commun. 12:15892021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu Y, Wang Y, Liu J, Kang R and Tang D:
Interplay between MTOR and GPX4 signaling modulates
autophagy-dependent ferroptotic cancer cell death. Cancer Gene
Ther. 28:55–63. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Muhoberac BB and Vidal R: Iron, Ferritin,
hereditary ferritinopathy, and neurodegeneration. Front Neurosci.
13:11952019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Jung KH, Kim SE, Go HG, Lee YJ, Park MS,
Ko S, Han BS, Yoon YC, Cho YJ, Lee P, et al: Synergistic
renoprotective effect of melatonin and zileuton by inhibition of
ferroptosis via the AKT/mTOR/NRF2 signaling in kidney injury and
fibrosis. Biomol Ther (Seoul). 31:599–610. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hu Z and Li L, Li M, Zhang X, Zhang Y, Ran
J and Li L: miR-21-5p inhibits ferroptosis in hepatocellular
carcinoma cells by regulating the AKT/mTOR signaling pathway
through MELK. J Immunol Res. 2023:89295252023. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yamaguchi H, Hsu JL, Chen CT, Wang YN, Hsu
MC, Chang SS, Du Y, Ko HW, Herbst R and Hung MC:
Caspase-independent cell death is involved in the negative effect
of EGF receptor inhibitors on cisplatin in non-small cell lung
cancer cells. Clin Cancer Res. 19:845–854. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yagoda N, von Rechenberg M, Zaganjor E,
Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM,
Boniface JJ, et al: RAS-RAF-MEK-dependent oxidative cell death
involving voltage-dependent anion channels. Nature. 447:864–868.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun
X, Kang R and Tang D: Ferroptosis: Process and function. Cell Death
Differ. 23:369–379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Bebber CM, Thomas ES, Stroh J, Chen Z,
Androulidaki A, Schmitt A, Höhne MN, Stüker L, de Pádua Alves C,
Khonsari A, et al: Ferroptosis response segregates small cell lung
cancer (SCLC) neuroendocrine subtypes. Nat Commun. 12:20482021.
View Article : Google Scholar : PubMed/NCBI
|














