Introduction
Breast cancer is the most common malignancy in women
and is an imminent threat to the health and lives of women;
according to a World Health Organization report, 2.1 million women
are affected by breast cancer each year. It was reported that
627,000 women died due to breast tumors in 2018 (1,2).
Furthermore, it has the worst morbidity and mortality rates among
all female-associated malignancies worldwide, with the latest data
showing that breast cancer accounts for 24.5% of global female
cancer morbidities and 15.5% of associated mortalities (3).
Estrogen receptor (ER) positivity is present in 70%
of patients with breast cancer (4).
To suppress ER-mediated mitogenic estrogen signaling, tamoxifen
(TAM), a particular ER modulator, competes with estrogen for its ER
ligand-binding domain (1). TAM has
known therapeutic effects against ER+ breast carcinoma
(1,5). TAM (an endocrine therapy) mitigates
the recurrence rate of early-stage ER+ breast cancer by
~40%; nevertheless, TAM resistance develops in ~30% of such
patients after ongoing therapy, leading to tumor recurrence and
metastasis (6). TAM resistance is
the primary cause contributing to treatment failure in patients who
have ER+ breast carcinoma; however, few treatment
options are currently available (7). Therefore, investigating efficacious
strategies for TAM-resistant breast cancer is imperative.
Breast cancer resistance to TAM abnormally enhances
malignant phenotypes, such as metastasis, proliferation, invasion,
resistance to apoptosis and chemotherapy tolerance (7–9).
Through the PI3K/AKT/mTOR signaling process, ER+ breast
tumor cells have been shown to bypass the inhibitory properties of
TAM to develop additional malignant phenotypes. Growth factors
stimulate the PI3K/AKT/mTOR signaling pathway by activating growth
factor receptors such as human epidermal growth factor receptor 2,
epidermal growth factor receptor, vascular endothelial growth
factor or insulin-like growth factor. These factors enable the
overexpression of anti-apoptotic genes [B-cell lymphoma
(Bcl)-extra-large and Bcl-2], genes linked to migration
[Snail, which induces epithelial-mesenchymal transition of
tumor metastasis by binding to the E-box in the E-cadherin promoter
region among epithelial tumors; Twist, which promotes the
metastasis process in human breast tumor and it promotes breast
cancer by downregulating E-cadherin and estrogen receptor; and
MMP9, which is crucial in protein lysis and extracellular
matrix remodelling, and is related to tumor metastasis, invasion
and regulation of the tumor microenvironment] and genes related to
the cell cycle (Cyclin D1, CDK4/6 and c-Myc), thereby
generating malignant phenotypes in the drug-resistant strain
(7,10–18).
Therefore, small-molecule drugs that inhibit cell proliferation by
interfering with cell growth factors may overcome the malignant
phenotype of MCF-7/TAMR cells.
Studies have reported that podophyllotoxin (PPT) is
derived from different species of PPT rhizomes, which are lignans
isolated from plants of the genus Podophyllum. PPT and its
derivatives are pharmacologically active (19,20).
The widespread interest in lignans stems from their potent
antiviral and antineoplastic properties (21). Microtubule protein polymerization is
known to be blocked by PPT and its derivatives, which have
anticancer characteristics, thereby halting the cell cycle at the
G2/M phase (22,23). This has attracted attention for
pharmacological studies in recent decades with the invention of the
semi-synthetic anticancer compound etoposide and teniposide, which
have inspired long-term studies of this structural phenotype
(24). To date, the two epiPPT
derivatives have been developed as clinical therapeutic agents
(24); they are chemotherapeutic
agents in the treatment of myeloid leukemia, pancreatic malignancy,
testicular cancer, gastric tumors, lymphoma and small-cell lung
cancer (23,25).
Notably, the two medications have shown certain
inhibitory effects on breast cancer cells at the cellular level
(26,27). For example, to hinder cell
proliferation, etoposide inhibits DNA topoisomerase II and stops
the cell cycle at the S phase (19,21).
Certain phase II clinical studies have reported the effectiveness
of oral etoposide as a therapy for metastatic breast cancer.
However, its widespread clinical use is yet to be confirmed by
large-scale randomized controlled clinical studies (28). Etoposide is a genotoxic compound
that occasionally causes severe side effects, including secondary
leukemia, bone marrow suppression, gastrointestinal disorders,
neutropenia and peripheral neuropathy. Presently, etoposide therapy
for breast tumors is regarded as experimental. When etoposide is
applied, cells can evade apoptosis, continue proliferating and
become resistant to the drug (28–30).
Furthermore, teniposide is a potent broad-spectrum antineoplastic
drug that results in DNA damage during replication and the
induction of apoptosis (31). Its
minimal water solubility and unfavorable response to the marketed
dose induce anaphylaxis and restrict its clinical feasibility
(32). Anhydrous ethanol is often
applied to increase water solubility. This has undesirable effects
(anaphylaxis) and is not well tolerated by certain patients
(33). Conversely, teniposide is
free in VM-26, thus promoting swift clearance and broad tissue
dissemination involving cancerous tissues and healthy organs,
thereby decreasing its curative effectiveness and enhancing adverse
reactions. Despite successfully halting the expansion of MCF-7
transplanted tumor models in vivo, teniposide micelles are
not clinically useful (34,35).
Etoposide and teniposide often exhibit unexpected
toxicity at effective doses in clinical trials, thereby limiting
their clinical application. This has provided justified creation of
novel PPT derivatives with reduced harmful in vivo effects
and enhanced antineoplastic properties (21,36).
Under these circumstances, structural modifications of the PPT
backbone have been performed to synthesize novel, effective PPT
compounds, including tafluposide, NPF, NK-611, TOP-53 and
nitrogenous toxins. NK-611, which replaces the hydroxyl group with
dimethyl in the sugar group of etoposide, inhibits TOP2 and is more
effective than etoposide against some tumor cells in vitro.
TOP-53 is more likely to cause chromosome breakage, is more
effective than etoposide on non-small cell lung cancer cells, and
shows extremely different drug resistance, anti-tumor spectrum and
TOP2 inhibitory function from etoposide. NPF is a 100× more potent
than etoposide against all cancer cells and can be used as a lead
compound for further manufacturing of new antitumor drugs (21,25,37). A
previous study reported that a nitroxyl spin-labelled derivative of
podophyllotoxin (GP7) induced apoptosis in human leukemia cells
(38). GP7 showed potent inhibitory
effects on mouse and human osteosarcoma cells in vitro, and
GP7 arrested the cell cycle at the S phase (38). Studies on any of the aforementioned
PPT derivatives in the therapy of TAM resistance in breast cancer,
however, are lacking.
The present study aimed to screen one of the
derivatives, bromosulfonamidine amino-PPT (BSAPPT), that
effectively targets TAM-resistant breast cancer cells, and to
assess the role of BSAPPT in different types of tumor cell lines.
Aside from breast cancer, lung cancer ranks second in terms of
incidence among both men and women (11.4% of total cases), and it
is the main cause of cancer-related deaths worldwide (18.0% of
total cases). Therefore, it also warrants close attention (3,39). In
the present study, CCK-8, colony formation, apoptosis, cell cycle,
RT-qPCR and western blotting assays were used to detect the
proliferation inhibition, apoptotic and cell cycle effects, and the
molecular targets of BSAPPT on MCF-7, MCF-7/TAMR, A549 and
MDA-MB-231 cancer cells.
Materials and methods
Cells and drugs
MCF-7 (human breast adenocarcinoma), MCF-10A (human
normal mammary epithelial cells), A549 (human non-small cell lung
cancer) and MDA-MB-231 (human breast adenocarcinoma) cell lines
were purchased from the American Type Culture Collection Cell Bank.
All cell lines used in the current study were identified by
Shanghai Yihe Applied Biotechnology Co., Ltd., and matched exactly
to the corresponding cell lines. Bromosulfonamidine amino-PPT
(BSAPPT) was kindly donated by Dr Weiguang Yang of Guangdong
Medical University (Zhanjiang, China) and stored in the
laboratory.
Main reagents
FBS serum was purchased from Shanghai ExCell
Biology, Inc. Trypsin, RPMI-1640 medium and DMEM were purchased
from Gibco (Thermo Fisher Scientific, Inc.). 4-hydroxytamoxifen was
purchased from Sigma-Aldrich (Merck KGaA). The reverse
transcription kit (cat. no. A0010CGQ), SYBR Green Premix Dye
Real-Time Fluorescence Quantitative (q)PCR kits (cat. no. A0012-R2)
and the total intracellular RNA extraction reagent (cat. no.
B0004D) were purchased from EZBioscience. The kit for proliferation
(Cell Counting Kit-8; cat. no. C6030) was purchased from NCM
Biotechnology. Crystalline violet staining solution and 4%
paraformaldehyde fixative were purchased from Beyotime Institute of
Biotechnology. The assay kit for apoptosis (BD Pharmingen™ FITC
Annexin V Apoptosis Detection Kit; cat. no. 556547) was purchased
from BD Biosciences and the kit for the cell cycle test [Cell Cycle
Assay Kit (Red Fluorescence); cat. no. E-CK-A351] was purchased
from Elabscience Biotechnology, Inc. Mouse anti-human Caspase-9
antibodies (cat. no. 9508S; 1:1,000) were purchased from Cell
Signaling Technology, Inc., rabbit anti-human Bcl-2 (cat. no.
BA0412; 1:1,000) and cyclin B1 (CCNB1; cat. no. BA0766; 1:1,000)
antibodies were purchased from Wuhan Boster Biological Technology,
Ltd., rabbit anti-human polo like kinase (PLK)-1 antibodies (cat.
no. 10305-1-AP; 1:1,000) and targeting protein for Xklp2 (TPX2)
antibodies (cat. no. 11741-1-AP; 1:2,000) were purchased from
Proteintech Group, Inc., and the rabbit anti-human PLK-4 antibodies
(cat. no. A9863; 1:1,000) were purchased from ABclonal Biotech Co.,
Ltd. Mouse anti-GAPDH antibodies (cat. no. abs830030; 1:1,000) were
purchased from Absin Bioscience, Inc. The UltraSignal
hypersensitive ECL Chemiluminescence Substrate (cat. no.
4AW011-500) was purchased from Beijing 4A Biotech Co., Ltd. and the
PVDF membranes (cat. no. FFP33) were purchased from Beyotime
Institute of Biotechnology.
Cell culture
MCF-7 and MCF-7/TAMR cells were cultured in
RPMI-1640 media containing 10% FBS in a constant-temperature
incubator maintained at 37°C with 5% CO2. MCF-7/TAMR
cells were obtained by cultivating MCF-7 in an estrogen-deprived
environment with low-to-high TAM concentrations (100, 200, 500, 800
and 1,000 nM) for >6 months to ultimately form MCF-7/TAMR
resistant to TAM at a concentration of 1 µM (7). MCF-10A cells were cultured in a
specialized complete medium at 37°C with 5% CO2, whilst
A549 and MDA-MB-231 cells were cultured in DMEM and RPMI-1640 media
at 37°C with 5% CO2, respectively. Then these cells were
passaged at 3-day intervals.
Morphological observation of
cells
Cell death and floating conditions of MCF-7 and
MCF-7/TAMR cells (3×105) treated with or without BSAPPT
(10 µg/ml) at 37°C for 48 h were observed under an inverted optical
microscope using a 10X objective.
Reverse transcription-qPCR
Intracellular RNA was isolated from four types of
cells, MCF-7, MCF-7/TAMR, A549 and MDA-MB-231, treated or untreated
with BSAPPT, using the EZ-Press RNA Purification Kit (cat. no.
B0004D; EZBioscience). Moreover, reverse transcription was
performed using 1 µg total RNA to produce cDNA after determining a
specific concentration using the Color Reverse Transcription Kit
(with genomic DNA remover) (cat. no. A0010CGQ; EZBioscience).
Reverse transcription was performed at 42°C for 15 min and 95°C for
30 sec. The relative expression levels of Bcl-2, Caspase-9,
PLK1, PLK4, CCNB1, TPX2 and β-actin genes were detected
using SYBR Green dye. The qPCR procedure was as follows: 95°C for 5
min for one cycle, followed by 95°C for 10 sec and 60°C for 30 sec
for 40 cycles. The results were calculated using the
2−ΔΔCq (40) method to
obtain the ploidy of each target gene relative to β-actin. Table I lists the primer sequences.
 | Table I.Quantitative PCR primers. |
Table I.
Quantitative PCR primers.
| Primer | Direction | Sequence
(5′-3′) |
|---|
| PLK1 | F |
CGAGTTCTTTACTTCTGGCT |
|
| R |
TATTGAGGACTGTGAGGGGC |
| CCNB1 | F |
GCACTTTCCTCCTTCTCA |
|
| R |
CGATGTGGCATACTTGTT |
| Bcl-2 | F |
GGTGGGGTCATGTGTGTGG |
|
| R |
CGGTTCAGGTACTCAGTCATCC |
| Caspase-9 | F |
GCAGTAACCCCGAGCCAGATG |
|
| R |
CCGGAGGAAATTAAAGCAACCAG |
| PLK4 | F |
AGTGCTCCCTTTTTCCCAAT |
|
| R |
AGCAGCACTATGCATGACCA |
| TPX2 | F |
ATGGAACTGGAGGGCTTTTTC |
|
| R |
TGTTGTCAACTGGTTTCAAAGGT |
| Bax | F |
GGGGACGAACTGGACAGTAA |
|
| R |
CAGTTGAAGTTGCCGTCAGA |
| Cyt-C | F |
AGGCCCCTGGATACTCTTACACA |
|
| R |
TCTGCCCTTTCTTCCTTCTTCTTA |
| Apaf-1 | F |
AATGGCAGGCTGTGGGAAGTC |
|
| R |
TAAGTGGAAGCCTCTGGGAAAAAC |
| Caspase-3 | F |
TGGCGAAATTCAAAGGATG |
|
| R |
TAACCCGGGTAAGAATGTGC |
| β-actin | F |
CATGTACGTTGCTATCCAGGC |
|
| R |
CTCCTTAATGTCACGCACGAT |
Western blotting
To detect target proteins, MCF-7 and MCF-7/TAMR
cells were added to two separate six-well plates with 250,000 cells
per well, respectively. They were set up in two groups: One with
added BSAPPT and one without BSAPPT. After 48 h, three wet washes
at 4°C using pre-cooled PBS were performed on the cells. Next, the
mixed solution containing RIPA-strong lysis solution (Cowin Biotech
Co., Ltd) and proteinase inbitor (Biosharp) (RIPA lysis solution:
proteinase inhibitor, 100:1) was added to the cells, and the cells
were lysed on ice for 15 min. The BCA (cat. no. P0010; Beyotime
Institute of Biotechnology) assay was used to test the protein
concentrations. Cells were then centrifuged at 4°C and 16,099 × g
for 15 min. The supernatant was collected and the 5X SDS-PAGE
loading buffer (Beyotime Institute of Biotechnology) was added,
then the cells were incubated in a metal bath at 100°C for 10 min.
Proteins (15 µg/lane) were separated on 10% gels using SDS-PAGE.
The mixture was electrotransferred to a PVDF membrane and vibrated
at room temperature (25°C) for 1 h after being blocked with 5%
skimmed milk powder (Fujifilm Wako Pure Chemical Corporation) at
25°C for 1 h. Primary antibodies (GAPDH, Bcl-2, Caspase-9, PLK1,
PLK4, CCNB1 and TPX2) were then added with diluted 5% BSA and
refrigerated at 4°C overnight. A total of three TBST (0.05%
Tween-20) washes were performed on the membrane, 5 min each time,
and then the membrane was incubated with the corresponding
HRP-conjugated secondary antibodies (cat. no. BA1054; 1:5,000;
Boster Biological Technology) for 2 h at 4°C, followed by three
10-min washes. ECL reagent (4A Biotech, Co., Ltd.) was used for
visualization of protein bands in fluorescence and
chemiluminescence imaging systems (Clinx Science Instruments Co.,
Ltd.), and images were captured.
Cytotoxicity assay
MCF-7, MCF-7/TAMR, MCF-10A, A549 and MDA-MB-231
cells were each placed at an average density of 50,000 cells/well
in 24-well plates to produce the control and treatment groups. A
total of 500 µl complete media was added to each well. BSAPPT at a
drug concentration gradient of 1, 5, 10, 20 and 40 µg/ml was added
to the treatment group. MCF-7, MCF-7/TAMR and MCF-10A cells were
separately added to three 24-well plates at 50,000/well and treated
with etoposide at a concentration gradient of 1, 5, 10, 20, 40 and
80 µg/ml (MCF-7 and MCF-7/TAMR) or an etoposide concentration
gradient of 1, 2.5, 5, 10, 20 and 40 µg/ml (MCF-10A). Prior to the
assay, the old medium was removed from each well, washed with PBS
at room temperature (25°C), and refilled with fresh 380 µl media.
Following the addition of 38 µl of CCK-8 solution, the culture
plate was gently shaken. The mixture was incubated for 30 min at
37°C in an incubator. Using a microplate reader, the absorbance of
each well was measured at 450 nm.
Colony formation assay
A total of 500 MCF-7 and MCF-7/TAMR cells/well were
added to two separate 6-well cell culture plates. BSAPPT was added
at increasing dosages (1, 5, 10, 20 and 40 µg/ml). The medium was
changed every three days during the incubation period at 37°C of
7–14 days to monitor the condition of the cells. Under a inverted
microscope, the number of colonies was counted, and when it reached
an average of >50 cells in a colony, the cells were fixed for 15
min in 1 ml 4% polyformaldehyde at room temperature (25°C) before
being stained for an additional 15 min in 500 µl crystal violet
staining solution (0.1%) at room temperature (25°C). Colony counts
were then performed by ImageJ software (version 1.8.0; National
Institutes of Health).
Cell apoptosis assay
A total of 250,000 MCF-7, MCF7/TAMR, A549 and
MDA-MB-231 cells were separately inserted in each well of four
six-well plates. The control and experimental groups were set up
concurrently. When cell growth reached 50–60%, a total of 5 µg/ml
BSAPPT was separately added to the 4 types of cells in the
experimental group at 37°C for 48 h before the cell supernatant was
collected; at the same time, the control group was replaced with
fresh culture medium. After 48 h of cell culture, the cell
supernatant of the control and treatment groups was collected.
After trypsin digestion, all cells were collected and the six-well
plates were rinsed with cold PBS, and after removing the
supernatant by centrifugation at 800 × g and room temperature
(25°C) for 3 min, they were then washed twice using cold PBS before
cell precipitates were resuspended with 500 µl 1X binding buffer
([10X Annexin V Binding Buffer, 0.1 M Hepes/NaOH (pH 7.4), 1.4 M
NaCl and 25 mM CaCl2] and mixed gently for 5 min).
Annexin V binds to this antigen (Phosphatidylserine) on the
apoptotic cell surface and fluorochromes (Annexin V-FITC and PI: BD
Pharmingen™ FITC Annexin V Apoptosis Detection Kit; cat. no.
556547; BD Biosciences) were used to differentiate between
apoptotic and non-viable cells. A total of 5 µl Annexin V-FITC and
5 µl PI was added to the mixture. After 15 min of incubation at
ambient temperature (25°C) in the darkness, the cells were shaken
evenly, and within 1 h, flow cytometry was performed to assess cell
apoptosis by sorting flow cytometer (BD FACSAria II instrument and
computer table; BD Biosciences). Finally, data analysis was
conducted using FlowJo software (v10.8.1; FlowJo LLC).
Cell cycle assay
After seeding MCF-7 and MCF-7/TAMR cells in two
separate six-well plates at a density of 250,000 cells/well, the
control and experimental groups were set up simultaneously. A total
of 5 µg/ml BSAPPT was added to the cells in the experimental group.
After 8 h, all the cells were collected, centrifuged at 300 × g and
25°C for 5 min, and the liquid that was left over was discarded.
Before the precipitate was centrifuged (300 × g, 25°C, 5 min), it
was washed with 1 ml PBS. The cells were then fully mixed after
adding 0.3 ml PBS and 1.2 ml 100% ethanol at −20°C, and placed in a
refrigerator at −20°C for 1 h. Centrifugation at 25°C and 300 × g
for 5 min was performed to remove the remaining supernatant, and
after reconstitution in 1 ml PBS for 15 min at ambient temperature
(25°C), the cells were centrifuged at 25°C and 300 × g for 5 min to
separate the remaining fluid. Subsequently, the cells were
resuspended in 100 µl RNase A reagent and incubated for 30 min at
37°C. A total of 400 µl PI reagent (50 µg/ml) was then added,
thoroughly combined, and incubated at 2–8°C in a pitch-black
environment for 30 min. The specimens were immediately evaluated on
the sorting flow cytometer (BD FACSAria II instrument and computer
table; BD Biosciences), and red fluorescence was recorded at the
excitation wavelength of 488 nm.
Statistical analysis
Data are presented as mean ± standard deviation.
GraphPad Prism 8 software (Dotmatics) was used for analysis and
plotting of the data. To assess differences between groups in the
present study, unpaired Student's t-tests, one-way ANOVA and
regression analyses were used. Tukey's post hoc multiple
comparisons test was used following ANOVA. FlowJo (v10.8.1; FlowJo
LLC) and ImageJ (1.8.0; National Institutes of Health) software
were used to process the apoptosis, cell cycle and colony formation
images. P<0.05 was considered to indicate a statistically
significant difference.
Results
BSAPPT strongly inhibits the
proliferation of TAM-resistant breast cancer
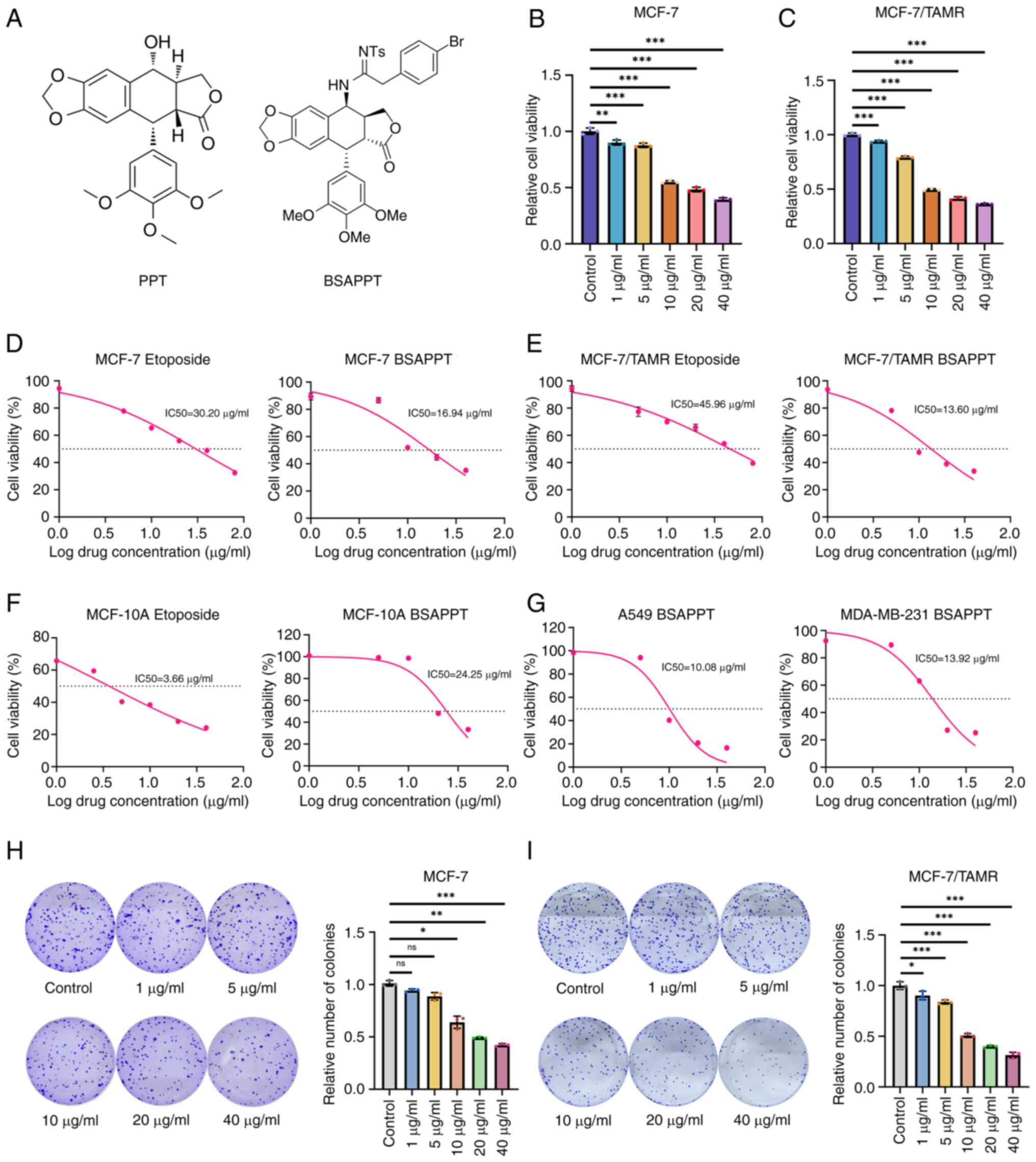
The molecular formula of PPT is
C22H22O8 (19), and BSAPPT is a previously unstudied
agent that was obtained from natural products containing PPTs
(Fig. 1A). To determine how BSAPPT
impacts MCF-7/TAMR cell proliferation, the present study assessed
the implications of different dosages (1, 5, 10, 20 and 40 µg/ml)
of BSAPPT on the viability of MCF-7, MCF-7/TAMR and MCF-10A cells.
The findings demonstrated that BSAPPT significantly inhibited cell
proliferation in a concentration-dependent manner after 48 h. The
cell viability of MCF-7 and MCF7/TAMR cells treated with 5 µg/ml
BSAPPT were 87.40 and 79.05%, respectively, relative to the control
group. Compared with the MCF7/TAMR cells, the cell viability of
BSAPPT-treated MCF-7 cells was 1.11× stronger (both P<0.05;
Fig. 1B and C). Furthermore, the
cell viability of MCF-7 and MCF-7/TAMR cells treated with 10 µg/ml
BSAPPT was 55.09 and 49.55%, respectively, relative to the control
group. The cell viability of BSAPPT-treated MCF-7 cells was
1.11-fold greater than that of the MCF7/TAMR cells (both P<0.05;
Fig. 1B and C). Moreover, the cell
vitality of MCF-7 and TAMR cells treated with 20 µg/ml BSAPPT was
48.32 and 41.31%, respectively, relative to the control group.
Finally, the cell viability of BSAPPT-treated MCF-7 cells was
1.17-fold greater than that of the MCF7/TAMR cells (both P<0.05;
Fig. 1B and C).
Half-maximal inhibitory concentration
(IC50) analysis demonstrated that, in the presence of
BSAPPT, the IC50 of MCF-7 cells was 16.94 µg/ml (22.18
µM; Fig. 1D), the IC50
of MCF7/TAMR cells was 13.60 µg/ml (17.81 µM; Fig. 1E), and the IC50 of
MCF-10A cells was 24.25 µg/ml (31.76 µM; Fig. 1F). The IC50 of MCF-10A
cells was 1.43× that of MCF-7 cells, and 1.78× that of MCF7/TAMR
cells. These results demonstrate that the degree of injury caused
by BSAPPT in normal mammary gland cells (MCF-10A) was notably less
than that in MCF-7 and MCF-7/TAMR cells. In the presence of
etoposide (control), the IC50 of the MCF-7 cells was
30.20 µg/ml (51.31 µM) (Fig. 1D);
the IC50 of MCF7/TAMR cells was 45.96 µg/ml (78.09 µM;
Fig. 1E); and the IC50
of MCF-10A cells was 3.66 µg/ml (6.22 µM; Fig. 1F). This demonstrates that in MCF-7
and MCF-7/TAMR cells, the IC50 in the etoposide samples
were notably higher than that of the BSAPPT-treated samples; whilst
in MCF-10A cells, the IC50 of the etoposide group was
markedly less than that of the BSAPPT-treated group. These findings
indicate that the effective injury concentration of BSAPPT in MCF-7
and MCF-7/TAMR cells was notably lower than that of etoposide,
whilst that of BSAPPT in normal cells was markedly greater than
that of etoposide. Furthermore, the IC50 of
BSAPPT-treated A549 and MDA-MB-231 cells was 10.08 and 13.92 µg/ml,
respectively (Fig. 1G), thereby
confirming that BSAPPT exerted good inhibitory effects on other
cancer cell lines.
The role of BSAPPT was subsequently assessed using a
colony formation assay, with the aim of determining its effect on
the proliferation of the two tumor cell lines, MCF-7 and
MCF-7/TAMR. It was demonstrated that 1, 5, 10, 20 and 40 µg/ml
BSAPPT resulted in MCF-7 cell relative colony numbers of 91.1,
87.5, 68.6, 48.0 and 40.6, respectively, relative to the normal
group (P<0.05; Fig. 1H).
Furthermore, the MCF-7/TAMR cell relative colony numbers were
90.21, 83.8, 50.79, 39.9 and 31.44, respectively, relative to the
control group (P<0.05; Fig. 1I).
The number of colonies formed by MCF-7 was 1.35, 1.20 and 1.29×
that of the MCF-7/TAMR group when the concentrations were 10, 20,
and 40 µg/ml, respectively. These data showed that BSAPPT could
induce a reduction in both MCF-7 and MCF-7/TAMR colony formation,
and that the reduction in colony formation was greater in
MCF-7/TAMR than in MCF-7. The aforementioned findings indicate that
the process of colony formation was concentration-dependent; that
is, the number of colonies formed decreased gradually with
increasing drug concentration. Moreover, the results revealed that
at doses of 10, 20 and 40 µg/ml, the MCF-7/TAMR cells produced
significantly fewer colonies than the MCF-7 cells. The
proliferation of TAM-resistant strains was thus significantly more
inhibited by BSAPPT than the wild type cells.
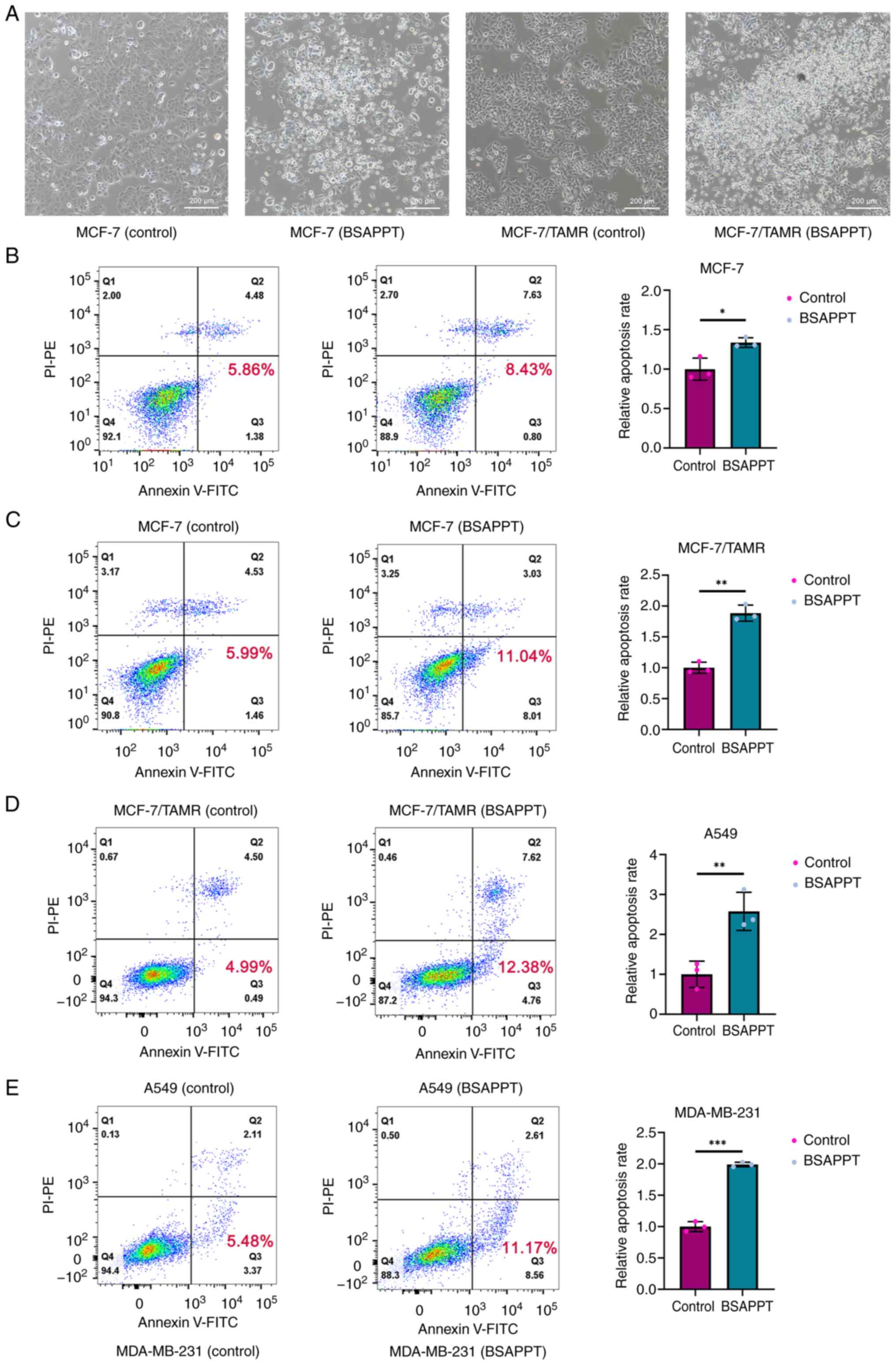
BSAPPT exhibits enhanced pro-apoptotic
effects in MCF-7/TAMR cells
Many physiologic mechanisms depend on apoptosis,
which associated with the onset of several diseases, including
cancer, autoimmune disorders and neurological problems (41). To assess whether BSAPPT induces
apoptosis in MCF-7 and drug-resistance cells, apoptotic assays were
performed. Following a 48-h incubation with 5 µg/ml BSAPPT to MCF-7
and MCF7/TAMR cells, the morphological alterations and
characteristics of the cells were evaluated under a light
microscope. It was demonstrated that MCF-7 and drug-resistance
cells treated with BSAPPT had notably more floating dead cells than
those in the control group. Furthermore, it was observed that a
markedly greater proportion of cells showed characteristics of
apoptosis, such as cytoplasmic condensation, cell division and the
formation of apoptotic bodies (Fig.
2A). This indicates that BSAPPT can induce the death of MCF-7
and MCF7/TAMR cells, and PPT derivatives have been known to cause
human tumor cell death (38).
Moreover, the apoptosis rates of MCF-7 and MCF7/TAMR cells were
assessed using flow cytometry. This was following the cells being
left untreated or treated with 5 µg/ml BSAPPT for 48 h to further
evaluate the ability of the drug to induce apoptosis in MCF-7 and
MCF7/TAMR cells. The results demonstrated that the apoptosis rate
of the BSAPPT-treated MCF-7 cell group was 1.44-fold greater in
comparison with that of the MCF-7 control group (Fig. 2B). However, the apoptosis rate in
the MCF-7/TAMR group receiving treatment was 1.84× higher than that
of the MCF-7/TAMR control group (Fig.
2C). In comparison with the MCF-7 cell sample, the apoptosis
rate in the MCF-7/TAMR group was 1.28× greater, indicating that the
MCF-7/TAMR cells had a notably higher rate of apoptosis following
treatment than the MCF-7 cells, which was in concordance with the
IC50 results. Furthermore, the average apoptotic rate of
the MCF-7 group receiving treatment was 1.34× greater than that of
the untreated group in three repeated experiments (both P<0.05),
whilst it was 1.89-fold greater in the MCF-7/TAMR treatment group
than in the control group (both P<0.05). Therefore, BSAPPT
promoted the apoptosis in the MCF-7/TAMR cells by 1.41× more than
that in the MCF-7 cells. This indicates that BSAPPT more
effectively induced apoptosis in MCF-7/TAMR cells than in MCF-7
cells. Moreover, the apoptosis rate of BSAPPT-treated A549 cells
was 2.48× greater than that of the A549 control group (Fig. 2D), and the apoptosis rate of the
BSAPPT-treated MDA-MB-231 cells was 2.04-fold greater than that of
the MDA-MB-231 control cells (Fig.
2E). The average apoptotic rate of the A549 group receiving
treatment was 2.58× greater than that of the untreated group in
three repeated experiments (both P<0.05), whilst it was
1.99-fold greater in the MDA-MB-231 treatment sample than in the
control sample (both P<0.05). This indicates that BSAPPT also
effectively promoted apoptosis in other cancer cells. Thus, BSAPPT
may cause apoptosis in other cancer cell lines and induce a greater
rate of apoptosis in MCF-7/TAMR cells than in MCF-7 cells.
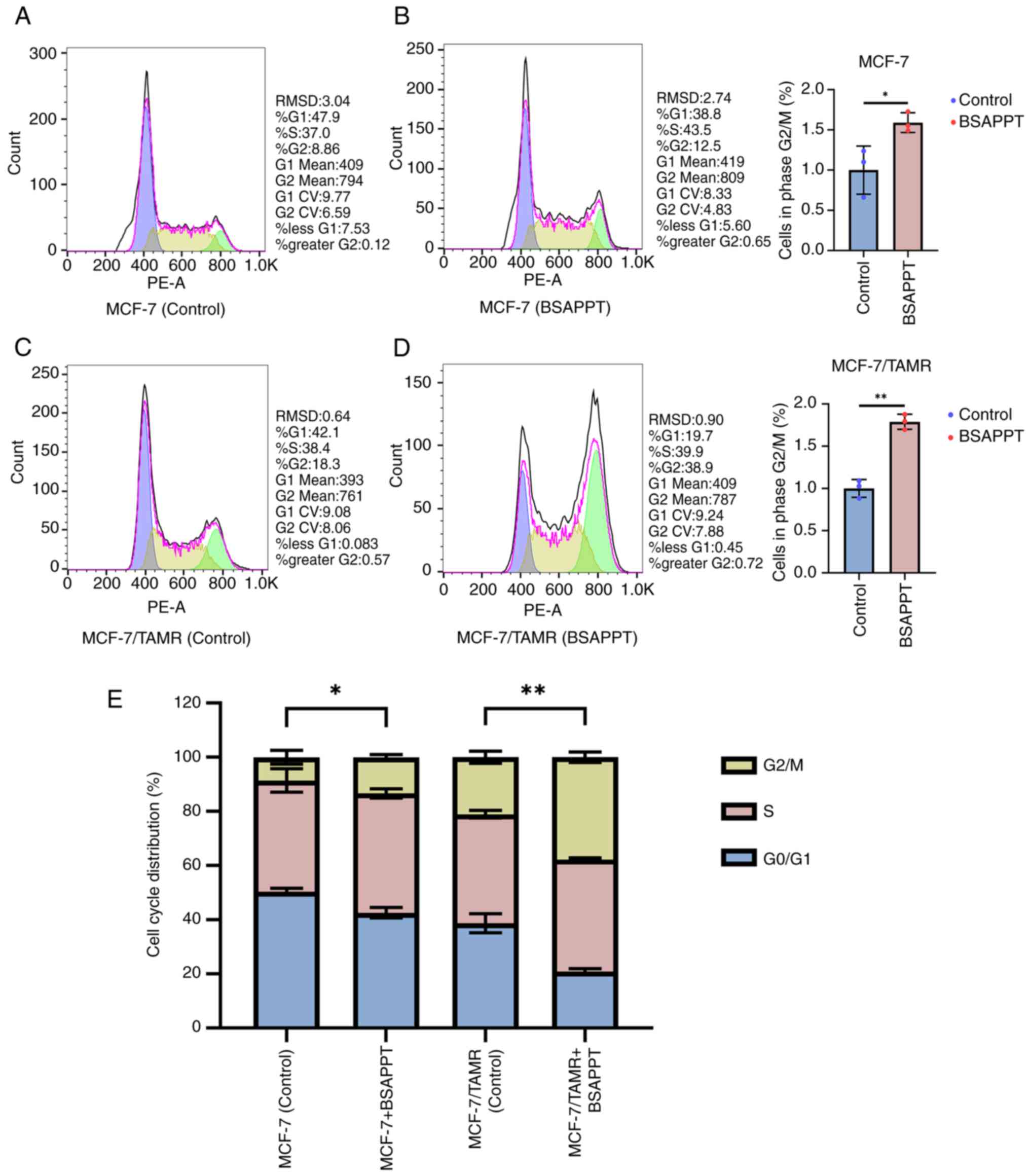
BSAPPT induces a more significant cell
cycle arrest among drug-resistant breast cancer cells
PPT-induced microtubule breakage results in cell
cycle arrest during the G2/M phase, triggering apoptosis
(22). Cell cycle evaluation tests
were performed to assess the BSAPPT-induced cycle modifications in
MCF-7 and MCF-7/TAMR. It was demonstrated that the proportion of
MCF-7 in the G2 phase after 8 h with the addition of 5
µg/ml BSAPPT was 1.41-fold greater than the MCF-7 normal group
(Fig. 3A and B). However, after 8
h, the proportion of the MCF-7/TAMR treated with 5 µg/ml BSAPPT
during the G2 phase was 2.13× greater than the
MCF-7/TAMR group without treatment (Fig. 3C and D). Furthermore, the proportion
of the BSAPPT-treated MCF-7/TAMR group in the G2 phase
was 1.5-fold greater than that of the MCF-7 group. The average
ratio of the MCF-7 group in the G2/M phase was 1.59-fold
higher than that of the untreated control group, compared with
1.79-fold greater for the MCF-7/TAMR group receiving treatment
compared with the untreated sample (both P<0.05). The mean
proportion of the G2/M phase induced by BSAPPT in
MCF-7/TAMR cells was 1.13-fold higher than that in MCF-7 cells.
This indicates that BSAPPT more effectively arrests the cell cycle
in MCF-7/TAMR cells than in MCF-7 cells, indicating that BSAPPT
inhibits cell cycle progression in MCF-7/TAMR cells.
By comparing the untreated group and the
drug-treated group, it was demonstrated that the interkinesis of
the two untreated groups was markedly longer than that of the
division phase; the G0/G1 phase
>G2/M phase (Fig.
3E), However, the G0/G1 phase interval of
the two drug treatment groups was reduced, and BSAPPT induced a
1.6× shortening of G0/G1 phase in MCF-7/TAMR cells compared with
MCF-7 cells; consequently, the G0/G1 phase
was notably shorter in the MCF-7/TAMR cells. Meanwhile, the peak
G2/M phase was markedly higher in the two drug-treated
groups, and the G2/M interval in the MCF-7-treated group
was 1.59× greater than the untreated group (P<0.05). The
G2/M interval among the MCF-7/TAMR-treated group was
1.79× greater than that of the control sample (P<0.05). The
G2/M phases of the MCF-7 control sample and the
treatment sample differed significantly, and the G2/M
phases of the MCF-7/TAMR control sample and the treatment sample
also differed significantly, indicating a cell cycle arrest role of
BSAPPT in both the MCF-7 and MCF-7/TAMR cells. The M phase is the
spindle assembly checkpoint (42),
and the peak G2/M phase was notably elevated in the
drug-treated MCF-7/TAMR group compared with that in the
drug-treated MCF-7 group (Fig. 3E).
This indicates that BSAPPT may inhibit tubulin binding in both
cells. Moreover, BSAPPT may more effectively disrupt the
microtubule polymerization of MCF-7/TAMR cells, promoting an
increase in the number of cells in the G2/M state. The
cell cycles of both cell lines were inhibited by BSAPPT at the
G2/M period, and the level of G2/M arrest in
the MCF-7/TAMR cells was greater than that in the MCF-7 cells. This
indicates a higher degree of inhibition of the drug to MCF-7/TAMR
cells. These results reveal that BSAPPT mainly inhibits the
G2/M period in the cell cycle, which consequently leads
to the apoptosis of breast cancer cells and inhibits MCF-7/TAMR
cell development.
BSAPP regulates the expression of
apoptosis and cycle-related genes
The effect of BSAPPT on the expression levels of
genes linked to the cell cycle and apoptosis (Bcl-2, Caspase-9,
PLK1, PLK4, CCNB1 and TPX2) were assessed using qPCR to gain
insight into the molecular mechanisms behind the apoptosis and
proliferation restrictions associated with treatment with 10 µg/ml
BSAPPT in the MCF-7 and MCF-7/TAMR groups. The results demonstrated
that the levels of Bcl-2 expression were significantly upregulated
in TAM-resistant cells compared with the MCF-7 group (2.1-fold
higher than the original value) and significantly downregulated in
MCF-7 and MCF-7/TAMR cells after BSAPPT administration compared
with the respective untreated group (82.17 and 70.34%,
respectively). Bcl-2 expression reduction was 1.17× greater in
MCF-7/TAMR cells than in MCF-7 cells, so the effect was
significantly more pronounced in MCF-7/TAMR cells. This indicates
that BSAPPT exerted pro-apoptotic effects by inhibiting Bcl-2
expression (Fig. 4A). In contrast,
the TAM-resistant cells exhibited a significant reduction in the
expression of caspase-9 compared with the MCF-7 group, the gene
that initiates apoptosis (43) (76%
of the MCF-7 group). Moreover, BSAPPT treatment of MCF-7 and
MCF-7/TAMR cells was associated with a significantly upregulated
mRNA expression level of caspase-9, which was 1.46× and 1.61×
greater, respectively, than in the respective untreated control
cells. Moreover, the upregulation of caspase-9 in MCF-7/TAMR cells
was 1.10× that in MCF-7 cells. The effect was significantly more
pronounced in MCF-7/TAMR cells, indicating that BSAPPT exerts
pro-apoptotic effects by promoting the expression of caspase-9
(Fig. 4A).
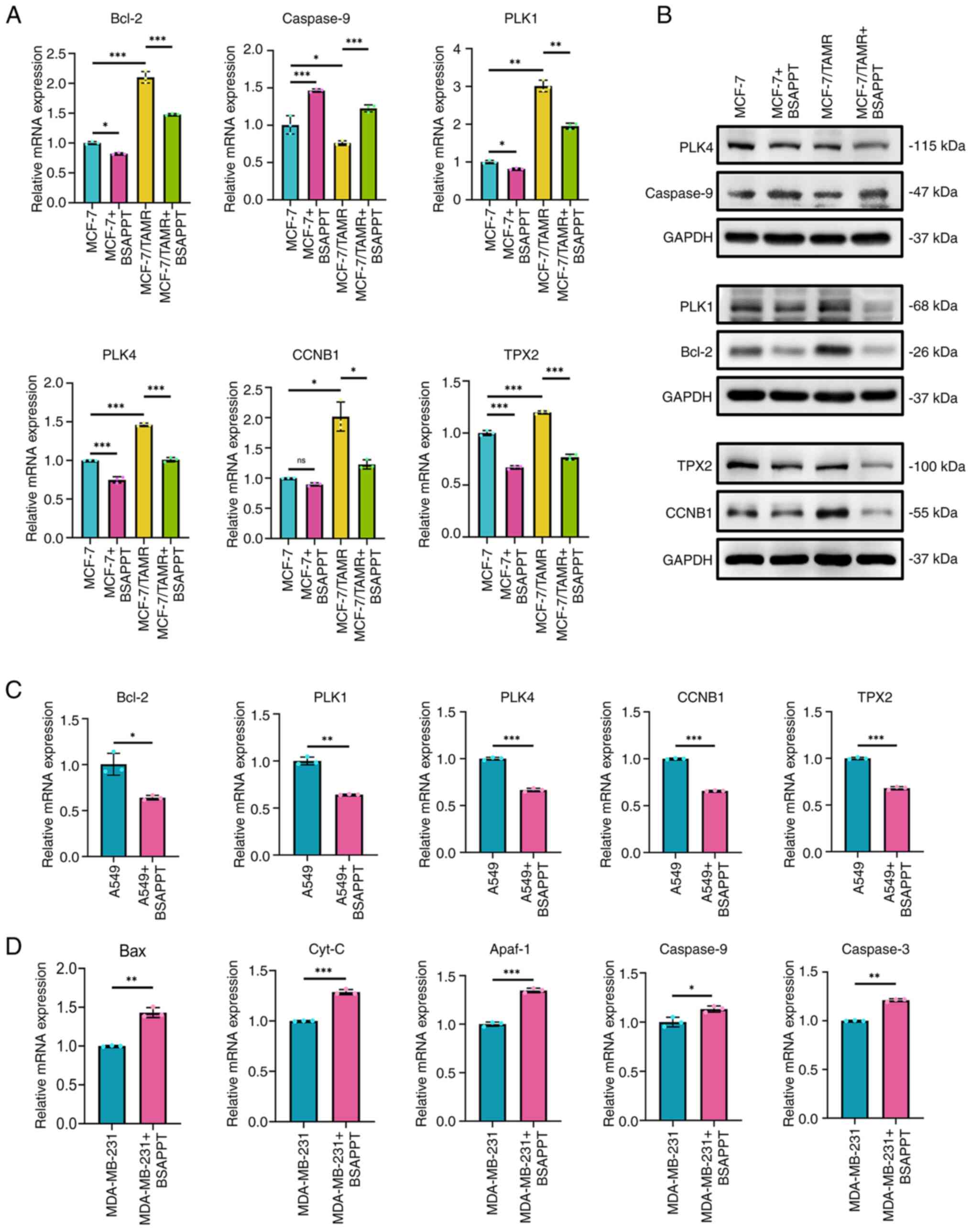 | Figure 4.Effect of BSAPPT on apoptosis and
cycle-related gene or protein expression in MCF-7, MCF7/TAMR and
other cancer cells. (A) mRNA expression levels of genes linked to
the cell cycle and apoptosis were measured by qPCR both before and
after MCF-7 and MCF7/TAMR cells were treated with 10 µg/ml BSAPPT.
(B) Western blotting detection of apoptotic and cycle-related
protein expression variations in MCF-7 and MCF7/TAMR cells before
and after using 10 µg/ml BSAPPT. Results of qPCR analysis that
assessed differences in the level of expression of genes linked to
the cell cycle and apoptosis before and after (C) A549 and (D)
MDA-MB-231 cells were treated with 10 µg/ml BSAPPT. *P<0.05;
**P<0.01; ***P<0.001. BSAPPT, bromosulfonamidine
amino-podophyllotoxin; qPCR, quantitative PCR; Bcl-2, B-cell
lymphoma 2; Caspase, cysteine aspartic acid-specific protease; PLK,
polo like kinase; CCNB1, cyclin B1; TPX2, targeting protein for
Xklp2; Bax, Bcl-2 associated X; Cyt-C, cytochrome c; Apaf-1,
apoptotic protease activating factor 1. |
PLK1, PLK4, CCNB1 and TPX2 are all related genes
that regulate the cell cycle: PLK1 is essential for the start,
progression and termination of mitosis (44); PLK4 regulates centromere replication
during the cell cycle (45); the
protein that CCNB1 encodes needs to exist for the proper regulation
of the G2/M transition point during the cell cycle
(46); and TPX2 is selectively
present in the nucleus throughout the S and G2 periods
inside the cell cycle and is regarded as a critical component
connected with cell mitosis and spindle construction (46). These cycle-related genes were
significantly more expressed in MCF-7/TAMR cells compared with
MCF-7 cells, and both MCF-7 and MCF-7/TAMR cells demonstrated a
significant reduction in the expression levels of these genes with
BSAPPT administration compared with the respective untreated
control group. Nonetheless, the reduced expression of these genes
was significantly more pronounced in MCF-7/TAMR cells than MCF-7
cells (Fig. 4A).
Subsequently, the influence of BSAPPT on the protein
levels of Bcl-2, Caspase-9, PLK1, PLK4, CCNB1, and TPX2 were
assessed using western blotting. Following 48 h of BSAPPT
administration, it was revealed that the protein expression degree
of the apoptosis-inhibiting gene, Bcl-2, was markedly increased in
MCF-7/TAMR cells compared with that in MCF-7 cells, and in both the
MCF-7 and MCF7/TAMR cells, BSAPPT notably reduced Bcl-2 protein
expression in treated cells compared with that in respective
untreated cells (Fig. 4B);
Caspase-9 protein expression was reduced in MCF-7/TAMR cells
compared with that in MCF-7 cells, and BSAPPT administration was
associated with a marked increase in expression of Caspase-9 among
the treated group compared with that in the respective untreated
group in both the MCF-7 and MCF-7/TAMR cells (Fig. 4B); MCF-7/TAMR cells demonstrated
notably elevated levels of PLK1 and CCNB1 protein expression
compared with MCF-7 cells, and BSAPPT treatment was associated with
a notably reduced levels of PLK1, PLK4, CCNB1 and TPX2 protein
expression in treated cells compared with that in respective
untreated cells in both MCF-7 and MCF7/TAMR cells (Fig. 4B).
Upon assessing the molecular mechanism of BSAPPT in
other tumor cell lines, it was observed that BSAPPT administration
was associated with a significant reduction in the relative gene
expression of Bcl-2, PLK1, PLK4, CCNB1 and TPX2 in A549 cells to
63.77, 64.29, 66.60, 65.82 and 68.00, respectively, compared with
untreated groups (all P<0.05; Fig.
4C). Bcl-2 associated X (Bax) is an apoptosis-promoting gene
and a water-soluble related protein homolog of Bcl-2, belonging to
the Bcl-2 gene family. It undermines the protective function of
Bcl-2, often resulting in cell death and the emergence of
cytochrome c (Cyt C) (47).
Cyt-C is a key molecule involved in apoptosis. When cells are
stimulated by apoptotic signals, released Cyt-C from the
mitochondria forms an apoptotic complex with apoptotic protease
activating factor (Apaf)-1 protein with the assistance of
deoxyadenosine triphosphate and the caspase-recruiting structural
domain at the amino-terminus of Apaf-1 in the apoptotic complex
will recruit Pro-Caspase-9 to form activated Caspase-9. Apoptosis
is induced by Caspase-3, which is activated by Caspase-9 (43,48,49).
In the present study, BSAPPT administration was associated with a
significant increase in the relative levels of gene expression of
Caspase-9, Cyt-C, Apaf-1, Caspase-3 and Bax in MDA-MB-231 cells by
1.13-, 1.29-, 1.35-, 1.21- and 1.43-fold, respectively, compared
with untreated cells (all P<0.05; Fig. 4D).
Discussion
Most patients with ER+ breast cancer
receive endocrine therapy, including TAM, and although certain
patients respond well to TAM, the risk of resistance and recurrence
remains (6). PPT has been of
interest for its broad-spectrum and highly potent antitumor,
antiviral, antibacterial and immunosuppressive activities, as well
as its antiproliferative activity in many types of cancers
(36), but no studies related to
PPT drugs targeting MCF-7/TAMR cells have been reported, to the
best of our knowledge. Although semi-synthetic derivatives of PPT
have antineoplastic properties, their clinical use is limited due
to their high toxicity, increased cancer cell resistance,
uncontrolled release, poor water solubility, low bioavailability
and increased myelosuppression and cytotoxicity to normal human
cells (50,51). To improve the efficiency and lower
the side effects of PPT, more PPT-based derivatives were developed
(52). The present study
investigated a completely new, unstudied derivative, BSAPPT, and
the demonstrated that BSAPPT reverses the malignant phenotype of
MCF-7/TAMR cells by inhibiting their proliferation.
It was revealed that BSAPPT exhibited a
concentration-dependent effect on the inhibition of MCF-7 and
MCF-7/TAMR cell development, and the degree of injury to MCF-10A
cells was less than that in MCF-7 and MCF-7/TAMR cells.
Furthermore, the results demonstrated that the extent of injury to
MCF-10A cells by etoposide was greater than that by BSAPPT.
Therefore, the small degree of injury caused by BSAPPT to normal
human cells is advantageous. In both MCF-7 and MCF-7/TAMR cells,
BSAPPT effectively increased the rate of apoptosis, with a greater
degree of apoptosis in the MCF-7/TAMR cells. This concurs with a
previous report stating that the main molecular mechanism
underlying the antineoplastic effect of ghrelin included the
trigger of apoptosis (20), which
is characterized by nuclear chromatin condensation and
fragmentation, dense cytoplasmic organelles, endoplasmic reticulum
expansion and phagocytosis of apoptotic cells (43). Moreover, the G2/M stage
of the MCF-7/TAMR cell cycle was inhibited by BSAPPT, thereby
inducing the apoptosis of MCF-7/TAMR cells and inhibiting the
development of MCF-7/TAMR, which agrees with results from a
previous study that reported that, in a dose-dependent manner, PPT
may stop SGC-7901 cells from proliferating and trigger their
apoptosis, resulting in cell cycle block during the G2/M
phase (53). The inhibition by
BSAPPT in the MCF-7 cell cycle was not as significant as that in
the MCF-7/TAMR cells. Additionally, it was demonstrated that BSAPPT
notably decreased the rate of cell division by promoting apoptosis
in several types of cells, including MDA-MB-231 and A549 cells.
The Bcl-2-regulated apoptotic pathway is associated
with carcinogenesis and could be a useful target for future
medication progress (47). The
present research demonstrated that drug-resistant cells express
Bcl-2 at high levels, and the downregulation of Bcl-2 promoted
apoptosis in MCF-7 and MCF7/TAMR cells after BSAPPT therapy. This
agrees with the findings of a previous study that reported that a
critical variation in the acquisition of TAM resistance in
MCF-7/TAMR is the promotion of proliferation and reduction of
apoptosis through Bcl-2 regulation (54). The results are also in concordance
with a previous study that reported that overexpression of the
Bcl-2 protein was associated with several malignancies, including
breast cancer (55). Furthermore,
the present research demonstrated that MCF-7/TAMR cells expresses
caspase-9 at low levels, and the activation of caspase-9 can
promote apoptosis in MCF-7 and MCF-7/TAMR cells with BSAPPT
treatment. This agrees with the findings of a previous study which
reported that inhibition of caspase-9 led to attenuation of the
apoptosis, resulting in increased migration, proliferation and
invasion of breast cancer cells (56). Lastly, the activation of the caspase
family and effector caspases is caused by certain
apoptosis-inducing stimuli (57).
Caspase-9 of the intrinsic or mitochondrial apoptotic pathway is a
protease that initiates apoptosis and is triggered by a
multi-protein triggering platform, which later induces apoptotic
signals through the activation of caspase-3 and −7, and then their
role triggers apoptosis in many cell lines (43).
Cell cycle proteins are vital for regulating the
length of the cell cycle (58).
According to results of the present research, MCF-7/TAMR cells
displayed increased protein expressions of PLK1 and CCNB1. The
decreased protein expressions of PLK1, CCNB1, PLK4 and TPX2 were
associated with the action of BSAPPT, which is consistent with the
findings of a previous study that reported that the overexpression
of PLK1 and CCNB1 was associated with a reduced survival rate of
patients with breast cancer, indicating their potentiality as
prognostic markers (59).
Meanwhile, a mitogenic protein, CCNB1, is an essential cell cycle
regulator at the G2/M checkpoint and serves a role in
the oncogene route of several malignancies, including colon cancer
and breast cancer (46). A previous
study indicated that the level of expression of PLK1 and PLK4 was
greater within breast cancer tissue compared with healthy normal
tissues, and increased levels of PLK1 and PLK4 in breast malignant
tissue induced the stimulation of breast cancer carcinogenesis;
however, their association with breast cancer-resistant cells was
not studied (60). The level of
expression of TPX2 is tightly regulated by the cell cycle (61); however, a previous study reported
that TPX2 was markedly increased in different stages of breast
cancer, and the low overall survival of many patients with cancer
is associated with its excessive expression (46). These cycle proteins block the
progression of the cell cycle of MCF-7 and MCF-7/TAMR cells,
leading to proliferation inhibition of MCF-7 and MCF-7/TAMR cells.
Otherwise, the degree of blockage in the MCF-7/TAMR cells was
greater than that in the MCF-7 cells. Moreover, the present study
revealed that the mRNA levels of Bcl-2, PLK1, PLK4, CCNB1 and TPX2
in A549 cells were significantly decreased following BSAPPT
administration, which is consistent with the trends of mRNA
expression within MCF-7 and MCF-7/TAMR cells, when treated cells
are compared with untreated cells. However, MDA-MB-231 cells had
higher mRNA levels of Bax, Cyt-C, Apaf-1, Caspase-9 and Caspase-3
in drug-treated cells compared with untreated cells, which may be
because the MDA-MB-231 cells underwent apoptosis due to an
alteration in the Cyt-C/Apaf-1/Caspase-9/Caspase-3 route
activation. This agrees with a previous study, which reported that
within human multiple myeloma cells, the
Cyt-C/Apaf-1/Caspase-9/Caspase-3 signaling cascade caused cell
death (48). Overall, the results
of the present research indicate that BSAPPT causes apoptosis and
G2/M stage cell cycle block, which therefore suppresses
the growth of MCF-7/TAMR cells in a dose-dependent manner.
In summary, the creation of a novel class of
PPT-derived compounds with enhanced cytotoxicity and selectivity
against cancer cells is motivated by the adverse effects and
broad-spectrum anticancer capabilities of PPT as an antineoplastic
drug (36). The present study
demonstrated that BSAPPT promotes apoptosis in TAM-resistant breast
cancer cells by downregulating Bcl-2, upregulating Caspase-9 and
inhibiting the cell cycle of MCF-7/TAMR cells through the
downregulation of PLK1, PLK4, CCNB1 and TPX2, which inhibits the
development of breast cancer tumors. This indicates that BSAPPT
exerts a suppressive role in TAM resistance in breast tumors,
thereby revealing potential for the inhibition and control of
breast cancer involving TAM resistance as well as providing
strategies for the therapy of other cancers; however, more in-depth
investigations are needed to verify this. Moreover, it is uncertain
if BSAPPT acts on the colchicine binding site on microtubule
proteins to prevent microtubule proteins from assembling into
mitotic spindle microtubules. This is a major limitation of the
present study, and further investigations should be performed
related to microtubule proteins.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Guangzhou and Dongguan
Joint Fund Cultivation Project (grant nos. 2021B1515140002 and
2023A1515140033) and the PhD Research Start-Up Fund Project (grant
no. DBBS2023001).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YZ conceived and designed the study. FL performed
the drug screening and cycle experiments. JW was the primary
contributor to the composition of the manuscript, whilst also
performing all other experiments and data analysis. XL and BZ
participated in the design of the study and reviewed the work
critically for important intellectual content. JW, XL and BZ
confirm the authenticity of all the raw data. All authors have read
and approved the final version of the manuscript. All authors
agreed to be accountable for all aspects of the work in ensuring
that questions related to the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shen Y, Zhong J, Liu J, Liu K, Zhao J, Xu
T, Zeng T, Li Z, Chen Y, Ding W, et al: Protein arginine
N-methyltransferase 2 reverses tamoxifen resistance in breast
cancer cells through suppression of ER-α36. Oncol Rep.
39:2604–2612. 2018.PubMed/NCBI
|
|
2
|
Mishra A, Srivastava A, Pateriya A, Tomar
MS, Mishra AK and Shrivastava A: Metabolic reprograming confers
tamoxifen resistance in breast cancer. Chem Biol Interact.
347:1096022021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jin ML, Kim YW, Jin HL, Kang H, Lee EK,
Stallcup MR and Jeong KW: Aberrant expression of SETD1A promotes
survival and migration of estrogen receptor α-positive breast
cancer cells. Int J Cancer. 143:2871–2883. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ring A and Dowsett M: Mechanisms of
tamoxifen resistance. Endocr Relat Cancer. 11:643–658. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shi Q, Li Y, Li S, Jin L, Lai H, Wu Y, Cai
Z, Zhu M, Li Q, Li Y, et al: LncRNA DILA1 inhibits Cyclin D1
degradation and contributes to tamoxifen resistance in breast
cancer. Nat Commun. 11:55132020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhu Y, Liu Y, Zhang C, Chu J, Wu Y, Li Y,
Liu J, Li Q, Li S, Shi Q, et al: Tamoxifen-resistant breast cancer
cells are resistant to DNA-damaging chemotherapy because of
upregulated BARD1 and BRCA1. Nat Commun. 9:15952018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lüönd F, Sugiyama N, Bill R, Bornes L,
Hager C, Tang F, Santacroce N, Beisel C, Ivanek R, Bürglin T, et
al: Distinct contributions of partial and full EMT to breast cancer
malignancy. Dev Cell. 56:3203–3221.e11. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tufail M, Cui J and Wu C: Breast cancer:
Molecular mechanisms of underlying resistance and therapeutic
approaches. Am J Cancer Res. 12:2920–2949. 2022.PubMed/NCBI
|
|
10
|
Yin L, Zhang XT, Bian XW, Guo YM and Wang
ZY: Disruption of the ER-α36-EGFR/HER2 positive regulatory loops
restores tamoxifen sensitivity in tamoxifen resistance breast
cancer cells. PLoS One. 9:e1073692014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hosford SR and Miller TW: Clinical
potential of novel therapeutic targets in breast cancer: CDK4/6,
Src, JAK/STAT, PARP, HDAC, and PI3K/AKT/mTOR pathways.
Pharmgenomics Pers Med. 7:203–215. 2014.PubMed/NCBI
|
|
12
|
Li D, Ji H, Niu X, Yin L, Wang Y, Gu Y,
Wang J, Zhou X, Zhang H and Zhang Q: Tumor-associated macrophages
secrete CC-chemokine ligand 2 and induce tamoxifen resistance by
activating PI3K/Akt/mTOR in breast cancer. Cancer Sci. 111:47–58.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mansouri S, Farahmand L, Teymourzadeh A
and Majidzadeh AK: Clinical evidence on the magnitude of change in
growth pathway activity in relation to tamoxifen resistance is
required. Curr Cancer Drug Targets. 18:668–676. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Viedma-Rodriguez R, Baiza-Gutman L,
Salamanca-Gomez F, Diaz-Zaragoza M, Martinez-Hernandez G, Ruiz
Esparza-Garrido R, Velázquez-Flores MA and Arenas-Aranda D:
Mechanisms associated with resistance to tamoxifen in estrogen
receptor-positive breast cancer (review). Oncol Rep. 32:3–15. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gao A, Sun T, Ma G, Cao J, Hu Q, Chen L,
Wang Y, Wang Q, Sun J, Wu R, et al: LEM4 confers tamoxifen
resistance to breast cancer cells by activating cyclin D-CDK4/6-Rb
and ERα pathway. Nat Commun. 9:41802018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cai F, Xiao H, Sun Y, Wang D and Tang J:
Expression of Snail and E-cadherin in Drug-resistant MCF-7/ADM
breast cancer cell strains. J Coll Physicians Surg Pak. 29:240–244.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vesuna F, Bergman Y and Raman V: Genomic
pathways modulated by Twist in breast cancer. BMC Cancer.
17:522017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Joseph C, Alsaleem M, Orah N, Narasimha
PL, Miligy IM, Kurozumi S, Ellis IO, Mongan NP, Green AR and Rakha
EA: Elevated MMP9 expression in breast cancer is a predictor of
shorter patient survival. Breast Cancer Res Treat. 182:267–282.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shah Z, Gohar UF, Jamshed I, Mushtaq A,
Mukhtar H, Zia-Ui-Haq M, Toma SI, Manea R, Moga M and Popovici B:
Podophyllotoxin: History, recent advances and future prospects.
Biomolecules. 11:6032021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hong WG, Cho JH, Hwang SG, Lee E, Lee J,
Kim JI, Um HD and Park JK: Chemosensitizing effect of
podophyllotoxin acetate on topoisomerase inhibitors leads to
synergistic enhancement of lung cancer cell apoptosis. Int J Oncol.
48:2265–2276. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guerram M, Jiang ZZ and Zhang LY:
Podophyllotoxin, a medicinal agent of plant origin: Past, present
and future. Chin J Nat Med. 10:161–169. 2012. View Article : Google Scholar
|
|
22
|
Xiao J, Gao M, Sun Z, Diao Q, Wang P and
Gao F: Recent advances of podophyllotoxin/epipodophyllotoxin
hybrids in anticancer activity, mode of action, and
structure-activity relationship: An update (2010–2020). Eur J Med
Chem. 208:1128302020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ma Y, Fang S, Li H, Han C, Lu Y, Zhao Y,
Liu Y and Zhao C: Biological evaluation and molecular modelling
study of podophyllotoxin derivatives as potent inhibitors of
tubulin polymerization. Chem Biol Drug Des. 82:12–21. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang X, Rakesh KP, Shantharam CS,
Manukumar HM, Asiri AM, Marwani HM and Qin HL: Podophyllotoxin
derivatives as an excellent anticancer aspirant for future
chemotherapy: A key current imminent needs. Bioorg Med Chem.
26:340–355. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Renouard S, Lopez T, Hendrawati O, Dupre
P, Doussot J, Falguieres A, Ferroud C, Hagege D, Lamblin F, Laine E
and Hano C: Podophyllotoxin and deoxypodophyllotoxin in Juniperus
bermudiana and 12 other Juniperus species: Optimization of
extraction, method validation, and quantification. J Agric Food
Chem. 59:8101–8107. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yu X, Xu T, Su B, Zhou J, Xu B, Zhang Y,
Zhu Y, Jiang N and He Z: The novel role of etoposide in inhibiting
the migration and proliferation of small cell lung cancer and
breast cancer via targeting Daam1. Biochem Pharmacol.
210:1154682023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Orr MS, Fornari FA, Randolph JK and
Gewirtz DA: Transcriptional down-regulation of c-myc expression in
the MCF-7 breast tumor cell line by the topoisomerase II inhibitor,
VM-26. Biochim Biophys Acta. 1262:139–145. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sledge GW Jr: Etoposide in the management
of metastatic breast cancer. Cancer. 67 (Suppl 1):S266–S270. 1991.
View Article : Google Scholar
|
|
29
|
Cabel L, Carton M, Cheaib B, Pierga JY,
Dalenc F, Mailliez A, Levy C, Jacot W, Debled M, Leheurteur M, et
al: Oral etoposide in heavily pre-treated metastatic breast cancer:
Results from the ESME cohort and comparison with other chemotherapy
regimens. Breast Cancer Res Treat. 173:397–406. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Alpsoy A, Yasa S and Gündüz U: Etoposide
resistance in MCF-7 breast cancer cell line is marked by multiple
mechanisms. Biomed Pharmacother. 68:351–355. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hartmann JT and Lipp HP: Camptothecin and
podophyllotoxin derivatives: Inhibitors of topoisomerase I and
II-mechanisms of action, pharmacokinetics and toxicity profile.
Drug Saf. 29:209–230. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Allen TM and Cullis PR: Drug delivery
systems: Entering the mainstream. Science. 303:1818–1822. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Carstensen H, Nolte H and Hertz H:
Teniposide-induced hypersensitivity reactions in children. Lancet.
2:551989. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chu B, Shi S, Li X, Hu L, Shi L, Zhang H,
Xu Q, Ye L, Lin G, Zhang N and Zhang X: Preparation and evaluation
of teniposide-loaded polymeric micelles for breast cancer therapy.
Int J Pharm. 513:118–129. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nielsen D, Boas J, Engelholm SA, Hansen OP
and Dombernowsky P: Teniposide in advanced breast cancer. A phase
II trial in patients with no prior chemotherapy. Ann Oncol.
3:377–378. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zi CT, Yang L, Xu FQ, Dong FW, Yang D, Li
Y, Ding ZT, Zhou J, Jiang ZH and Hu JM: Synthesis and anticancer
activity of dimeric podophyllotoxin derivatives. Drug Des Devel
Ther. 12:3393–3406. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pujol MD, Romero M and Sánchez I:
Synthesis and biological activity of new class of dioxygenated
anticancer agents. Curr Med Chem Anticancer Agents. 5:215–237.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang TM, Qi SN, Zhao N, Yang YJ, Yuan HQ,
Zhang B and Jin S: Induction of apoptosis through
caspase-independent or caspase-9-dependent pathway in mouse and
human osteosarcoma cells by a new nitroxyl spin-labeled derivative
of podophyllotoxin. Apoptosis. 18:727–738. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Siegel RL, Miller KD, Wagle NS and Jemal
A: Cancer statistics, 2023. CA Cancer J Clin. 73:17–48. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Han HW, Lin HY, He DL, Ren Y, Sun WX,
Liang L, Du MH, Li DC, Chu YC, Yang MK, et al: Novel
podophyllotoxin derivatives as potential tubulin inhibitors:
Design, synthesis, and antiproliferative activity evaluation. Chem
Biodivers. 15:e18002892018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Qin X, Zhang Y, Yu H and Ma L: Progress in
the Study of spindle assembly checkpoint in lung cancer. Zhongguo
Fei Ai Za Zhi. 26:310–318. 2023.(In Chinese). PubMed/NCBI
|
|
43
|
Kim B, Srivastava SK and Kim SH: Caspase-9
as a therapeutic target for treating cancer. Expert Opin Ther
Targets. 19:113–127. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu Z, Sun Q and Wang X: PLK1, A potential
target for cancer therapy. Transl Oncol. 10:22–32. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhao Y and Wang X: PLK4: A promising
target for cancer therapy. J Cancer Res Clin Oncol. 145:2413–2422.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen G, Yu M, Cao J, Zhao H, Dai Y, Cong Y
and Qiao G: Identification of candidate biomarkers correlated with
poor prognosis of breast cancer based on bioinformatics analysis.
Bioengineered. 12:5149–5161. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yamaguchi H, Paranawithana S, Lee M, Huang
Z, Bhalla K, Wang HG. Yamaguchi H, Paranawithana SR, Lee MW, Huang
Z, et al: Epothilone B analogue (BMS-247550)-mediated cytotoxicity
through induction of Bax conformational change in human breast
cancer cells. Cancer Res. 62:466–471. 2002.PubMed/NCBI
|
|
48
|
Li Z, Guo D, Yin X, Ding S, Shen M, Zhang
R, Wang Y and Xu R: Zinc oxide nanoparticles induce human multiple
myeloma cell death via reactive oxygen species and
Cyt-C/Apaf-1/Caspase-9/Caspase-3 signaling pathway in vitro. Biomed
Pharmacother. 122:1097122020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yadav N, Gogada R, O'Malley J, Gundampati
RK, Jayanthi S, Hashmi S, Lella R, Zhang D, Wang J, Kumar R, et al:
Molecular insights on cytochrome c and nucleotide regulation of
apoptosome function and its implication in cancer. Biochim Biophys
Acta Mol Cell Res. 1867:1185732020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li M, Zhao Y, Sun J, Chen H, Liu Z, Lin K,
Ma P, Zhang W, Zhen Y and Zhang S and Zhang S: pH/reduction
dual-responsive hyaluronic acid-podophyllotoxin prodrug micelles
for tumor targeted delivery. Carbohydr Polym. 288:1194022022.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Paidakula S, Nerella S, Vadde R, Kamal A
and Kankala S: Design and synthesis of
4β-Acetamidobenzofuranone-podophyllotoxin hybrids and their
anti-cancer evaluation. Bioorg Med Chem Lett. 29:2153–2156. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhao W, Cong Y, Li HM, Li S, Shen Y, Qi Q,
Zhang Y, Li YZ and Tang YJ: Challenges and potential for improving
the druggability of podophyllotoxin-derived drugs in cancer
chemotherapy. Nat Prod Rep. 38:470–488. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ji CF and Ji YB: Apoptosis of human
gastric cancer SGC-7901 cells induced by podophyllotoxin. Exp Ther
Med. 7:1317–1322. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Jeong D, Ham J, Kim HW, Kim H, Ji HW, Yun
SH, Park JE, Lee KS, Jo H, Han JH, et al: ELOVL2: A novel tumor
suppressor attenuating tamoxifen resistance in breast cancer. Am J
Cancer Res. 11:2568–2589. 2021.PubMed/NCBI
|
|
55
|
Dawson SJ, Makretsov N, Blows FM, Driver
KE, Provenzano E, Le Quesne J, Baglietto L, Severi G, Giles GG,
McLean CA, et al: BCL2 in breast cancer: A favourable prognostic
marker across molecular subtypes and independent of adjuvant
therapy received. Br J Cancer. 103:668–675. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhang L, Zhang X, Wang X, He M and Qiao S:
MicroRNA-224 promotes tumorigenesis through downregulation of
caspase-9 in triple-negative breast cancer. Dis Markers.
2019:73789672019.PubMed/NCBI
|
|
57
|
Frenzel A, Labi V, Chmelewskij W, Ploner
C, Geley S, Fiegl H, Tzankov A and Villunger A: Suppression of
B-cell lymphomagenesis by the BH3-only proteins Bmf and Bad. Blood.
115:995–1005. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Goodger NM, Gannon J, Hunt T and Morgan
PR: Cell cycle regulatory proteins-an overview with relevance to
oral cancer. Oral Oncol. 33:61–73. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Fang L, Liu Q, Cui H, Zheng Y and Wu C:
Bioinformatics analysis highlight differentially expressed CCNB1
and PLK1 genes as potential anti-breast cancer drug targets and
prognostic markers. Genes (Basel). 13:6542022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Jiawei W, Xiajun B, Tian S, Xuzheng G and
Zhenwang Z: Comprehensive analysis of PLKs expression and prognosis
in breast cancer. Cancer Genet. 268–269. 83–92. 2022.PubMed/NCBI
|
|
61
|
Ma S, Rong X, Gao F, Yang Y and Wei L:
TPX2 promotes cell proliferation and migration via PLK1 in OC.
Cancer Biomark. 22:443–451. 2018. View Article : Google Scholar : PubMed/NCBI
|