Introduction
Copy number alterations (CNAs) refer to the gain or
loss of sections of genetic material, which can contribute to
disease development by modifying gene dosage and functionality
(1,2). In non-small cell lung cancer (NSCLC),
CNAs are common and diverse, affecting critical biological
processes that promote tumorigenesis (3,4).
Genome-wide copy number (CN) analysis has been proposed as a
diagnostic, pathological and prognostic tool for NSCLC (5–7).
The investigation of focal CNAs in programmed death
ligand 1 (PD-L1) and EGFR genes has gained interest due to their
close association with targeted therapy. Notably, alterations in
PD-L1 CN have been shown to influence PD-L1 immunopositivity, tumor
proportion score (TPS) and patient survival (8–10).
Meanwhile, variations in EGFR CN have been found to influence the
overall survival of NSCLC patients with EGFR-mutated tumors and can
predict lung cancer metastasis to the brain (11,12).
These findings have indicated the importance of CNA analysis in
understanding lung cancer pathophysiology and predicting treatment
outcomes in patients with lung cancer. However, CNAs are complex
and dynamic, and the landscape of CN perturbations in NSCLC remains
largely unexplored.
Previously, we reported that the lineage-survival
oncogene NK2 homeobox 1 (NKX2-1), also known as thyroid
transcription factor 1 (TTF-1), may upregulate the expression of
oncogenic proteins, and could influence the signaling pathways
associated with EGFR and PD-L1 (13). Additionally, NKX2-1 CN is
significantly correlated with NKX2-1 protein expression (14), which justifies the interest in
studying the prognostic implications of focal CN amplification
(14,15). However, NKX2-1 CN is variable in
lung cancer and only ~15% of cases of NSCLC have amplifications
(16). Thus, the elucidation and
comparison of tumor biology in NSCLC with focal loss and gain of
NKX2-1 CN remain poorly investigated in lung cancer. Additionally,
the clinical significance of NKX2-1 CNAs and their association with
targeted therapy remain elusive. The present study reports on a
comprehensive investigation involving the molecular features
associated with focal alterations in NKX2-1 CN, their prognostic
significance in patient survival, association with chromosomal
instability and implications for targeted therapy.
Materials and methods
Study design and cohorts
The present study was conducted using clinical data
from the Clinical Proteomics for Cancer Initiative program
described elsewhere (Filipino cohort) (13,17,18);
lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC)
data from The Cancer Genome Atlas (TCGA cohort) (19); and longitudinal lung cancer
single-cell RNA-sequencing (scRNA-seq) project data (Bivona cohort)
(20). All participants were
confirmed as having NSCLC with either adenocarcinoma or squamous
histology. A total of 45, 49 and 1,130 enrolled participants were
included in the analyses from the Filipino, Bivona and TCGA
cohorts, respectively. The tumor molecular data of TCGA cohort were
obtained from 501 patients with LUSC and 629 with LUAD. The tumor
sequencing data from the Bivona cohort had 1,654 unique scRNA-seq
profiles. In the Filipino cohort, NKX2-1/TTF-1 and PD-L1 expression
levels were analyzed using immunohistochemistry (IHC), whereas EGFR
mutational frequency was determined using PCR. In both TCGA and
Bivona cohorts, expression levels, mutational frequencies and CNs
were retrieved using the curated databases of UCSC Xena platform
(21) and GDC Data Portal (22). The study involving the Filipino
cohort was approved by The Institutional Ethics Review Board
(approval no. LCP-CS-001-2019) of the Lung Center of the
Philippines (Quezon City, Philippines). All patients provided
written informed consent for genetic testing, as well as for the
use of their clinical data. The clinicodemographic and pathological
deidentified data of TCGA and Bivona cohorts were described in each
study publication. The clinicopathological data of the Filipino
cohort are described in Table
SI.
PCR analysis and IHC
In accordance with the status as a multicenter
study, the protocol for specimen collection was approved by The
Single Joint Research Ethics Board of the Department of Health,
Manila, Philippines (approval no. SJREB-2020-97). Blood and tissues
samples were collected from recruited Filipino patients with NSCLC
and were immediately processed for plasma testing,
histopathological examination or cryopreservation at −80°C, as
previously described (18). The
specimens were tested for the presence of EGFR-sensitizing
mutations using PCR, and assayed for PD-L1 and TTF-1 protein
expression using IHC. EGFR mutations were assessed using the AmoyDx
EGFR 29 Mutations Detection Kit (designed to detect mutations in
EGFR exons 19–21; Amoy Diagnostics Co., Ltd.) as previously
described (23). EGFR without
driver mutations, with single and co-mutations were scored as 0, 1
and 2, respectively. Expression of PD-L1 and NKX2-1/TTF-1 in NSCLC
were evaluated by IHC using the pharmDx 22C3 kit (cat no. SK006;
Agilent Technologies, Inc.) (24)
and clone 8G7G3/1 (cat no. M3575; Agilent Technologies, Inc.)
(25), respectively. After
pathology confirmation, expression was classified as negative
(scored 0) if the TPS was <1%, or positive (scored as 1) if TPS
was ≥1%. The representative histopathological data are shown in
Fig. S1.
Analysis of tumor molecular
profiles
Gene expression, copy number variations (CNVs),
mutational frequencies and treatment modalities of patients from
TCGA and Bivona cohorts were obtained through the UCSC Xena
platform (https://xenabrowser.net). Gene
expression units in TCGA cohort (LUAD and LUSC) were expressed in
log2(TPM+1,) while expression units in the Bivona cohort were
equalized to the depth of sequencing per cell, where log-normalized
counts were scaled by linear regression against the number of reads
(scaled expression) (20). CNVs in
TCGA and Bivona cohorts were expressed as log2-transformed
(https://xenabrowser.net/datapages/)
and raw proportions of tumor to normal counts (https://singlecell.xenabrowser.net/datapages/),
respectively. CNAs were synchronized between cohorts by deriving
log2-transformed values from raw proportion data, or vice versa.
List of driver genes, type of driver mutations, presence of
secondary mutations and treatment interventions of participants in
the Bivona cohort were queried using the ‘Phenotypic Data’ field,
whereas whole genome CNV profile was queried using the ‘Analytic
Data’ field. Tumor mutational architecture (TMA) in chromosomes 7,
X, Y, 15 and 14 were analyzed using tumor profiles in TCGA cohort
by querying ‘Somatic Mutation Dataset’ with chromosomal length
according to hg19 assembly. Tumor mutational burden (TMB) of LUAD
and LUSC tumors in TCGA cohort were extracted from the Data
Exploration of GDC Data Portal (https://portal.gdc.cancer.gov/exploration). Proteome
and phosphoproteome data of tumor samples were retrieved from
Proteomic Data Commons (https://proteomic.datacommons.cancer.gov/pdc/) with
IDs PDC000153, PDC000149 and PDC000149.
Survival analysis
Survival of patients with NSCLC (LUAD and LUSC) in
TCGA cohort were analyzed using the Kaplan Meier Plot through the
Xena platform (21) and survival
analysis in Gene Expression Profiling Interactive Analysis
(26). Median cutoff was used to
assess the survival of patients with high and low CN or gene
expression. Quartile cutoff was used to assess the survival
associated with CN gain and loss. Binary grouping was used to
compare certain subgroups with the rest of the cohort. Log-rank
P-values were reported for each survival analysis with P<0.05
being considered significant.
Gene set operations and clustering
analysis
Mutational signatures associated with NKX2-1 CN gain
(>0.1875; n=251) and loss (<-0.0767; n=251) were assessed by
comparing the TMB of patients with CN gain and loss with the
overall TMB of participants in TCGA cohort (n=1,130). Unique
mutational features of the two prognostic groups were identified by
performing ‘Set Operations Analysis’ in the GDC Data Portal
(https://portal.gdc.cancer.gov/analysis). Mutational
clusters were presented as classic or Edwards’ constructed Venn
diagram generated using jvenn (27). Frequency of genes with commonly or
uniquely altered CN between the two groups was identified using the
set operation in jvenn (http://jvenn.toulouse.inra.fr). Symbols of genes that
were significantly mutated or had altered CNs in patients with
NKX2-1 CN gain and loss were plotted as a word cloud using
WordClouds (https://www.wordclouds.com/).
Enrichment analysis
Functional annotation of molecular, cellular and
biological processes that were potentially affected by genetic
mutations and alteration of CNs was performed using Enrichr
(28,29). Affected pathways were enriched using
BioPlanet (30) and Kyoto
Encyclopedia of Genes and Genomes (31), while perturbed processes were
enriched using Gene Ontology (32).
Chromosomal locations (hg19) were enriched using the sequence
annotation of UCSC Genome Browser (33). Subcellular enrichment was performed
using COMPARTMENTS (34), and
cellular enrichments were performed using CellMarker (35) and Human Gene Atlas in BioGPS
(36) curations. Enrichments with
P<0.05 were considered significant.
Tumor-infiltrating lymphocyte (TIL)
gene marker analysis
The expression signature of 53 widely used gene
markers for TILs (20,37,38)
from TCGA cohort was analyzed against NKX2-1 CNVs. Significantly
correlating genes that were common or unique in patients with
NKX2-1 CN gain and loss were identified and visualized using jvenn.
Direction of linear correlation (positive or negative) and
labelling of significant gene markers was visualized as volcano
plots using VolcaNoseR (39).
Heatmap analysis
Expression levels of JAK, STAT and TIL marker genes
were plotted against NKX2-1 expression or CNV without data labels.
Unsupervised clustering analysis was performed through Heatmapper
using Euclidian average linkage (40). Cluster dendrograms were applied to
data columns.
TIL proportion estimation
Immune infiltration estimation using the gene
expression profiles of 53 TIL markers was performed using CIBERSORT
(41), CIBERSORT-ABS (41), MCPCounter (42) and TIMER (43) algorithms through TIMER 2.0
(http://timer.cistrome.org/). Proportions
of estimated lymphocytes were compared between NKX2-1 CN gain and
loss. Overlapping immune estimation was consolidated based on the
highest absolute significance or lowest P-value.
Statistical analysis
Data are presented as the mean ± standard deviation
using box charts, percentages as pie charts and
frequencies/distributions as bar graphs. Frequency statistics were
used to describe each result of IHC and PCR tests of participants
in the Filipino cohort, NKX2-1 CNV in TCGA cohort, as well as
treatment modalities and genome-wide CNV differences between
patient groups in the Bivona cohort. Pearson's correlation analysis
was used to compare the trend and significance of two variables in
one or two groups. One sample t-test was used to identify the
variability of NKX2-1 CN among NSCLC tumors. Independent sample
t-test was used to determine the statistical difference of two
samples, while one-way ANOVA (without post hoc test) was used to
determine the statistical difference of three or more samples.
P-values and different levels of significance were reported in the
figures and/or data legends. All statistical analysis was performed
using JASP version 0.16.3 (University of Amsterdam). P<0.05 was
considered to indicate a statistically significant difference.
Results
NKX2-1 CNAs have a stronger
correlation with combined EGFR and PD-L1 status of NSCLC tumors
than expression
The correlation of NKX2-1/TTF-1 expression with the
co-occurrence of EGFR-sensitizing mutations and PD-L1 co-expression
in the Filipino cohort was first examined. Immunohistochemical
analysis revealed that most tumors expressed NKX2-1/TTF-1 (98%) and
more than half expressed PD-L1 (51%). The majority of participants
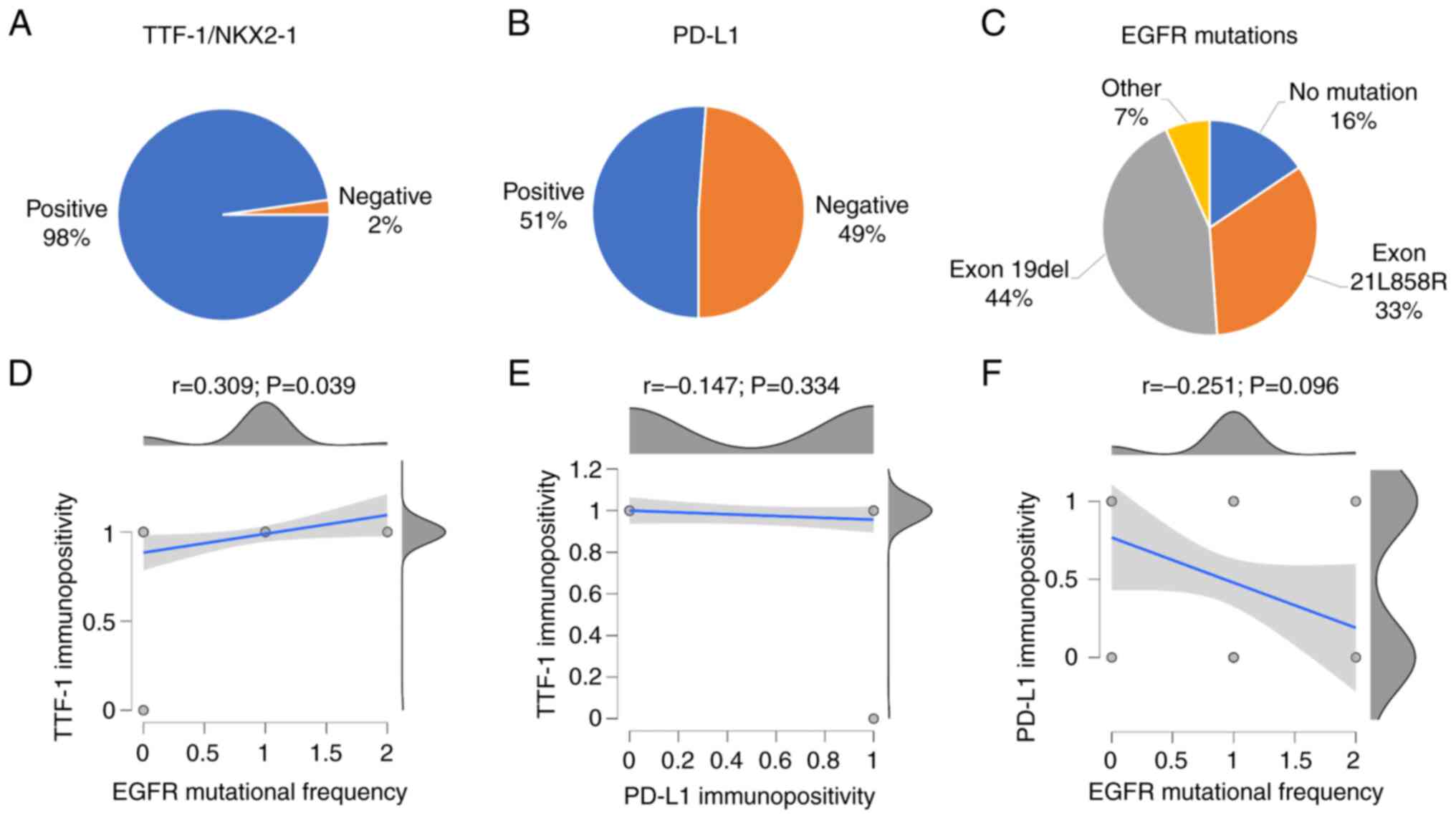
had exon19del (44%) and exon21L858R (33%) mutations (Fig. 1A-C). Pearson's correlation revealed
that NKX2-1/TTF-1 expression was significantly associated with EGFR
mutations (P=0.039). By contrast, PD-L1 positivity had no
significant correlation with NKX2-1/TTF-1 expression (P=0.334) or
EGFR mutation (P=0.096) (Fig.
1D-F). This observation was in agreement with previous reports
(44,45) and may justify their independent
prognostic value.
A previous expression-based analysis in
adenocarcinoma proposed the potential involvement of NKX2-1 in EGFR
and PD-L1 signaling through the PI3K-STAT pathway (13). In vitro experiments found
that NKX2-1 transactivates the receptor tyrosine kinase-like orphan
receptor 1 (ROR1), which in turn promotes EGFR-induced
ERBB3-dependent activation of the PI3K pathway (46). ERBB3 also regulates the expression
of PD-L1 through the activation of the PI3K/PDK1/RSK/CREB signaling
axis (47). In addition, ROR1
activates the proto-oncogene tyrosine-protein kinase Src (c-Src),
which in turn activates the PI3K pathway (46) and constitutively activates STAT
signaling (48). STAT signaling is
known to regulate the expression of PD-L1 (49). This NKX2-1-mediated ROR1-dependent
PI3K-STAT signaling activation could link the influence of NKX2-1
in the combined EGFR and PD-L1 status of lung tumors. Notably, a
previous study partially revealed the crosstalk between EGFR and
PD-L1 pathways through interleukin-mediated PI3K and STAT signaling
(50,51). Expression analysis in NSCLC showed
that ROR1 transcript levels were significantly correlated with the
upregulation of NKX2-1 (r=0.499; P<0.001), and ROR1 protein
levels were positively associated with ERBB3 (r=0.221; P<0.01),
PIK3s (r=0.222–0.266; P<0.01), STATs (r=0.282–0.306; P<0.001)
and c-SRC (r=0.186; P<0.01) protein expression (Fig. S2A and B). Phosphoproteome analysis
of LUAD tumors retrieved from Proteomic Data Commons revealed that
the phosphorylation of ERBB3 was positively associated with
phosphoactivation of c-SRC at different serine, threonine and
tyrosine residues (Fig. S2C).
Additionally, ERBB3 phosphorylation was positively associated with
the phosphorylation of EGFR and PIK3 proteins (Fig. S2D), confirming the contribution of
ERBB3-dependent activation of the PI3K pathway in NSCLC. Lastly,
c-SRC phosphorylation was also positively associated with the
phosphorylation of PIK3 and STAT proteins (Fig. S2E), confirming the c-SRC-mediated
activation of PI3K and STAT signaling in NSCLC. Collectively, these
findings support the involvement of NKX2-1 in influencing the
combined EGFR and PD-L1 status in tumors through ROR1
signaling.
Next, the expression levels of PIK3 and STAT genes
in NSCLC tumors from TCGA cohort were examined. Pearson's
correlation analysis revealed that NKX2-1 expression was
significantly correlated with the positive regulation of PIK3CG,
PIK3CD, PIK3R5, PIK3R6, PIK3C2A/B, STAT3, STAT4, STAT5A/B and STAT6
(all P<0.001). Meanwhile, CNV had a positive correlation with
PIK3C2A and STAT4 only (both P<0.01; Fig. S2F and G), despite a strong positive
correlation detected between NKX2-1 CNV and expression (P<0.001;
Fig. 2A). The variability of NKX2-1
CN among NSCLC tumors was significantly high (P<0.001; Fig. 2B). Subgrouping according to NKX2-1
CN gain and loss revealed that CN loss, but not gain, was
significantly correlated with the positive regulation of PIK3CG,
PIK3CD, PIK3R5, PIK3R6, STAT4, STAT5A (all P<0.001) and STAT5B
(P<0.05; Fig. S2H and I).
Unsupervised heatmap analysis revealed the close association
between NKX2-1 gene expression and CN loss (Fig. S2J). These results signify that CNAs
may influence the combined EGFR and PD-L1 profiles of patients with
lung cancer.
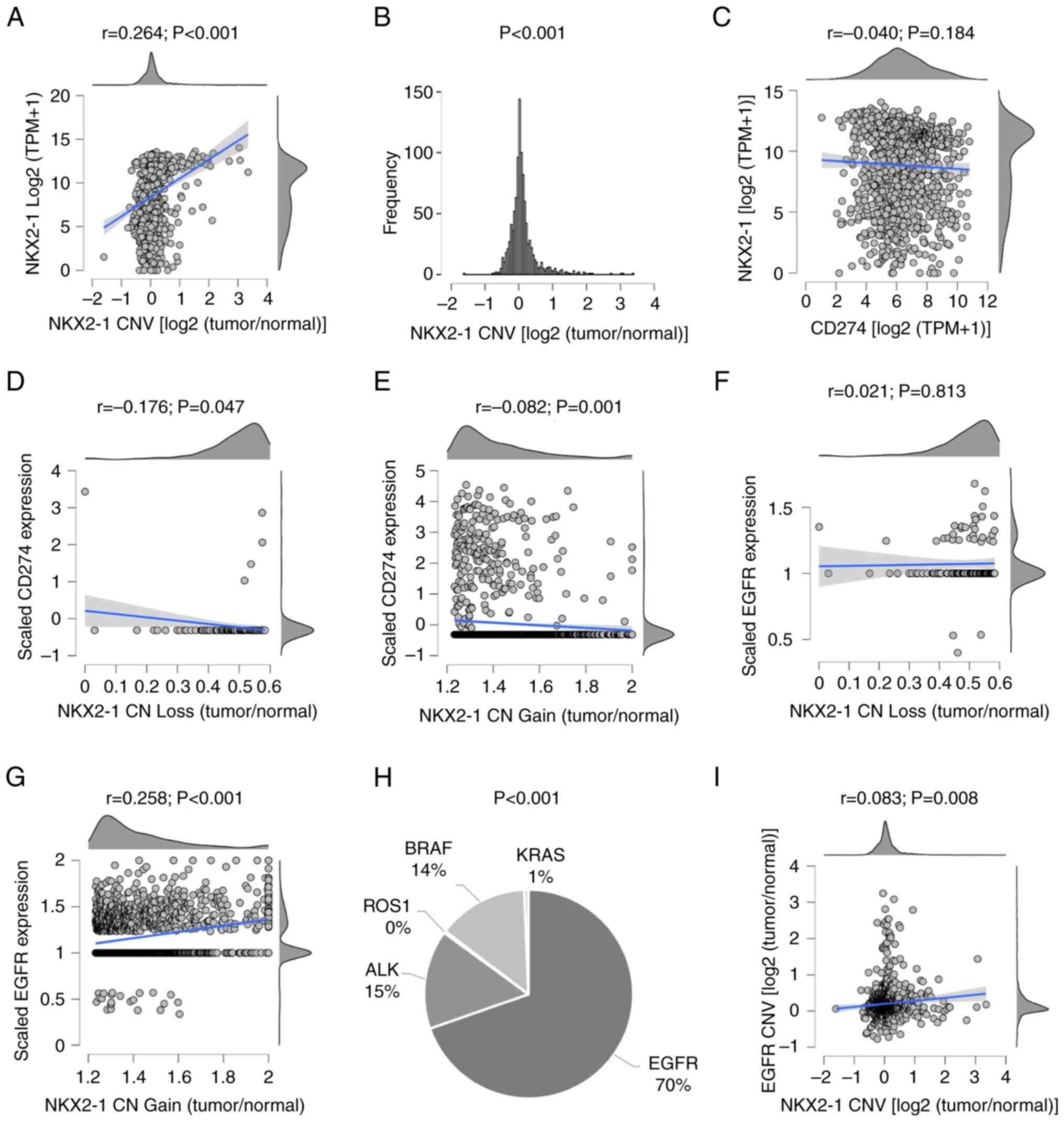 | Figure 2.NKX2-1, PD-L1 and EGFR status, and
types of driver genes in tumors of patients with NSCLC from The
Cancer Genome Atlas and the Bivona cohorts. (A) Intertumoral
variations in NKX2-1 CN and correlation with expression, (B)
frequency of CNV, and (C) correlation between NKX2-1 and CD274
expression. Consequences of (D) loss or (E) gain of NKX2-1 CN on
CD274 expression. Consequence of (F) loss or (G) gain of NKX2-1 CN
on EGFR expression. (H) Clonal heterogeneity in driver mutations in
NSCLC with NKX2-1 CNAs. (I) Co-alteration in EGFR and NKX2-1 CNVs.
Pearson's coefficient was used to analyze correlations, one-sample
t-test was used to analyze variability of CNVs and one-way ANOVA
was used to analyze the difference in driver mutations. NKX2-1, NK2
homeobox 1; PD-L1, programmed death ligand 1; CN, copy number; CNV,
CN variation; CNA, CN alteration. |
To test this hypothesis, the molecular profiles of
patients with NSCLC from TCGA and Bivona cohorts were explored.
Consistent with the IHC results, NKX2-1 transcript levels did not
correlate with CD274 (PD-L1) expression (P=0.184; Fig. 2C). However, NKX2-1 CNAs were
significantly correlated with CD274 downregulation (P=0.047 in
loss; P=0.001 in gain) and EGFR upregulation (P<0.001 in gain),
at the clonal level (Fig. 2D-G).
EGFR-driver mutations in tumors with NKX2-1 CNAs were significantly
higher compared to other oncogenes such as ALK, BRAF and KRAS (70
vs. 15, 14 and 1%, respectively; P<0.001; Fig. 2H). Additionally, NKX2-1 and EGFR
CNVs were found co-altered in NSCLC (P=0.008; Fig. 2I). These results suggested that the
EGFR and PD-L1 profiles of NSCLC tumors may be strongly associated
with NKX2-1 CNAs rather than expression.
NKX2-1 CNAs differentially prognose
the survival of patients with NSCLC
Germline and somatic CNVs may influence the survival
of patients with lung cancer (7).
Therefore, the prognostic value of NKX2-1 CNAs, with and without
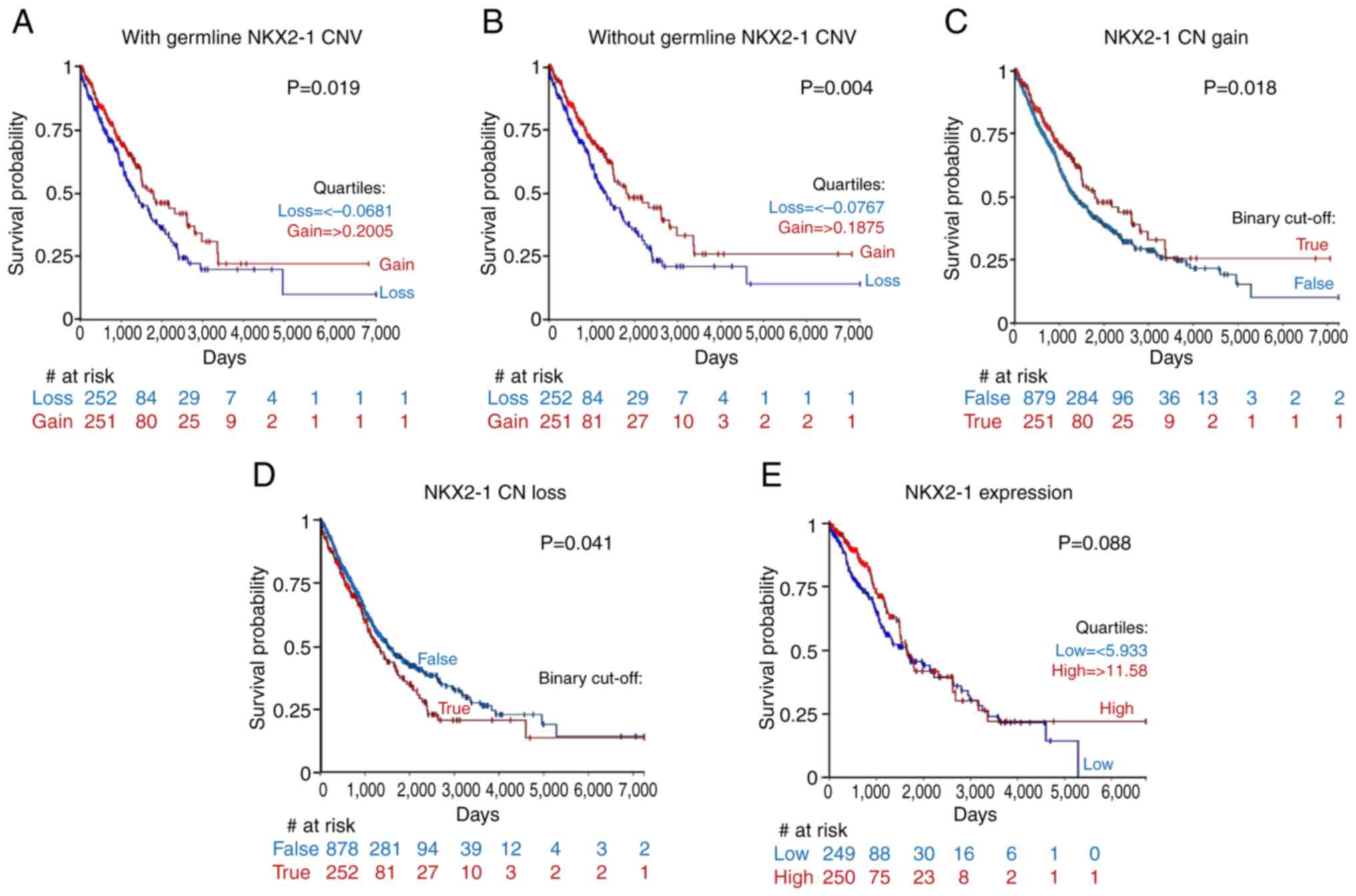
germline CNVs was examined. The results showed that NKX2-1 CN with
and without germline CNVs had positive and significant correlation
(r=0.996; P<0.001), which suggested that NKX2-1 CNAs were most
likely acquired somatically (Fig.
S3A). Survival analysis based on high and low NKX2-1 CN yielded
comparable survival curves, regardless of whether they were with or
without germline CNVs (P=0.079 and P=0.066, respectively; Fig. S3B and C). Quartile subgrouping by
NKX2-1 CNAs showed that patients with NKX2-1 CN loss had
significantly shorter survival than those with CN gain, with
(P=0.019) or without germline CNVs (P=0.004; Fig. 3A and B).
The present study aimed to identify the contribution
of NKX2-1 CNAs that developed somatically and defined the threshold
of NKX2-1 CN gain [>0.1875 log2(tumor/normal)] and NKX2-1 CN
loss [<-0.0767 log2(tumor/normal)]. When the survival of these
two patient subgroups were compared with the rest of the NSCLC
cohort, it was found that patients with NKX2-1 CN gain had
significantly longer survival than the rest of the cohort (P=0.018;
Fig. 3C). While patients with CN
loss had significantly shorter survival than the rest of the cohort
(P=0.041; Fig. 3D). EGFR mutational
subgrouping showed that the survival of patients with EGFR mutant
and wildtype tumors had no statistical difference, regardless of
NKX2-1 CN gain or loss (Fig. S3D and
E). This is potentially due to the small sample size of
participants with EGFR driver mutations (e.g., T90M, L858R and
19del). Furthermore, NKX2-1 CN gain significantly prognosed shorter
progression-free interval in patients with EGFR driver mutations
than those with the wildtype sequence (P=0.037), while subgrouping
by CN loss did not show a significant difference in the survival of
patients with EGFR mutant and wildtype tumors (Fig. S3F and G). Lastly, quartile
subgrouping by NKX2-1 expression failed to provide significant
prognostic value (P=0.088; Fig.
3E). Collectively, these results suggested that NKX2-1 CNAs
differentially influence patient survival, and CN gain may stratify
a prognostically favorable subgroup, while CN loss may prognose
worse survival in patients with NSCLC. Additionally, the prognostic
value of NKX2-1 CNAs may be more superior than expression. Further
studies are needed to verify the prognostic value of NKX2-1 CN gain
and loss in patients with EGFR driver mutations and wildtype
sequence.
Mutational burden in chromosome Y may
differentiate the mutational architecture of tumors with NKX2-1
CNAs
Next, the present study aimed to determine the
unique molecular features of tumors from the two prognostic groups
(CN gain and CN loss) to identify theragnostic markers and to
understand the physiologies associated with different survival
outcomes. The two groups were characterized with high EGFR
mutational frequencies (Fig. 2H)
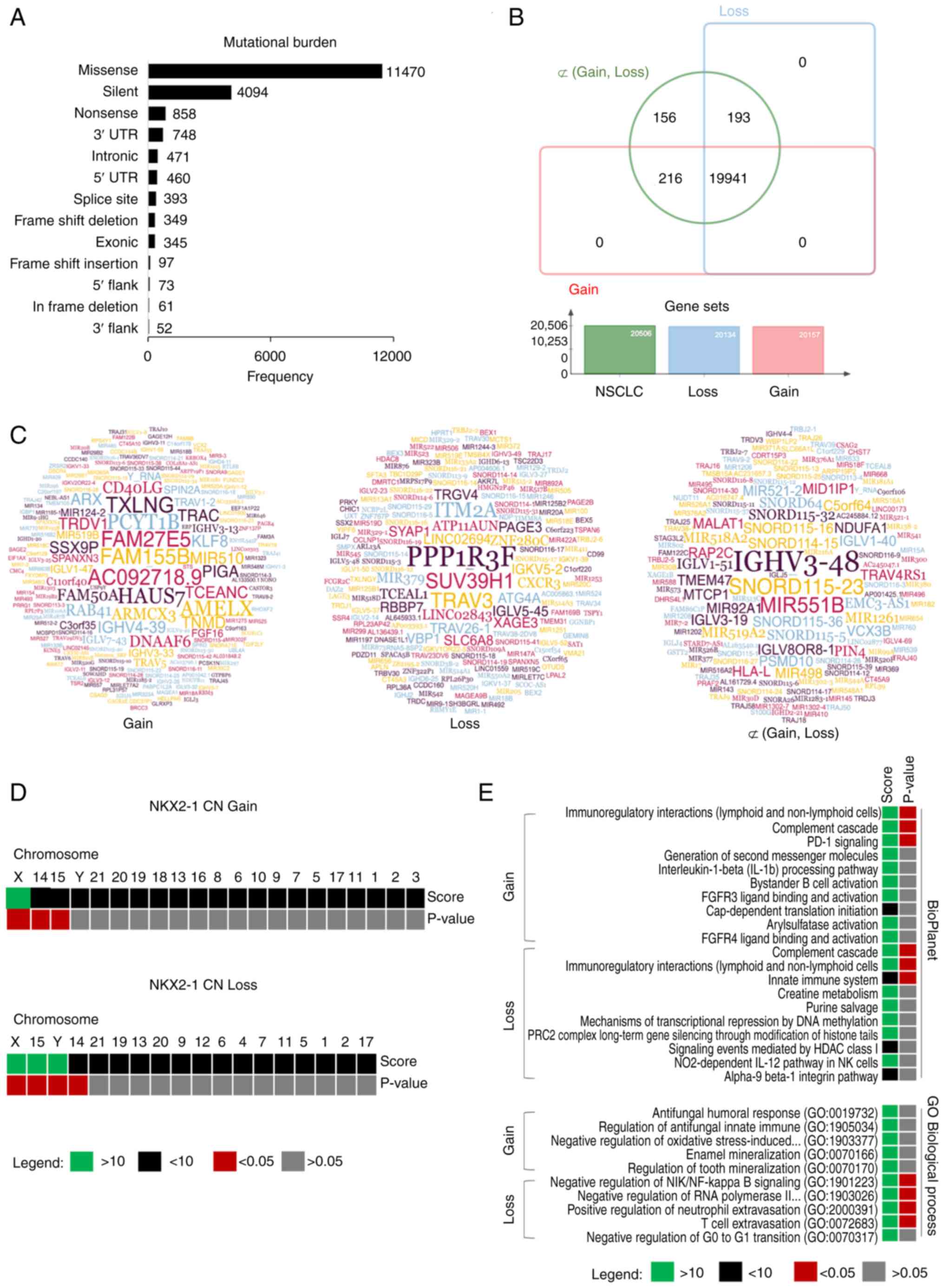
and high mutational burden in chromosome 7 (Fig. 4A), suggesting culprits in their
tumor mutational architectures (TMAs). Genome-wide mutational
analysis revealed that among the reported 20,506 mutated genes in
NSCLC from TCGA cohort, there were 20,134 and 20,157 mutations
associated with NKX2-1 CN loss and gain, respectively. A total of
~19,941 (97.24%) synonymous mutations were found. There were 216
(1.05%) and 193 (0.94%) uniquely mutated genes associated with CN
gain and loss, respectively (Fig.
4B). Gene set analysis revealed that most affected genes were
found in adenocarcinoma, rather than in squamous carcinomas
(Fig. S4A).
Enrichment analysis showed that uniquely mutated
genes (Figs. 4C and S4B) associated with NKX2-1 CNAs were
found significantly distributed in chromosomes X, 15 and 14
(P<0.05) for both CN gain and loss. Additionally, Y-specific
mutational signatures were found in patients with CN loss
(P<0.05; Fig. 4D). BioPlanet
enrichment analysis revealed that the mutated genes were found to
play a significant role in a number of immune processes. Genes
involved in PD-1 signaling were preferentially affected in CN gain;
whereas, genes of the innate immune system were found enriched for
CN loss. No significant GO biological process enrichment was found
for CN gain, but mutated genes in CN loss were found involved in
immunoregulation (Fig. 4E). These
results suggested that NKX2-1 CNAs are closely associated with
mutational clusters in chromosomes X, 14 and 15. Y-specific
mutations may differentiate the two prognostic groups. TMA in
tumors with NKX2-1 CNAs are potentially linked to dysregulation of
antitumor activity of the immune system.
NKX2-1 CNAs are associated with high
frequency of chromosomal instability
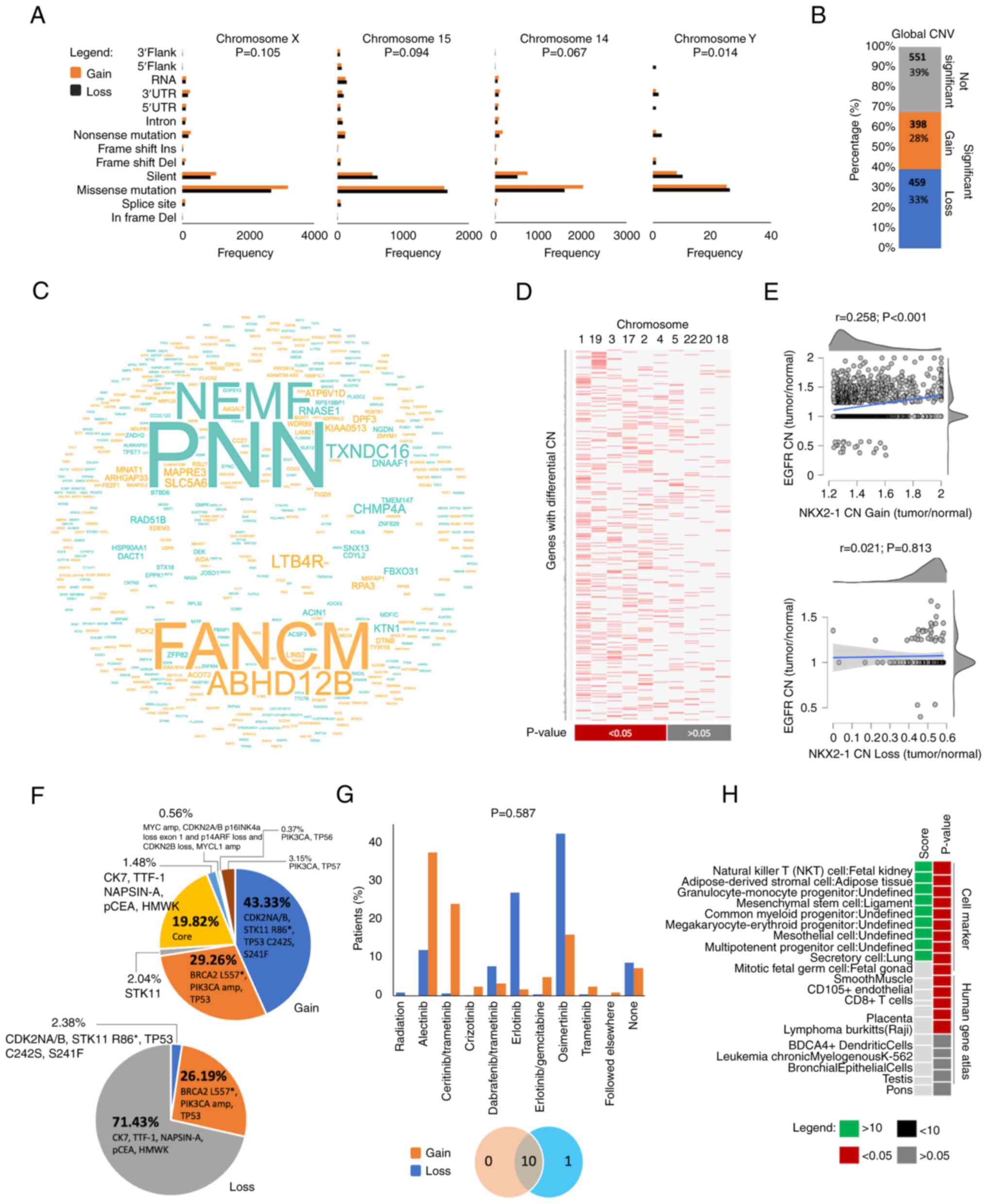
Y-specific mutations were significantly higher in
tumors with NKX2-1 CN loss (P=0.014) but the number of mutational
frequencies in chromosomes X, 15 and 14 had no significant
differences between the two prognostic groups (P>0.05; Fig. 5A). Additionally, a number of
biological processes were found to be dysregulated, which are
independent of genes located from these chromosomes (Fig. S4C), suggesting that other critical
tumorigenic drivers exist. Thus, the present study next examined
the degree of clonal chromosomal instability of tumors with NKX2-1
CNAs from the Bivona cohort. A total of 1,408 affected genes in
clones with CNAs were identified, and 61% of these genes
significantly differentiated the clonal features of the two
prognostic groups (Fig. 5B). Genes
with altered CN were found to be higher in clones with NKX2-1 CN
loss (33%) than in gain (28%). Genes with altered CN were found
significantly enriched in chromosomes 1, 19, 3, 17, 2 and 4
(Fig. 5C and D). This suggested
that chromosomal instability may also influence the disease
outcomes of patients with NXK2-1 CNAs.
Focal alteration in EGFR CN was positively
correlated with NKX2-1 CN gain (Fig.
5E), which may justify the sensitization of cancer cells with
therapeutic interventions leading to favorable outcomes (Fig. 3B). Consistently, patients with
NKX2-1 CN gain had a higher frequency of developing secondary
mutations compared to those with CN loss (Fig. 5F), which may suggest that NKX2-1 CN
gain could serve as biomarker to stratify patients with higher
chromosomal instability that could predict better survival outcomes
using tyrosine kinase inhibitors (TKIs; Fig. 5G). Genes with significantly altered
CNs were also found enriched for immune processes (Figs. 5H, S4D
and E).
NKX2-1 CNAs can influence tumor
infiltration by lymphocytes
To gain deeper insights into how NKX2-1 CNAs could
influence the immune microenvironment, the expression of the CD2
marker in tumors from the Bivona and TCGA cohorts was measured. CD2
expression was statistically comparable between tumors with NKX2-1
CN gain and loss (P=0.866; Fig.
S5A). However, CD2 expression was significantly correlated with
NKX2-1 CN loss (P<0.001; Fig.
S5B), which indicated a dynamic lymphocyte infiltration in
tumors with NKX2-1 CNAs.
The expression of 53 gene markers of TILs in NSCLC
with NKX2-1 CNAs was next compared. Unsupervised clustering
analysis showed that the expression of TIL markers was heterogenous
in CN gain, whereas, some markers strongly clustered with CN loss
(Fig. 6A). There were 37 markers
that were significantly correlated with NKX2-1 CN loss, while four
were correlated with CN gain (Figs.
6B and S5C). Excluding DPP4,
which was common for both, the higher expression of three (NGF,
SCN3A and GPNMB) unique TIL signatures in CN gain was prognostic of
longer disease-free survival than those with lower expression
(P=0.005; Fig. 6C). By contrast,
the higher expression of 36 TIL marker genes (associated with
NKX2-1 CN loss) had no significant effect on the survival of
patients with lower expression (P=0.89). These results suggested
that NKX2-1 CNAs could be linked to differential TIL profiles of
NSCLC tumors, and that the immune infiltration was prognostic of
survival (52).
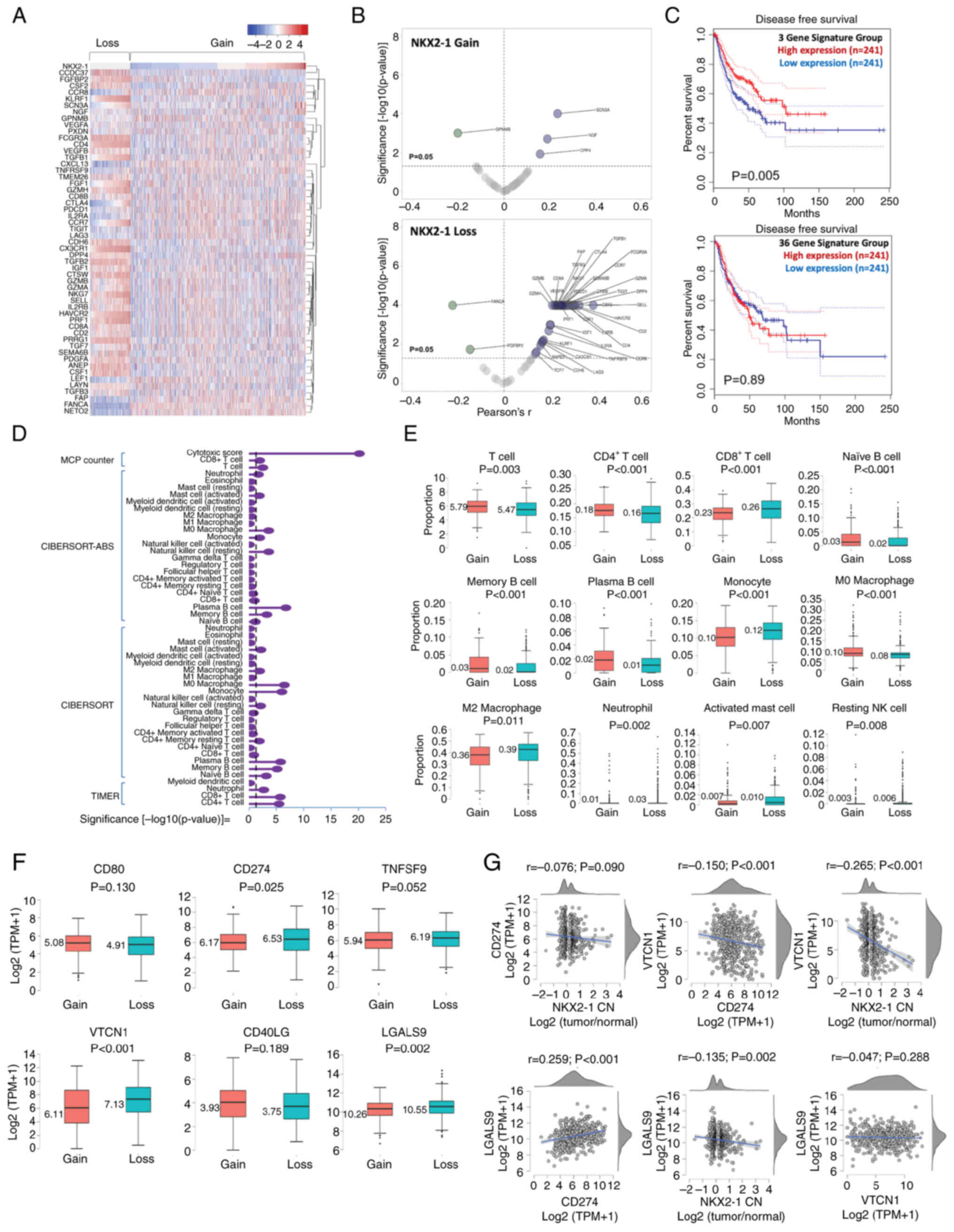 | Figure 6.Immune profile of non-small cell lung
cancer tumors with NKX2-1 CN gain and loss. (A) Clustering analysis
of the expression of 53 TIL marker genes in tumors with NKX2-1
CNAs. (B) Volcano plot of gene expression profiles of TIL markers
in tumors that were significantly correlated with NKX2-1 CN gain
and loss. (C) Prognostic value of the TIL gene signatures
associated with NKX2-1 CNAs. (D) Comparison of the proportion of
immune infiltrates in tumors with NKX2-1 CN gain and loss;
estimation was performed using MCPCounter, CIBERSORT-ABS, CIBERSORT
and TIMER 2.0. The broken line represents the P-value threshold of
0.05, proportions on the right of the line mark a significant
difference between CN gain and loss. (E) Immune cells with
significantly different proportions between tumors with NKX2-1 CN
gain and loss. (F) Comparison of the expression of immune
checkpoint proteins CD80, CD274, TNFSF9, VTCN1, CD40LG and LGALS9
in tumors with NKX2-1 CN gain and loss. (G) Correlation of NKX2-1
CN with immune checkpoint proteins and co-expression correlation.
Comparison of the two groups were performed using Student's t-test,
and correlation analysis was performed using Pearson's coefficient.
NKX2-1, NK2 homeobox 1; TIL, tumor-infiltrating lymphocyte; CN,
copy number; CNA, CN alteration. |
The multivariate correlations of the 53 genes varied
significantly, which may link to differences in lymphocyte
proportions (Fig. S5D). There were
significant differences in TIL estimates between tumors with focal
gain and loss of NKX2-1 CN (Fig.
6D). T-cell count was generally higher in tumors with CN gain
(P=0.003) and was dominated by the CD4+ subtype
(P<0.001). Meanwhile, CD8+ T cells were found higher
in CN loss (P<0.001). B cell (naïve, memory and plasma)
infiltrations were significantly higher in CN gain (P<0.001).
Monocyte, neutrophil, activated mast cell and resting natural
killer cell levels were significantly higher in CN loss
(P<0.001, P=0.002, P=0.007 and P=0.008, respectively). M0
macrophage levels were higher in CN gain (P<0.001), whereas the
tumor-promoting M2 macrophage levels were found higher in CN loss
(P=0.011; Fig. 6E). The proportion
of regulatory T cells did not significantly differ between the two
groups (P=0.948; Fig. S6A).
Patients with NKX2-1 CN loss previously associated with a higher
degree of chromosomal instability (Fig.
4D) had a significantly higher CD8+ infiltration
(P=0.011; Fig. S6B) and higher
cytotoxicity score (P<0.001; Fig.
S6C) than those with CN gain. This suggested a potential
ongoing battle between immune evolution and clonal tumor
heterogeneity, and that the loss of NKX2-1 CN may be indicative of
tumor maintenance (53). Whereas
NKX2-1 CN gain may represent co-perturbation events that constrain
the evolution of NSCLC subclones, justifying their favorable
outcomes (Fig. 3C). These results
suggested that NKX2-1 CNAs may be associated with immunologically
dynamic tumors.
NKX2-1 CNAs as biomarkers for
NSCLC-targeted therapy
The extent to which co-alteration in NKX2-1 and EGFR
CNVs (Fig. 2I) is significant for
EGFR-targeted therapy was unclear. Therefore, the survival of
patients with NKX2-1 CNAs who did and did not receive targeted
therapy was compared. The survival of patients with CN gain
(P=0.3006) and loss (P=0.7019) was found to be statistically
comparable. However, patients with NKX2-1 CN gain had a relatively
longer survival than those with CN loss who had received targeted
therapy (Fig. S3H and I). Analysis
of driver genes in those two patient groups showed that there was a
higher frequency of EGFR-sensitizing mutation (exon19 del and
exon21L858R) in CN gain than in loss (Fig. S3J). These results signified that
NKX2-1 CN gain may be associated with higher occurrence of
EGFR-targetable mutations and could be linked to EGFR-targeted
stratification, which was not observed previously because past
reports failed to compare CN amplification with deletion (14,15).
Notably, NKX2-1 CN loss had a negative and
significant correlation with CD274 expression (Fig. 2D) and associated with infiltration
of M2 macrophages (Fig. 6E). Thus,
the expression-based association of immune checkpoint proteins with
NKX2-1 CNAs was explored. It was found that the positive regulation
of CD80 and CD40LG, and the negative regulation of TNFSF9, VTCN1
and LGALS9 were significantly correlated with NKX2-1 CNVs (Fig. S7). The expression of CD274, VTCN1
and LGALS9 were significantly higher in CN loss than in gain. The
expression of CD80 and CD40LG had no significant difference between
the two groups (Fig. 6F). The
higher expression of immune checkpoint proteins provided additional
evidence that NSCLC with NKX2-1 CN loss may be associated with a
‘cold’ immune microenvironment, which may justify their poor
prognostic outcomes.
Further correlation analysis showed that CD274 may
not be the dominant immune checkpoint in NSCLC tumors with NKX2-1
CN loss. The immunoexpression of VTCN1 and LGALS9 were more
significant (P<0.001 and P=0.002, respectively) than CD274
(P=0.090). Co-expression analysis revealed that VTCN1 was
significantly correlated with the negative regulation of CD274
(P<0.001), whereas LGALS9 was significantly correlated with the
positive regulation of CD274 (P<0.001). No significant
correlation was observed between the expression of LGALS9 and VTCN1
(P=0.288; Fig. 6G). These results
suggested that NKX2-1 CNAs could be associated with differential
immune checkpoint profiles in NSCLC tumors.
Discussion
Alterations in NKX2-1 CN are one of the most
frequently observed consequences of genomic instability in lung
cancer (16). However, beyond
amplification, little is known about the clinical significance of
NKX2-1 CNVs, the disease burden associated with focal CNAs and
their implications for targeted therapy. In the present study, it
was reported that NKX2-1 CNAs had a stronger correlation with the
combined EGFR and PD-L1 status of tumors than RNA and protein
expression. Furthermore, it was shown that focal loss and gain in
NKX2-1 CN were prognostic in NSCLC. CN gain may represent a
prognostically favorable subgroup, whereas CN loss may be
associated with poor survival. With the enigma surrounding TTF-1
expression studies, including inconsistencies in patient
stratification and immunostaining specificities (13), the prognostic implication of CNAs
could be superior to expression. In the present study, the
prognostic threshold associated with NKX2-1 CN gain was defined as
>0.1875 and loss was defined as <-0.0767
[log2(tumor/normal)].
The present analysis revealed that NKX2-1 and EGFR
CNs were significantly co-altered. Consistently, tumors with NKX2-1
CNAs had a high frequency of missense mutations in chromosome 7
where the EGFR gene is located, suggesting that NKX2-1 CNAs may
serve as a biomarker for EGFR-directed therapy. Additionally,
patients with NKX2-1 CN gain and loss had 97.25% synonymous
mutations, with 216 and 193 distinct mutational signatures,
respectively. The mutational burden in the Y-chromosome was found
to be significantly higher in tumors with NKX2-1 CN loss,
indicating that the unique Y-specific mutational cluster associated
with CN loss tends to negatively impact survival. The findings of
the present study justify the foundational basis for exploring the
Y-specific contribution of tumor mutational signatures in NSCLC and
how sex could influence treatment outcomes in lung cancer (54).
The results of the present study also revealed the
difference in the degree of genome-wide CNAs in cancer clones with
focal gain and loss in NKX2-1 CN. Out of 1,408 genes with CNAs,
more than half (61%) were significantly co-altered with NKX2-1.
Most genome-wide alterations were found in clones with NKX2-1 CN
loss than in gain (33 vs. 28%, respectively). The majority of the
affected genes were enriched in chromosomes 1, 19, 3, 17, 2 and 4,
which suggested that NKX2-1 CNAs are closely linked to a high
frequency of chromosomal instability.
A recent study showed that chromosomal instability
could predict treatment outcomes of patients with NSCLC receiving
TKIs (55). The researchers showed
that chromosomal instability resulting in Chr 1p13.3-p13.1 gain
could predict poor response to EGFR-TKI. However, CN gain in Chr
14q31.1-q31.3 and Chr 7q31.1-q31.31, and CN loss in Chr
8p23.3-p23.1 and Chr 10q21.2-q22.1 were found to have favorable
survival outcomes in patients receiving TKIs. This suggests that
contrary to the general assumption that high chromosomal
instability could negatively impact survival, the chromosomal
location and the affected genes have a more significant impact on
patient outcomes. The results of the present study showed that
NKX2-1 CN gain may be associated with chromosomal instability that
resulted in the development of more secondary mutations, which
could confer a better response to TKI treatments. Additionally, the
present study showed that the unique genomic CN perturbations and
instabilities in chromosomes 1, 19, 3, 17, 2 and 4 among patients
with NSCLC with NKX2-1 CN gain could predict better treatment
outcomes with TKIs.
Genes with perturbed CN and uniquely mutated in
tumors with NKX2-1 CNAs were found enriched for a number of
immunological processes, suggesting a close association between
NKX2-1 CNAs and immune microenvironment remodeling. The results of
the present study showed that the TIL profiles of patients with
NKX2-1 CN gain and loss were different. TIL estimation analysis
showed that total T-cell and B-cell proportions were higher in
tumors with CN gain than loss. This suggested that tumors with
NKX2-1 CN gain had higher adaptive infiltration and robust humoral
responses that resulted in better survival. Tumors with CN loss
were found to have a ‘colder’ immune environment due to
significantly higher M2 macrophage infiltrates and immune
checkpoint protein expression.
Currently, there is a limited number of effective
biomarkers that may accurately prognose whether patients will
benefit, develop resistance or experience severe side effects from
therapy, particularly using TKIs and immune checkpoint inhibitors
(ICIs). Subgrouping by PD-L1 positivity, EGFR mutational status and
NKX2-1 expression produced insufficient results (56–58).
The findings of the present study revealed that patients with
NKX2-1 CN gain may benefit from either EGFR-TKIs or ICIs due to a
higher frequency of EGFR-driver mutations and higher adaptive
infiltrates.
Consequently, enrichment of EGFR-targetable
mutations in CN gain could also present a negative impact due to a
higher tendency of developing resistant mutations (e.g., T790M) and
co-mutations. Indeed, NKX2-1 CN amplification prognosed a poor
response to EGFR-TKIs in patients with recurrent tumors after
surgical treatment (59). However,
this study stratified patients as NKX2-1 CN amplified or
unamplified only, and patients designated as unamplified could have
harbored CN deletions, which could radically change the survival
outcomes of the cohort. Also, the study failed to correlate CN
amplification with other genomic alterations that may confound with
the prognostic value of NKX2-1 CN amplification. Lastly, the
detection of NKX2-1 CNAs remains unstandardized at present, and
studies utilizing fluorescence in situ hybridization and PCR
are normally hampered by variations and complexities of DNA
breakpoints in analyzing CNVs (14,60).
Thus, the dynamic and complete changes in CN are often not captured
from studies utilizing these techniques.
A previous paper that utilized next generation
sequencing to analyze EGFR-TKI outcomes in patients with lung
cancer with concurrent genomic alterations identified a lower
frequency of NKX2-1 CN amplification in tumors that developed
resistance to EGFR-TKI than those that were naïve (11 vs. 15%), and
reported that acquired T790M mutation, co-mutation in TP53 and
co-amplification of EGFR had higher frequencies in EGFR-TKI
resistant tumors than NKX2-1 CN amplification alone (61). In fact, there was only a small
frequency of T790M mutations in TCGA cohort with NKX2-1 CN gain.
Thus, there is a need for a more quantitative approach in
determining the extent to which NKX2-1 CN gain could be predictive
for patients receiving EGFR-TKIs. With the advent of more
sophisticated tools, such as sequencing platforms, the development
of quantitative CNV subgrouping is now possible. To the best of our
knowledge, the present study is the first to provide retrospective
data and propose a quantitative approach to stratifying patients as
having NKX2-1 CN gain with measurable CNVs [>0.1875
log2(tumor/normal)] that may benefit from EGFR-TKIs. However, this
hypothesis requires further clinical analysis.
Additionally, previous studies failed to account
NKX2-1 CN deletion, and thus a comprehensive and quantitative
comparison of the prognostic value of NKX2-1 CNAs is still elusive.
The present analysis also showed that NKX2-1 CN loss with
measurable CNVs [<-0.0767 log2(tumor/normal)] was associated
with lower TMB, higher genome-wide CNV perturbations, reduced
adaptive infiltrations and increased expression of
immunosuppressive molecules. All these qualifications have been
linked to poor responses that may be unlikely to benefit from
combinatorial TKI–ICI therapy. However, these hypotheses require
further clinical validations. Lastly, the present findings
emphasized the need to develop other target approaches, other than
CD274, LGALS9 and VTCN1, that could be potential targets in
NSCLC.
Limitations of the present study include the small
sample size, particularly in the Filipino cohort, which may impact
correlation analysis. Additionally, subgrouping by EGFR mutation
did not reach significant survival probabilities due to the small
sample size and requires further study. Second, the involvement of
ROR1 in the combined EGFR and PD-L1 status needs to be further
explored through knock-in and knock-out mouse models, supported by
protein-protein interaction experiments and functional mice
studies. Third, the CNV data from TCGA and the Bivona cohorts were
detected using Affymetrix SNP 6.0 arrays and RNA sequencing,
respectively. Although CNV was normalized to tumor-normal ratio,
difference in methods may have contributed to variations in
measurements. Fourth, the expression data in TCGA and the Bivona
cohorts were scaled differently, which may have affected
statistical analyses, although data from each cohort were
interpreted independently. Lastly, the analysis of TILs depended on
53 select gene markers, which may have influenced the estimation
analysis.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank Dr. Neil Andrew
Bascos (National Institute of Molecular Biology and Biotechnology,
University of the Philippines, Quezon City, Philippines) for his
insights and contributions in the study.
Funding
This study was funded by the Philippine Council for Health
Research and Development and the Grants-In-Aid program of The
Department of Science and Technology (grant no. IC-21-02421).
Availability of data and materials
The datasets used and/or analyzed during the current
study available from the corresponding author on reasonable
request.
Authors' contributions
HGCL, MSI, NJ, KVH, TMS, GCL, SSN and MIDLS
conceived and designed the study; HGCL, DM and JS provided
administrative support; HGCL, MSI, NJ, KVH, TMS, GCL, SSN and MIDLS
provided study materials or patients; SMAG, MB, KJD, BBB, MIDLS,
VML, JS and DM performed collection and assembly of the data; HGCL,
SMAG, MB, KJD, BBB, MIDLS and VML performed data analysis and
interpretation; and HGCL, MSI, NJ, KVH, TMS, GCL, SSN, SMAG, MB,
KJD, BBB, MIDLS, VML, DM and JS wrote the manuscript. MIDLS and
HGCL confirm the authenticity of all the raw data. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
The study was conducted in accordance with The
Declaration of Helsinki (as revised in 2013). The study was
approved by The Institutional Ethics Review Board (approval no.
LCP-CS-001-2019) of the Lung Center of the Philippines (Quezon
City, Philippines). The protocol for specimen collection was
approved by The Single Joint Research Ethics Board (approval no.
SJREB-2020-97) of the Department of Health, Manila, Philippines.
All patients provided written informed consent for genetic testing,
as well as the use of their clinical data.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Feuk L, Carson AR and Scherer SW:
Structural variation in the human genome. Nat Rev Genet. 7:85–97.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Steele CD, Abbasi A, Islam SMA, Bowes AL,
Khandekar A, Haase K, Hames-Fathi S, Ajayi D, Verfaillie A, Dhami
P, et al: Signatures of copy number alterations in human cancer.
Nature. 606:984–991. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bjaanæs MM, Nilsen G, Halvorsen AR,
Russnes HG, Solberg S, Jørgensen L, Brustugun OT, Lingjærde OC and
Helland Å: Whole genome copy number analyses reveal a highly
aberrant genome in TP53 mutant lung adenocarcinoma tumors. BMC
Cancer. 21:10892021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shao X, Lv N, Liao J, Long J, Xue R, Ai N,
Xu D and Fan X: Copy number variation is highly correlated with
differential gene expression: A pan-cancer study. BMC Med Genet.
20:1752019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Heo Y, Heo J, Han SS, Kim WJ, Cheong HS
and Hong Y: Difference of copy number variation in blood of
patients with lung cancer. Int J Biol Markers. 36:3–9. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Qiu ZW, Bi JH, Gazdar AF and Song K:
Genome-wide copy number variation pattern analysis and a
classification signature for non-small cell lung cancer. Genes
Chromosomes Cancer. 56:559–569. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen S, Lu L, Xian J, Shi C, Chen J, Rao
B, Qiu F, Lu J and Yang L: Prognostic value of germline copy number
variants and environmental exposures in Non-small cell lung cancer.
Front Genet. 12:681852021.
|
|
8
|
Aujla S, Aloe C, Vannitamby A, Hendry S,
Rangamuwa K, Wang H, Vlahos R, Selemidis S, Leong T, Steinfort D
and Bozinovski S: Programmed Death-Ligand 1 Copy number loss in
NSCLC associates with reduced programmed Death-Ligand 1 tumor
staining and a cold immunophenotype. J Thorac Oncol. 17:675–687.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Alden S, Ricciuti B, Spurr L, Gupta H,
Lamberti G, Li Y, Sholl L, Cherniack A and Awad A: P33.04
programmed death-ligand 1 (PD-L1) changes in non-small-cell lung
cancer (NSCLC): Clinical, pathologic, and genomic correlates. J
Thorac Oncol. 16:S406–S407. 2021. View Article : Google Scholar
|
|
10
|
Inoue Y, Yoshimura K, Mori K, Kurabe N,
Kahyo T, Mori H, Kawase A, Tanahashi M, Ogawa H, Inui N, et al:
Clinical significance of PD-L1 and PD-L2 copy number gains in
non-small-cell lung cancer. Oncotarget. 7:32113–32128. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wei J, Meng P, Terpstra MM, van Rijk A,
Tamminga M, Scherpen F, Ter Elst A, Alimohamed MZ, Johansson LF,
Stigt J, et al: Clinical Value of EGFR Copy number gain determined
by amplicon-based targeted next generation sequencing in patients
with EGFR-Mutated NSCLC. Target Oncol. 16:215–226. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Peng D, Liang P, Zhong C, Xu P, He Y, Luo
Y, Wang X, Liu A and Zeng Z: Effect of EGFR amplification on the
prognosis of EGFR-mutated advanced non-small-cell lung cancer
patients: A prospective observational study. BMC Cancer.
22:13232022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gloriane C Luna H, Severino Imasa M, Juat
N, Hernandez KV, May Sayo T, Cristal-Luna G, Marie Asur-Galang S,
Bellengan M, John Duga K, Brian Buenaobra B, et al: Expression
landscapes in non-small cell lung cancer shaped by the thyroid
transcription factor 1. Lung Cancer. 176:121–131. 2023. View Article : Google Scholar
|
|
14
|
Yoshimura K, Inoue Y, Mori K, Iwashita Y,
Kahyo T, Kawase A, Tanahashi M, Ogawa H, Inui N, Funai K, et al:
Distinct prognostic roles and heterogeneity of TTF1 copy number and
TTF1 protein expression in non-small cell lung cancer. Genes
Chromosomes Cancer. 56:570–581. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li X, Wan L, Shen H, Geng J, Nie J, Wang
G, Jia N, Dai M and Bai X: Thyroid transcription Factor-1
amplification and expressions in lung adenocarcinoma tissues and
pleural effusions predict patient survival and prognosis. J Thorac
Oncol. 7:76–84. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Clarke N, Biscocho J, Kwei KA, Davidson
JM, Sridhar S, Gong X and Pollack JR: Integrative genomics
implicates EGFR as a downstream mediator in NKX2-1 amplified
non-small cell lung cancer. PLoS One. 10:e01420612015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Luna HG, Prieto E, Dimayacyac-Esleta BR,
Imasa MS, Juat N, Hernandez KV, Sayo TM, Cristal-Luna GR,
Asur-Galang SM, Bellengan M, et al: 342P Prognostic implications of
PD-L1 co-expression among Filipino EGFR MT mNSCLC. Ann Oncol. 33
(Suppl):S15762022. View Article : Google Scholar
|
|
18
|
Luna HGC, Imasa MS, Juat N, Hernandez KV,
Sayo TM, Cristal-Luna G, Asur-Galang SM, Bellengan M and Duga KJ:
The differential prognostic implications of PD-L1 expression in the
outcomes of Filipinos with EGFR-mutant NSCLC treated with tyrosine
kinase inhibitors. Transl Lung Cancer Res. 12:1896–1911. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cancer Genome Atlas Research Network, .
Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA,
Ellrott K, Shmulevich I, Sander C and Stuart JM: The cancer genome
atlas pan-cancer analysis project. Nat Genet. 45:1113–1120. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Maynard A, McCoach CE, Rotow JK, Harris L,
Haderk F, Kerr DL, Yu EA, Schenk EL, Tan W, Zee A, et al:
Therapy-Induced evolution of human lung cancer revealed by
single-cell RNA sequencing. Cell. 182:1232–1251.e22. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Goldman MJ, Craft B, Hastie M, Repečka K,
McDade F, Kamath A, Banerjee A, Luo Y, Rogers D, Brooks AN, et al:
Visualizing and interpreting cancer genomics data via the Xena
platform. Nat Biotechnol. 38:675–678. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Grossman RL, Heath AP, Ferretti V, Varmus
HE, Lowy DR, Kibbe WA and Staudt LM: Toward a shared vision for
cancer genomic data. N Engl J Med. 375:1109–1112. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Luna HGC, Imasa MS, Juat N, Hernandez V,
Sayo MT, Cristal-Luna G, Asur-Galang SM, Bellengan M, Duga KJ,
Buenaobra BB, et al: Prognostic Value of PD-L1 in Metastatic NSCLC
with EGFR-Sensitizing Mutations: A Benchmark Filipino Cohort Study.
SSRN. 2022.doi: 10.2139/ssrn.4286198. View Article : Google Scholar
|
|
24
|
Torous VF, Rangachari D, Gallant BP, Shea
M, Costa DB and VanderLaan PA: PD-L1 testing using the clone 22C3
pharmDx kit for selection of patients with non-small cell lung
cancer to receive immune checkpoint inhibitor therapy: Are cytology
cell blocks a viable option? J Am Soc Cytopathol. 7:133–141. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gu K, Shah V, Ma C, Zhang L and Yang M:
Cytoplasmic immunoreactivity of thyroid transcription Factor-1
(Clone 8G7G3/1) in hepatocytestrue positivity or cross-reaction? Am
J Clin Pathol. 128:382–388. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bardou P, Mariette J, Escudié F, Djemiel C
and Klopp C: Jvenn: An interactive Venn diagram viewer. BMC
Bioinformatics. 15:2932014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kuleshov MV, Jones MR, Rouillard AD,
Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM,
Lachmann A, et al: Enrichr: A comprehensive gene set enrichment
analysis web server 2016 update. Nucleic Acids Res. 44:W90–W97.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xie Z, Bailey A, Kuleshov M V, Clarke DJB,
Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML,
Kropiwnicki E, Jagodnik KM, et al: Gene set knowledge discovery
with enrichr. Curr Protoc. 1:e902021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huang R, Grishagin I, Wang Y, Zhao T,
Greene J, Obenauer JC, Ngan D, Nguyen DT, Guha R, Jadhav A, et al:
The NCATS BioPlanet-An integrated platform for exploring the
universe of cellular signaling pathways for toxicology, systems
biology, and chemical genomics. Front Pharmacol. 10:4452019.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kanehisa M, Furumichi M, Sato Y,
Ishiguro-Watanabe M and Tanabe M: KEGG: Integrating viruses and
cellular organisms. Nucleic Acids Res. 49:D545–D551. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gene Ontology Consortium: The Gene
Ontology resource: Enriching a GOld mine. Nucleic Acids Res.
49:D325–D334. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lee BT, Barber GP, Benet-Pagès A, Casper
J, Clawson H, Diekhans M, Fischer C, Gonzalez JN, Hinrichs AS, Lee
CM, et al: The UCSC Genome Browser database: 2022 update. Nucleic
Acids Res. 50:D1115–D1122. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Binder JX, Pletscher-Frankild S, Tsafou K,
Stolte C, O'Donoghue SI, Schneider R and Jensen LJ: COMPARTMENTS:
Unification and visualization of protein subcellular localization
evidence. Database (Oxford). 2014:bau0122014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang X, Lan Y, Xu J, Quan F, Zhao E, Deng
C, Luo T, Xu L, Liao G, Yan M, et al: CellMarker: A manually
curated resource of cell markers in human and mouse. Nucleic Acids
Res. 47:D721–D728. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wu C, MacLeod I and Su AI: BioGPS and
MyGene.info: Organizing online, gene-centric information. Nucleic
Acids Res. 41:D561–D565. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tang Z, Kang B, Li C, Chen T and Zhang Z:
GEPIA2: An enhanced web server for large-scale expression profiling
and interactive analysis. Nucleic Acids Res. 47:W556–W560. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wu M, Mei F, Liu W and Jiang J:
Comprehensive characterization of tumor infiltrating natural killer
cells and clinical significance in hepatocellular carcinoma based
on gene expression profiles. Biomed Pharmacother. 121:1096372020.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Goedhart J and Luijsterburg MS: VolcaNoseR
is a web app for creating, exploring, labeling and sharing volcano
plots. Sci Reports. 10:205602020.PubMed/NCBI
|
|
40
|
Babicki S, Arndt D, Marcu A, Liang Y,
Grant JR, Maciejewski A and Wishart DS: Heatmapper: Web-enabled
heat mapping for all. Nucleic Acids Res. 44:W147–W153. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Newman AM, Liu CL, Green MR, Gentles AJ,
Feng W, Xu Y, Hoang CD, Diehn M and Alizadeh AA: Robust enumeration
of cell subsets from tissue expression profiles. Nat Methods.
12:453–457. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Becht E, Giraldo NA, Lacroix L, Buttard B,
Elarouci N, Petitprez F, Selves J, Laurent-Puig P, Sautès-Fridman
C, Fridman WH and de Reyniès A: Estimating the population abundance
of tissue-infiltrating immune and stromal cell populations using
gene expression. Genome Biol. 17:2182016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li T, Fan J, Wang B, Traugh N, Chen Q, Liu
JS, Li B and Liu XS: TIMER: A web server for comprehensive analysis
of tumor-infiltrating immune cells. Cancer Res. 77:e108–e110. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Galland L, Le Page AL, Lecuelle J, Bibeau
F, Oulkhouir Y, Derangère V, Truntzer C and Ghiringhelli F:
Prognostic value of thyroid transcription Factor-1 expression in
lung adenocarcinoma in patients treated with anti PD-1/PD-L1.
Oncoimmunology. 10:19576032021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lamberti G, Spurr LF, Li Y, Ricciuti B,
Recondo G, Umeton R, Nishino M, Sholl LM, Meyerson ML, Cherniack AD
and Awad MM: Clinicopathological and genomic correlates of
programmed cell death ligand 1 (PD-L1) expression in nonsquamous
non-small-cell lung cancer. Ann Oncol. 31:807–814. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yamaguchi T, Yanagisawa K, Sugiyama R,
Hosono Y, Shimada Y, Arima C, Kato S, Tomida S, Suzuki M, Osada H
and Takahashi T: NKX2-1/TITF1/TTF-1-Induced ROR1 is required to
sustain EGFR survival signaling in lung adenocarcinoma. Cancer
Cell. 21:348–361. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Li Y, Liu Z, Zhao Y, Yang J, Xiao TS,
Conlon RA and Wang Z: PD-L1 expression is regulated by ATP-binding
of the ERBB3 pseudokinase domain. Genes Dis. 10:1702–1713. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hu X, Li J, Fu M, Zhao X and Wang W: The
JAK/STAT signaling pathway: From bench to clinic. Signal Transduct
Target Ther. 6:4022021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jahangiri A, Dadmanesh M and Ghorban K:
STAT3 inhibition reduced PD-L1 expression and enhanced antitumor
immune responses. J Cell Physiol. 235:9457–9463. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zegeye MM, Lindkvist M, Fälker K, Kumawat
AK, Paramel G, Grenegård M, Sirsjö A and Ljungberg LU: Activation
of the JAK/STAT3 and PI3K/AKT pathways are crucial for IL-6
trans-signaling-mediated pro-inflammatory response in human
vascular endothelial cells. Cell Commun Signal. 16:552018.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhang N, Zeng Y, Du W, Zhu J, Shen D, Liu
Z and Huang JA: The EGFR pathway is involved in the regulation of
PDL1 expression via the IL-6/JAK/STAT3 signaling pathway in
EGFR-mutated non-small cell lung cancer. Int J Oncol. 49:1360–1368.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Federico L, McGrail DJ, Bentebibel SE,
Haymaker C, Ravelli A, Forget MA, Karpinets T, Jiang P, Reuben A,
Negrao MV, et al: Distinct tumor-infiltrating lymphocyte landscapes
are associated with clinical outcomes in localized non-small-cell
lung cancer. Ann Oncol. 33:42–56. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Jamal-Hanjani M, Wilson GA, McGranahan N,
Birkbak NJ, Watkins TBK, Veeriah S, Shafi S, Johnson DH, Mitter R,
Rosenthal R, et al: Tracking the evolution of non-small-cell lung
cancer. N Engl J Med. 376:2109–2121. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Pinto JA, Vallejos CS, Raez LE, Mas LA,
Ruiz R, Torres-Roman JS, Morante Z, Araujo JM, Gómez HL, Aguilar A,
et al: Gender and outcomes in non-small cell lung cancer: An old
prognostic variable comes back for targeted therapy and
immunotherapy? ESMO Open. 3:e0003442018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
He H, Ma H, Chen Z, Chen J, Wu D, Lv X and
Zhu J: Chromosomal copy number variation predicts EGFR-TKI response
and prognosis for patients with non-small cell lung cancer.
Pharmgenomics Pers Med. 16:835–846. 2023.PubMed/NCBI
|
|
56
|
Hong L, Dibaj S, Negrao MV, Reuben A,
Roarty E, Rinsurongkawong W, Lewis J, Gibbons DL, Sepesi B,
Papadimitrakopoulou V, et al: Spatial and temporal heterogeneity of
PD-L1 and its impact on benefit from immune checkpoint blockade in
non-small cell lung cancer (NSCLC). J Clin Oncol. 37:90172019.
View Article : Google Scholar
|
|
57
|
De-Rui Huang D and Chih-Hsin Yang J:
Checkpoint inhibitor combined with tyrosine kinase inhibitor-the
end or beginning? J Thorac Oncol. 15:305–307. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Nakahama K, Kaneda H, Osawa M, Izumi M,
Yoshimoto N, Sugimoto A, Nagamine H, Ogawa K, Matsumoto Y, Sawa K,
et al: Association of thyroid transcription factor-1 with the
efficacy of immune-checkpoint inhibitors in patients with advanced
lung adenocarcinoma. Thorac Cancer. 13:2309–2317. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lee JS, Kim HR, Lee CY, Shin M and Shim
HS: EGFR and TTF-1 gene amplification in surgically resected lung
adenocarcinomas: Clinicopathologic significance and effect on
response to EGFR-tyrosine kinase inhibitors in recurred cases. Ann
Surg Oncol. 20:3015–3022. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Pös O, Radvanszky J, Styk J, Pös Z, Buglyó
G, Kajsik M, Budis J and Nagy B and Nagy B: Copy number variation:
Methods and clinical applications. Appl Sci. 11:8192021. View Article : Google Scholar
|
|
61
|
Yu HA, Suzawa K, Jordan E, Zehir A, Ni A,
Kim R, Kris MG, Hellmann MD, Li BT, Somwar R, et al: Concurrent
alterations in EGFR-mutant lung cancers associated with resistance
to EGFR kinase inhibitors and characterization of MTOR as a
mediator of resistance. Clin Cancer Res. 24:3108–3118. 2018.
View Article : Google Scholar : PubMed/NCBI
|