Introduction
Intrahepatic cholangiocarcinoma (ICC) is a malignant
tumor originating from the epithelium of the secondary bile duct
and its branches. Chronic hepatitis and cirrhosis, inflammatory
diseases of the biliary tract and hepatobiliary flukes increase the
risk of ICC (1). Notably, 5–30% of
all primary liver cancer cases are ICC, the incidence of which has
increased in the past 30 years (2).
ICC is associated with fatal outcomes in most patients. Regarding
treatment of ICC, 20–30% of patients are eligible for surgical
resection, which is currently the best treatment option; after
surgical resection, capecitabine is typically used as an adjuvant
therapy, and the median survival time among these patients is
reported to be 53 months (3). For
70–80% of patients with local infiltration or distant metastasis,
systemic treatment may delay disease progression, but once
metastasis has occurred, survival is still limited to ~1 year
(3). Therefore, it is crucial to
understand the drivers of metastasis in ICC.
The E26 transformation-specific sequence (ETS)
family is a large group of transcription factors, which includes
ETS variant 4 (ETV4). This particular variant belongs to the
polyomavirus enhancer activator 3 subfamily, which serves a crucial
role in tumor advancement and spread by controlling various
cellular processes, such as the cell cycle, apoptosis,
epithelial-mesenchymal transition (EMT), cell migration, invasion,
tumor stem cell characteristics and resistance to chemotherapy
(4). ETV4 is upregulated in a
number of tumor tissues; for example, ETV4 is highly expressed in
malignant tumors, such as pancreatic ductal adenocarcinoma (PDAC)
(5) and prostate cancer (6), and can act as a pro-carcinogenic
factor. In comparison to other tumors, research on ETV4 in ICC is
limited (7).
Epithelial cells undergo a transformation known as
EMT, enabling them to acquire mesenchymal traits that promote
migration and invasion into surrounding cells and the extracellular
matrix. Dysregulated signaling pathways, hypoxia and interactions
within the tumor microenvironment trigger the initiation and
progression of EMT; this results in the disruption of epithelial
cell polarity and intercellular adhesion, and ultimately enhances
migratory capabilities towards adjacent cells and penetration into
the extracellular matrix (8). EMT
is characterized by rearrangement of the cytoskeleton (such as
actin recombination), cellular motility and invasiveness, increased
cell-related protein hydrolysis activity and reprogramming of gene
expression (9). EMT leads to in
situ invasion or distant metastasis of tumors, and is
considered a key mechanism for epithelial cells to assume a
malignant phenotype. Given that it is considered the primary driver
of tumor metastasis and recurrence, inhibiting EMT is considered
important in the development of chemotherapeutic drugs for treating
malignant tumors (10,11).
TGF-β1 is a multifunctional regulator responsible
for stimulating EMT. Smad2/3 and Smad4 form a trimeric complex as
transcription factors that participate in regulating the expression
of EMT-related genes (12),
inhibiting E-cadherin synthesis, promoting the expression of
interstitial cytokines, such as Vimentin and N-cadherin, reducing
cell polarity, weakening epithelial features of tumor cells and
enhancing interstitial features, all of which are promoters of
tumor invasion and metastasis (13). EMT is also common in
cholangiocarcinoma with high invasiveness and recurrence (14–16).
The role of ETV4 in ICC cells and its connection to EMT warrants
additional investigation. The present study aimed to investigate
the biological impact of ETV4 in ICC and the underlying mechanism
by which ETV4 influences EMT.
Materials and methods
Cell culture and cell lines
The human ICC Hccc9810 cell line was obtained from
iCell Bioscience, Inc. The cells were cultured in RPMI-1640 medium
(Dalian Meilun Biology Technology Co., Ltd.) supplemented with 10%
fetal bovine serum [FBS; Cyagen Biosciences (Guangzhou) Inc.] at
37°C in a 5% CO2 cell incubator.
Lentivirus infection
The lentivirus packaging process was completed using
the 2nd generation self-inactivating lentiviral packaging system
developed by Shanghai GeneChem Co., Ltd. The human ETV4 sequence
(NM_001986) was obtained from the cDNA library of Shanghai GeneChem
Co., Ltd. The following primers were used for PCR amplification of
the target gene from the template plasmid to obtain gene fragments:
ETV4, forward
5′-AGGTCGACTCTAGAGGATCCCGCCACCATGGAGCGGAGGATGAAAGC-3′, reverse
5′-TCCTTGTAGTCCATACCGTAAGAGTAGCCACCCTTGGGGC-3′. The GV492
(Ubi-MCS-3FLAG-CBh-gcGFP-IRES-puromycin) lentiviral vector plasmid
(Shanghai GeneChem Co., Ltd.) and the ETV4 gene sequence were
digested using AgeI and NheI restriction enzymes, and
complete cloning was performed using the in-fusion recombination
method. The recombinant vector was detected by Sanger sequencing.
293T cells (American Type Culture Collection) were used as the
interim cell line in the present study. Lentiviral plasmids were
transfected with GeneChem transfection reagent (Shanghai GeneChem
Co., Ltd.) at the following quantities into 293T cells in a 10-cm
cell culture dish: Empty GV492 vector plasmid (negative control) or
GV492 vector plasmid containing ETV4, 20 µg; PHelper 1.0 vector
plasmid (Shanghai GeneChem Co., Ltd.), 15 µg; PHelper 2.0 vector
plasmid (Shanghai GeneChem Co., Ltd.), 10 µg. The 293T cells were
placed in a 37°C cell culture incubator with 5% CO2. After
culturing for 6–8 h, the culture medium (DMEM; Corning, Inc.)
containing the transfection system mixture was discarded. PBS
solution (Beijing Solarbio Science & Technology Co., Ltd.) was
used to wash the cells, followed by the addition of 20 ml cell
culture medium containing 10% FBS (Beijing Diyi Biotechnology Co.,
Ltd.). The cells were then cultured at 37°C for 48 h in an
incubator containing 5% CO2. After 48 h, the supernatant of the
293T cells was collected and was centrifuged at 4,000 × g for 10
min at 4°C to remove cell debris and impurities. Subsequently, the
supernatant was filtered with a 0.45-µm filter and centrifuged at
90,000 × g for 2 h at 4°C. The supernatant was discarded, and any
remaining liquid on the tube wall was removed. Virus preservation
solution (Shanghai GeneChem Co., Ltd.) was added, gently
resuspended, and centrifuged at 14,000 × g for 5 min at 4°C. The
supernatant was then collected as a sample for safety and titer
testing.
Lentiviral vectors expressing a short hairpin RNA
(shRNA) targeting the ETV4 gene sequence
(5′-GTCCCTTTGTCCCACTTGGAT-3′) or the negative control
(5′-TTCTCCGAACGTGTCACGT-3′) were synthesized using the GV493
(hU6-MCS-CBh-gcGFP-IRES- puromycin) vector (Shanghai GeneChem Co.,
Ltd.); the recombinant vector was detected by Sanger sequencing.
293T cells were used as the interim cell line in the present study.
Lentiviral plasmids were transfected with GeneChem transfection
reagent at the following quantities into 293T cells in a 10 cm cell
culture dish: GV493 vector plasmid containing shRNA, 20 µg; PHelper
1.0 vector plasmid, 15 µg; PHelper 2.0 vector plasmid, 10 µg. The
293T cells were placed in a 37°C cell culture incubator with 5%
CO2. After culturing for 6 h, the culture medium containing the
transfection system mixture was discarded, and the cells were
washed with PBS solution. Subsequently, 20 ml cell culture medium
containing 10% FBS was slowly added, and the cells were cultured at
37°C for 48 h in an incubator containing 5% CO2. A total of 48 h
post-transfection, the supernatant of the 293T cells was collected.
The supernatant was centrifuged at 4,000 × g for 10 min at 4°C to
remove cell debris and impurities. Subsequently, the supernatant
was filtered through a 0.45-µm filter and then centrifuged at
90,000 × g for 2 h at 4°C. The supernatant was discarded, and the
remaining liquid on the tube wall was removed. Virus preservation
solution was added to the tube, gently resuspended, and centrifuged
at 14,000 × g for 5 minutes at 4°C. The supernatant was then taken
as a sample for safety and titer testing.
For Hccc9810 cell infection, the multiplicity of
infection for the overexpression group was 30, and that for the
knockdown group was 10. The cells were cultured in an incubator
with 5% CO2 at 37°C. The medium was changed 16 h after infection
with the virus stock solution, and the infection efficiency was
observed at 96 h. Stable cell lines were screened and created using
a puromycin working solution of 2 µg/ml, with a maintenance
concentration of 1 µg/ml puromycin. The overexpression group was
named LV-ETV4 and its negative control group (empty vector) was
named CON335. The knockdown group was named LV-ETV4-RNAi and its
negative control group was named CON313. The non-transfected group
comprised normal Hccc9810 cells.
Cell proliferation assay
Cell proliferation was assessed using the Cell
Counting Kit-8 (CCK-8) assay (Dalian Meilun Biology Technology Co.,
Ltd.) according to the manufacturer's instructions. Briefly, the
cell suspension was inoculated into a 96-well plate at a density of
5×103 cells/well and cultured overnight at 37°C in a 5% CO2 cell
incubator. Subsequently, 10 µl CCK-8 solution was added to each
well, followed by a 2-h incubation period. The optical density of
the solution in each well was then measured using a microplate
reader at 450 nm at 0, 24, 48, 72 and 96 h (17).
Cell migration and invasion
assays
Cell migration was evaluated using Transwell plates
(pore size, 8 µm) that were not coated in Matrigel, whereas the
cell invasion assay was carried out on Transwell plates that were
precoated with Matrigel (cat. no. 354480; Corning, Inc.).
Transfected cells were digested and centrifuged at 100 × g for 5
min at room temperature before being suspended in serum-free medium
and added to the upper chamber of the Transwell plate at a density
of 1×105 cells/chamber (migration) or 8×104 cells/chamber
(invasion). The lower chamber was filled with 500 µl medium
containing 10% FBS. After a 24-h incubation period at 37°C in a 5%
CO2 cell incubator, cells that had migrated or invaded through the
membrane were fixed with methanol solution (>99.5%; Chengdu
Chemical Co., Ltd.) for 30 min and stained with 1% crystal violet
solution (Beijing Solarbio Science & Technology Co., Ltd.) for
60 min at room temperature, and images were captured and
semi-quantified under a light inverted microscope (12,18).
Flow cytometric detection of cell
cycle progression and apoptosis
According to the manufacturer's instructions, the
Cell Cycle Staining Kit [cat. no. CCS012; Multi Sciences (Lianke)
Biotech Co., Ltd.] was used to assess cell cycle progression.
Briefly, infected cells underwent digestion with EDTA-free trypsin
(Beijing Solarbio Science & Technology Co., Ltd.) and
centrifugation at 100 × g for 5 min at room temperature to prepare
a cell suspension with a concentration of 1×106 cells/ml, and were
then washed with phosphate-buffered saline (PBS). Subsequently,
cells were centrifuged at 100 × g for 5 min at room temperature and
the supernatant was discarded; 1 ml DNA staining solution, which
contained PI and RNase A, and 10 µl permeabilization solution were
then applied to the cells, accompanied by vortex oscillation and a
30-min incubation at room temperature. Flow cytometric analysis (BD
FACSVerse; BD Biosciences) of the cell cycle was conducted
utilizing the lowest loading speed configuration. FlowJo (10.8.1;
FlowJo, LLC) was used to analyze cell cycle progression.
According to the manufacturer's instructions, the
Annexin V-allophycocyanin (APC)/propidium iodide (PI) cell
apoptosis detection kit (Jiangsu Kaiji Biotechnology Co., Ltd.) was
used to assess apoptosis. After digestion of the cells with
EDTA-free trypsin, they were centrifuged at 100 × g for 5 min at
room temperature to prepare a cell suspension with a concentration
of 5×105 cells/ml, and washed twice with PBS. Subsequently, cells
were centrifuged at 100 × g for 5 min at room temperature and the
supernatant was discarded; 500 µl binding buffer was then added to
the cells, and single cell suspension samples were obtained, which
were incubated with 5 µl Annexin V-APC (0.49 µg/ml) and 5 µl PI
(0.98 µg/ml) in the dark at room temperature for 5–10 min prior to
flow cytometric analysis (BD FACSVerse). FlowJo (10.8.1) was used
to analyze apoptosis.
Reverse transcription-quantitative
(RT-qPCR)
According to the manufacturers' instructions,
RT-qPCR analysis of gene transcription levels in the cells was
performed. Total RNA was extracted from the cells using the RNA
Easy RNA extraction kit (Beyotime Institute of Biotechnology), and
a cDNA RT kit (PrimeScript™ RT Master Mix Perfect Real-time; Takara
Biotechnology Co., Ltd.) was used to convert RNA into cDNA. cDNA
was then amplified by fluorescence qPCR using specific primers
(Nanning GenSys Biotechnology Co., Ltd.) and a qPCR kit (TB
Green® Premix Ex Taq™ Tli RnaseH Plus; Takara
Biotechnology Co., Ltd.). The qPCR program included preheating at
95°C for 30 sec; followed by 40 cycles of heating at 95°C for 5
sec, and cooling and extension at 60°C for 34 sec; and final
dissolution curve procedures (95°C for 15 sec, 60°C for 60 sec and
95°C for 15 sec). The relative mRNA expression levels were
calculated using the 2-ΔΔCq method (19). The primer sequences used for qPCR
analysis are listed in Table I, and
GAPDH was used as the reference gene (20,21).
 | Table I.List of primer sequences. |
Table I.
List of primer sequences.
| Gene | Sequence,
5′-3′ |
|---|
| ETV4 | F:
GGTGGCTGGTGAGCGTTA |
|
| R:
CCAAGTGGGACAAAGGGA |
| E-cadherin | F:
AATCTGAAAGCGGCTGATACTG |
|
| R:
GCGATTGCCCCATTCGTT |
| N-cadherin | F:
TGACTCCAACGGGGACTGC |
|
| R:
CAAACATCAGCACAAGGATAAGC |
| Vimentin | F:
GCCAGGCAAAGCAGGAGTC |
|
| R:
CACGAAGGTGACGAGCCATT |
| TGF-β1 | F:
GGGTGGCTCACGCCTGTAA |
|
| R:
GCTGGTCTCAAATGCCTGGAT |
| Smad4 | F:
ATGAGGTTATGGTTCTGGGTGG |
|
| R:
GAATGAGGTCTTACGGTGGGTG |
| Smad7 | F:
TGGTGCGTGGTGGCATACT |
|
|
R:AAGCCATTCCCCTGAGGTAGA |
| GAPDH | F:
AAGGTCGGAGTCAACGGAT |
|
| R:
CCTGGAAGATGGTGATGGG |
Inhibitor amygdalin and activator
SRI-011381 hydrochloride treatment
Cells (5×105 cells/ml) were seeded in a 6-well plate
(2 ml/well) and allowed to reach ~80% confluence. Subsequently, the
TGF-β/Smad inhibitor amygdalin (MedChemExpress), at a working
concentration of 400 µg/ml (22),
was added to the LV-ETV4 group, forming the LV-ETV4 + amygdalin
group. In addition, the TGF-β activator SRI-011381 hydrochloride
(MedChemExpress) solution, at a working concentration of 10 µM
(23), was added to the
LV-ETV4-RNAi group, creating the LV-ETV4-RNAi + SRI-011381 group.
These experimental groups, along with cells from the CON335,
LV-ETV4, CON313 and LV-ETV4-RNAi groups, were then placed in a 95%
humidity and 5% CO2 incubator at 37°C for 24 h.
Western blot analysis
The cells were rinsed with PBS, and were incubated
with a mixture of RIPA lysis buffer and PMSF (both from Beijing
Solarbio Science & Technology Co., Ltd.) on ice for 20 min.
Subsequently, the cell lysates were centrifuged at 12,000 × g for
20 min at 4°C, and the total protein concentration was quantified
by ultraviolet spectrophotometry. The samples containing protein
(50 µg protein/lane) were then subjected to SDS-PAGE on 8% gels in
protein loading buffer and were heated to 95–100°C for
electrophoresis. The separated proteins were transferred to a PVDF
membrane. Blocking was performed using 5% skim milk powder at room
temperature for 1 h, prior to incubation with the following primary
antibodies at 4°C for 18 h: Anti-Vimentin (cat. no. ab92547;
1:1,000), anti-TGF-β1 (cat. no. ab215715; 1:1,000), anti-Smad4
(cat. no. ab40759; 1:1,000), anti-GAPDH (cat. no. ab181603;
1:5,000) (all from Abcam), anti-E-cadherin (cat no. 20874-1-AP;
1:5,000), anti-N-cadherin (cat no. 22018-1-AP; 1:2,000) (both from
Wuhan Sanying Biotechnology), anti-ETV4 (cat. no. PA5-99226;
1:1,000) and anti-Smad7 (cat. no. 701940; 1:1,000) (both from
Invitrogen; Thermo Fisher Scientific, Inc.). Subsequently, the
membrane was washed three times with 1X Tris-buffered saline-0.1%
Tween 20 buffer (TBST) and incubated with a fluorescent DyLight™
800-conjugated secondary antibody (anti-rabbit; cat. no. 5151;
1:10,000; Cell Signaling Technology, Inc.) for 1 h at room
temperature. The membrane was washed a further three times with
TBST and protein bands were detected using an Odyssey membrane
scanner (LI-COR Biosciences). ImageJ (1.46; National Institutes of
Health) was used to semi-quantify band staining intensities, and
the relative expression of these proteins was determined (17). GAPDH was used as an internal
reference protein.
Statistical analysis
All experiments were performed at least three times,
and the data are presented as the mean ± standard deviation. The
data were analyzed using SPSS Statistics 27 (IBM Corporation).
Unpaired Student's t-test was used for comparing the mean between
two groups, whereas one-way ANOVA followed by Tukey's post-hoc test
was used for comparisons among multiple groups. The Wilcoxon
rank-sum test was used to compare two groups that did not conform
to normal distribution. Bar plots were prepared using GraphPad
Prism 8.0 (Dotmatics). P<0.05 considered to indicate a
statistically significant difference.
Results
Overexpression of ETV4 promotes
Hccc9810 cell proliferation, migration and invasion
Lentiviral infection was used to construct stable
ETV4-overexpressing cells, and the infection efficiency was
verified through RT-qPCR and western blot analysis (Fig. 1A and B). Notably, the ETV4 antibody
is a polyclonal antibody; the thicker band located at 54 kDa was
used for western blot analysis of ETV4. No significant variation
was observed in ETV4 expression between Hccc9810 cells and the
negative control group CON335 (Fig.
1B-D). Subsequently, the CON335 and LV-ETV4 groups underwent
subsequent experiments. For the CCK-8 experiment, cells were
incubated for 24, 48, 72 and 96 h. The LV-ETV4 group showed an
increase in cell proliferation compared with that in the CON335
group, thus suggesting that ETV4 overexpression may promote cell
proliferation (Fig. 1E). In the
Transwell experiment, after incubating cells for 24 h, the invasion
and migration of cells in the LV-ETV4 group were increased compared
with those in the CON335 group (Fig.
1F).
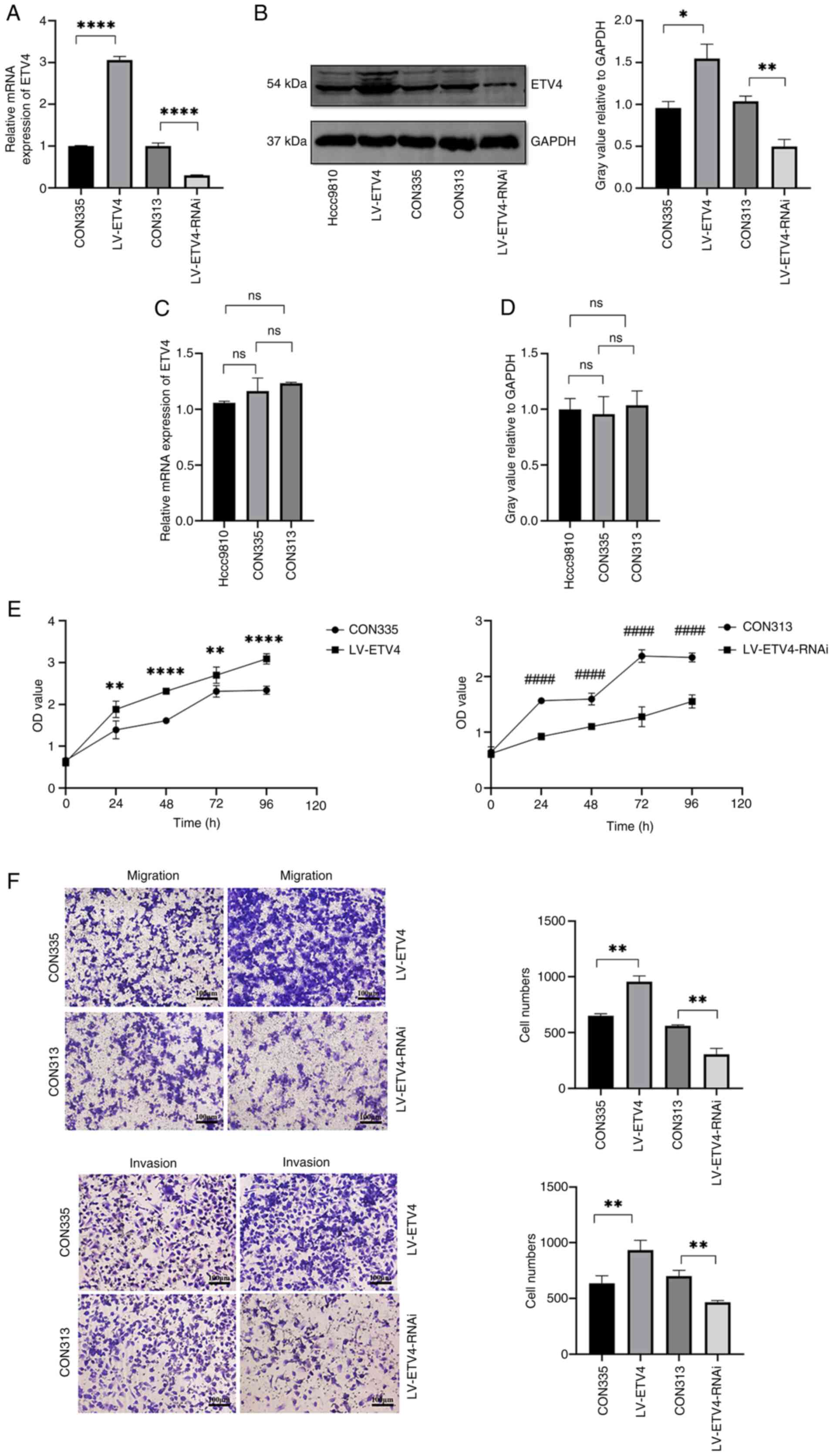 | Figure 1.Effects of overexpression and
knockdown of ETV4 on the proliferation, migration and invasion of
Hccc9810 cells. Verification of the lentiviral infection efficiency
of LV-ETV4 and LV-ETV4-RNAi using (A) RT-qPCR and (B) western blot
analysis. (C) RT-qPCR and (D) western blot analysis showed no
difference in the expression levels of ETV4 mRNA and protein
expression among the Hccc9810, CON335 and CON313 groups. (E)
LV-ETV4 promoted cell proliferation, whereas LV-ETV4-RNAi inhibited
cell proliferation. (F) LV-ETV4 promoted cell migration and
invasion, whereas LV-ETV4-RNAi inhibited cell migration and
invasion (scale bar, 100 µm). *P<0.05, **P<0.01,
****P<0.0001 vs. CON335 or as indicated; ####P<0.0001 vs.
CON313. ETV4, E26 transformation-specific sequence variant 4;
LV-ETV4, ETV4 overexpression; LV-ETV4-RNAi, ETV4 knockdown; ns, not
significant; ns, not significant; RT-qPCR, reverse
transcription-quantitative PCR. |
Knocking down ETV4 inhibits Hccc9810
cell proliferation, migration and invasion
Lentiviral infection was also used to construct
stable cells with ETV4 knockdown, and the infection efficiency was
verified through RT-qPCR and western blot analysis (Fig. 1A and B). No significant variation
was observed in ETV4 expression between Hccc9810 cells and the
negative control group CON313 (Fig.
1B-D). Subsequently, the CON313 and LV-ETV4-RNAi groups
underwent subsequent experiments. For the CCK-8 experiment, cells
were incubated for 24, 48, 72 and 96 h. The LV-ETV4-RNAi group
exhibited lower cell proliferation than the CON313 group,
indicating that knocking down ETV4 may inhibit cell proliferation
(Fig. 1E). In the Transwell
experiment, after incubating cells for 24 h, the invasion and
migration of cells in the LV-ETV4-RNAi group were decreased
compared with those in the CON313 group (Fig. 1F).
Effects of overexpression and
knockdown of ETV4 on cell cycle progression and apoptosis
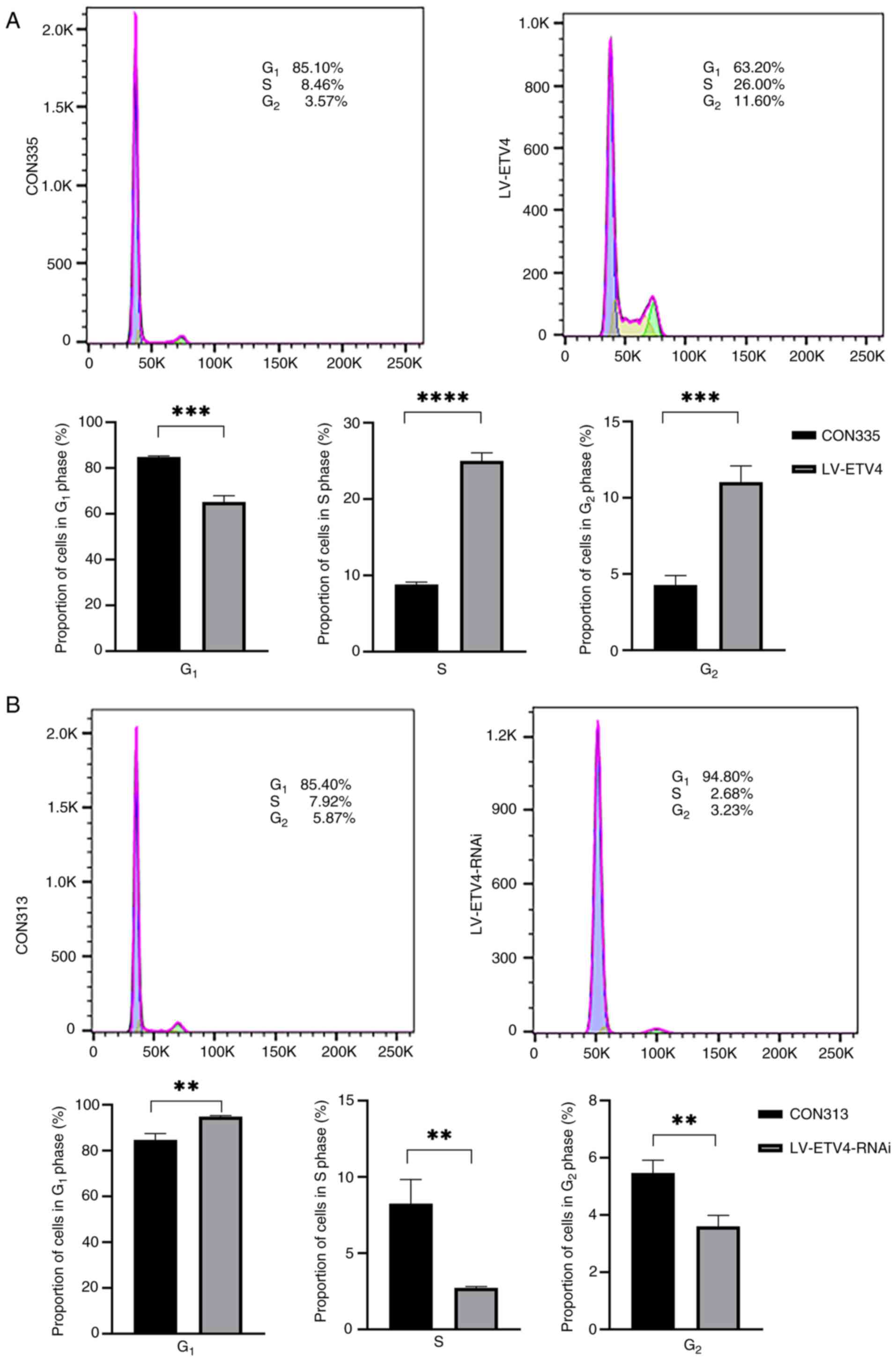
Flow cytometry was used to analyze the effects of
overexpression and knockdown of ETV4 on cell cycle progression. The
results showed that there were more cells in the S (25.1 vs. 8.88%)
and G2 phases (11.7 vs. 4.70%), and fewer in the G1 phase (64.1 vs.
85.2%) of the cell cycle in the LV-ETV4 group compared with in the
CON335 group (Fig. 2A). This
finding suggested that overexpression of ETV4 promoted cell
differentiation i.e., transition from the G1 to S phase, and
accelerated cell proliferation. By contrast, the LV-ETV4-RNAi group
exhibited a significant increase in the number of cells in the G1
phase of the cell cycle (94.8 vs. 85.4%), whereas the proportion of
cells in the S (2.68 vs. 7.92%) and G2 phases (3.23 vs. 5.87%) was
decreased compared with in the CON313 group (Fig. 2B). This finding suggested that the
cells in the LV-ETV4-RNAi group were arrested in the G1 phase,
leading to cell cycle arrest and reduced cell proliferation. In
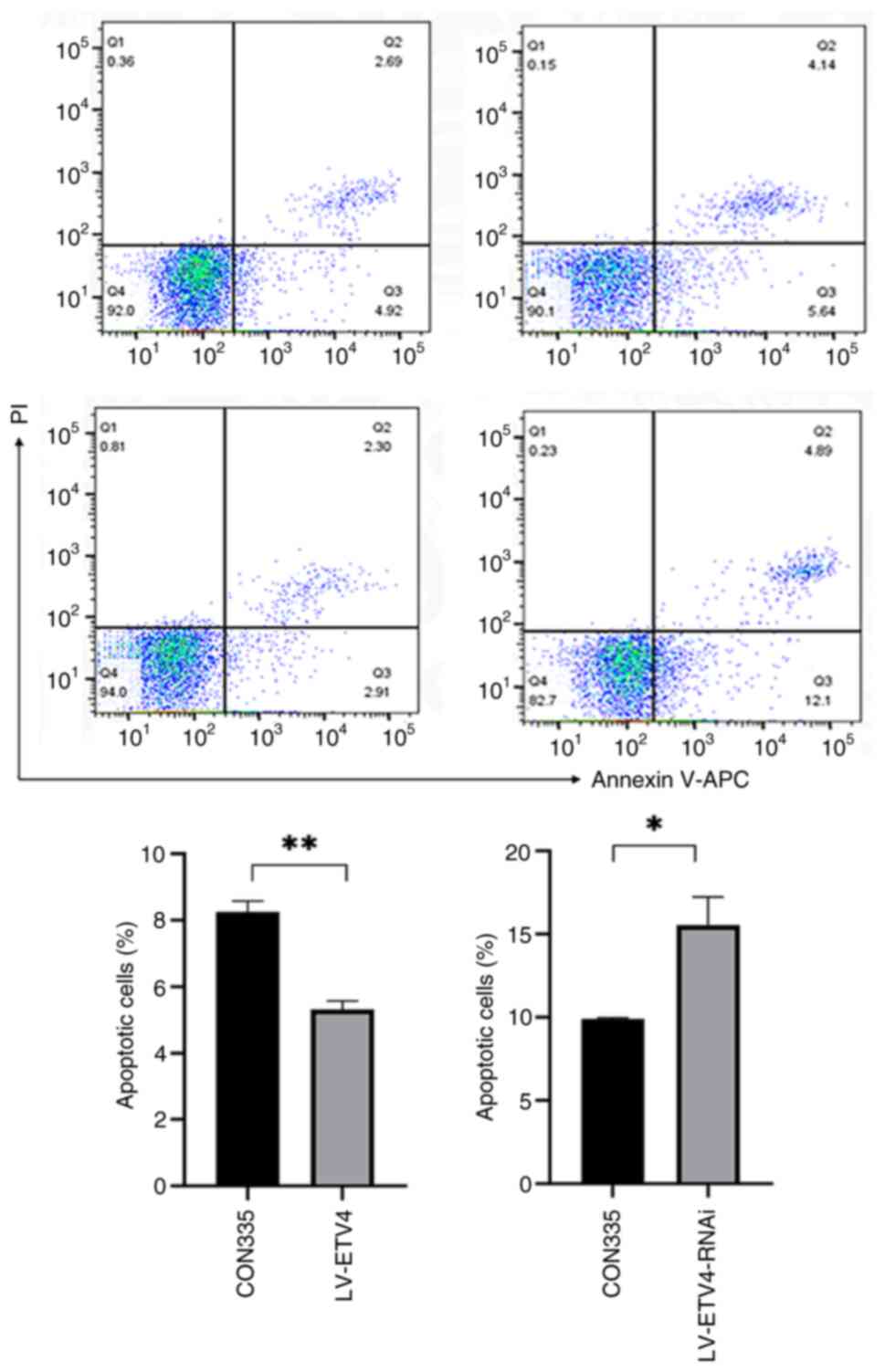
addition, the LV-ETV4 group exhibited reduced apoptosis compared
with that in the CON335 group (5.21 vs. 7.61%), whereas the
LV-ETV4-RNAi group exhibited increased apoptosis compared with that
in the CON313 group (16.99 vs. 9.78%) (Fig. 3).
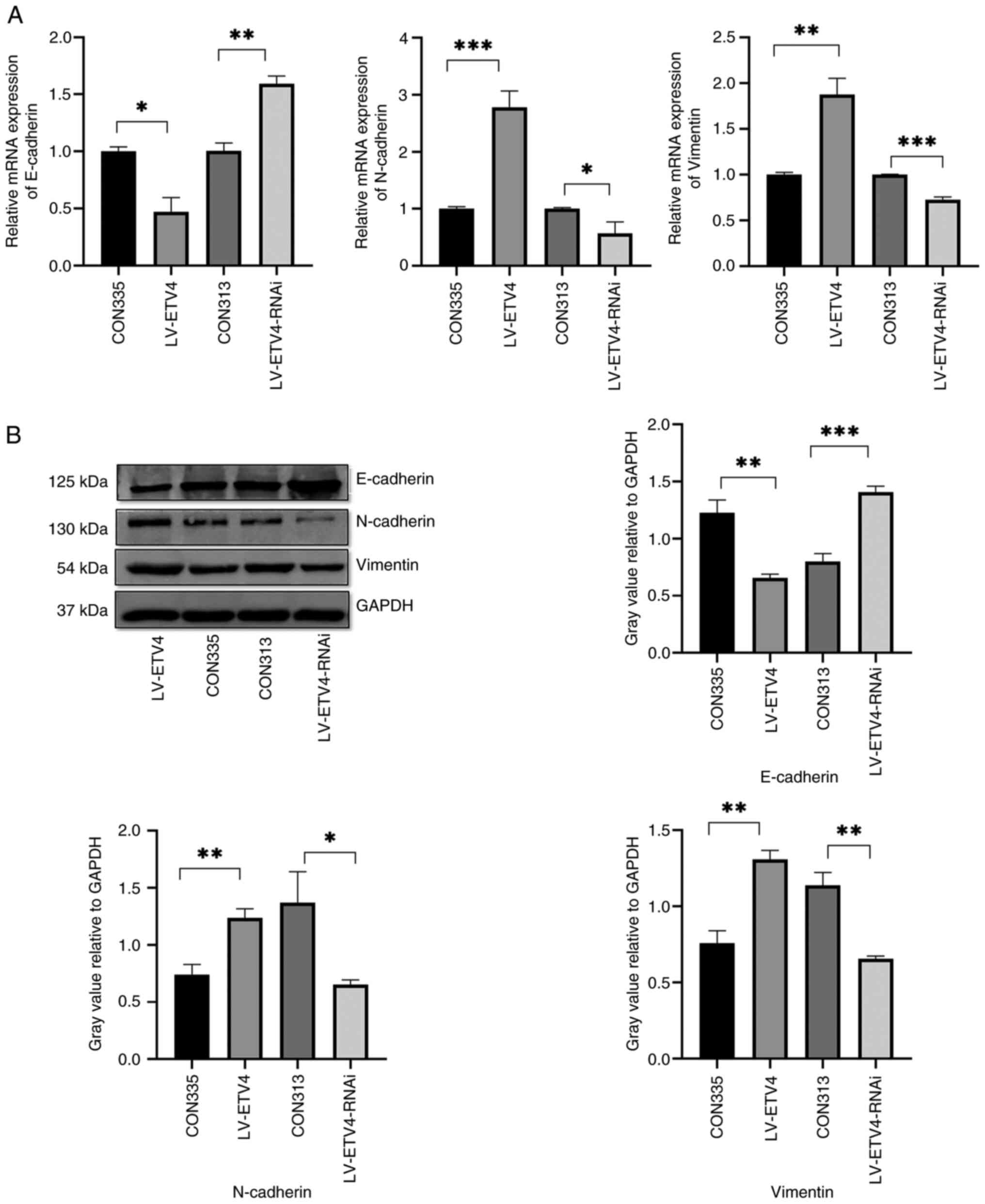
ETV4 overexpression promotes EMT,
whereas ETV4 knockdown inhibits EMT
The LV-ETV4 group exhibited reduced mRNA and protein
expression levels of E-cadherin, and increased mRNA and protein
expression levels of N-cadherin and Vimentin compared with those in
the CON335 group, thus indicating that EMT was stimulated (Fig. 4A and B). By contrast, the
LV-ETV4-RNAi group exhibited significantly increased mRNA and
protein expression levels of E-cadherin, and reduced mRNA and
protein expression levels of N-cadherin and Vimentin compared with
those in the CON313 group, thus suggesting that EMT was inhibited
(Fig. 4A and B). These findings
indicated that overexpression and knockdown of ETV4 had an impact
on EMT.
ETV4 regulates EMT through the
TGF-β1/Smad signaling pathway
The results of RT-qPCR showed that, in the LV-ETV4
group, the mRNA expression levels of TGF-β1 and Smad4 were
increased, whereas those of Smad7 were decreased compared with
those in the CON335 group (Fig.
5A). By contrast, in the LV-ETV4-RNAi group, the mRNA
expression levels of TGF-β1 and Smad4 were decreased, whereas those
of Smad7 were increased compared with those in the CON313 group
(Fig. 5A). Similarly, western blot
analysis showed upregulated protein expression levels of TGF-β1 and
Smad4, and downregulation of Smad7 in the LV-ETV4 group compared
with those in the CON335 group (Fig.
5B). In addition, in the LV-ETV4-RNAi group, the protein
expression levels of TGF-β1 and Smad4 were decreased, whereas those
of Smad7 were increased compared with those in the CON313 group
(Fig. 5B). Subsequently, treatment
with an inhibitor (amygdalin) and an activator (SRI-011381
hydrochloride) of the TGF-β1/Smad signaling pathway was used to
validate the regulatory effect of ETV4 on EMT through western blot
analysis.
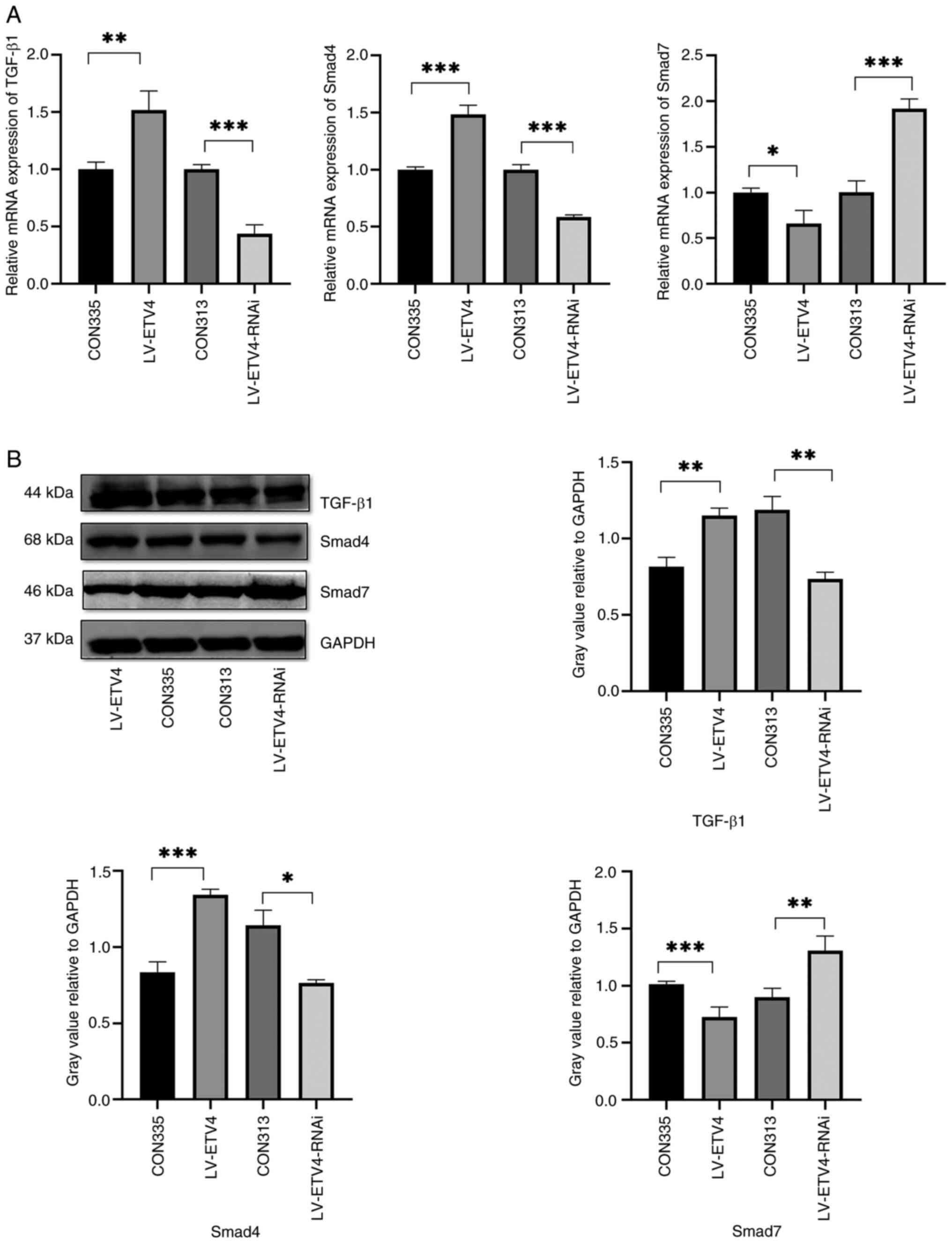 | Figure 5.LV-ETV4 activates the TGF-β1/Smad
signaling pathway, whereas LV-ETV4-RNAi inhibits the TGF-β1/Smad
signaling pathway. (A) Reverse transcription-quantitative PCR
results showed that in the LV-ETV4 group, the mRNA expression
levels of TGF-β1 and Smad4 were increased, whereas the mRNA
expression levels of Smad7 were decreased. In the LV-ETV4-RNAi
group, the mRNA expression levels of TGF-β1 and Smad4 were
decreased, whereas those of Smad7 were increased. (B) Western
blotting showed similar results. *P<0.05, **P<0.01,
***P<0.001. ETV4, E26 transformation-specific sequence variant
4; LV-ETV4, ETV4 overexpression; LV-ETV4-RNAi, ETV4 knockdown. |
In the LV-ETV4 group, E-cadherin expression was
decreased, whereas N-cadherin and Vimentin expression levels were
increased compared with in the CON335 group (Fig. 6A). Conversely, in the LV-ETV4 +
amygdalin group, E-cadherin expression was increased, while
N-cadherin and Vimentin expression was decreased in comparison to
the LV-ETV4 group. Similarly, in the LV-ETV4-RNAi group, E-cadherin
expression was increased, while N-cadherin and Vimentin expression
was decreased compared with in the CON313 group. Finally, in the
LV-ETV4-RNAi + SRI-011381 group, E-cadherin expression was
decreased, while N-cadherin and Vimentin expression was increased
compared with in the LV-ETV4-RNAi group. Furthermore, in the
LV-ETV4 group, the expression levels of TGF-β1 and Smad4 were
increased, whereas the expression of Smad7 was decreased in
comparison to the CON335 group (Fig.
6B). Conversely, in the LV-ETV4 + amygdalin group, the
expression of TGF-β1 and Smad4 was decreased, while the expression
of Smad7 was increased compared with in the LV-ETV4 group.
Similarly, in the LV-ETV4-RNAi group, the expression of TGF-β1 and
Smad4 was decreased, while the expression of Smad7 was increased
compared with in the CON313 group. Finally, in the LV-ETV4-RNAi +
SRI-011381 group, the expression of TGF-β1 and Smad4 was increased,
while the expression of Smad7 was decreased compared with in the
LV-ETV4-RNAi group. These findings indicated that ETV4 may regulate
EMT through the TGF-β1/Smad signaling pathway.
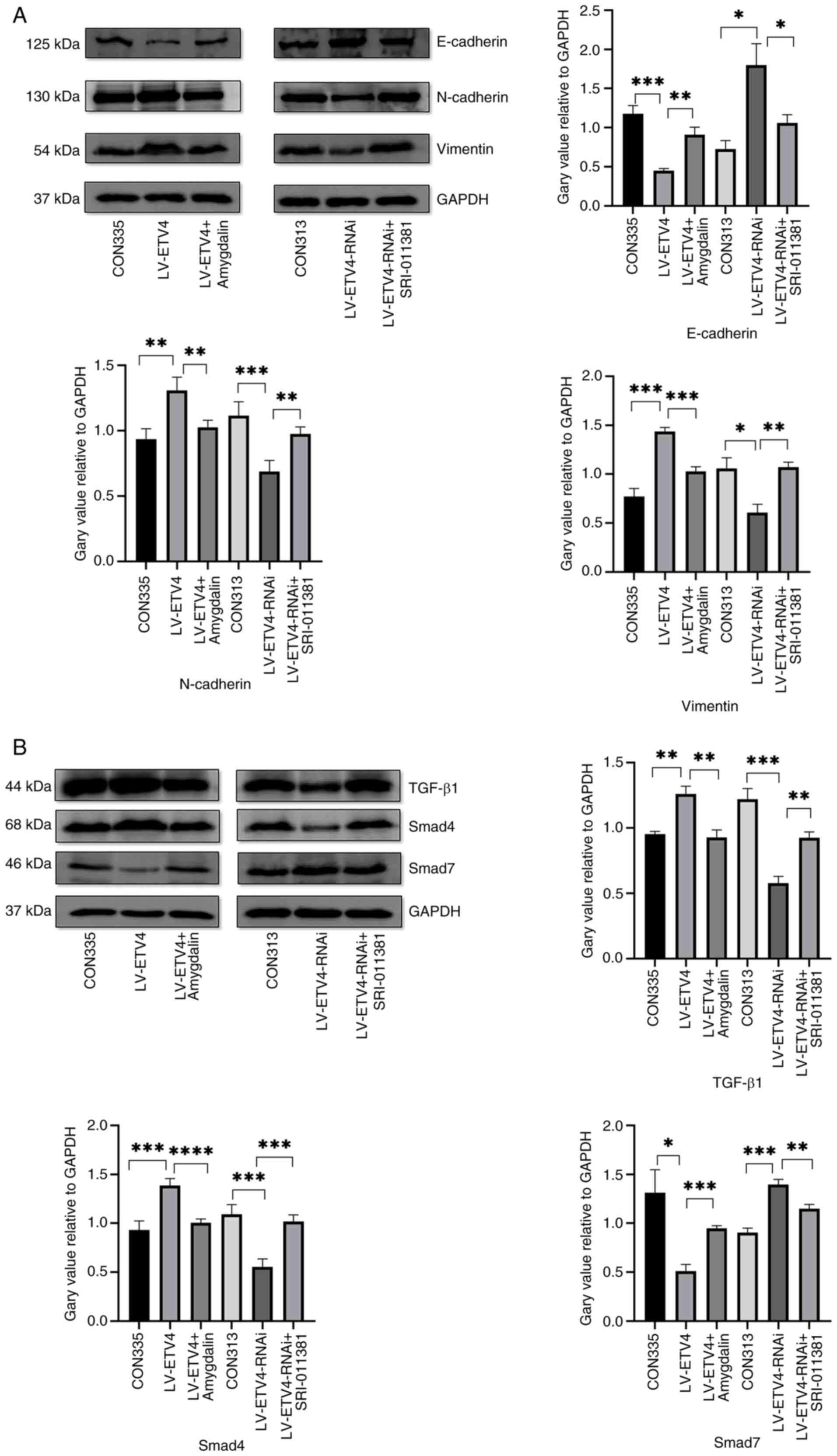 | Figure 6.Validation of the results using the
TGF-β1/Smad signaling pathway inhibitor amygdalin and activator
SRI-011381 hydrochloride. (A) In the LV-ETV4 group, the expression
levels of E-cadherin were decreased, whereas those of N-cadherin
and Vimentin were increased compared with those in the CON335
group. In the LV-ETV4 + amygdalin group, E-cadherin expression was
increased, whereas N-cadherin and Vimentin expression was decreased
compared with those in the LV-ETV4 group. In the LV-ETV4-RNAi
group, E-cadherin expression was increased, whereas N-cadherin and
Vimentin expression was decreased compared with those in the CON313
group. In the LV-ETV4-RNAi + SRI- 011381 group, E-cadherin
expression was decreased, while N-cadherin and Vimentin expression
was increased compared with those in the LV-ETV4-RNAi group. (B) In
LV-ETV4 group, the expression of TGF-β1 and Smad4 was increased,
while the expression of Smad7 was decreased compared with those in
the CON335 group. In the LV-ETV4 + amygdalin group, the expression
of TGF-β1 and Smad4 was decreased, while the expression of Smad7
was increased compared with those in the LV-ETV4 group. In the
LV-ETV4-RNAi group, the expression of TGF-β1 and Smad4 was
decreased, while the expression of Smad7 was increased compared
with those in the CON313 group. In the LV-ETV4-RNAi + SRI-011381
group, the expression of TGF-β1 and Smad4 was increased, while the
expression of Smad7 was decreased compared with those in the
LV-ETV4-RNAi group. *P<0.05, **P<0.01, ***P<0.001,
****P<0.0001. ETV4, E26 transformation-specific sequence variant
4; LV-ETV4, ETV4 overexpression; LV-ETV4-RNAi, ETV4 knockdown. |
Discussion
The present study investigated the effects of ETV4
on proliferation, migration, invasion, cell cycle progression,
apoptosis and EMT activation in Hccc9810 ICC cells. The findings
indicated that ETV4 modulated the EMT process through acting on the
TGF-β1/Smad signaling pathway.
As a member of the ETS transcription factor family,
ETV4 can target related genes, such as dishevelled segment polarity
protein 2, and matrix metalloproteinases (MMP1, MMP2, MMP7, MMP9,
MMP13, MMP24 and MMP25), thus influencing disease progression in
numerous types of cancer (24).
Studies have shown that ETV4 enhances the proliferation,
infiltration and metastasis of tumors. Overexpression of ETV4 in
hepatitis B-related liver cancer has been reported to promote the
invasion and migration of cancer cells, whereas knocking down ETV4
can weaken the ability of these cancer cells to invade and migrate
(25). Similarly, knocking down
ETV4 in glioblastoma multiforme cells was found to reduce the
transcriptional activation of epithelial membrane protein 1,
thereby inhibiting PI3K/AKT/mTOR signaling and promoting autophagy
and apoptosis (26). Silencing ETV4
in ASPC1 and Colo357 pancreatic cancer cell lines has been reported
to decrease cell proliferation by 55.3 and 38.9%, respectively.
Conversely, overexpression of ETV4 in BXPC3 human pancreatic
adenocarcinoma cells was shown to result in a 46.8% increase in
cell proliferation compared with control cells. Further mechanistic
investigations revealed that ETV4 specifically controls cyclin D1
expression, facilitating a swift transition from the G1 to S phase
of the cell cycle and enhancing cell proliferation (27). However, to the best of our
knowledge, the function of ETV4 in ICC has not been fully
elucidated. The present study revealed that ETV4 overexpression in
ICC increased cell proliferation, migration and invasion, and
reduced apoptosis. By contrast, ETV4 knockdown had the opposite
effect. Apoptosis was detected in ICC cells transfected with
LV-ETV4-RNAi; however, no sub-G1 fraction was observed by flow
cytometry in these cells. This discrepancy may be attributed to the
removal of the supernatant containing apoptotic cells during cell
collection, as well as the possibility that cell cycle arrest did
not necessarily coincide with apoptosis. It is important to note
that apoptosis and cell cycle progression can occur concurrently,
or that cell cycle arrest may precede apoptosis. Further
experiments are required to validate these findings. Conversely,
the LV-ETV4 group, which exhibited reduced apoptosis, did not show
a sub-G1 peak. Given the evidence of the cancer regulatory activity
of ETV4 (25–27), it appears that ETV4 may act as a
pro-carcinogenic factor in ICC and a key driver of ICC infiltration
or metastasis.
EMT is characterized by the absence of the
epithelial marker E-cadherin and the elevated expression of the
mesenchymal marker Vimentin (28).
This transition is associated with the loss of epithelial cell
polarity, disruption of epithelial cell contacts, alterations in
cell adhesion molecules and cytoskeletal components, as well as the
degradation of certain extracellular matrix and basement membrane
constituents (29). Through EMT,
tumor cells are able to breach the histological barriers, enabling
invasion, separation and metastasis from primary sites to adjacent
or distant normal tissues. E-cadherin serves a crucial role in
facilitating this process (30).
The absence of E-cadherin in cancer leads to metastasis and
diffusion (31), and activation of
several EMT transcription factors, such as Twist and β-catenin
(32). A significant characteristic
of the EMT is the occurrence of ‘cadherin alteration’, in which
there is a decrease in E-cadherin expression and an increase in
N-cadherin expression (33).
N-cadherin serves as a marker for active EMT, and its abnormal
expression is strongly linked to multiple facets of malignant tumor
advancement, including transformation, adhesion, apoptosis,
angiogenesis, invasion and metastasis (34). Vimentin is another indicator of the
mesenchymal transition within the EMT (35), and it serves a crucial role in
regulating the cellular mechanics necessary for coordinating
mechanical sensing, transduction, signaling pathways, motility and
inflammatory responses (36), as
well as for maintaining cell integrity and combating cellular
stress. Overexpression of Vimentin has been observed in numerous
epithelial malignancies, such as prostate cancer, gastrointestinal
tumors, central nervous system tumors, breast cancer and malignant
melanoma. The excessive levels of Vimentin in cancer have been
strongly associated with the proliferation and invasion of tumors
(37). In the present study,
markers of EMT, including E-cadherin, N-cadherin and Vimentin, were
detected. The results from both RT-qPCR and western blot analysis
revealed the downregulation of E-cadherin, along with the
upregulation of N-cadherin and Vimentin, in the group
overexpressing ETV4 compared with in the negative control group.
These findings suggested that the overexpression of ETV4 may have
the potential to trigger EMT. Conversely, a knockdown in ETV4 led
to an increase in E-cadherin expression, and a decrease in
N-cadherin and Vimentin levels in comparison to the respective
negative control group, suggesting that ETV4 knockdown has the
ability to suppress the EMT process. These findings indicated that
ETV4 may promote EMT in ICC, leading to peritumoral infiltration or
distant metastasis. ETV4 has been reported to activate B3GNT3 and
mediate TGF-β1 signal transduction, thereby promoting EMT in liver
cancer (38). In colon
adenocarcinoma, knocking out ETV4 has been reported to reduce EMT
(39), which is consistent with the
present study.
TGF-β signaling promotes EMT in cancer cells,
facilitating cancer growth and advancement in later stages
(40). Upon activation of the TGF-β
signaling pathway, cancer cells undergo EMT, resulting in the loss
of their epithelial characteristics, such as polarity and cell-cell
contact, and the gain of migratory and invasive capabilities to
adopt a mesenchymal phenotype (41). These transformed cells are able to
penetrate nearby tissues, lymphatic vessels and blood vessels,
leading to distant metastasis. Consequently, TGF-β
signaling-induced EMT serves a critical role in the progression of
tumor metastasis. In the canonical TGF-β1/Smad pathway, Smad2/3
binds to Smad4 to form a heteromeric complex, which then enters the
nucleus to regulate gene transcription (42). At the same time, TGF-β1 is also
regulated by inhibitory Smads, such as Smad7 (43).
As a vital element in the TGF-β1/Smad signaling
pathway, the absence of Smad4 on its own does not initiate tumor
formation. Notably, it can promote tumor progression initiated by
other genes or act as a critical initiator by interrupting the
cellular DNA damage response and repair mechanisms, thereby
increasing genomic instability, suggesting its distinct roles in
different types of cancer (44). In
a mouse liver cancer model, knocking down Smad4 significantly
inhibited tumor development, and the expression levels of
EMT-inducible factors Snail and Twist1 were also significantly
reduced (45). Knocking down Smad4
in NMuMG breast epithelial cells has been shown to effectively
block TGF-β-induced EMT, restore E-cadherin levels, and hinder the
induction of N-cadherin and morphological changes (46). Furthermore, knocking down Smad4 by
95% was revealed to effectively block TGF-β-induced EMT (47). Spirulina phycocyanin extract and its
active components can suppress the EMT process in endometrial
cancer via targeting the TGF-β1/Smad4 signaling pathway (48). The TGF-β/Smad4 signal can also
inhibit the transcription of Kruppel-like factor 5 (KLF5) through
Snail; when KLF5 levels are low, sex determining region Y box
protein 4 shifts from being a tumor promoter to a suppressor.
Consequently, TGF-β/Smad4 effectively induces lethal EMT in PDAC
(49). These reports have indicated
that the TGF-β signal can affect EMT in a Smad4-dependent manner
(44).
Smad7 acts as a potent endogenous inhibitor of TGF-β
signaling, which leads to the inhibition of downstream protein
activation in the TGF-β signaling pathway, disrupting signal
transduction (43). Smad7 competes
with Smad4 to associate with the receptor-regulated Smads and
recruits the NEDD4-like E3 ubiquitin protein ligase to activated
the receptor-regulated Smads, leading to their polyubiquitination
and proteasomal degradation (50).
The overexpression of microRNA-21 has been shown to promote
TGF-β-induced EMT by inhibiting Smad7/p-Smad7 and activating
Smad3/p-Smad3 (51). The results of
RT-qPCR and western blot analysis in the present study demonstrated
that, in the LV-ETV4 group, there was an increase in the expression
levels of TGF-β1 and Smad4, alongside a decrease in Smad7
expression. Conversely, the LV-ETV4-RNAi group displayed a decrease
in the expression levels of TGF-β1 and Smad4, with a corresponding
increase in Smad7 expression. These results suggested that
activation of ETV4 may trigger the TGF-β1/Smad signaling pathway,
whereas suppressing ETV4 hinders it, thus aligning with an earlier
liver cancer study (38).
Furthermore, the evaluation of E-cadherin, N-cadherin and Vimentin
levels through RT-qPCR and western blot analysis revealed that ETV4
may mediate EMT via the TGF-β1/Smad pathway. Application of the
TGF-β1/Smad pathway inhibitor amygdalin or activator SRI-011381
hydrochloride aided in confirming the observations regarding the
regulatory impact of ETV4 on EMT.
The present study mainly focused on the effects and
mechanisms of ETV4 on ICC cells, providing novel insights into ICC
progression and providing a basis for further clinical research on
ETV4. EMT is a complex process involving numerous signaling
pathways. However, it should be noted that the present study has
certain limitations. The findings regarding TGF-β1/Smad
signaling-induced EMT were based only on in vitro
experiments. Additional data on the expression of ETV4 in clinical
patient tissue, and validation studies using animal models are
needed. There is also a lack of validation in other ICC cell lines
and the effects on other downstream genes/proteins of ETV4,
including cell cycle-related proteins such as cyclin D1, and
apoptosis-related genes such as Bax/Bcl-2. Our forthcoming studies
will be centered on comprehensive exploration, using both clinical
and animal studies, to unravel the downstream genes/proteins such
as cyclin D1 and Bax/Bcl-2, regulated by ETV4. In addition, we aim
to elucidate the synergistic or antagonistic mechanisms involving
the TGF-β1/Smad signaling pathway and other pathways.
In conclusion, the present study showed that ETV4
may act as a cancer inducer in ICC, which could enhance the
proliferation, migration and invasion, and inhibit the apoptosis of
Hccc9810 cells. Notably, ETV4-induced increases in N-cadherin and
Vimentin expression, along with the decrease in E-cadherin, may
initiate EMT via the TGF-β1/Smad signaling pathway.
Acknowledgements
Not applicable.
Funding
This research was supported by the National Natural Science
Foundation of China (grant no. 81970558).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
GT contributed to the conception of the study. LL
performed the experiments, contributed to the data analyses and
wrote the manuscript. YF, XX and MX helped perform the experiments
and the analysis with constructive discussions. GT and LL confirm
the authenticity of all the raw data. All authors read and approved
the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
El-Diwany R, Pawlik TM and Ejaz A:
Intrahepatic cholangiocarcinoma. Surg Oncol Clin N Am. 28:587–599.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee AJ and Chun YS: Intrahepatic
cholangiocarcinoma: The AJCC/UICC 8th edition updates. Chin Clin
Oncol. 7:522018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Moris D, Palta M, Kim C, Allen PJ, Morse
MA and Lidsky ME: Advances in the treatment of intrahepatic
cholangiocarcinoma: An overview of t-he current and future
therapeutic landscape for clinicians. CA Cancer J Clin. 73:198–222.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Qi T, Qu Q, Li G, Wang J, Zhu H, Yang Z,
Sun Y, Lu Q and Qu J: Function and regulation of the PEA3 subfamily
of ETS transcription factors in cancer. Am J Cancer Res.
10:3083–3105. 2020.PubMed/NCBI
|
|
5
|
Gao X, Jiang M, Chu Y, Han Y, Jin Y, Zhang
W, Wang W, Yang S, Li W, Fan A, et al: ETV4 promotes pancreatic
ductal adenocarcinoma metastasis through activation of the
CXCL13/CXCR5 signaling axis. Cancer Lett. 524:42–56. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cosi I, Pellecchia A, De Lorenzo E, Torre
E, Sica M, Nesi G, Notaro R and De Angioletti M: ETV4 promotes late
development of prostatic intraepithelial neoplasia and cell
proliferation through direct and p53-mediated downregulation of
p21. J Hematol Oncol. 13:1122020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Singsuksawat E, Thuwajit C, Charngkaew K
and Thuwajit P: Increased ETV4 expression correlates with
estrogen-enhanced proliferation and invasiveness of
cholangiocarcinoma cells. Cancer Cell Int. 18:252018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ang HL, Mohan CD, Shanmugam MK, Leong HC,
Makvandi P, Rangappa KS, Bishayee A, Kumar AP and Sethi G:
Mechanism of epithelial mesenchymal transition in cancer and its
regulation by natural compounds. Med Res Rev. 43:1141–1200. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Derynck R and Weinberg RA: EMT and cancer:
More than meets the eye. Dev Cell. 49:313–316. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Navas T, Kinders RJ, Lawrence SM,
FerryGalow KV, Borgel S, Hollingshead MG, Srivastava AK, Alcoser
SY, Makhlouf HR, Chuaqui R, et al: Clinical evolution of
epithelial-mesenchymal transition in human carcinomas. Cancer Res.
80:304–318. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Williams ED, Gao D, Redfern A and Thompson
EW: Controversies around epithelial-mesenchymal plasticity in
cancer metastasis. Nat Rev Cancer. 19:716–732. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lim W, Jeon BN, Kim YJ, Kim KH and Ko H:
FBI-1 inhibits epithelial-to-mesenchymal transition, migration, and
invasion in lung adenocarcinoma A549 cells by downregulating
transforming growth factor-β1 signaling pathway. J Cell Biochem.
123:644–656. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Singh A and Settleman J: EMT, cancer stem
cells and drug resistance: An emerging axis of evil in the war on
cancer. Oncogene. 29:4741–4751. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sawanyawisuth K, Sashida G and Sheng G:
Epithelial-mesenchymal transition in liver fluke-induced
cholangiocarcinoma. Cancers (Basel). 13:7912021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vaquero J, Guedj N, Clapéron A,
Ho-Bouldoires TH, Paradis V and Fouassier L: Epithelial-mesenchymal
transition in cholangiocarcinoma: From clinical evidence to
regulatory networks. J Hepatol. 66:424–441. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Clapéron A, Mergey M, Ho-Bouldoires TH,
Vignjevic D, Wendum D, Chrétien Y, Merabtene F, Frazao A, Paradis
V, Housset C, et al: EGF/EGFR axis contributes to the progression
of cholangiocarcinoma through the induction of an
epithelial-mesenchymal transition. J Hepatol. 61:325–332. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen X, Huang Y, Liu J, Lin W, Chen C,
Chen Y, Ding Y, Yang Y, Chen Y, Wang H and Teng L: EXOSC5 promotes
proliferation of gastric cancer through regulating AKT/STAT3
signaling pathways. J Cancer. 13:1456–1467. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xiang C, Sun WH, Ke Y, Yu X and Wang Y:
CDCA8 contributes to the development and progression of thyroid
cancer through regulating CDK1. J Cancer. 13:2322–2335. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xia XL, Xue D, Xiang TH, Xu HY, Song DK,
Cheng PG and Wang JQ: Overexpression of long non-coding RNA CRNDE
facilitates epithelial-mesenchymal transition and correlates with
poor prognosis in intrahepatic cholangiocarcinoma. Oncol Lett.
15:4105–4112. 2018.PubMed/NCBI
|
|
21
|
Vermani L, Kumar R and Kumar NS: GAPDH and
PUM1: Optimal housekeeping genes for quantitative polymerase chain
reaction-based analysis of cancer stem cells and
epithelial-mesenchymal transition gene expression in rectal tumors.
Cureus. 12:e120202020.PubMed/NCBI
|
|
22
|
Zhang AN, Li N, Chen ZC, Guo YL, Tian CJ,
Cheng DJ, Tang XY and Zhang XY: Amygdalin alleviated TGF-β-induced
epithelial-mesenchymal transition in bronchial epithelial cells.
Chem Biol Interact. 369:1102352023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang H, Huang Y, Wen Q, Li Y, Guo L and
Ge N: INHBA gene silencing inhibits proliferation, migration, and
invasion of osteosarcoma cells by repressing TGF-β signaling
pathway activation. J Orthop Surg Res. 18:8482023. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jiang W, Xu Y, Chen X, Pan S and Zhu X:
E26 transformation-specific variant 4 as a tumor promotor in human
cancers through specific molecular mechanisms. Mol Ther Oncolytics.
22:518–527. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zheng C, Liu M, Ge Y, Qian Y and Fan H:
HBx increases chromatin accessibility and ETV4 expression to
regulate dishevelled-2 and promote HCC progression. Cell Death Dis.
13:1162022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang J, Sun C, Li J, Jiang H, Qiu Y and
Gong M: Knockdown of ETV4 promotes autophagy-dependent apoptosis in
GBM cells by reducing the transcriptional activation of EMP1. Oncol
Lett. 23:412022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tyagi N, Deshmukh SK, Srivastava SK, Azim
S, Ahmad A, AlGhadhban A, Singh AP, Carter JE, Wang B and Singh S:
ETV4 facilitates cell-cycle progression in pancreatic cells through
transcriptional regulation of cyclin D1. Mol Cancer Res.
16:187–196. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pastushenko I and Blanpain C: EMT
transition states during tumor progression and metastasis. Trends
Cell Biol. 29:212–226. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lu W and Kang Y: Epithelial-mesenchymal
plasticity in cancer progression and metastasis. Dev Cell.
49:361–374. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bure IV, Nemtsova MV and Zaletaev DV:
Roles of E-cadherin and Noncoding RNAs in the
epithelial-mesenchymal transition and progression in gastric
cancer. Int J Mol Sci. 20:28702019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rosso M, Majem B, Devis L, Lapyckyj L,
Besso MJ, Llauradó M, Abascal MF, Matos ML, Lanau L, Castellví J,
et al: E-cadherin: A determinant molecule associated with ovarian
cancer progression, dissemination and aggressiveness. PLoS One.
12:e01844392017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Onder TT, Gupta PB, Mani SA, Yang J,
Lander ES and Weinberg RA: Loss of E-cadherin promotes metastasis
via multiple downstream transcriptional pathwa-ys. Cancer Res.
68:3645–3654. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ali AN, Ghoneim SM, Ahmed ER, Salam LO and
Saleh SM: Cadherin switching in oral squamous cell carcinoma: A
clinicopathological study. J Oral Biol Craniofac Res. 13:486–494.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cao ZQ, Wang Z and Leng P: Aberrant
N-cadherin expression in cancer. Biomed Pharmacother.
118:1093202019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Serrano-Gomez SJ, Maziveyi M and Alahari
SK: Regulation of epithelial-mesenchymal transition through
epigenetic and post-translational modifications. Mol Cancer.
15:182016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ridge KM, Eriksson JE, Pekny M and Goldman
RD: Roles of vimentin in health and disease. Genes Dev. 36:391–407.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Satelli A and Li S: Vimentin in cancer and
its potential as a molecular target for cancer therapy. Cell Mol
Life Sci. 68:3033–3046. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhou Z, Wu B, Chen J, Shen Y, Wang J, Chen
X, Fei F and Li L: ETV4 facilitates proliferation, migration, and
invasion of liver cancer by mediating TGF-β signal transduction
through activation of B3GNT 3. Genes Genomics. 45:1433–1443. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yao D, Bao Z, Qian X, Yang Y and Mao Z:
ETV4 transcriptionally activates HES1 and promotes Stat3
phosphorylation to promote malignant behaviors of colon
adenocarcinoma. Cell Biol Int. 45:2129–2139. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hao Y, Baker D and Dijke PT:
TGF-β-mediated epithelial-mesenchymal transition and cancer
metastasis. Int J Mol Sci. 20:27672019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Katsuno Y, Lamouille S and Derynck R:
TGF-β signaling and epithelial-mesenchymal transition in cancer
progression. Curr Opin Oncol. 25:76–84. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang J, van Dinther M, Thorikay M,
Gourabi BM, Kruithof BPT and Ten Dijke P: Opposing USP19 splice
variants in TGF-β signaling and TGF-β-induced
epithelial-mesenchymal transition of breast cancer cells. Cell Mol
Life Sci. 80:432023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hu HH, Chen DQ, Wang YN, Feng YL, Cao G,
Vaziri ND and Zhao YY: New insights into TGF-β/Smad signaling in
tissue fibrosis. Chem Biol Interact. 292:76–83. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhao M, Mishra L and Deng CX: The role of
TGF-β/SMAD4 signaling in cancer. Int J Biol Sci. 14:111–123. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Moon H, Ju HL, Chung SI, Cho KJ, Eun JW,
Nam SW, Han KH, Calvisi DF and Ro SW: Transforming growth factor-β
promotes liver tumorigenesis in mice via up-regulation of snail.
Gastroenterology. 153:1378–1391. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Deckers M, van Dinther M, Buijs J, Que I,
Löwik C, van der Pluijm G and ten Dijke P: The tumor suppressor
Smad4 is required for transforming growth factor beta-induced
epithelial to mesenchymal transition and bone metastasis of breast
cancer cells. Cancer Res. 66:2202–2209. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Thuault S, Valcourt U, Petersen M,
Manfioletti G, Heldin CH and Moustakas A: Transforming growth
factor-beta employs HMGA2 to elicit epithelial-mesenchymal
transition. J Cell Biol. 174:175–183. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen HY, Chiang YF, Huang CY, Shieh TM,
Kao C, Chang FK, Huang TC, Ali M, Chang HY, Hong YH and Hsia SM:
Spirulina phycocyanin extract and its active components suppress
epithelial-mesenchymal transition process in endometrial cancer via
targeting TGF-beta1/SMAD4 signaling pathway. Biomed Pharmacother.
152:1132192022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
David CJ, Huang YH, Chen M, Su J, Zou Y,
Bardeesy N, Iacobuzio-Donahue CA and Massagué J: TGF-β tumor
suppression through a lethal EMT. Cell. 164:1015–1030. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yan X, Liao H, Cheng M, Shi X, Lin X, Feng
XH and Chen YG: Smad7 protein interacts with receptor-regulated
smads (R-Smads) to inhibit transforming growth factor-β
(TGF-β)/Smad Signaling. J Biol Chem. 291:382–392. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang JY, Gao YB, Zhang N, Zou DW, Wang P,
Zhu ZY, Li JY, Zhou SN, Wang SC, Wang YY and Yang JK: miR-21
overexpression enhances TGF-β1-induced epithelial-to-mesenchymal
transition by target smad7 and aggravates renal damage in diabetic
nephropathy. Mol Cell Endocrinol. 392:163–172. 2014. View Article : Google Scholar : PubMed/NCBI
|