Introduction
T-cell acute lymphoblastic leukemia (T-ALL) is a
disease characterized by the uncontrolled proliferation of mature
or immature T cells (1). T-ALL is
more common in adults than in children, although the incidence
starts to diminish with age after 10 years old. Notably, T-ALL
accounts for 15% of childhood and 25% of adult cases of ALL
(2). The outcomes of patients with
T-ALL have improved as novel therapies have been identified and
existing therapies have improved; however, the 5-year survival rate
has remained poor, with event-free and overall survival rates of
<25% for relapsed disease (3).
In addition, treatment resistance, disease recurrence,
treatment-related deaths and long-term harmful side effects of
chemotherapy in cancer survivors remain serious issues that need to
be addressed. Therefore, discovering more effective but less toxic
strategies for treating T-ALL is a priority.
According to statistics, >50% of modern clinical
antitumor drugs are directly or indirectly derived from natural
products and their derivatives (4).
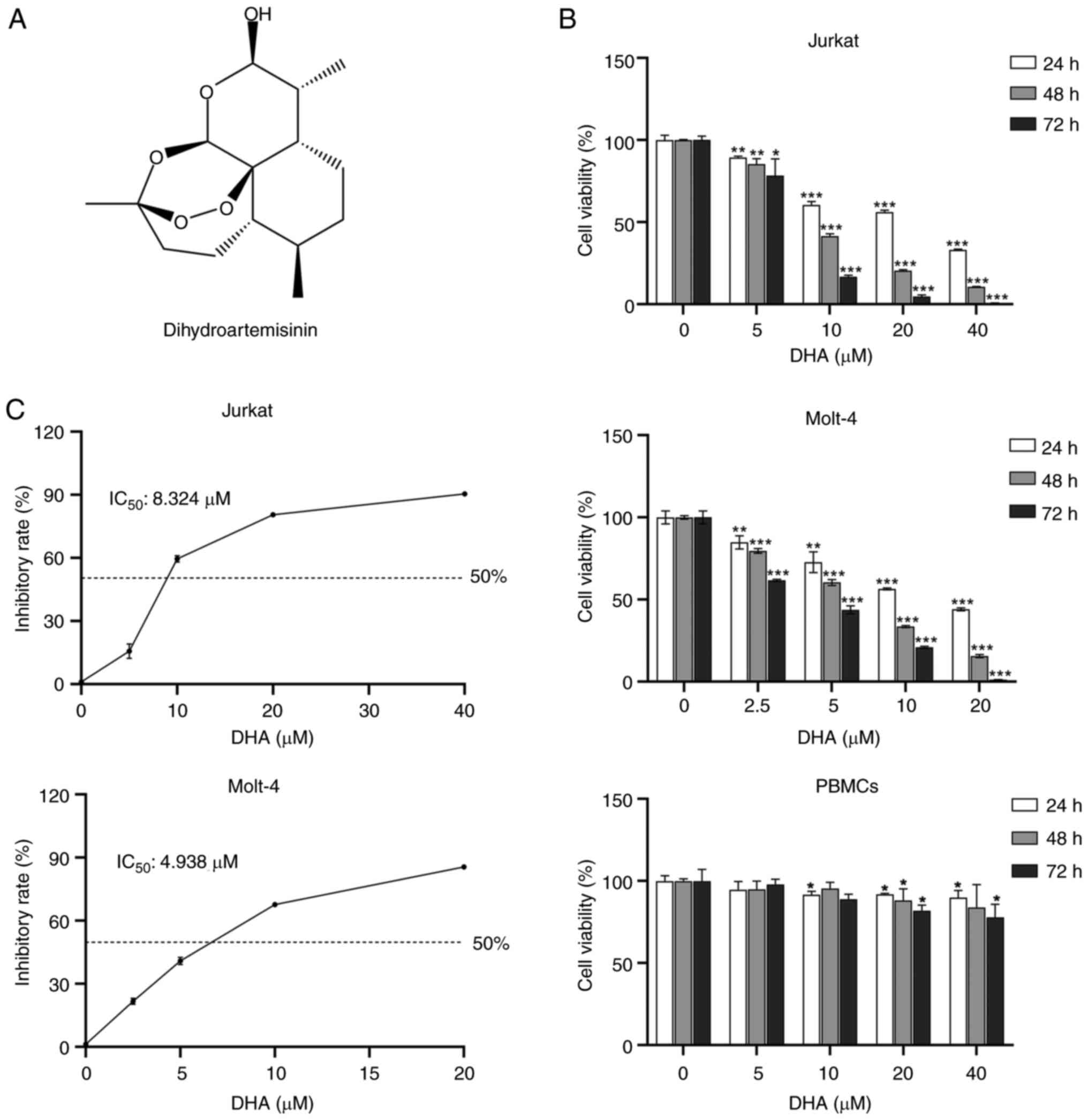
Dihydroartemisinin (DHA; Fig. 1A),
a water-soluble semi-synthetic derivative of artemisinin, is a
sesquiterpene lactone with a peroxide moiety that has long been an
essential component of anti-malarial therapy (5). Increasing evidence has demonstrated
that DHA possesses multiple pharmacological actions, including, but
not limited to, anti-viral, anti-inflammatory and anticancer
effects (6). Based on the safety
profile of DHA, a number of studies have assessed the antitumor
effects of DHA on glioma (7), liver
cancer (8) and breast malignancies
(9).
DHA has been shown to possess a number of antitumor
mechanisms, including induction of apoptosis- and
autophagy-mediated cell death, adjustment of the tumor immune
microenvironment, and suppression of metastasis and angiogenesis
(10,11). DHA has also been reported to
suppress the invasion and migration of bladder cancer cells via the
downregulation of histone demethylase KDM3A expression and
induction of p21 expression (12).
The role of DHA in inducing apoptosis in T-ALL cells was first
reported by Sun et al (13).
A recent study has revealed that the anticancer effects of DHA are
highly reliant on the cleavage of the endoperoxide bridge within
its molecular structure and subsequent reactive oxygen species
(ROS) generation (14). In
addition, the selective cytotoxic effects of DHA on some cancer
cells are associated with ferroptosis, which is a non-apoptotic
type of cell death (15,16).
Ferroptosis is characterized by the accumulation of
intracellular soluble and lipid ROS, which can be counteracted by
the glutathione (GSH)-dependent activity of the GSH peroxidase 4
(GPX4) enzyme (17). System
Xc−, also named cystine-glutamate antiporter, is
composed of the light chain SLC7A11 (also commonly known as xCT)
linked via a disulfide bridge to the heavy chain SLC3A2. It is an
essential intracellular antioxidant element and functions as a
regulator for GSH synthesis (18).
Suppressing system Xc− triggers an excess of ROS and
lipid peroxidation, ultimately resulting in cell death and diseases
such as neurodegenerative and cardiovascular diseases (19–21).
In addition, Dixon et al (22) proposed that the inhibition of system
Xc− leads to endoplasmic reticulum (ER) stress,
evidenced by transcriptional upregulation of genes associated with
the ER stress response, and posited a close correlation between ER
stress elevation and erastin-induced ferroptosis.
To the best of our knowledge, no study has
determined whether DHA can affect ferroptosis in T-ALL cells.
Therefore, the present study aimed to investigate the regulatory
mechanisms of DHA for ferroptosis in T-ALL, and to develop novel
approaches for treating T-ALL.
Materials and methods
Reagents and antibodies
DHA and ferrostatin-1 (Fer-1), purchased from Glpbio
Technology, Inc., were dissolved in DMSO and stored at −20°C. In
all experiments, the final DMSO concentration was 0.1% (v/v), and
DMSO alone had no demonstrable effect on cultured cells. DMSO
(0.1%) served as the vehicle control. RPMI-1640 medium, fetal
bovine serum (FBS), penicillin (5,000 U/ml), streptomycin (5,000
mg/ml) and SparkZol reagent were purchased from Shandong Sparkjade
Biotechnology Co., Ltd. The propidium iodide (PI)/RNase staining
buffer and the fluorescein isothiocyanate (FITC) Annexin V
apoptosis detection kit were purchased from BD Biosciences.
Anti-SLC7A11/xCT antibody [cat. no. ab175186; 1:2,000 for western
blotting (WB)] was purchased from Abcam; anti-GPX4 antibody (cat.
no. 67763-1-Ig; 1:1,000 for WB) and anti-GAPDH antibody (cat. no.
60004-1-Ig; 1:10,000 for WB) were purchased from Wuhan Sanying
Biotechnology; anti-ATF4 antibody (cat. no. A18687; 1:1,000 for WB)
and anti-CHOP antibody (cat. no. A21902; 1:1,000 for WB) were
purchased from ABclonal Biotech Co., Ltd. HRP Goat Anti-Mouse IgG
(cat. no. AS003; 1:10,000 for WB) and HRP Goat Anti-Rabbit IgG
(cat. no. AS014; 1:10,000 for WB) were also purchased from ABclonal
Biotech Co., Ltd.
Cell lines and cell culture
Jurkat T-ALL cells were acquired from Leibniz
Institute DSMZ-German Collection of Microorganisms and Cell
Cultures GmbH, and Molt-4 T-ALL cells were purchased from The Cell
Bank of Type Culture Collection of The Chinese Academy of Sciences.
Both cell lines were cultured in RPMI-1640 medium supplemented with
10% FBS and 1% penicillin-streptomycin solution. Cells were
cultured in a humidified incubator at 37°C with 5%
CO2.
Isolation of peripheral blood
mononuclear cells (PBMCs) from cord blood
Cord blood samples were collected from three healthy
mothers who gave birth at the Affiliated Hospital of Weifang
Medical University (Weifang, China) in March 2024 after providing
written informed consent. Briefly, ~20 ml cord blood was collected
from each sample into EDTA anticoagulant tubes and diluted with 20
ml PBS. Subsequently, 20 ml diluted cord blood was layered onto 15
ml Ficoll-Paque (Beijing Solarbio Science & Technology Co.,
Ltd.) in a 50-ml conical tube and centrifuged at 400 × g for 30 min
at 18°C. The PBMC fraction was then transferred to another tube and
washed twice with PBS. Cell viability was required to exceed 90% of
total cells determined by trypan blue assay for each sample. In
brief, the cell suspension was mixed with 0.4% Trypan Blue solution
(cat. no. 93595, Sigma-Aldrich, Beijing, China) in a 9:1 ratio, mix
gently and allow to stain for 3–5 min. The stained cell suspension
was loaded onto the hemocytometer, the number of viable (unstained,
clear) and non-viable (stained, blue) cells were counted in the
gridded area of the hemocytometer using a microscope. Cell
viability was calculated as a percentage using the formula:
Viability=(Number of viable cells/Total number of cells) ×100%.
Cell viability and proliferation
assay
To evaluate the viability and proliferation of T-ALL
cells, the Cell Counting Kit-8 (CCK-8; Beijing Solarbio Science
& Technology Co., Ltd.) was used. Jurkat and Molt-4 cells were
seeded in 96-well plates at a density of 5,000 cells/well and were
treated with DHA at the indicated concentrations (0, 2.5, 5, 10 and
20 µM for Molt-4; 0, 5, 10, 20 and 40 µM for Jurkat) for 24, 48 and
72 h at 37°C. Cytotoxic effects of DHA on PBMCs isolated from cord
blood were evaluated PBMCs were seeded into 96-well plates
(1×105 cells per well) and treated with various DHA
concentrations (0, 5, 10, 20 and 40 µM) for 24, 48 and 72 h at
37°C. Furthermore, Jurkat and Molt-4 cell lines were treated with
DHA (10 µM), Fer-1 (1 µM) or both for 48 h at 37°C. Following the
treatment, 10 µl CCK-8 solution was added to each well and
incubated for 3 h, according to the manufacturer's protocol. The
optical density (OD) was measured at a wavelength of 450 nm using a
microplate reader (Multiskan GO; Thermo Fisher Scientific, Inc.).
Cell viability was calculated as the OD value of treated cells/OD
value of control cells. The IC50 for each cell line was
calculated based on the OD value corresponding to 50% inhibition by
DHA.
Cell apoptosis assay
Jurkat and Molt-4 cells were plated in 6-well plates
at a density of 5×105 cells/well and were treated with
DHA (0, 5, 10, and 20 µM) for 48 h at 37°C. In addition, cells
(5×105/ml) were treated with DHA (10 µM) in the presence
or absence of Fer-1 (1 µM) for 48 h at 37°C. Subsequently, the
cells were collected, washed twice in cold 1X PBS, and resuspended
in 300 µl binding buffer containing 5 µl PI and 5 µl Annexin V-FITC
for 30 min in the dark at room temperature. The percentage of
apoptotic cells was determined using a Flow Cytometer (DxFLEX;
Beckman Coulter, Biotechnology (Suzhou) Co., Ltd.). The data were
analyzed using CytExpert software for DxFLEX (version 2.0.0.283,
Beckman Coulter, Inc.) and FlowJo software (version 10.8.1, Becton,
Dickinson & Company).
Cell cycle analysis
A PI/RNase Staining Solution kit was used to assess
cell cycle distribution. After 48 h of treatment at 37°C with
various doses of DHA (Jurkat cells: 5, 10, and 20 µM; Molt-4 cells:
2.5, 5, and 10 µM) or vehicle control, both cells were then
harvested and washed twice with pre-cooled PBS. The cells were then
fixed with 70% cold ethanol at −20°C overnight. Subsequently, the
cells were then stained with 500 µl PI/RNase buffer at room
temperature in the dark for 15 min, according to the manufacturer's
instructions. The cell cycle was examined using flow cytometry
(DxFLEX; Beckman Coulter, Inc.) and data were analyzed using FlowJo
software (version 10.8.1; Becton, Dickinson & Company).
ROS analysis
ROS levels in cells were measured using
2′,7′-dichlorofluorescin diacetate (DCFH-DA; Beijing Solarbio
Science & Technology Co., Ltd.). Jurkat and Molt-4 cells were
seeded in 6-well plates at a density of 1×106
cells/well. The cells were treated with DHA at the indicated
concentrations (0, 5, 10, 20, and 40 µM for Jurkat; 0, 2.5, 5, 10,
and 20 µM for Molt-4) for 48 h at 37°C. Furthermore, both cell
lines were treated with DHA (10 µM), Fer-1 (1 µM), or a combination
of both for 48 h at 37°C. After the indicated treatments, cells
were harvested and washed twice with PBS and suspended in
serum-free culture medium containing DCFH-DA at a final
concentration of 10 µmol/l at 37°C for 30 min. Finally,
fluorescence intensity was determined by flow cytometry (DxFLEX;
Beckman Coulter, Biotechnology (Suzhou) Co., Ltd.) and the results
were analyzed using FlowJo software.
GSH and malondialdehyde (MDA)
assays
Intracellular GSH levels were detected using a
Reduced GSH Content Assay Kit (Beijing Solarbio Science &
Technology Co., Ltd.). MDA, as an end product of lipid
peroxidation, was evaluated using the MDA Content Assay Kit
(Beijing Solarbio Science & Technology Co., Ltd.). All assays
were performed in strict accordance with the kit protocol.
WB
Jurkat and Molt-4 (5×105 cells/per well)
were cultured in 6-well plates with 10, 20, 40 µM and 5, 10, 20 µM
DHA for 48 h at 37°C, respectively. Pretreated cells were lysed on
ice with RIPA buffer (Shandong Sparkjade Biotechnology Co., Ltd.)
containing 0.1% protease inhibitor and 1% phenylmethylsulfonyl
fluoride. The soluble fraction was isolated by centrifugation at
12,000 × g for 10 min at 4°C, and the supernatant was transferred
to a new tube. Subsequently, a BCA kit was used to determine
protein concentration. Equal amounts of protein from each sample
(20 µg) were separated by SDS-PAGE on 12.5 or 15% gels, and were
then transferred to 0.22-µm polyvinylidene fluoride membranes,
which were sealed with 5% nonfat dry milk in Tris-buffered
saline-0.1% Tween-20 (TBST) for 2 h at room temperature. The
membranes were then incubated overnight at 4°C with primary
antibodies against GAPDH, SLC7A11, GPX4, ATF4 and CHOP, followed by
incubation with secondary antibodies for 1 h at room temperature,
before being washed with TBST three times. Finally, an Ultra High
Sensitivity ECL Kit (cat. no. GK10008, Glpbio Technology) was used
to visualize the proteins on the membranes using an Amersham Imager
600 (GE Healthcare Bio-Sciences), and Image J software version
1.53t (National Institutes of Health) was subsequently employed for
semi-quantitative analysis of the obtained images.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the control and
DHA-treated, Fer-1-treated, and combination-treated Jurkat and
Molt-4 cells using SparkZol reagent and was quantified using an
ultra-micro spectrophotometer (NanoDrop OneC; Thermo Fisher
Scientific, Inc.). According to manufacturer's protocol, RNA (1 µg)
was reverse transcribed into cDNA using the Evo M-MLV RT Premix
(Hunan Aikeru Bioengineering Co., Ltd.). The mRNA expression levels
of ATF4, CHOP and GAPDH were measured by qPCR in 96-well plates
with cDNA as the template using a 7500 Fast Real-Time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.) and 2X
SYBR® Green Premix Pro TaqHS qPCR Kit (Hunan Aikeru
Bioengineering Co., Ltd.). The reaction conditions for
amplification were set according to the manufacturer's
instructions: Initial denaturation, 95°C for 30 sec, followed by 40
cycles of denaturation (95°C for 10 sec), annealing (55°C for 20
sec) and extension (72°C for 30 sec). The relative gene expression
levels were calculated using the 2−ΔΔCq method (23). GAPDH was used as the internal
control. The primer sequences were as follows: ATF4, forward
5′-AAGCCTAGGTCTCTTAGATG-3′, reverse 5′-TTCCAGGTCATCTATACCCA-3′;
CHOP, forward 5′-GGAAACAGAGTGGTCATTCCC-3′, reverse
5′-CTGCTTGAGCCGTTCATTCTC-3′; and GAPDH, forward
5′-ACAACTTTGGTATCGTGGAAGG-3′ and reverse
5′-GCCATCACGCCACAGTTTC-3′.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 8.0 software (Dotmatics). All data are presented as the mean
± SD and experiments were performed in triplicate. The differences
between two groups were determined using the unpaired two-tailed
Student's t-test. Comparisons among multiple groups were analyzed
by one-way ANOVA followed by Tukey's multiple comparisons test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
DHA treatment suppresses T-ALL cell
viability in vitro
To assess whether DHA can inhibit the proliferation
of T-ALL cells in vitro, the CCK-8 assay was used to detect
the effects of DHA on the viability of T-ALL cell lines. Jurkat and
Molt-4 cells were treated with DHA at various concentrations,
ranging from 0 to 40 µM for 24, 48 and 72 h. As demonstrated in
Fig. 1B, the results showed that at
the same dosages used to treat the T-ALL cell lines Jurkat and
Molt-4, DHA displayed minimum toxicity in PBMCs. Compared with in
the control groups, the viability of the two T-ALL cell lines after
DHA treatment was significantly decreased in a time- and
dose-dependent manner. DHA at 2.5 and 5 µM significantly reduced
the viability of Molt-4 and Jurkat cells at 24 h compared to the
control group, respectively. When the DHA concentration was ≥20 µM,
DHA exhibited a certain level of toxicity towards PBMCs. However,
in comparison to PBMCs, 20 µM DHA was more inclined to induce cell
death in T-ALL cells. After 48 h of incubation, the IC50
of DHA was 8.324 µM (Jurkat) and 4.938 µM (Molt-4) (Fig. 1C). Molt-4 cells were more sensitive
than Jurkat cells to DHA. For the selection of DHA doses in
subsequent experiments, the concentrations were based on
preliminary CCK-8 experiments as specified above. These experiments
helped determine the appropriate range of DHA concentrations that
would not cause excessive cell death while still eliciting a
biological response.
DHA induces apoptosis in T-ALL
cells
The present study subsequently evaluated the effects
of DHA treatment on the induction of cell death in T-ALL cells.
Jurkat and Molt-4 cells were treated with different doses of DHA
(5, 10 and 20 µM) for 48 h, and cell death was detected by
FITC-Annexin V staining. As depicted in Fig. 2A and B, following a 48-h exposure of
the two T-ALL cell lines to DHA, there was a dose-dependent
increase in cell death. Cells treated with DHA were predominantly
clustered in the right upper quadrant of the scatter plots (Annexin
V- and PI-positive cells), indicating late apoptotic cells. In the
presence of 10 µM DHA, the proportion of early and late apoptotic
Molt-4 and Jurkat cells were 28.41±0.67 and 16.33±0.93%,
respectively. Molt-4 cells treated with 20 µM DHA exhibited a 52.6%
proportion of late apoptotic or dead cells. However, the proportion
of cells in the right upper quadrant decreased after Fer-1
treatment compared with that in the DHA group. The proportion of
late apoptotic or dead cells in the DHA + Fer-1-treated Jurkat and
Molt-4 cells decreased by 6.7 and 8.9%, respectively, compared with
the DHA-treated group (Fig. 2C and
D). Initially, the selection of DHA concentrations was guided
by the CCK-8 data, focusing on those concentrations that were close
to the IC50. Notably, when treated with 20 µM DHA, there
was a marked increase in cell apoptosis, particularly in Molt-4
cells, where the apoptosis rate exceeded 50%. In order to ensure
comparability between Jurkat and Molt-4 cells, the present study
ultimately opted for a DHA concentration of 10 µM when performing
co-treatments with Fer-1. The present findings suggested that DHA
can induce apoptosis in T-ALL cells; however, the results of
Annexin V/PI staining pointed towards a mechanism that may be
involved in ferroptosis other than apoptosis.
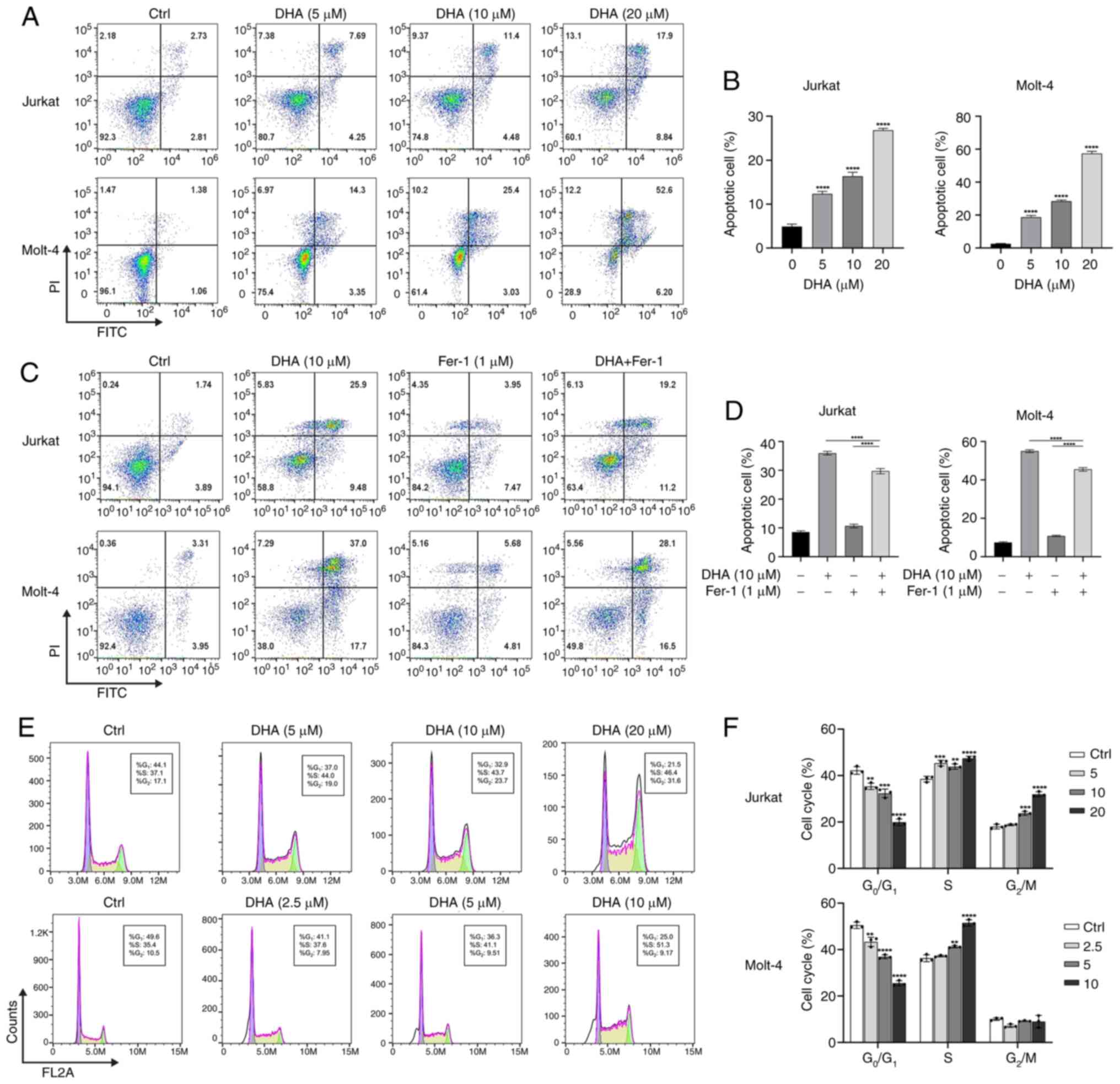 | Figure 2.Changes in cell cycle progression and
cell death induced by DHA in Jurkat and Molt-4 cells. (A) Cells
were treated with DHA (0, 5, 10 and 20 µM) for 48 h, followed by
Annexin V/FITC and PI double staining, and flow cytometric analysis
of apoptosis. (B) Quantification of the number of apoptotic cells
in (A). (C) Flow cytometric analysis of Annexin V/FITC and
PI-stained T-cell acute lymphoblastic leukemia cell lines incubated
with DHA (10 µM) for 48 h with or without Fer-1 (1 µM). (D)
Quantification of the number of dead cells in (C). (E) Jurkat cells
were treated with DHA (5, 10 and 20 µM) and Molt-4 cells were
treated with DHA (2.5, 5 and 10 µM) for 48 h, followed by PI
staining and flow cytometric analysis of cell cycle distribution.
(F) Cell cycle distribution analysis of (E). **P<0.01,
***P<0.001, ****P<0.0001 vs. 0 µM DHA. DHA,
dihydroartemisinin; Fer-1, ferrostatin-1; FITC, fluorescein
isothiocyanate; PI, propidium iodide. |
DHA induces cycle arrest in T-ALL cell
lines
To determine whether the decreased viability of
DHA-treated T-ALL cells was attributable to the induction of cell
cycle arrest, flow cytometry was applied to analyze the effect of
DHA on cell cycle distribution. By comparing the cell proportions
at each phase in response to different doses of DHA, it was
revealed that Jurkat cells began to accumulate in the S phase and
G2/M phase in a dose-dependent manner following
treatment with DHA for 48 h. In Molt-4 cells, the proportion of
cells in G0/G1 phase gradually decreased from
49.6 to 25%, while the percentage of cells in the S phase increased
from 35.4 to 51.3%, with increasing concentrations of DHA (Fig. 2E and F). Therefore, DHA may
facilitate the transition of T-ALL cells from the G1
phase to S phase, subsequently inducing cell cycle arrest in the S
and G2/M phases, weakening their proliferative capacity
and reducing cell viability.
The present study also performed the experiments
after prolonged treatment of Jurkat and Molt-4 cells with DHA for
72 h. The results showed that DHA arrested the Jurkat cell cycle in
S and G2/M phases, and the Molt-4 cell cycle in
G0/G1 phase (Fig. S1B), which may hinder cell
proliferation and was positively associated with the results of
apoptosis analysis. For example, when Jurkat cells were treated
with 10 µM DHA for 72 h, the cell cycle was arrested in S and G2/M
phases, and apoptotic cells increased by 35.84% (Fig. S1A). As Molt-4 cells exhibited
higher sensitivity to DHA, prolonged treatment of the Molt-4 cells
with DHA for 72 h resulted in the majority of the cells exiting the
cell cycle and ceasing proliferation, thus resulting in an
increased proportion of cells in the G0/G1
phase. These results suggested that the distinct mechanism for cell
cycle arrest serves a crucial role in ensuring an appropriate
response to DNA damage over time.
DHA induces ferroptosis in T-ALL
cells
Ferroptosis is a type of programmed cell death
driven by the accumulation of ROS and lipid peroxides closely
related to oxidative stress and cystine metabolism (17). To investigate whether ferroptosis is
associated with DHA-induced cell death in T-ALL cells, cytoplasmic
ROS was quantified by flow cytometry using DCFH-DA probe staining.
As shown in Fig. 3A, cytoplasmic
ROS levels were markedly increased in Jurkat and Molt-4 cells after
DHA treatment. Following treatment with 10 and 20 µM DHA,
respectively, the FITC-A subsets of Jurkat cells were 15.0 and
35.4, and those of Molt-4 cells were 38.0 and 55.7, which were
consistent with the previous results, indicating the higher
sensitivity of Molt-4 cells to DHA. The present study subsequently
examined the protein expression levels of SLC7A11 and GPX4 after 48
h of treatment with different doses of DHA. Compared with those in
the control group, the expression levels of SLC7A11 and GPX4 were
significantly downregulated in the DHA-treated groups (Jurkat cells
with 20, 40 µM DHA; Molt-4 cells with 10, 20 µM DHA), as indicated
in Fig. 3B. A previous study
revealed that lipid peroxides are typically removed catalytically
by the antioxidant enzyme GPX4, a process that requires GSH as a
cofactor (24). Therefore, the
present study examined MDA and GSH levels in the T-ALL cells
treated with DHA. As shown in Fig.
3C, in response to treatment with 5 µM DHA, there was no
discernible impact on the MDA content in either Jurkat or Molt-4
cells. At 10 µM DHA, the MDA content in Jurkat and Molt-4 cells was
significantly increased compared with that in the control group. In
response to 20 µM DHA, the MDA content doubled in both Jurkat and
Molt-4 cells compared to the control group, indicating prolonged
oxidative stress. Similarly, DHA administration reduced GSH levels
in T-ALL cells in a dose-dependent manner compared with in the
control cells, indicating an impaired antioxidative response, and
the GSH content was decreased even in response to a low
concentration of DHA (5 µM) (Fig.
3D). The effects of 40 µM DHA on MDA and GSH in Jurkat and
Molt-4 cells are not shown because not enough cells obtained
following high dose DHA treatment. These results indicated that DHA
may trigger ferroptosis in T-ALL cells.
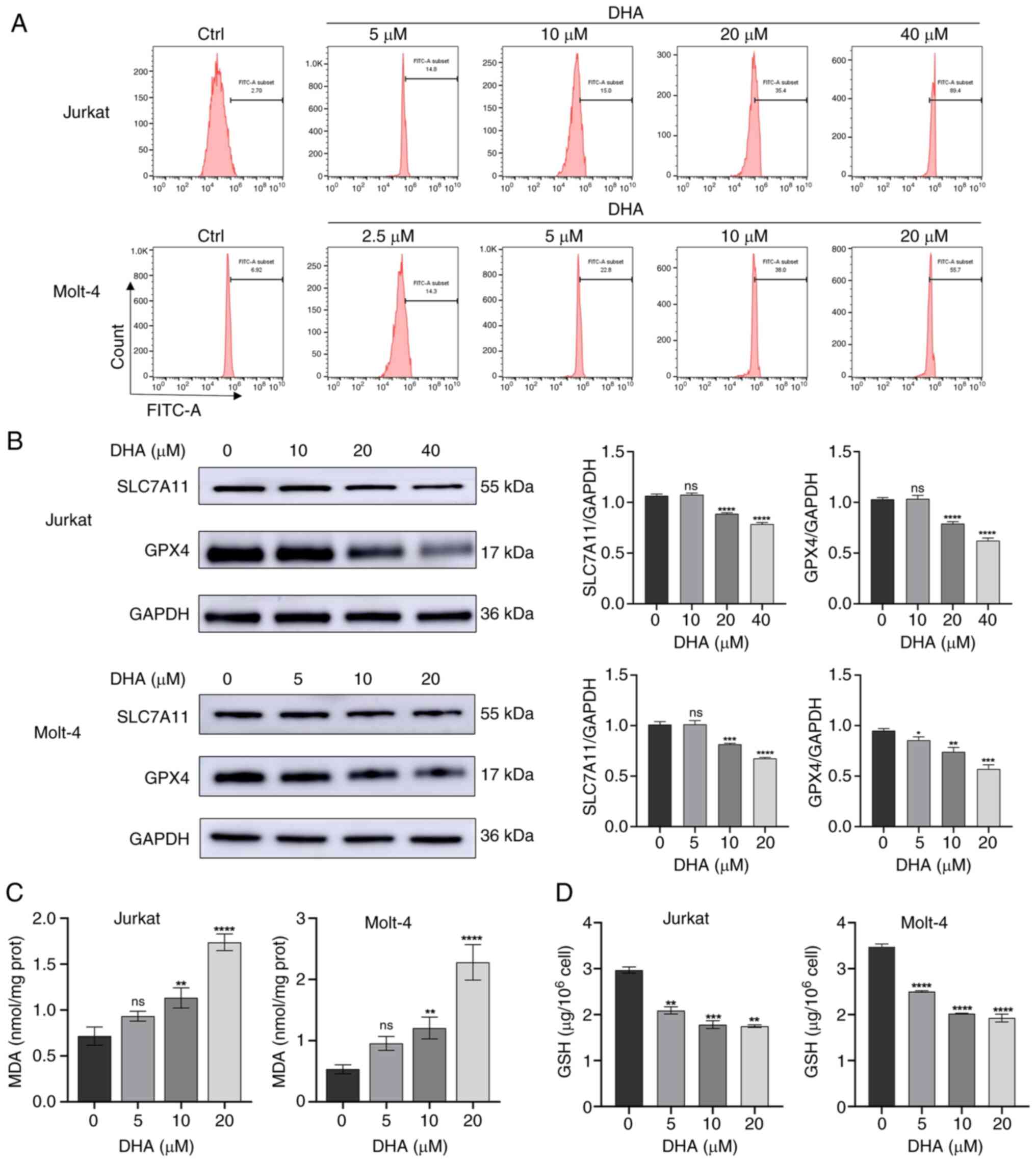 | Figure 3.DHA induces ferroptosis in T-ALL cell
lines. (A) Jurkat and Molt-4 cells were treated with DHA for 48 h,
and cell ROS levels were measured using flow cytometry. (B) Jurkat
and Molt-4 cells were treated with DHA for 48 h, and western
blotting was used to detect the protein expression levels of
SLC7A11 and GPX4 in T-ALL cells. (C) MDA and (D) GSH levels were
determined in T-ALL cells exposed to 5, 10 or 20 µM DHA. All the
data are representative of three independent experiments.
*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs. 0 µM
DHA. DHA, dihydroartemisinin; FITC, fluorescein isothiocyanate;
GPX4, glutathione peroxidase 4; MDA, malondialdehyde; ns, no
significance; T-ALL, T-cell acute lymphoblastic leukemia. |
Fer-1 partially attenuates the
ferroptosis of T-ALL cells induced by DHA
Fer-1, a specific inhibitor of ferroptosis, has been
reported to inhibit lipid peroxidation, thereby protecting cells
from lipid peroxidation-induced cellular damage (25). To further assess the effect of DHA
on the regulation of ferroptosis, Fer-1 was applied as a
ferroptosis inhibitor in the present study. The results revealed
that the combined treatment of DHA and Fer-1 inhibited viability
compared to the control group; however, cell viability was higher
in the DHA + Fer-1 group than in the DHA alone group (Fig. 4A). In addition, the expression
levels of SLC7A11 and GPX4 were upregulated in the DHA + Fer-1
group compared with those in the DHA group, suggesting that Fer-1
limited DHA-induced damage to the antioxidant system (Fig. 4B). Subsequently, the effects of
Fer-1 pretreatment on ROS, MDA and GSH levels after DHA treatment
were examined. As shown in Fig.
4C-E, Fer-1 attenuated the increase in ROS and MDA levels, and
rescued the DHA-induced reduction in GSH. These results suggested a
rescue role of Fer-1 in DHA-induced cell injury by blocking
ferroptosis. The results of WB indicated that there were no
statistically significant differences in protein expression levels
of SLC7A11, GPX4, ATF4, and CHOP between Jurkat cells treated with
5 and 10 µM DHA (Figs. 3B and
5B). Similarly, Molt-4 cells
treated with 2.5 µM DHA did not exhibit significant differences in
protein expression of SLC7A11, GPX4, ATF4, and CHOP compared with
those treated with 5 µM DHA (Figs.
3B and 5B). Therefore, 10 µM
for Jurkat cells and 5 µM for Molt-4 cells were selected as the
minimum concentrations for further WB experiments. In the fer-1
rescue experiment, both cell lines were treated with 10 µM of DHA,
as shown in Figs. 4B and 5D.
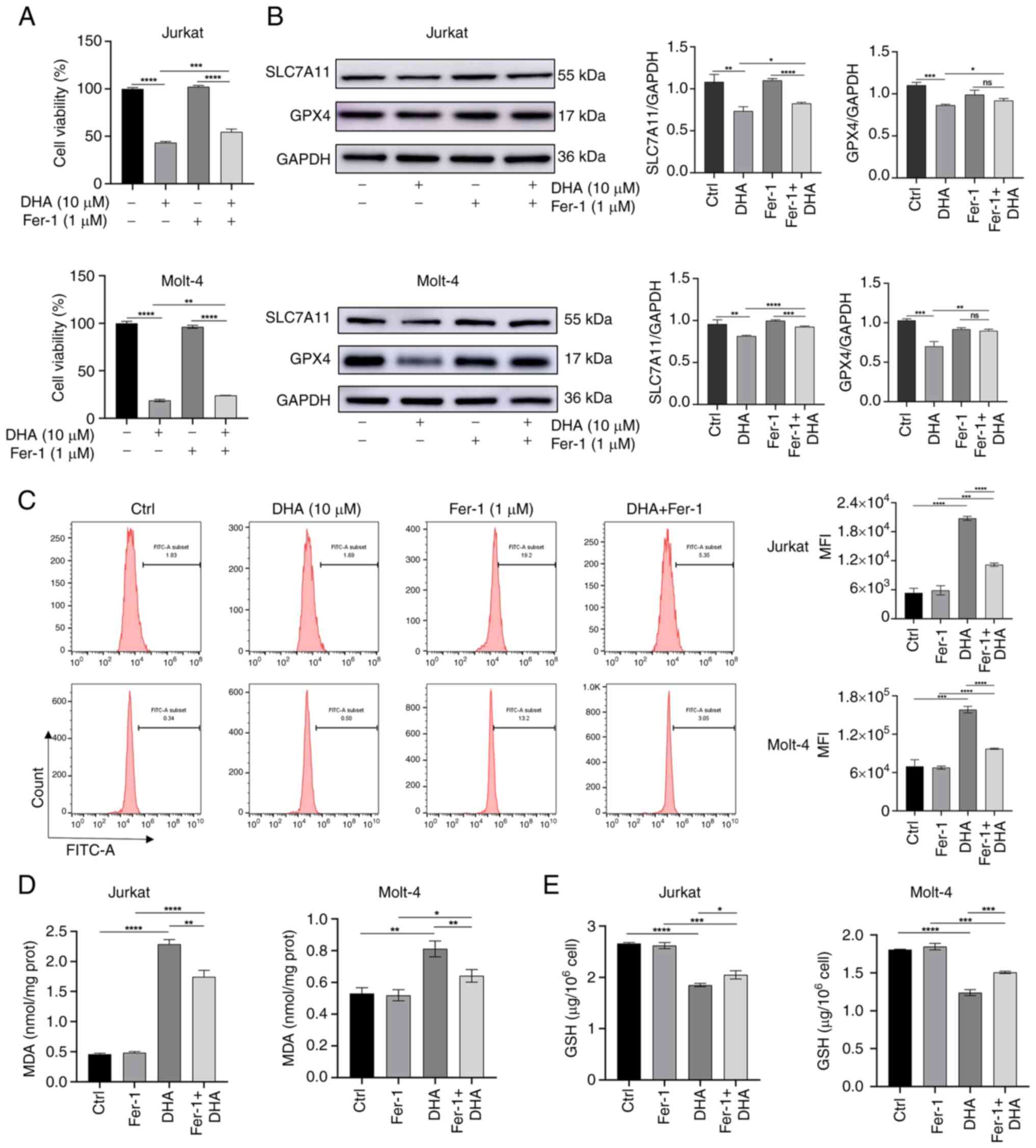 | Figure 4.Inhibition of ferroptosis can prevent
DHA-induced T-ALL cell death. T-ALL cells were treated with 1 µM
Fer-1 and 10 µM DHA for 48 h. (A) Cell Counting Kit-8 was used to
assess the viability of T-ALL cells. (B) Western blot analysis
measured the protein expression levels of SLC7A11 and GPX4 in
different groups of T-ALL cells, with histograms constructed based
on their respective relative grayscale values. (C) ROS levels in
cells were measured using flow cytometry, followed by assessment of
MFI. (D) MDA content was measured. (E) GSH content was measured
using a GSH assay kit. *P<0.05, **P<0.01, ***P<0.001,
****P<0.0001. DHA, dihydroartemisinin; Fer-1, ferrostatin-1;
FITC, fluorescein isothiocyanate; GPX4, glutathione peroxidase 4;
MDA, malondialdehyde; MFI, mean fluorescence intensity; ns, no
significance; T-ALL, T-cell acute lymphoblastic leukemia. |
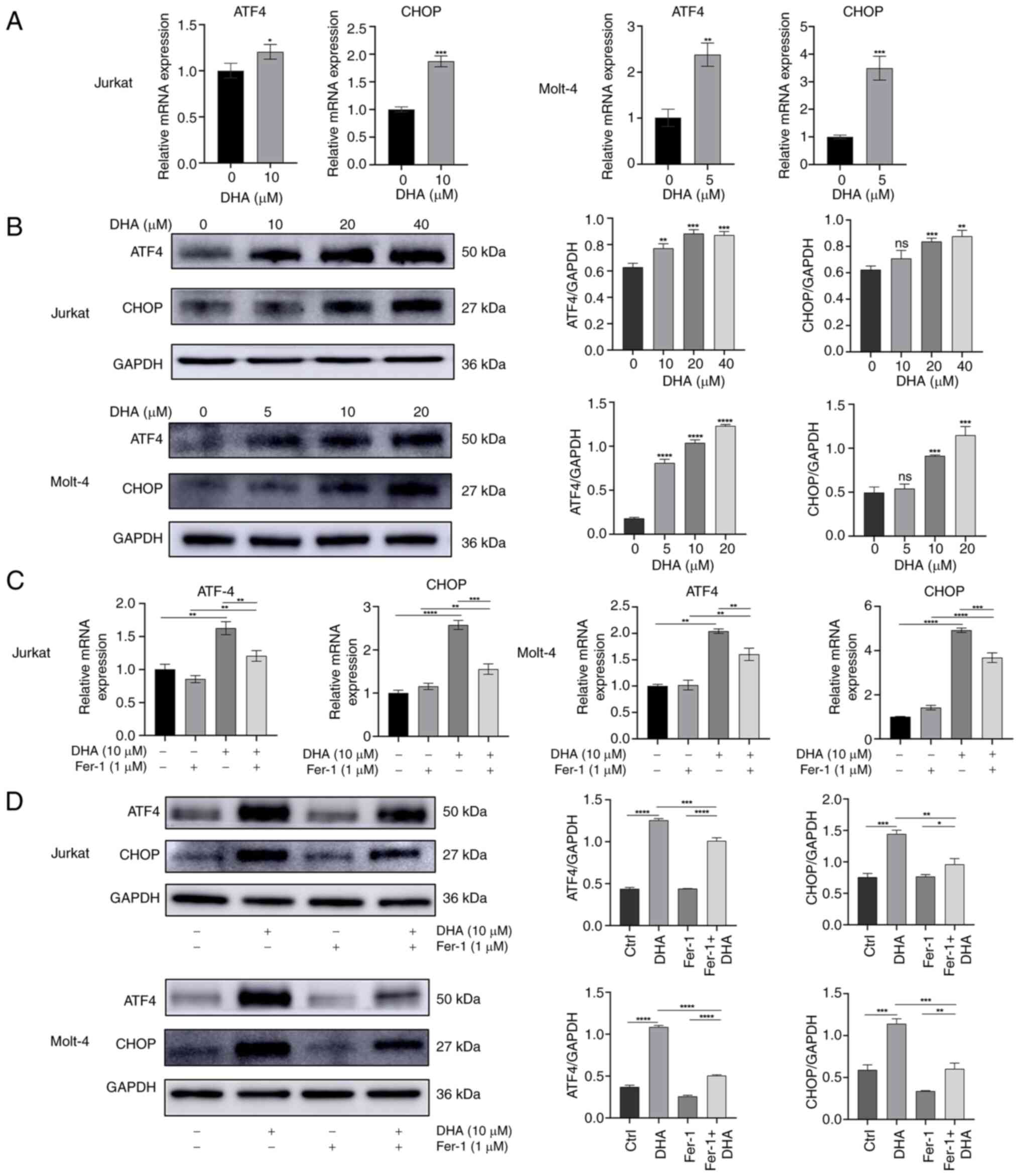 | Figure 5.DHA upregulates the expression of ER
stress-related genes. (A) Jurkat and Molt-4 cells were cultured in
10 and 5 µM DHA for 48 h, respectively. RT-qPCR was performed to
measure the mRNA expression levels of ATF4 and CHOP. (B) Jurkat and
Molt-4 cells were cultured in DHA for 48 h. The protein expression
levels of ATF4 and CHOP were assessed via western blot analysis,
followed by the generation of associated grayscale histograms. The
T-cell acute lymphoblastic leukemia cells were treated with 1 µM
Fer-1 and 10 µM DHA for 48 h. (C) mRNA expression levels of ATF4
and CHOP were assessed by RT-qPCR. (D) Protein expression levels of
ATF4 and CHOP were measured by western blotting, with histograms
constructed based on relative grayscale values. *P<0.05,
**P<0.01, ***P<0.001, ****P<0.0001 vs. 0 µM DHA or as
indicated. DHA, dihydroartemisinin; Fer-1, ferrostatin-1; ns, no
significance; RT-qPCR, reverse transcription-quantitative PCR. |
DHA upregulates the expression of ER
stress-related genes
Boelens et al (26) studied ER stress in cells using a
synthesized cytotoxic artemisinin compound conjugated with a
fluorescent dansyl moiety, and revealed that the ER is the main
site of its accumulation by organelle-specific dye co-localization.
ER stress can be triggered by anticancer chemicals, including
pro-ferroptotic reagents such as erastin (27). Based on the results of previous
study (22), the present study
examined the transcription of ER stress-related genes. The results
demonstrated that DHA, an emerging inducer of ferroptosis, induced
ER stress in T-ALL cells, as shown by elevated mRNA expression
levels of ATF4 and CHOP (Fig. 5A).
Compared with in the control group treated with DMSO, DHA also
upregulated the protein expression levels of ATF4 and CHOP in a
dose dependent manner in T-ALL cells (Fig. 5B). The present study further
investigated the effect of DHA with or without Fer-1 on the
expression levels of ATF4 and CHOP in Jurakt and Molt-4 cells. As
expected, the enhancement induced by DHA in Jurkat and Molt-4 cells
was attenuated by Fer-1 co-treatment (Fig. 5C and D). These results suggested
that ER stress may serve an important role in DHA-induced
ferroptosis.
Discussion
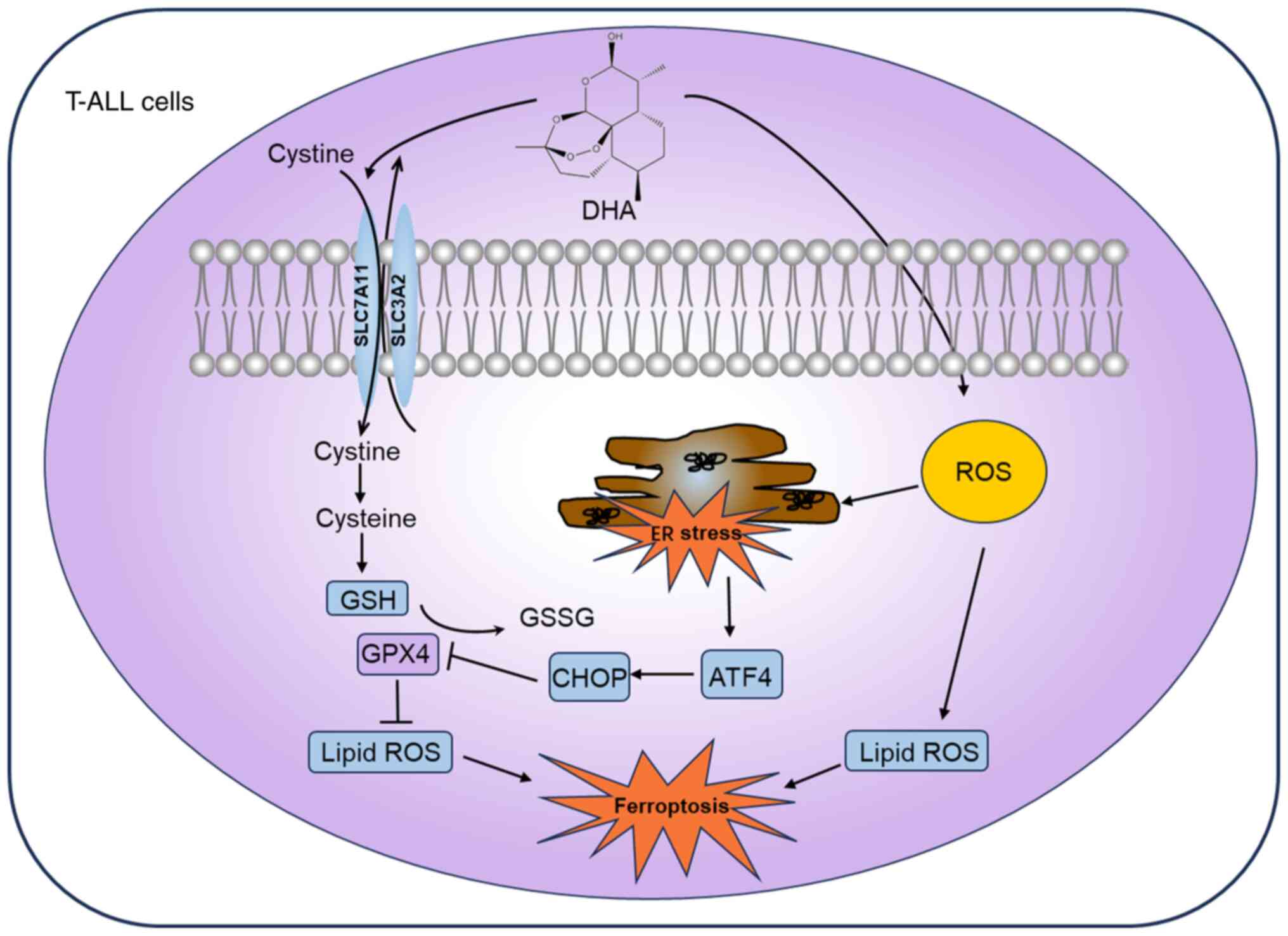
The present study investigated the antitumor effect
of DHA on T-ALL cells and identified the mechanisms involved. The
results demonstrated that DHA significantly decreased T-ALL cell
viability, and induced cell cycle arrest at S or G2/M
phase and apoptosis. Furthermore, in T-ALL cells treated with DHA,
ferroptosis was markedly induced, as evidenced by elevated levels
of MDA and ROS, coupled with decreased GSH levels. Additionally,
DHA triggered a significant ER stress response in T-ALL cells.
Notably, Fer-1 administration partially recovered the viability of
T-ALL cells, enhancing the protein expression levels of SLC7A11 and
GPX4, while lowering those of ATF4 and CHOP. These results further
suggested the critical role of ferroptosis in the suppressive
effects of DHA on T-ALL biological functions, whereby DHA triggers
ferroptosis in T-ALL cells by inhibiting SLC7A11 expression and
activating the ATF4-CHOP signaling pathway (Fig. 6).
In addition to its well-known anti-malarial
applications, DHA is currently being evaluated for its potential to
treat various types of cancer because of its high potency, low
toxicity and short half-life (28),
as well as its documented safety in patients with malaria. In the
present study, the anti-leukemic effects of DHA were assessed in
T-ALL cells. Initially, through the CCK-8 assay, it was
demonstrated that DHA inhibited the viability of T-ALL cells,
accompanied by a minor impact on normal human PBMCs. Cell cycle
arrest is considered a key element in the antitumor activity of
artemisinin derivatives (29).
Consistent with the findings of Jin et al (30), it was observed that DHA induced cell
cycle arrest at S phase and G2/M phase, thereby
inhibiting the viability of T-ALL cells. Apoptosis has been
regarded as the primary form of cell death (31,32).
The present study observed that, regardless of the concentration of
DHA employed, dead cells were predominantly clustered in the right
upper quadrant (Annexin V- and PI-positive) indicating late
apoptotic cells. In addition, ROS accumulation was present in
DHA-induced T-ALL cells. These findings suggested that, apart from
apoptosis, there are other types of cell death induced by DHA in
T-ALL cells.
Ferroptosis is a form of cell death that is
dependent on ROS (33). The role of
DHA in inducing ferroptosis in cancer cells was first reported in
head and neck cancer cells (34),
followed by acute myeloid leukemia cells (35). Ferroptosis is mainly associated with
iron accumulation and lipid peroxidation (36). In the present study, the increased
levels of the lipid peroxidation marker MDA and the decreased
levels of GSH in the cells indicated a pronounced induction of
ferroptosis in DHA-treated T-ALL cells. To further assess the
contribution of ferroptosis to the anti-leukemic effects of DHA,
Fer-1 was employed to rescue cellular viability. Fer-1 is a
ferroptosis inhibitor, extensively utilized both in vitro
and in vivo (37,38). Functioning as an antioxidant, the
efficacy of Fer-1 in the inhibition of ferroptosis is primarily
dependent on its suppression of lipid peroxidation (39). The results of the present study
indicated that the co-treatment with DHA and Fer-1 exhibited a
rescuing effect. It was observed that Fer-1 treatment significantly
attenuated the function of DHA in decreasing the viability of T-ALL
cells, and the proportion of late apoptotic or dead cells in the
right upper quadrant of flow cytometry plots was decreased.
Notably, Fer-1 treatment reduced the levels of ROS and MDA in the
DHA group, while increasing the GSH levels. This may be related to
the inhibition of lipid peroxidation by Fer-1; however, the
underlying mechanisms still require further research.
Recently, ferroptosis has been recognized as an
adaptive trait contributing to cancer cell destruction (40). Lipid ROS accumulation is a critical
factor in triggering ferroptosis (41). Inactivation of the cellular
antioxidant system is an important route leading to ROS generation.
SLC7A11 and GPX4 are considered central regulatory elements in
ferroptosis, with GPX4 acting as the primary defensive enzyme
against ferroptosis. The deficiency of GSH inhibits GPX4 activity,
thereby promoting ferroptosis (42). In addition, it has been shown that
system Xc− transports cystine from the extracellular
space into the intracellular space, promptly converting it to
cysteine, thus furnishing the requisite materials for GSH
synthesis. Inhibition of the system Xc− can impede
cysteine uptake, leading to decreased GSH levels and, consequently,
insufficient GPX4 capacity to eliminate lipid ROS, ultimately
inducing cell death (43). In the
present study, DHA treatment reduced the expression levels of
SLC7A11 and GPX4 in T-ALL cells. Furthermore, administration of
Fer-1 increased the protein expression levels of SLC7A11 and GPX4
in Jurkat and Molt-4 cells. These findings suggested that
ferroptosis serves a pivotal role in DHA-mediated inhibition of
T-ALL, and the ferroptosis induced by DHA may be achieved through
the inhibition of system Xc−.
Excessive ROS is a crucial stimulating factor in the
ER stress response. Lipid accumulation and oxidation are associated
with disturbances in ER protein homeostasis, known as ER stress
(44). Disturbances in ER
homeostasis involve a series of stress response signaling pathways,
collectively called the unfolded protein response (UPR). Although
the UPR is an adaptive protective mechanism, in the presence of
unresolvable ER stress, activation of ER stress by the UPR mediates
cell death in tumor cells along with the ER stress prolongation and
accumulation (45). ER stress has
been reported to be a regulator of the progression of ferroptosis
and one of the mechanisms by which DHA exerts its antitumor effects
(10). A previous study reported
that DHA kills protoscoleces through ER stress and the CHOP pathway
(46). Dixon et al (22) demonstrated that erastin induces
ferroptosis in various cellular environments by specifically
inhibiting system Xc−, and that small molecule
inhibitors of system Xc− trigger ER stress via the UPR.
As an emerging inducer of ferroptosis, the present study observed
that DHA can activate the ATF4-CHOP signaling pathway and induce ER
stress in Jurkat and Molt-4 cells, leading to ferroptosis.
Administration of Fer-1 decreased the protein expression levels of
ATF4 and CHOP in Jurkat and Molt-4 cells. A previous study has
shown that ATF4 protects against ferroptosis due to its ability to
activate xCT transcription (47).
It transcriptionally regulates membrane transporter proteins and
enzymes required for GSH biosynthesis in cancer cells involved in
chemoresistance (48).
Nevertheless, ER stress has been shown to promote HMOX1-mediated
ferroptosis caused by BAY11-7085, a IκBα inhibitor (49). A previous study evaluating the
effects of artesunate on Burkitt lymphoma cells supported the
findings of the present study (50).
The present study showed that DHA may activate
ATF4/CHOP-mediated ER stress, which may be related to antioxidant
system disturbances and excessive ROS accumulation. The
upregulation of ATF4 and CHOP may lead to the degradation of GSH,
thereby inducing ferroptosis. However, the current results do not
exclude the regulatory role of other types of ROS, such as the
increased ROS mediated by iron accumulation in the Fenton reaction,
on DHA-induced ferroptosis in T-ALL cells. Further study is
required to better understand the role of the ER stress-xCT axis in
DHA-induced ferroptosis in T-ALL cells. Given the potential
interconnection between ER stress and ferroptosis, a limitation of
the present study lies in the need for deeper exploration to
comprehensively grasp the intricate mechanisms underlying
ferroptosis and the supplementary impacts elicited by DHA in the
treatment of T-ALL. Our future studies would benefit from a
combined approach that leverages both flow cytometry and molecular
assays to provide a more complete understanding of the apoptotic
mechanism induced by DHA in T-ALL cells. Additionally, the absence
of in vivo animal experiments constitutes another constraint
of the present study.
In conclusion, the results of the present study
demonstrated that DHA may induce ferroptosis in different types of
T-ALL cells and elicit a significant ER stress response in tumor
cells. These findings may improve understanding of the antitumor
potential of DHA and provide novel insights for the development of
drugs for the treatment of T-ALL.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Professor Ying Song
(Weifang Medical University) for critically reading the manuscript,
and Dr Linda Hu (Upstate Medical University, New York, NY, USA) for
critically reading and language editing the manuscript.
Funding
The present study was supported by the Shandong Provincial
Natural Science Foundation of China (grant nos. ZR2020QH096 and
ZR2020KC016).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
NT and XL designed and performed experiments,
analyzed the data and wrote the manuscript. YL, HW and YZ helped
perform specific experiments and data analysis. HW and ZH designed
and supervised the project, acquired funding and revised the
manuscript. YL and HW confirm the authenticity of all the raw data.
All authors have read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
This present study was reviewed and approved by the
Ethics Committee of the Affiliated Hospital of Weifang Medical
University (approval no. wfmc-2023-ky-042). The participants
provided their written informed consent to participate in this
study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Belver L and Ferrando A: The genetics and
mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer.
16:494–507. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vadillo E, Dorantes-Acosta E, Pelayo R and
Schnoor M: T cell acute lymphoblastic leukemia (T-ALL): New
insights into the cellular origins and infiltration mechanisms
common and unique among hematologic malignancies. Blood Rev.
32:36–51. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Raetz EA and Teachey DT: T-cell acute
lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program.
2016:580–588. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Newman DJ, Cragg GM and Snader KM: Natural
products as sources of new drugs over the period 1981–2002. J Nat
Prod. 66:1022–1037. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Neill US: From branch to bedside: Youyou
Tu is awarded the 2011 Lasker~DeBakey Clinical Medical Research
Award for discovering artemisinin as a treatment for malaria. J
Clin Invest. 121:3768–3773. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cheong DHJ, Tan DWS, Wong FWS and Tran T:
Anti-malarial drug, artemisinin and its derivatives for the
treatment of respiratory diseases. Pharmacol Res. 158:1049012020.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lemke D, Pledl HW, Zorn M, Jugold M, Green
E, Blaes J, Löw S, Hertenstein A, Ott M, Sahm F, et al: Slowing
down glioblastoma progression in mice by running or the
anti-malarial drug dihydroartemisinin? Induction of oxidative
stress in murine glioblastoma therapy. Oncotarget. 7:56713–56725.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang CZ, Zhang H, Yun J, Chen GG and Lai
PBS: Dihydroartemisinin exhibits antitumor activity toward
hepatocellular carcinoma in vitro and in vivo. Biochem Pharmacol.
83:1278–1289. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li Y, Zhou X, Liu J, Gao N, Yang R, Wang
Q, Ji J, Ma L and He Q: Dihydroartemisinin inhibits the
tumorigenesis and metastasis of breast cancer via downregulating
CIZ1 expression associated with TGF-β1 signaling. Life Sci.
248:1174542020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dai X, Zhang X, Chen W, Chen Y, Zhang Q,
Mo S and Lu J: Dihydroartemisinin: A potential natural Anti-Cancer
drug. Int J Biol Sci. 17:603–622. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li S, Huang P, Gan J, Ling X, Du X, Liao
Y, Li L, Meng Y, Li Y and Bai Y: Dihydroartemisinin represses
esophageal cancer glycolysis by down-regulating pyruvate kinase M2.
Eur J Pharmacol. 854:232–239. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang T, Luo R, Li W, Yan H, Xie S, Xiao W,
Wang Y, Chen B, Bai P and Xing J: Dihydroartemisinin suppresses
bladder cancer cell invasion and migration by regulating KDM3A and
p21. J Cancer. 11:1115–1124. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sun WD, Yu XX, An YH, Wang X, Wang Y and
Tong XM: Dihydroartemisinin induces apoptosis of human acute T
lymphocytic leukemia cells by activating oxidative stress. Zhongguo
Shi Yan Xue Ye Xue Za Zhi. 28:753–757. 2020.(In Chinese).
PubMed/NCBI
|
|
14
|
Wong KH, Yang D, Chen S, He C and Chen M:
Development of nanoscale drug delivery systems of
dihydroartemisinin for cancer therapy: A review. Asian J Pharm Sci.
17:475–490. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shi H, Xiong L, Yan G, Du S, Liu J and Shi
Y: Susceptibility of cervical cancer to dihydroartemisinin-induced
ferritinophagy-dependent ferroptosis. Front Mol Biosci.
10:11560622023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Du J, Wang X, Li Y, Ren X, Zhou Y, Hu W,
Zhou C, Jing Q, Yang C, Wang L, et al: DHA exhibits synergistic
therapeutic efficacy with cisplatin to induce ferroptosis in
pancreatic ductal adenocarcinoma via modulation of iron metabolism.
Cell Death Dis. 12:7052021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cao JY and Dixon SJ: Mechanisms of
ferroptosis. Cell Mol Life Sci. 73:2195–2209. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tu H, Tang LJ, Luo XJ, Ai KL and Peng J:
Insights into the novel function of system Xc-in regulated cell
death. Eur Rev Med Pharmacol Sci. 25:1650–1662. 2021.PubMed/NCBI
|
|
19
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of non-apoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Patanè GT, Putaggio S, Tellone E, Barreca
D, Ficarra S, Maffei C, Calderaro A and Laganà G: Ferroptosis:
Emerging role in diseases and potential implication of bioactive
compounds. Int J Mol Sci. 24:172792023. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Qin Y, Qiao Y, Wang D, Tang C and Yan G:
Ferritinophagy and ferroptosis in cardiovascular disease:
Mechanisms and potential applications. Biomed Pharmacother.
141:1118722021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dixon SJ, Patel DN, Welsch M, Skouta R,
Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS,
et al: Pharmacological inhibition of Cystine-glutamate exchange
induces endoplasmic reticulum stress and ferroptosis. Elife.
3:e025232014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ursini F, Maiorino M, Valente M, Ferri L
and Gregolin C: Purification from pig liver of a protein which
protects liposomes and biomembranes from peroxidative degradation
and exhibits glutathione peroxidase activity on phosphatidylcholine
hydroperoxides. Biochim Biophys Acta. 710:197–211. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Miotto G, Rossetto M, Di Paolo ML, Orian
L, Venerando R, Roveri A, Vučković AM, Bosello Travain V, Zaccarin
M, Zennaro L, et al: Insight into the mechanism of ferroptosis
inhibition by ferrostatin-1. Redox Biol. 28:1013282020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Boelens J, Lust S, Offner F, Bracke ME and
Vanhoecke BW: Review. The endoplasmic reticulum: A target for new
anti-cancer drugs. In Vivo. 21:215–226. 2007.PubMed/NCBI
|
|
27
|
Zhu S, Zhang Q, Sun X, Zeh HJ III, Lotze
MT, Kang R and Tang D: HSPA5 regulates ferroptotic cell death in
cancer cells. Cancer Res. 77:2064–2077. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dai X, Zhang X, Chen W, Chen Y, Zhang Q,
Mo S and Lu J: Dihydroartemisinin: A potential natural anticancer
drug. Int J Biol Sci. 17:6032021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kiani BH, Kayani WK, Khayam AU, Dilshad E,
Ismail H and Mirza B: Artemisinin and its derivatives: A promising
cancer therapy. Mol Biol Rep. 47:6321–6336. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jin H, Jiang AY, Wang H, Cao Y, Wu Y and
Jiang XF: Dihydroartemisinin and gefitinib synergistically inhibit
NSCLC cell growth and promote apoptosis via the Akt/mTOR/STAT3
pathway. Mol Med Rep. 16:3475–3481. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ketelut-Carneiro N and Fitzgerald KA:
Apoptosis, pyroptosis, and Necroptosis-Oh my! the many ways a cell
can die. J Mol Biol. 434:1673782022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hänggi K and Ruffell B: Cell death,
therapeutics, and the immune response in cancer. Trends Cancer.
9:381–396. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tang D, Chen X, Kang R and Kroemer G:
Ferroptosis: Molecular mechanisms and health implications. Cell
Res. 31:107–125. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lin R, Zhang Z, Chen L, Zhou Y, Zou P,
Feng C, Wang L and Liang G: Dihydroartemisinin (DHA) induces
ferroptosis and causes cell cycle arrest in head and neck carcinoma
cells. Cancer Lett. 381:165–175. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Du J, Wang T, Li Y, Zhou Y, Wang X, Yu X,
Ren X, An Y, Wu Y, Sun W, et al: DHA inhibits proliferation and
induces ferroptosis of leukemia cells through autophagy dependent
degradation of ferritin. Free Radic Biol Med. 131:356–369. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao
N, Sun B and Wang G: Ferroptosis: Past, present and future. Cell
Death Dis. 11:882020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lai K, Song C, Gao M, Deng Y, Lu Z, Li N
and Geng Q: Uridine alleviates Sepsis-Induced acute lung injury by
inhibiting ferroptosis of macrophage. Int J Mol Sci. 24:50932023.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu GZ, Xu XW, Tao SH, Gao MJ and Hou ZH:
HBx facilitates ferroptosis in acute liver failure via EZH2
mediated SLC7A11 suppression. J Biomed Sci. 28:672021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Miotto G, Rossetto M, Di Paolo ML, Orian
L, Venerando R, Roveri A, Vučković AM, Bosello Travain V, Zaccarin
M, Zennaro L, et al: Insight into the mechanism of ferroptosis
inhibition by ferrostatin-1. Redox Biol. 28:1013282020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li L, Qiu C, Hou M, Wang X, Huang C, Zou
J, Liu T and Qu J: Ferroptosis in ovarian cancer: A Novel
therapeutic strategy. Front Oncol. 11:6659452021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cheung EC and Vousden KH: The role of ROS
in tumour development and progression. Nat Rev Cancer. 22:280–297.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Seibt TM, Proneth B and Conrad M: Role of
GPX4 in ferroptosis and its pharmacological implication. Free Radic
Biol Med. 133:144–152. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lee N, Carlisle AE, Peppers A, Park SJ,
Doshi MB, Spears ME and Kim D: xCT-Driven expression of GPX4
determines sensitivity of breast cancer cells to ferroptosis
inducers. Antioxidants (Basel). 10:3172021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang Z, Zhang L, Zhou L, Lei Y, Zhang Y
and Huang C: Redox signaling and unfolded protein response
coordinate cell fate decisions under ER stress. Redox Biol.
25:1010472019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bhardwaj M, Leli NM, Koumenis C and
Amaravadi RK: Regulation of autophagy by canonical and
non-canonical ER stress responses. Semin Cancer Biol. 66:116–128.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ma R, Qin W, Xie Y, Han Z, Li S, Jiang Y
and Lv H: Dihydroartemisinin induces ER stress-dependent apoptosis
of Echinococcus protoscoleces in vitro. Acta Biochim Biophys Sin
(Shanghai). 52:1140–1147. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chen D, Fan Z, Rauh M, Buchfelder M,
Eyupoglu IY and Savaskan N: ATF4 promotes angiogenesis and neuronal
cell death and confers ferroptosis in a xCT-dependent manner.
Oncogene. 36:5593–5608. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen D, Rauh M, Buchfelder M, Eyupoglu IY
and Savaskan N: The oxide-metabolic driver ATF4 enhances
temozolomide chemo-resistance in human gliomas. Oncotarget.
8:51164–51176. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chang LC, Chiang SK, Chen SE, Yu YL, Chou
RH and Chang WC: Heme oxygenase-1 mediates BAY 11-7085 induced
ferroptosis. Cancer Lett. 416:124–137. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wang N, Zeng GZ, Yin JL and Bian ZX:
Artesunate activates the ATF4-CHOP-CHAC1 pathway and affects
ferroptosis in Burkitt's Lymphoma. Biochem Biophys Res Commun.
519:533–539. 2019. View Article : Google Scholar : PubMed/NCBI
|