Introduction
Kidney cancer has been one of the top 10 most common
types of cancer worldwide for a number of years (1). The incidence of malignant kidney
tumors accounts for ~2% of all cancer cases worldwide. Due to
various histological and genetic mutations, there are several
subtypes of renal cancer, and kidney renal clear cell carcinoma
(KIRC) is the most prevalent subtype, accounting for 80% of adult
clinical cases worldwide (2,3). The
most typical scenario in KIRC is VHL gene mutation or methylation.
The tumor suppressor gene VHL encoded by chromosomal arm 3p is
widely lost in KIRC (4,5). Patients with VHL mutations have a
greater lifetime risk of recurrent cancer, along with an increased
risk of tumor development in several organs (2). Notably, to the best of our knowledge,
no genetic biomarker has been identified as a viable predictor of
KIRC prognosis or treatment outcome; therefore, efforts should be
made to develop effective biomarkers.
The mediator complex (MED) gene family forms a
multiprotein complex that connects transcription factors to RNA
polymerase II, which extensively participates in the proliferation,
differentiation and migration of cells (6). Notably, the MED is a protein complex
that has evolved to mediate specific protein-protein interactions
(PPIs) (7). The MED is
evolutionarily conserved and has a similar structure in both yeast
and humans, containing a head, middle, tail and kinase module
(8). In general, the MED is
considered to be a transcriptional coactivator; however, it was
previously identified as a genetic suppressor of the Ras/MAP kinase
pathway (9). Mediator subunits
serve a role in cellular signaling pathways, including EGFR
signaling, Wnt signaling and ERK/MAPK signaling (10). Furthermore, as a highly conserved
member of the MED family, MED21 can affect PPARA-related gene
expression and metabolism (11). In
addition, loss of MED7 has been demonstrated to have a substantial
impact on numerous aspects of cellular function, including
metabolism (12). Notably, there is
a strong relationship between the MED family and cancer biology;
for example, a mechanistic investigation indicated that MED19 is
highly expressed in breast cancer tissues, and is significantly
related to larger tumors, high-grade malignant characteristics and
poor prognosis (13). Another study
also showed that most uterine leiomyomas have mutations in MED12
exon 2 (14). Furthermore, it has
been revealed that MED19 disruption prevents tongue cancer cells
from proliferating and migrating (15). In addition, MED7 has been linked to
improved long-term survival outcomes and favorable prognostic
traits in patients with breast cancer, and its expression is
increased in hepatocellular carcinoma (HCC) and is related to the
progression of HCC (16,17). Moreover, high MED21 expression
levels are associated with poor HCC prognosis, according to a
comprehensive analysis (18). The
nuclear transcription-related protein NF-κB is involved in cell
adhesion, proliferation and differentiation, and is often active in
tumor cells; according to a previous study, transcription of a
reporter gene (firefly luciferase) controlled by NF-κB can be
inhibited by MED21 RNA interference (RNAi) (19).
Although a number of research studies have been
conducted to investigate the underlying mechanisms involved in
various malignancies, to the best of our knowledge, no
investigations have focused on the therapeutic and prognostic value
of the MED family in patients with KIRC or other types of kidney
cancer. Therefore, the present study aimed to analyze the
expression and prognostic significance of MED in KIRC. The present
study used bioinformatics to examine the predictive value of the
MED gene family in KIRC and created a risk score based on public
databases. The present findings may offer novel insights into
selecting MED genes as appropriate prognostic biomarkers in
KIRC.
Materials and methods
Data acquisition and analysis
mRNA expression data and relevant clinical
information of 532 patients were collected from The Cancer Genome
Atlas (TCGA)-KIRC dataset (https://portal.gdc.cancer.gov/), and the acquisition
and application methodology followed the corresponding guidelines
and policies. R (version 4.0.3) software (https://www.r-project.org/) and R Bioconductor
packages (http://bioconductor.org/) were
utilized to examine the data. There were no ethical concerns
because these data were available online. Additionally, not all of
the information, such as overall survival data, was available for
all of the data parameters.
Construction of the PPI
The MED is a protein complex that has evolved to
mediate specific PPIs (7). PPI
network analysis was used to examine the potential interactions
between the MED genes. The Search Tool for the Retrieval of
Interacting Genes/Proteins (version 11.5) (https://cn.stringdb.org/) was used to construct a PPI
network. A total of 30 MED genes were entered into the search box.
The active interaction sources selected included text mining,
experiments, databases, co-expression, neighborhood, gene fusion
and co-occurrence; the minimal needed interaction score was set at
0.4.
Correlation analysis
For TCGA-KIRC cohort, RNA-sequencing expression
profiles and associated clinical data were obtained from TCGA
dataset. The R software package ggstatsplot (https://indrajeetpatil.github.io/ggstatsplot/)
was used to construct a two-gene correlation map. The correlation
between quantitative variables was assessed using Spearman's
correlation analysis for non-normally distributed data. P<0.05
was considered to indicate a statistically significant difference,
and correlation coefficients >0.3 were considered to indicate a
correlation. The website Assistant for Clinical Bioinformatics
(https://www.aclbi.com/static/index.html#/), which
integrates all of these features, was used for these analyses.
Functional and pathway enrichment
analyses
Using the clusterProfiler R package (https://www.bioconductor.org/packages/release/bioc/html/clusterProfiler.html),
Gene Ontology (GO) analysis was carried out to determine the
putative biological roles of the 30 MED genes. Thresholds of
P<0.05 and q<1 were set for the GO enrichment analyses.
Identification of molecular
subtypes
All of these analyses were carried out using the web
application Assistant for Clinical Bioinformatics (https://www.aclbi.com/static/index.html#/). TCGA data
were used to obtain raw RNA-sequencing data and related clinical
data, and a cluster analysis of 30 MED genes in 532 patients with
KIRC was performed. In addition, ConsensusClusterPlus (version
1.60.0) was used for consistency analysis. The number of clusters
was limited to six, and 80% of the entire sample was analyzed 100
times. Clustering heatmaps were created using the Pheatmap package
(version 1.0.12). The survival ROC program was used to construct
survival curves after analyzing prognostic differences among
distinct subgroups (groups C1, C2 and C3).
Construction of the risk assessment
model
All these analyses were performed using the web tool
called ASSISTANT for Clinical Bioinformatics that combines all
these functions (https://www.aclbi.com/static/index.html#/). The
logistic LASSO model is a shrinkage technique that positively
chooses variables from a sizable and potentially multicollinear set
in the regression, producing a set of predictors that is pertinent
and understandable. Using LASSO regression analysis, a predictive
signature was created that divided 532 patients into high-risk and
low-risk groups by calculating the customized risk score with
coefficients, and the cut-off value was the median risk score.
Kaplan-Meier survival analysis with log-rank test was then
performed to analyze the differences in survival between the
aforementioned two groups. Additionally, to compare the prediction
accuracy of each gene and risk score, a TimeROC (v 0.4) analysis
was performed.
Survival prognostic analysis
The overall survival (OS) data for MED genes in
TCGA-KIRC cohort were obtained and processed using GEPIA 2
(http://gepia2.cancer-pku.cn/). The
expression thresholds, cut-off high (50%) and cut-ff low (50%),
were used to divide 532 patients into high- and low-expression
cohorts, respectively. The log-rank test was used to analyze the OS
data. P<0.05 was considered to indicate a statistically
significant difference.
Establishment of a five-gene-based
prognostic gene signature
Univariate and multivariate analyses were performed
using the Cox regression model approach. These analyses can assess
whether the predictive gene signature could be a factor that does
not depend on other pathological or clinical factors, such as age,
sex, pathological tumor-node-metastasis (TNM) stage, and tumor
grade. P<0.05 was considered to indicate a statistically
significant difference. All independent prognostic factors
identified by multivariate Cox regression analysis were used to
construct a nomogram to explore the likelihood of 1-, 2-, 3- and
5-year OS in patients with KIRC. All of these analyses were carried
out using the Assistant for Clinical Bioinformatics web tool.
Immune infiltration analysis
The Tumor Immune Estimation Resource (TIMER;
http://timer.cistrome.org/) was used to
investigate the relationships between MED7, MED16, MED21, MED25 and
MED29 expression, and immune infiltration across TCGA-KIRC cohort.
CD8+ T cells, CD4+ T cells, B cells and
macrophages were chosen as the immune cells for analysis. The TIMER
method was used to estimate immunological infiltration. The
purity-adjusted Spearman's rank correlation test was used to
generate the estimated P-value to assess the relationships between
MED genes and invading immune cells.
Immunohistochemistry (IHC)
analysis
KIRC tissues and paired adjacent normal tissues from
10 patients (age, 37–77 years) were used for the IHC analysis. KIRC
tissue chips (cat. no. HKidE020PG01) fixed in 4% paraformaldehyde
at room temperature for 48 h and embedded in paraffin were
purchased from Shanghai Xinchao Biological Technology Co., Ltd. The
tissue chips were successively placed in xylene for 10 min,
absolute ethanol for 5 min and 75% alcohol for 5 min, and washed
with pure water. The tissue sections were placed in Tris-EDTA
antigen repair buffer (pH 9) in a microwave oven for antigen
repair. The solution was heated at medium heat for 8 min and kept
warm for 8 min before being transferred to medium and low heat for
7 min. After natural cooling, the glass slides were placed in PBS
(pH 7.2–7.4) and washed on a decolorization shaker three times for
5 min each, followed by incubation with 0.1% Triton X-100 at room
temperature for 10 min for permeabilization. The glass slides were
placed in PBS (pH 7.2–7.4) and washed on a decolorization shaker
three times for 5 min each. A total of 50 µl 5–10% normal goat
serum (cat. no. ab7481; Abcam) was added per chip for blocking
(1:19 fold dilution) at room temperature for 30 min.
Immunohistochemical staining of the paraffin-embedded tumor tissues
was performed using primary antibodies against MED7 (1;200; cat.
no. K107987P; Beijing Solarbio Science & Technology Co., Ltd.)
and MED21 (1;200; cat. no. K107312P; Beijing Solarbio Science &
Technology Co., Ltd.) at 4°C overnight, HRP-conjugated goat
anti-rabbit secondary antibodies (1:200; cat. no. GB23303; Wuhan
Servicebio Technology Co., Ltd.) at room temperature for 1 h, and
an ABC Elite immunoperoxidase kit (Wuhan Servicebio Technology Co.,
Ltd.) according to the manufacturer's instructions. Subsequently,
an optical microscope was used to examine all visible fields, and
the particles in the cell cytoplasm that were stained brown were
considered positive. IHC analysis of human samples was approved by
the Institutional Review Board of West China Second University
Hospital (ethics approval no. 2023-012; Chengdu, China). All
procedures complied with the applicable norms and regulations.
Cell culture and reagents
The 786-o cells (human clear cell adenocarcinoma
cells) were purchased from Procell Life Science & Technology
Co., Ltd., and were maintained in RPMI 1640 (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% FBS (Nanjing SenBeiJia
Biological Technology Co., Ltd.) at 37°C in an incubator containing
5% CO2.
Small interfering RNA (siRNA)
transfection and transfection efficiency verification
The corresponding siRNAs were designed to disrupt
the gene expression of MED7 and MED21 (Table I). Briefly, 786-o tumor cells
(6×105 per well) were seeded in 6-well plates the day
before, and when the cell density reached 80% on the 2nd day, 100
pmol siRNA and Lipofectamine® 2000 (cat. no. 100014469;
Invitrogen; Thermo Fisher Scientific, Inc.) mixture was added
according to the standard protocol. The culture medium was changed
after 6 h, and the plates were incubated at 37°C in an incubator
containing 5% CO2. After 48 h, other assays were performed and
TRIzol® (cat. no. 15596018CN; Invitrogen; Thermo Fisher
Scientific, Inc.) was used to extract the RNA. Subsequently, the
Evo M-MLV RT Mix Kit (cat. no. AG11728; Accurate Biology) was used
to perform reverse transcription PCR (RT-PCR) according to the
manufacturer's protocol, and then SsoFast™ EvaGreen®
Supermix (cat. no. 1725200; Bio-Rad Laboratories, Inc.) was used to
perform quantitative PCR (qPCR) according to the manufacturer's
instructions. The thermocycling conditions were as follows:
Pre-denaturation at 95°C for 5 min, followed by 40 cycles of
denaturation at 95°C for 10 sec, annealing at 60°C for 30 sec and
extension at 60°C for 30 sec. The results were analyzed using the
2−ΔΔCq method to verify the transfection efficiency
(20). The primer sequences were as
follows: MED7, forward 5′-TATTCAAGAAGGCTTAGCTCCC-3′, reverse
5′-TCATCACATTGGAACTGATTGC-3′; MED21, forward
5′-GCAGATCAGTTTTGTAATGCCA-3′, reverse 5′-AAGCAGCTGTAGATTCTTCACT-3′;
GAPDH, forward 5′-TGCACCACCAACTGCTTAGC-3′ and reverse
5′-GGCATGGACTGTGGTCATGAG-3′.
 | Table I.siRNA sequences against human MED7
and MED21 genes. |
Table I.
siRNA sequences against human MED7
and MED21 genes.
| siRNA | Sense, 5′-3′ | Antisense,
5′-3′ |
|---|
| MED7-siRNA1 |
CAAGGAAUAUACGGAUGAA(dT)(dT) |
UUCAUCCGUAUAUUCCUUG(dT)(dT) |
| MED7-siRNA2 |
CCUGGAACGAGUAAUUGAA(dT)(dT) |
UUCAAUUACUCGUUCCAGG(dT)(dT) |
| MED21-siRNA1 |
CAGGCUGCUAGCUUGUAUA(dT)(dT) |
UAUACAAGCUAGCAGCCUG(dT)(dT) |
| MED21-siRNA2 |
GGAGGAUGUUGUUUAUCGA(dT)(dT) |
UCGAUAAACAACAUCCUCC(dT)(dT) |
| siRNA-UNC |
UUCUCCGAACGUGUCACGU(dT)(dT) |
ACGUGACACGUUCGGAGAA(dT)(dT) |
Transwell assay
For the migration assay, 24-well Transwell inserts
(pore size, 8 µm; cat. no. 11310; BeiJing LABSELECT;) were used. To
the upper chamber of the Transwell inserts, 786-o tumor cells
(1×105) were plated in 200 µl FBS-free culture medium.
To the lower chamber, 600 µl culture medium containing 10% FBS was
added as a chemoattractant. After 48 h of culture in a 37°C
incubator, the cells that had migrated the lower surface of the
filters were fixed in 4% formaldehyde at room temperature for 10
min and stained with 0.1% crystal violet solution at room
temperature for 10 min, and observed under a light microscope.
Wound healing assay
Briefly, 786-o tumor cells (6×105 cells
per well) were seeded in 6-well plates the day before, and the
culture medium was supplemented with 2% FBS. When the cell
confluence reached nearly 100%, a wound was created by manually
scraping the cell monolayer with a 200-µl pipette tip. A light
microscope camera was used to visualize the changes in the wound at
0 and 24 h.
Statistical analysis
R version 4.0.3 was used for statistical analysis.
The Kaplan-Meier method was used to plot survival curves. Log-rank
tests, and multivariate and univariate Cox proportional hazards
regression analysis were used to generate P-values and hazard
ratios (HRs) with 95% confidence interval (CI) values for
Kaplan-Meier curves. Unpaired two-tailed Student's t-test was used
to analyze the data between two groups, and one-way ANOVA was used
to analyze the data among three or more groups, and Dunnett's test
was used as the post hoc multiple comparisons test. In addition,
Shapiro-Wilk test was used to assess data normality. Assays were
repeated three times. P<0.05 was considered to indicate a
statistically significant difference.
Results
Relationships within the MED gene
family
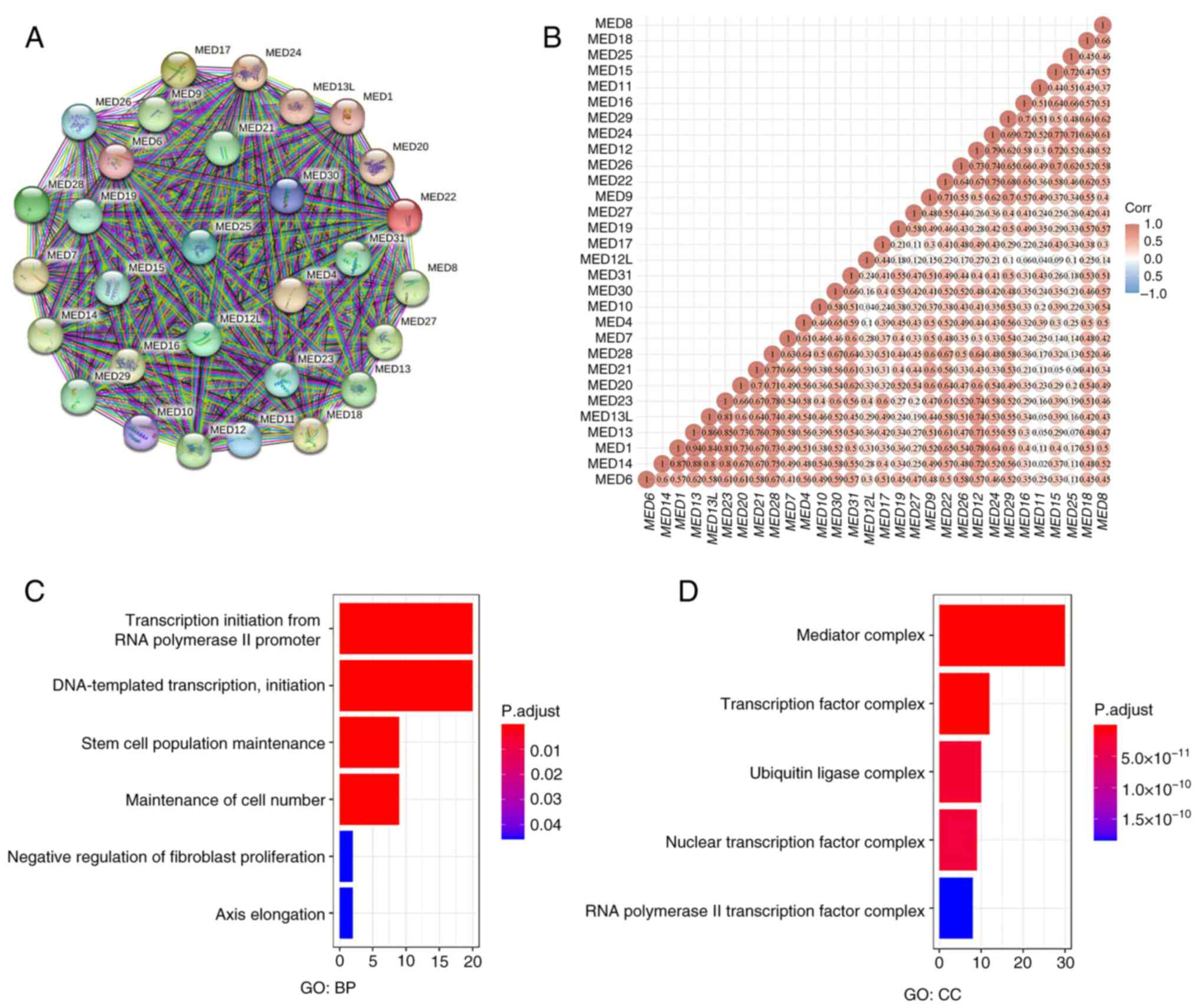
According to the PPI analysis results, the details
were as follows: Number of nodes, 30; number of edges, 435; mean
node degree, 29; PPI enrichment, P<1.0×10−16. These
numbers suggested that the MED family genes interacted strongly
(Fig. 1A). Additionally, the
relationships between MED family genes were determined by examining
their mRNA expression in KIRC with the R software package
ggstatsplot, which included Spearman's correlation analysis. The
findings revealed substantial correlations between genes, such as
between MED1 and MED13, between MED14 and MED13, and between MED13L
and MED23 (Fig. 1B). Functional
enrichment analysis revealed that ‘transcription initiation from
RNA polymerase II promoter’, ‘DNA-templated transcription,
initiation’, ‘stem cell population maintenance’ and ‘maintenance of
cell number’ were significantly related to MED family genes
(Fig. 1C). Additionally, ‘mediator
complex’ and ‘transcription factor complex’ were the most common
cellular components (Fig. 1D).
Molecular subtype of KIRC based on MED
family genes
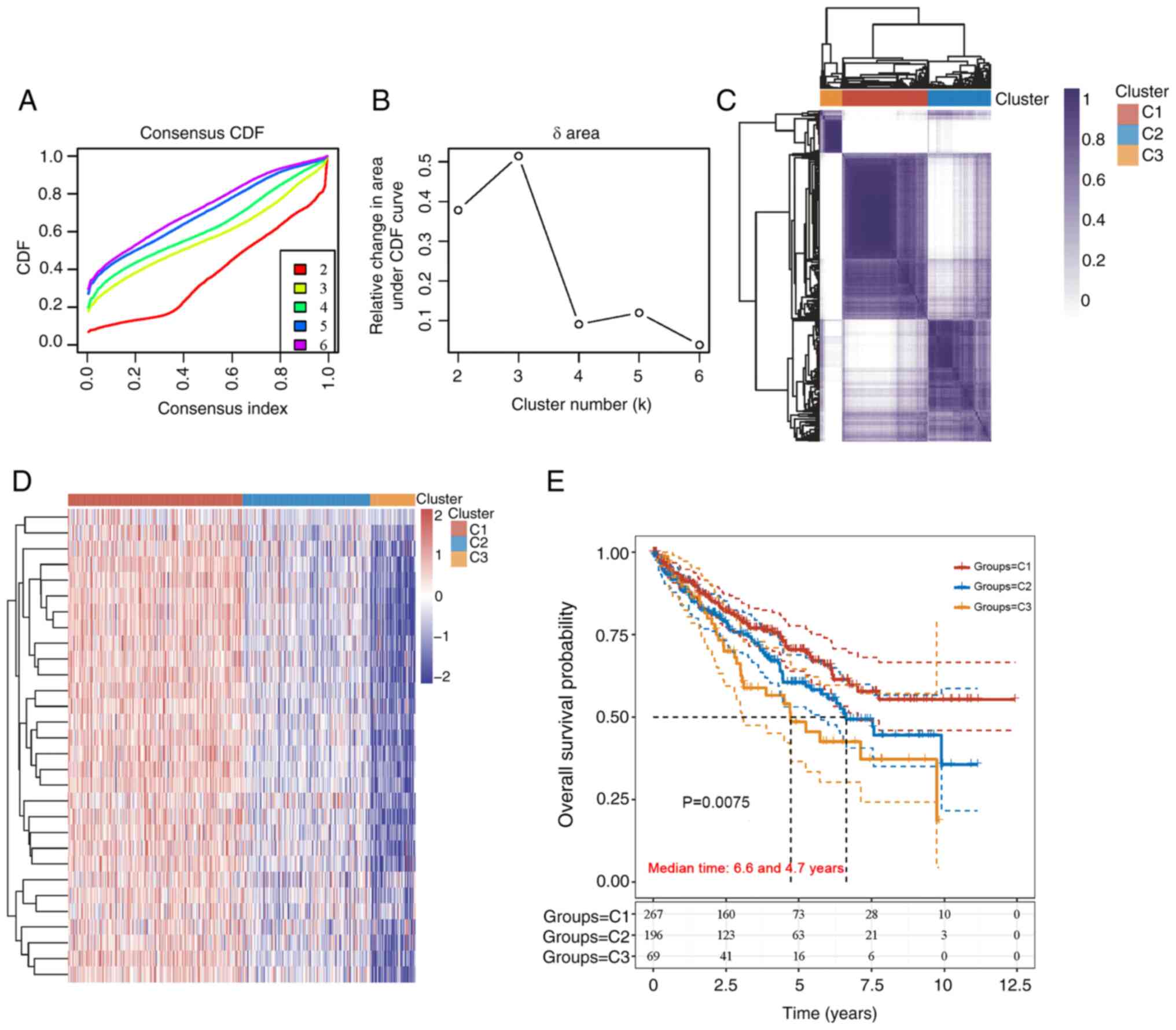
The unsupervised clustering of 532 samples from
patients with KIRC for MED family genes was carried out using
ConsensusClusterPlus software. The maximum number of clusters was
six (Fig. 2A), and the cumulative
distribution function curve of the MED family genes revealed that
k=3 was a good candidate for clustering (Fig. 2B). In addition, the heatmap of the
clustering results displayed in Fig.
2C demonstrated a relatively consistent distribution of samples
in the three clusters (C1, C2 and C3). As a result, patients with
KIRC were classified into C1, C2 and C3 subtypes. To examine the
expression of MED genes in the three subtypes, a heatmap was
created (Fig. 2D). Survival
analysis revealed a significant difference among the C1 (n=267), C2
(n=196) and C3 (n=69) subtypes. Compared with those in the C2 and
C3 subtypes, the prognosis of patients in the C1 subtype was better
(Fig. 2E).
Construction and validation of an
11-gene signature in patients with KIRC
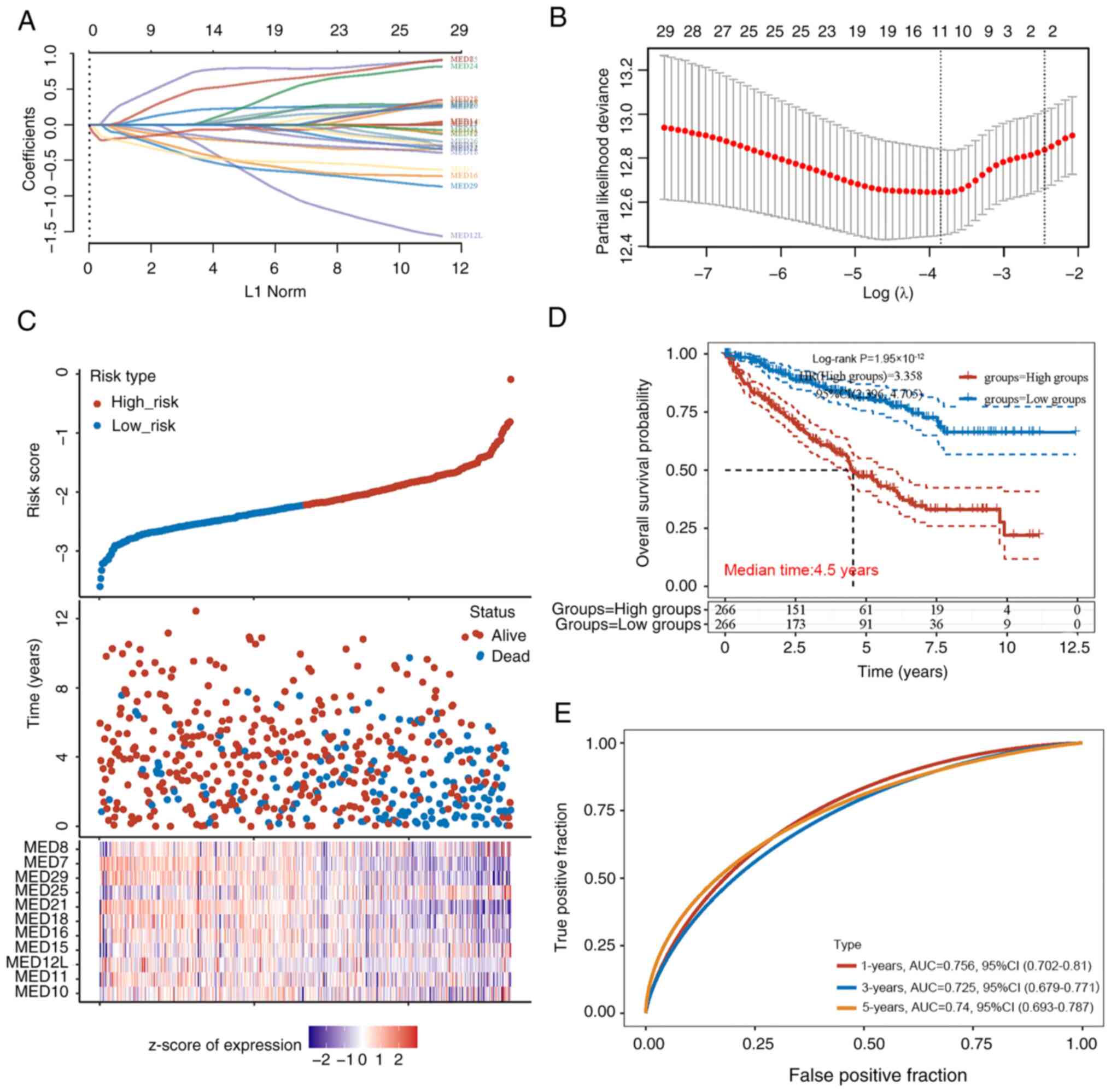
The most important prognostic genes within the MED
family were identified using the LASSO regression technique.
According to the independent variable change trajectory, there were
more independent variable coefficients with a tendency to zero as λ
steadily decreased. When variable coefficients reached zero, it
meant these variables contributed little to the model at this
point, and they can be excluded in the model (Fig. 3A). Based on this, the 10-fold
cross-validation approach was used to create a risk model and the
CI under each λ was examined (Fig.
3B). The risk model included 11 MED genes, and the details of
the formula used were as follows: Risk score=(0.2023) × MED10 +
(−0.1896) × MED11 + (−0.2343) × MED12L + (0.0053) × MED15 + (−0.38)
× MED16 + (−0.1433) × MED18 + (−0.0739) × MED21 + (0.7374) × MED25
+ (−0.4641) × MED29 + (−0.4498) × MED7 + (0.4838) × MED8. The risk
score of each patient with KIRC was calculated, and based on the
median risk score (cut-off value, −2.2), the patients were divided
into two groups: Low-risk (n=266) and high-risk (n=266). The
distribution of the 11 genes in all the samples showed that
patients in the high-risk group had higher levels of MED25
expression. The individuals in the low-risk group, however, were
more likely to express MED7 and MED21 (Fig. 3C). According to the Kaplan-Meier
analysis results of all patients, there was a marked difference
between the low-risk and high-risk groups. Notably, the prognosis
of the low-risk group was significantly better than that of the
high-risk group; the median survival time of the high-risk group
was 4.5 years (Fig. 3D). The area
under the curve values for the 11 genes in the survival assessment
model were 0.756 at 1 year, 0.725 at 3 years, and 0.74 at 5 years
of OS (Fig. 3E).
Clinical prognostic value of a
five-gene signature in KIRC patients
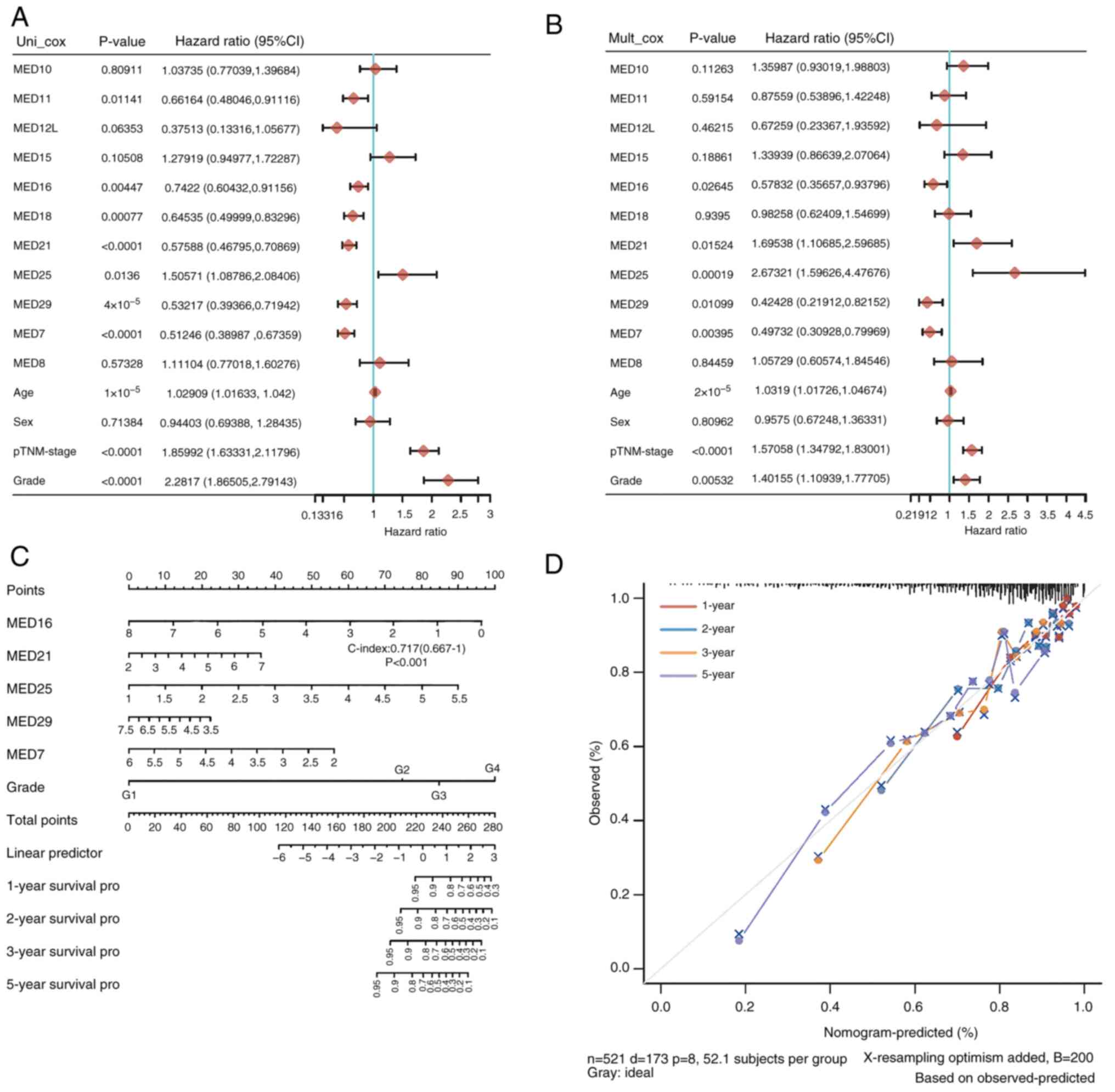
The prognostic role of MED family genes in KIRC was
subsequently explored. The findings showed that the prognosis of
patients with KIRC was strongly associated with MED11, MED16,
MED18, MED21, MED25, MED29 and MED7 expression. As shown in the
forest plots, of all the factors, MED11 (HR=0.66164), MED16
(HR=0.7422), MED18 (HR=0.64535), MED21 (HR=0.57588), MED25
(HR=1.50571), MED29 (HR=0.53217) and MED7 (HR=0.51246) were
significantly associated with the survival of patients with KIRC
(Fig. 4A). Subsequently,
multivariate Cox regression analysis was used to further evaluate
the 11 genes and other clinical characteristics in patients with
KIRC to identify the independent prognostic factors. MED16
(HR=0.57832), MED21 (HR=1.69538), MED25 (HR=2.67321), MED29
(HR=0.42428) and MED7 (HR=0.49732) were revealed to be independent
risk factors for the prognosis of patients with KIRC (Fig. 4B). A nomogram was constructed with
MED16, MED21, MED25, MED29 and MED7. The risk model based on MED16,
MED21, MED25, MED29, and MED7 had good performance in predicting
the prognosis of patients with KIRC, as indicated by the C-index of
this model, which was 0.717 (Fig.
4C). The anticipated survival rate was close to the actual
survival rate according to the 1-, 2-, 3- and 5-year nomograms
(Fig. 4D). These results indicated
that the five-gene signature comprising MED16, MED21, MED25, MED29
and MED7 may be useful for predicting the development of KIRC.
Prognostic features of mRNA
expression
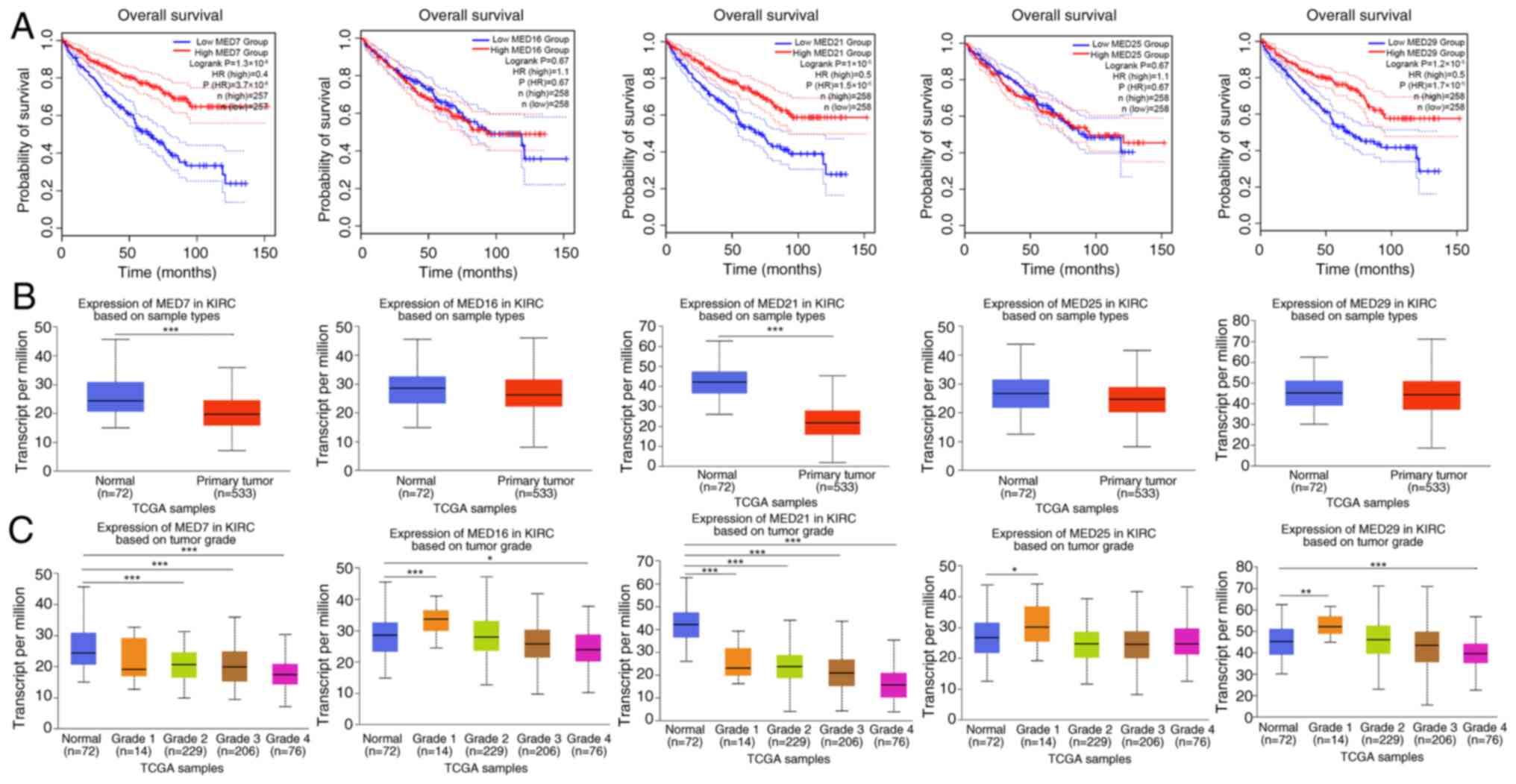
KIRC with high transcription levels of MED7, MED21
and MED29 was evidently related to a longer OS time, whereas MED16
and MED25 were not significantly associated with OS (Fig. 5A). Additionally, variations in the
expression levels of MED7, MED16, MED21, MED25 and MED29 were
assessed in KIRC tumor tissues (n=533) and adjacent normal tissues
(n=72). Since data for adjacent normal tissues were not available
for some patients, the number is unequal. The findings revealed
that the expression levels of MED7 and MED21 were higher in normal
tissues than in KIRC tumor tissues (Fig. 5B). Furthermore, the expression
levels of MED7, MED16, MED21, MED25 and MED29 were compared between
normal tissues and different KIRC tumor grade tissues. Compared
with in normal tissues, the expression levels of MED7 in grade 2,
grade 3 and grade 4 KIRC tissues were significantly lower. The
expression level of MED16 in grade 1 tissues was higher and that in
grade 4 KIRC tumor tissues was lower compared with that in normal
tissues. As for MED21 expression levels, tissues from all four
tumor stages, including grade 1, grade 2, grade 3 and grade 4,
exhibited lower expression compared with the normal control
tissues. The expression levels of MED25 in grade 1 KIRC tumor
tissues were significantly higher compared with those in normal
tissues. In addition, the expression level of MED29 in grade 1
tissues was higher and that in grade 4 KIRC tumor tissues was
significantly lower compared with those in normal tissues (Fig. 5C).
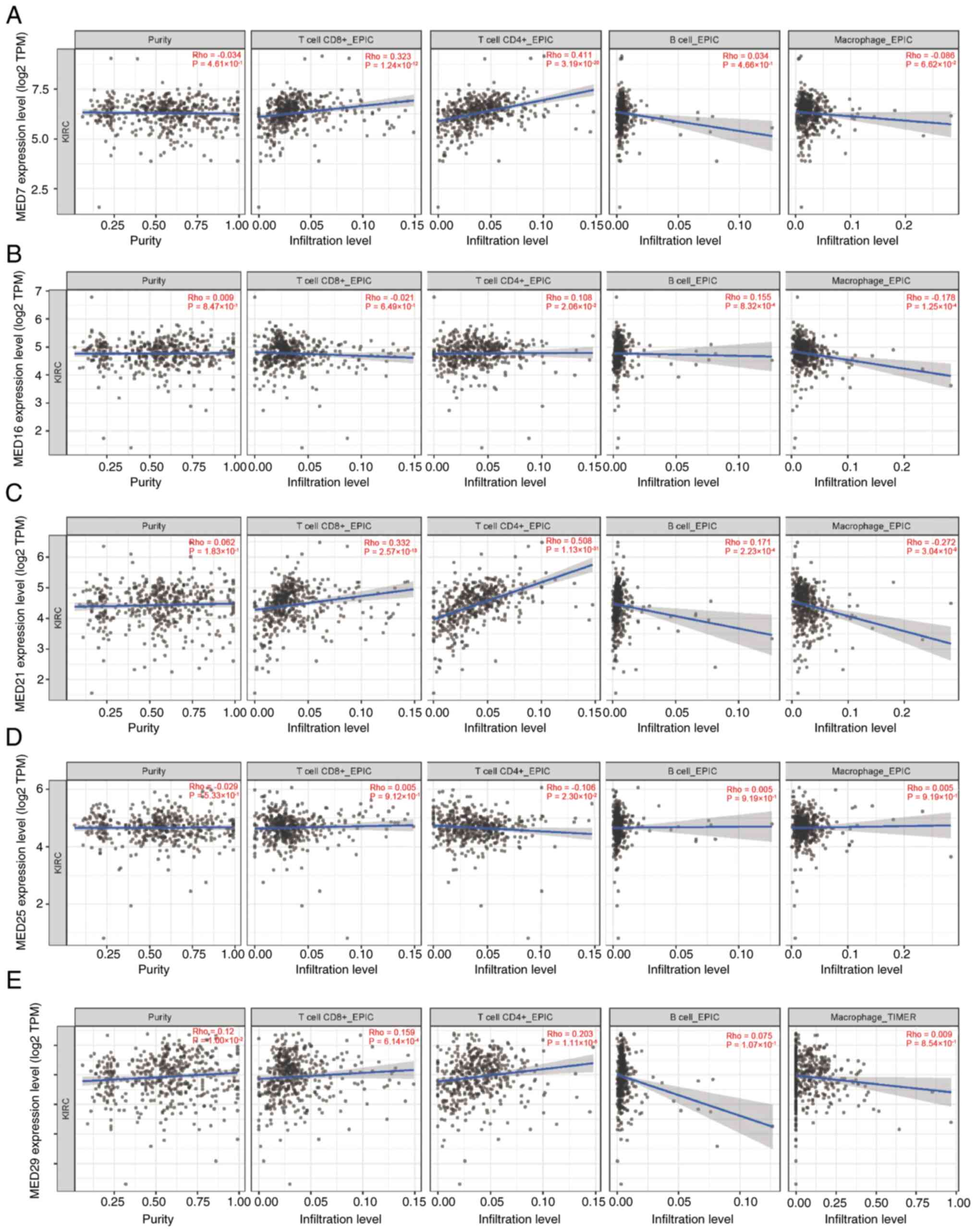
Relationships between MED genes and
tumor-infiltrating immune cells in KIRC
The occurrence, development and metastasis of cancer
are closely associated with infiltrating immune cells (21). Using the TIMER database, the
potential associations between the infiltration levels of distinct
immune cells and the five MED genes were explored. First, MED7
expression was strongly correlated with the infiltration of both
CD8+ T cells and CD4+ T cells in patients
with KIRC (Fig. 6A). The expression
of MED16 was not correlated with the infiltration of
CD4+ T cells, B cells and macrophages (Fig. 6B). There was also a link between
MED21 expression and the infiltration of CD8+ T cells
and CD4+ T cells (Fig.
6C). Additionally, the expression levels of MED25 and MED29
were not correlated with infiltrating immune cells (Fig. 6D and E).
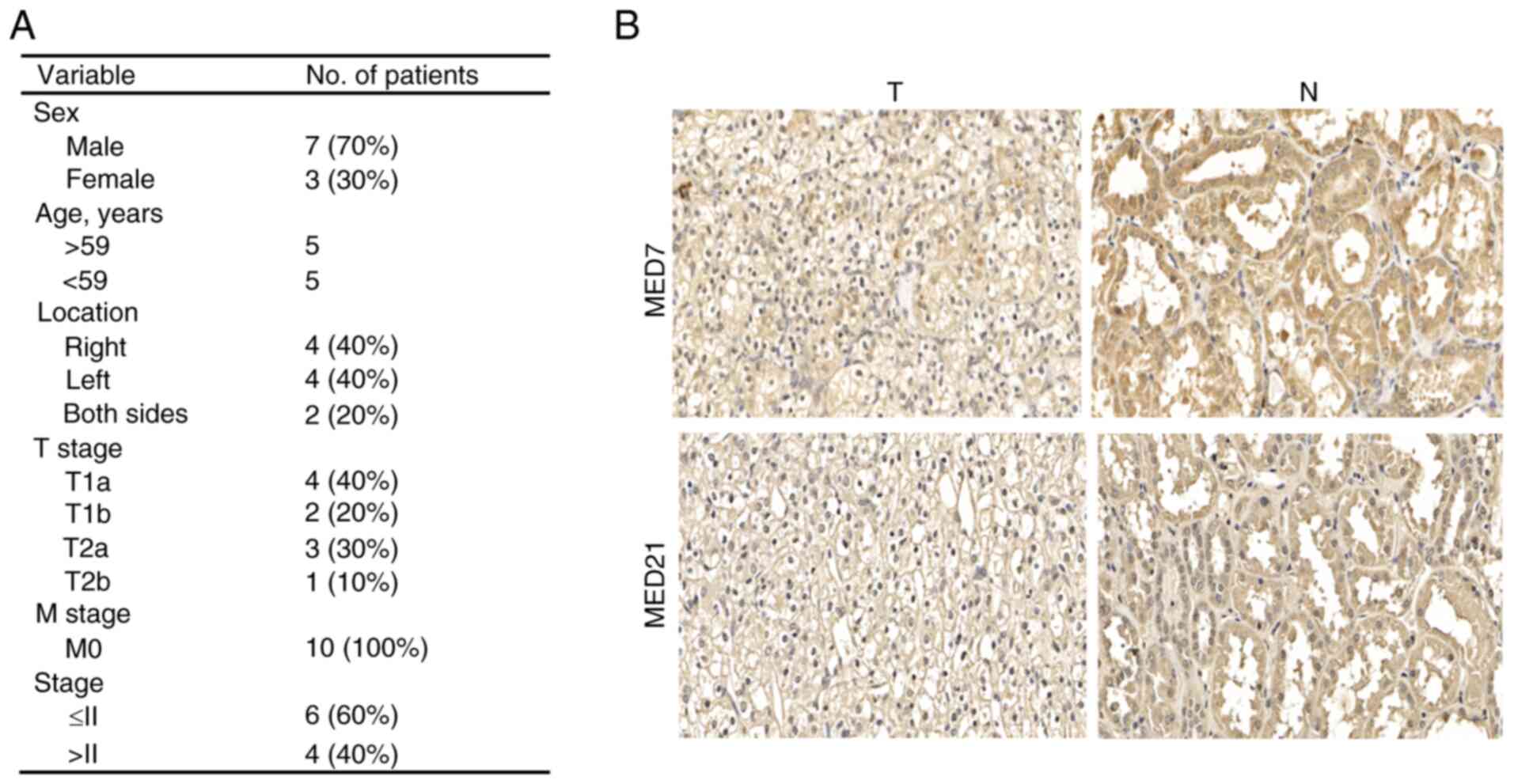
Verification of the relationship
between two MED genes and KIRC using IHC
To verify the relationship between MED genes and
KIRC, MED7 and MED21 were further examined. First, IHC was
performed to detect MED7 and MED21 protein expression in KIRC
tissues and paired adjacent normal tissues. The clinicopathological
information of the patients, including sex, age, pathological
grading, pathological region and TNM stage are listed in Fig. 7A. According to the staining results,
both MED7 and MED21 were more highly expressed in adjacent normal
tissues than in KIRC cancer tissues (Fig. 7B).
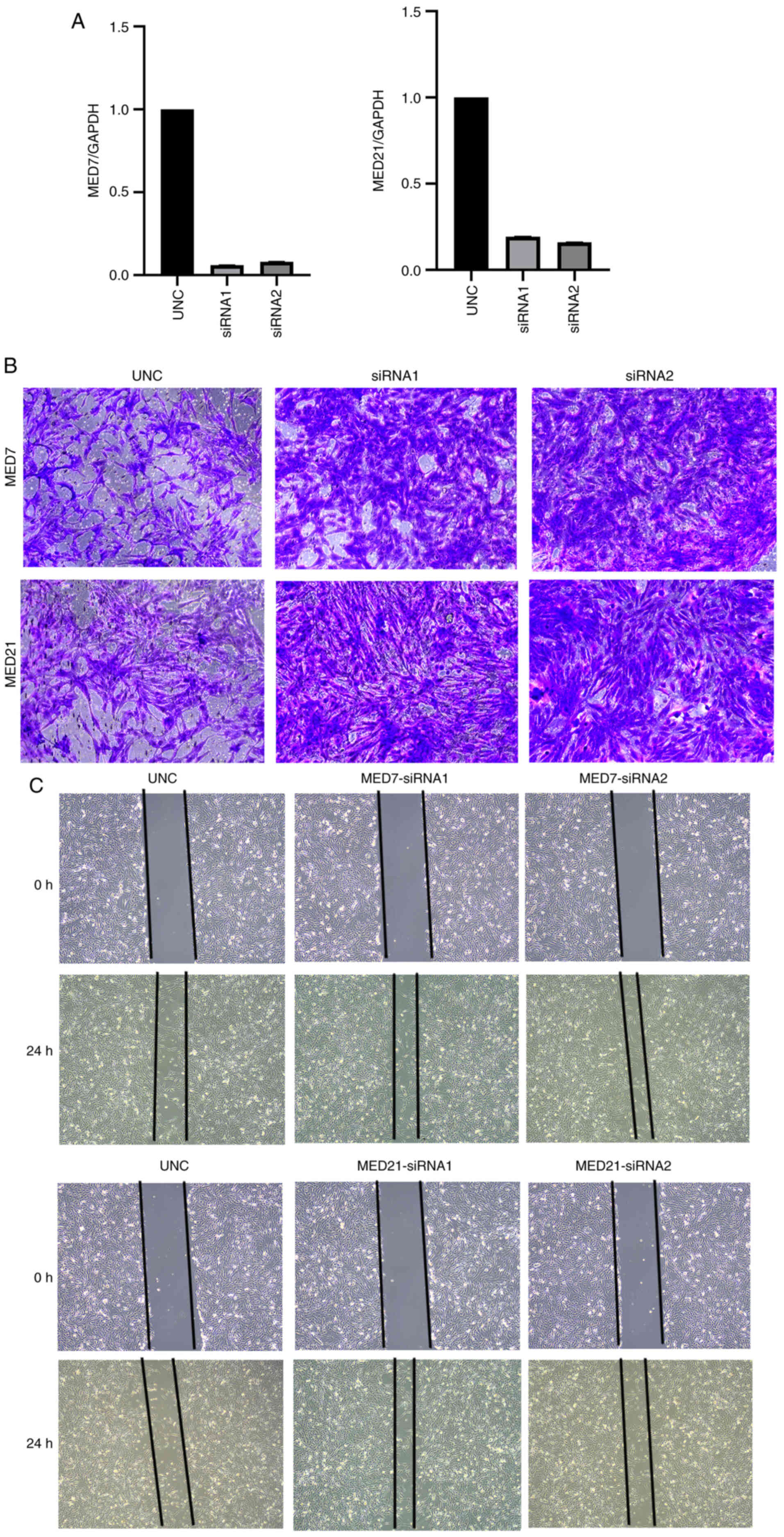
Verification of the relationship
between two MED genes and KIRC using siRNA transfection
Subsequently, corresponding siRNAs to interrupt MED7
and MED21 gene expression were designed and their effects were
observed. To assess the siRNA transfection efficiency, RT-qPCR
experiments were performed. All transfection efficiencies were
>70% (Fig. 8A). Moreover,
Transwell and wound healing assays were used to explore the
association between MED7 and MED21, and cell migration in KIRC. The
results revealed that MED7 and MED21 knockdown increased the
migration of KIRC cells in vitro (Fig. 8B and C).
Discussion
Renal cell carcinoma is a diverse category of
malignancies with various genetic and molecular changes
underpinning several described histological subtypes, and it is one
of the top 10 types of cancer with a rapidly increasing prevalence
(22,23). Traditional chemotherapy and
radiation therapy are ineffective in the treatment of kidney
cancer, and immunotherapy has replaced nonspecific immunological
treatments in some cases (24,25).
Cancer exists in a dynamic and complex environment, the components
of which change during different stages of cancer. The tumor
microenvironment (TME) may affect the evolution of cancer, and
there are currently several medicines, such as ipilimumab, that
specifically target the TME (26,27).
The MED family typically serves a key role as a regulatory element
in the progression of numerous types of cancer; for example, a
previous study showed that MED19 knockdown strongly hinders cell
proliferation, colony-forming ability and migration in
vitro, and downregulating MED19 can decrease the expression of
cyclin D1/cyclin B1 (28). In
addition, some preclinical studies have shown that MED12 is
associated with DNA damage repair and TGF-β receptor signaling
(29). Immune checkpoint inhibitors
have been shown to significantly increase survival in various types
of cancer, and another study revealed that MED12 mutation has the
potential to be a predictive biomarker for immune checkpoint
inhibitors in a variety of malignancies (30). Due to the immune regulatory
potential of the MED family, bioinformatics analysis was used in
the present study to explore the prognostic value of MED genes in
KIRC.
The present study identified the most significant
prognostic genes among the MED family using LASSO regression
analysis; 11 genes were selected. In addition, a prognostic
signature of the 11 genes was created, which performed well in
prognostic predictions for patients with KIRC. In addition, the
risk scores of patients with KIRC were computed and it was revealed
that patients with higher MED25 levels were in the high-risk group,
whereas patients with higher MED7 and MED21 expression were in the
low-risk group.
In addition, univariate and multivariate Cox
regression analyses were performed on the 11 genes in TCGA-KIRC
cohort. The findings showed that MED16, MED21, MED25, MED29 and
MED7 were independent risk factors for the prognosis of patients
with KIRC and were strongly associated with survival. These
findings suggested that the five MED genes may serve crucial roles
in the carcinogenesis and development of KIRC.
Notably, the present study revealed that higher
MED7, MED21 and MED29 transcript levels were closely related to a
longer OS time in patients with KIRC. Furthermore, a nomogram was
constructed based on MED16, MED21, MED25, MED29 and MED7. The
calibration map revealed that the current model performed well in
predicting the prognosis of patients with KIRC.
It has been demonstrated that immune infiltrates,
which are a significant factor of the TME, influence tumor
development and immunotherapy responses (31). Therefore, the present study analyzed
the relationships between MED genes and tumor-infiltrating immune
cells in patients with KIRC. The results showed that MED7 and MED21
were correlated with CD8+ T cells and CD4+ T
cells. In addition, to further verify the relationship between MED
genes and KIRC, cells in which MED7 and MED21 were knocked down
were subjected to IHC, Transwell and wound healing assays due to
their high expression levels in the high OS group and normal kidney
tissues. The binding of human MED to RNA polymerase II is dependent
on the integrity of a preserved ‘hinge’ in the intermediate module
of the MED21-MED7 heterodimer (32). MED7 is a highly conserved subunit,
and loss of MED7 has been shown to significantly affect cellular
functioning, including metabolic activity (12). As the most conserved MED subunit,
MED21 is essential to the viability of cells in yeast and mice, and
it can impact PPARA-related gene expression and metabolism
(11). Furthermore, as several
studies have demonstrated, MED7 and MED21 are associated with HCC
(17,18). However, to the best of our
knowledge, there is no research on the role of MED7 and MED21 in
KIRC or other types of kidney cancer. According to the present
study, the expression levels of MED7 and MED21 were greater in
normal tissues than in KIRC tumor tissues. In addition, when MED7
and MED21 were knocked down, the migration of KIRC cells was
increased in vitro.
In conclusion, a thorough analysis of the expression
and prognostic significance of the MED gene family was conducted in
KIRC. The results revealed that MED7, MED8, MED10, MED11, MED12L,
MED15, MED16, MED18, MED21, MED25 and MED29 may serve a potential
prognostic role in KIRC, and among the 11 MED genes, MED16, MED21,
MED25, MED29 and MED7 were shown to be closely related to the
prognosis of KIRC. These findings may aid in the improvement of the
survival and prognosis of patients with KIRC.
However, there are some limitations in the present
study. Firstly, since not all of the information from TCGA was
available for all of the data parameters, the number of patients
and controls is not totally equivalent. Secondly, the IHC results
cannot determine the relationship between target antigens and
survival prognosis, they can only verify the protein expression
levels of target antigens between KIRC cancer tissues and paired
adjacent normal tissues, due to the lack of some information about
patients, such as overall survival. Thirdly, immune cell
immunofluorescence staining was not performed because it was
difficult to obtain the target KIRC tissues at our hospital (West
China Second University Hospital, Chengdu, China), since this is a
children and women's hospital.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 82301923) and the crosstalk task of
Sichuan University (grant nos. 19H0125, 23H0221 and 23H0222).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
XL and JM designed and supervised the implementation
of the entire study. JM analyzed the data and completed the
immunohistochemistry experiment. MW completed the Transwell and
wound healing assays, and wrote the manuscript. MM analyzed the
data and organized the images. XL and JM confirm the authenticity
of all the raw data. All authors read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
The study was approved by the Institutional Review
Board of West China Second University Hospital (ethics approval no.
2023-012). All of the datasets were retrieved from the online
databases; therefore, it was confirmed that written informed
consent had already been obtained, which was also the case for the
purchased tissue microarray.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
MED
|
mediator complex
|
|
KIRC
|
kidney renal clear cell carcinoma
|
|
PPI
|
protein-protein interaction
|
|
TCGA
|
The Cancer Genome Atlas
|
|
GO
|
Gene Ontology
|
References
|
1
|
Owens B: Kidney cancer. Nature.
537:S972016. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Linehan WM, Schmidt LS, Crooks DR, Wei D,
Srinivasan R, Lang M and Ricketts CJ: The metabolic basis of kidney
cancer. Cancer Discov. 9:1006–1021. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ricketts CJ, De Cubas AA, Fan H, Smith CC,
Lang M, Reznik E, Bowlby R, Gibb EA, Akbani R, Beroukhim R, et al:
The cancer genome atlas comprehensive molecular characterization of
renal cell carcinoma. Cell Rep. 23:313–326.e315. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Turajlic S, Swanton C and Boshoff C:
Kidney cancer: The next decade. J Exp Med. 215:2477–2479. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Latif F, Tory K, Gnarra J, Yao M, Duh FM,
Orcutt ML, Stackhouse T, Kuzmin I, Modi W and Geil L:
Identification of the von Hippel-Lindau disease tumor suppressor
gene. Science. 260:1317–1320. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang Y, Qin P, Tian L, Yan J and Zhou Y:
The role of mediator complex subunit 19 in human diseases. Exp Biol
Med (Maywood). 246:1681–1687. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Napoli C, Schiano C and Soricelli A:
Increasing evidence of pathogenic role of the mediator (MED)
complex in the development of cardiovascular diseases. Biochimie.
165:1–8. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chadick JZ and Asturias FJ: Structure of
eukaryotic Mediator complexes. Trends Biochem Sci. 30:264–271.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Singh N and Han M: sur-2, a novel gene,
functions late in the let-60 ras-mediated signaling pathway during
Caenorhabditis elegans vulval induction. Gene Dev. 9:2251–2265.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Grants JM, Goh GY and Taubert S: The
mediator complex of caenorhabditis elegans: Insights into the
developmental and physiological roles of a conserved
transcriptional coregulator. Nucl Acids Res. 43:2442–2453. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bourbon HM: Comparative genomics supports
a deep evolutionary origin for the large, four-module
transcriptional mediator complex. Nucleic Acids Res. 36:3993–4008.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Koschubs T, Seizl M, Larivière L, Kurth F,
Baumli S, Martin DE and Cramer P: Identification, structure, and
functional requirement of the Mediator submodule Med7N/31. EMBO J.
28:69–80. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang X, Gao D, Fang K, Guo Z and Li L:
Med19 is targeted by miR-101-3p/miR-422a and promotes breast cancer
progression by regulating the EGFR/MEK/ERK signaling pathway.
Cancer Lett. 444:105–115. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mehine M, Mäkinen N, Heinonen HR, Aaltonen
LA and Vahteristo P: Genomics of uterine leiomyomas: Insights from
high-throughput sequencing. Fertil Steril. 102:621–629. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhu LJ, Yan WX, Chen ZW, Chen Y, Chen D,
Zhang TH and Liao GQ: Disruption of mediator complex subunit 19
(Med19) inhibits cell growth and migration in tongue cancer. World
J Surg Oncol. 11:1162013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Joseph C, Macnamara O, Craze M, Russell R,
Provenzano E, Nolan CC, Diez-Rodriguez M, Sonbul SN, Aleskandarany
MA, Green AR, et al: Mediator complex (MED) 7: A biomarker
associated with good prognosis in invasive breast cancer,
especially ER+ luminal subtypes. Br J Cancer. 118:1142–1151. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen ZL, Ma YY, Mou XZ and Zhang JG:
Upregulation of MED7 was associated with progression in
hepatocellular carcinoma. Cancer Biomark. 38:603–611. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tan W, Peng S, Li Z, Zhang R, Xiao Y, Chen
X, Zhu J, Li B and Lv X: Identification of therapeutic targets and
prognostic biomarkers among genes from the mediator complex family
in the hepatocellular carcinoma tumour-immune microenvironment.
Comput Math Methods Med. 2022:20216132022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sato S, Tomomori-Sato C, Tsai KL, Yu X,
Sardiu M, Saraf A, Washburn MP, Florens L, Asturias FJ, Conaway RC
and Conaway JW: Role for the MED21-MED7 hinge in assembly of the
mediator-RNA polymerase II holoenzyme. J Biol Chem.
291:26886–26898. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Steven A and Seliger B: The role of immune
escape and immune cell infiltration in breast cancer. Breast care
(Basel). 13:16–21. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shuch B, Amin A, Armstrong AJ, Eble JN,
Ficarra V, Lopez-Beltran A, Martignoni G, Rini BI and Kutikov A:
Understanding pathologic variants of renal cell carcinoma:
Distilling therapeutic opportunities from biologic complexity. Eur
Urol. 67:85–97. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Goyal R, Gersbach E, Yang XJ and Rohan SM:
Differential diagnosis of renal tumors with clear cytoplasm:
Clinical relevance of renal tumor subclassification in the era of
targeted therapies and personalized medicine. Arch Pathol and Lab
Med. 137:467–480. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Choueiri TK and Motzer RJ: Systemic
therapy for metastatic renal-cell carcinoma. N Eng J Med.
376:354–366. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Quail DF and Joyce JA: Microenvironmental
regulation of tumor progression and metastasis. Nat Med.
19:1423–1437. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Coussens LM, Zitvogel L and Palucka AK:
Neutralizing tumor-promoting chronic inflammation: A magic bullet?
Science. 339:286–291. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yu W, Zhang Z, Min D, Yang Q, Du X, Tang
L, Lin F, Sun Y, Zhao H, Zheng S, et al: Mediator of RNA polymerase
II transcription subunit 19 promotes osteosarcoma growth and
metastasis and associates with prognosis. Eur J Cancer.
50:1125–1136. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Huang S, Hölzel M, Knijnenburg T,
Schlicker A, Roepman P, McDermott U, Garnett M, Grernrum W, Sun C,
Prahallad A, et al: MED12 controls the response to multiple cancer
drugs through regulation of TGF-β receptor signaling. Cell.
151:937–950. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhou Y, Tan Y, Zhang Q, Duan Q and Chen J:
MED12 mutation as a potential predictive biomarker for immune
checkpoint inhibitors in pan-cancer. Eur J Med Re. 27:2252022.
View Article : Google Scholar
|
|
31
|
Zhang Y and Zhang Z: The history and
advances in cancer immunotherapy: Understanding the characteristics
of tumor-infiltrating immune cells and their therapeutic
implications. Cell Mol Immunol. 17:807–821. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Robinson PJ, Trnka MJ, Pellarin R,
Greenberg CH, Bushnell DA, Davis R, Burlingame AL, Sali A and
Kornberg RD: Molecular architecture of the yeast mediator complex.
eLife. 4:e087192015. View Article : Google Scholar : PubMed/NCBI
|