Introduction
The use of tyrosine kinase inhibitors (TKIs), i.e.,
imatinib, in the hematological malignancy chronic myeloid leukemia
(CML) is the most prominent model for successful targeted therapy.
This drug targets the disease-causing, constitutively active
BCR::ABL1 (breakpoint cluster region/Abelson tyrosine kinase 1)
kinase, which results from the reciprocal t(9;22)(q34;q11)
translocation forming the so-called Philadelphia chromosome and the
BCR::ABL1 fusion gene (1–3). With
a five-year survival rate of 83%, TKI treatment of CML is
tremendously successful, however, the handling of CML has become
more complex due to the development of TKI resistance (4,5). About
half of the TKI-resistant CML patients acquire sequence variants in
BCR::ABL1 that prevent TKI binding and sufficient inhibition
of downstream target phosphorylation (6–9). The
most prominent pathogenic variant is the so-called gatekeeper
mutation p.(Thr315Ile), which impairs the ATP binding pocket of
BCR::ABL1 leading to therapy failure for first and second
generation TKIs (10–12). In addition, the P-loop mutation
p.(Gly250Glu) or the activation loop mutation p.(His396Arg) also
lead to treatment failure (13).
For the other half of TKI-resistant CML patients, a variety of
resistance mechanisms are discussed, e.g., drug-drug interactions
via CYP3A4, pharmacogenetic polymorphisms, drug efflux transporter,
in particular overexpression of the drug efflux transporters of the
ATP binding cassette (ABC) family, epigenetics, microRNA
deregulation and activation of alternative signaling pathways, in
particular JAK/STAT, MAP kinases or PI3K/Akt signaling (12,14).
To analyze drug resistance mechanisms, genome-wide gene expression
and exome sequencing data of an in vitro-TKI-resistance CML
model were performed in a previous study, which identified
downregulation of the non-receptor kinase Bruton's Tyrosine Kinase
(BTK) expression and an acquired novel potentially
pathogenic dinucleotide BTK variant in imatinib resistance
[GSE227347 (15), PRJEB60564
(16)].
First identified in loss-of-function mutations
causing X-linked agammaglobulinemia (XLA)/Bruton syndrome, BTK is
central for B lymphocyte development (17–19).
BTK is known to play a crucial role in Toll-like receptor (TLR),
chemokine receptor, as well as B cell receptor (BCR) signaling
(20). Besides its essential role
in the pathogenesis of XLA, there are also several other
malignancies based on BTK dysregulation: Waldenström's
macroglobulinemia, e.g., is an indolent B cell lymphoma caused by
myeloid differentiation primary response 88-(MYD88)-dependent BTK
activation (21). In chronic
lymphocytic leukemia (CLL), a malignant neoplasm resulting in
overproduction of mature CD5+ B lymphocytes by a constitutively
activated BCR pathway, BTK is an essential component of signal
transmission (22). Another
malignancy also linked to the BTK-dependent BCR signaling pathway
is mantle cell lymphoma (MCL), an aggressive mature B cell
non-Hodgkin lymphoma characterized by t(11;14)(q13;q32)
translocation, leading to an upregulation of cyclin D1 and BCR
activation (23). For all
malignancies, in which BTK is a crucial component of pathogenesis,
BTK inhibition represents a possible treatment option.
Consequently, BTK became a potential druggable
candidate for targeted therapy, leading to the introduction of the
selective and FDA-approved BTK inhibitor (BTKi) ibrutinib, which is
used as a single agent or combinational treatment in various
cancers (17). Similar to other
tyrosine kinase inhibitors, relapses and treatment failure can be
observed during the treatment with imatinib. This problem, as well
as the occurrence of adverse events, led to the development of
second generation BTKIs, e.g., acalabrutinib, zanubrutinib and
tirabrutinib, with the goal to improve the tolerability by higher
selectivity to BTK and to overcome ibrutinib resistance (23–25).
The aim of the current study was to investigate the
role of BTK and the previously discovered novel dinucleotide
BTK variant in imatinib resistance in chronic myeloid
leukemia cells.
Materials and methods
Reagents and compounds
All chemicals and reagents not indicated
differently, were purchased from Sigma-Aldrich or Carl Roth.
Imatinib and ibrutinib were obtained from Sigma-Aldrich; Merck
KGaA. Imatinib was diluted in 10 mM aqueous stock solutions,
ibrutinib in 100 mM stock solutions in DMSO, both stored at
−20°C.
Cell culture
K-562 cells (RRID: CVCL 0004), retrieved from the
pleural effusion of a 53-year old woman (26), originate from the German Collection
of Microorganisms and Cell Cultures (DSMZ). Cells were cultivated
in RPMI-1640 (Thermo Fisher Scientific, Inc.) supplemented with 10%
v/v FCS (Bio&Sell), 1% v/v Penicillin/Streptomycin (Carl Roth)
and 1% v/v L-Alanyl-L-Glutamin. Imatinib-resistant cell lines were
generated as previously described (15). Authenticity of treatment-naïve as
well as cell lines resistant against 0.5 µM and 2 µM imatinib was
confirmed by short tandem repeats (STR) analysis using the
GenePrint 10 system (Promega). BCR::ABL1 mutations were
analyzed as described elsewhere (15). The cell lines showed no
BCR::ABL1 mutations.
Site-directed mutagenesis
Mutagenesis of an BTK-encoding plasmid (Gene
ID: 2335, ABIN3996197, antibodies-online, Aachen) was performed to
insert the BTK variants (NM_000061.3,) c.1699G>A
p.(Glu567Lys), c.1700A>G p.(GluE567Gly) and c1699_1700delinsAG
p.(GluE567Arg) using the primers BTK_G1699A_F
5′-GTCCCCACCGAAAGTCCTGAT-3′, BTK_G1699A_R
5′-CACCGGACTGGAAATTTGG-3′, BTK_A1700G_F
5′-TCCCCACCGGGAGTCCTGATG-3′, BTK_A1700G_R
5′-CCACCGGACTGGAAATTTGG-3′, BTK_GA1699-1700AG_F
5′-GTCCCCACCGAGAGTCCTGATG-3′ and BTK_GA1699-1700AG_R
5′-CACCGGACTGGAAATTTG-3′ obtained from Sigma-Aldrich. 25 ng of
template DNA, an annealing temperature of 67°C and the Q5
Site-Directed Mutagenesis Kit (New England Biolabs) were used
according to the manufacturer's protocol. Sequence identification
was confirmed using Sanger sequencing.
Isolation of nucleic acids
Total RNA isolation was performed using
1×106 K-562 cells and PeqGOLD TriFast (VWR) according to
the manufacturer's recommendation. Cell line DNA was purified using
Gentra Puregene Cell Kit (Qiagen) according to the manufacturer's
protocol.
Amplicon sequencing (MiSeq,
Illumina)
Generation of PCR products was performed using MyTaq
Polymerase (Bioline), DNA of treatment-naïve, 0.5 µM and 2 µM
imatinib-resistant cells, and the primers BTK_Intron_F
5′-TGACACTCTTGTGACCGTGC-3′ and BTK_Intron_R
5′-ACAGTAAGCACTCCCCAAGG-3′ with annealing at 54°C. PCR products
were purified using the GeneJET PCR Purification Kit (Thermo Fisher
Scientific, Inc.). After PCR amplicon preparation with the Nextera
XT Sequencing Kit (Illumina), Next Generation Sequencing (NGS) SBS
technology with Illumina MiSeq was performed as described elsewhere
(16,27).
RT-qPCR
According to the manufacturer's protocol, 1 µg total
RNA was reversely transcribed using random hexamer primers and the
High Capacity cDNA Synthesis Kit (Thermo Fisher Scientific, Inc.)
for 10 min at 25°C, 120 min at 37°C and 5 min at 85°C. cDNA was
stored at −80°C prior further usage. Reverse transcription
quantitative polymerase chain reaction (RT-qPCR) of target genes
was performed in triplicates using the TaqMan assays (Thermo
Fischer Scientific, Inc.) HPRT1 (Hs02800695_m1,
Chr.X:134460145-134500668), TBP (Hs00427620_m1,
Chr.6:170554333-170572870) and GAPDH (Hs0266705_g1,
Chr.12:6534405-653837) as internal controls, as well as BTK
(Hs00975865_m1, Chr.X:101349447-101390796), with Universal Master
Mix II without UNG (Thermo Fisher Scientific, Inc.) on the
QuantStudio 7 device (Thermo Fisher Scientific, Inc.) with default
cycling conditions. Relative gene expression was calculated using
the 2−ΔΔCt method (28).
Immunoblotting
Whole cell lysates and immunoblotting were performed
as described elsewhere (29–31).
20 µg of protein were loaded onto the membranes and blots were
probed with the following antibodies, obtained from Santa Cruz,
Cell Signaling Technology or LiCOR (Bad Homburg): BTK: Cat#
sc-28387, RRID: AB_626770, 1:1,000; p-BTK: Cat# 5082, RRID:
AB_10561017, 1:1,000; GAPDH: Cat# sc-47724, RRID: AB_627678,
1:1,000; anti-mouse: Cat# 926-68070, RRID: AB_10956588, all
1:10,000; anti-rabbit: Cat# 926-32211, RRID: AB_621843, all
1:10,000. Primary antibodies were diluted in Intercept/TBS blocking
solution (LiCOR), supplemented with 0.2% v/v Tween-20. Secondary
antibodies were diluted in TBS, supplemented with 0.1% v/v
Tween-20.
Plasmid and siRNA transfection
Plasmid and siRNA transfection were performed using
the Amaxa nucleofector Kit V and nucleofector 2 b device (Lonza).
For plasmid transfection, 2×106 cells were transfected
with 10 µg of the respective plasmid. 24 h after transfection,
cells were seeded onto cell plates for subsequent experiments. For
siRNA transfection, K-562 cells were transfected with 200 nM
negative control stealth (1295300) and stealth RNAi Pre-designed
BTK-siRNA 5′-GGAGUCAGGCUGAGCAACUGCUAAA-3′ (HSS101131, both
Thermo Fisher Scientific, Inc.). After transfection, cells were
seeded onto respective cell culture plates and exposed to 2 µM
imatinib for 48 h. Stable transfection was performed exposing the
cells to 800 mg/ml G-418 for 4 weeks.
Cellular fitness assays
Cell numbers were obtained by trypan blue staining
and performed after 48 h imatinib incubation as described elsewhere
(27). WST-1 assay (Sigma-Aldrich;
Merck KGaA) was performed after 48 h with 5×104 cells as
previously described (29).
Proliferation was analyzed using human MKI67 ELISA Kit
(MyBioSource) on 1×106 cells according to the
manufacturer's recommendation with 50 µg protein/well. Data were
analyzed normalizing imatinib-treated to non-treated samples.
Pathogenicity prediction and
statistical analysis
Pathogenicity prediction of BTK variants was
performed using the CADD score [CADD v1.6, (32)]. Statistical analysis was performed
using unpaired student's t-test or one-way ANOVA with subsequent
Dunnett's test using GraphPad prism software (Version 9.0 for
Windows).
Results
Reduction of BTK expression and gain
of a BTK variant in imatinib-resistant CML cells
In a previous study, an in vitro-TKI
resistance model of K-562 CML cells was obtained by stepwise
exposure to increasing imatinib concentrations to obtain cells
resistant against 0.5 and 2 µM imatinib (15). Analysis of genome-wide expression in
this model revealed a BTK downregulation in one of the
replicate cell lines of imatinib resistance [-3.7-fold,
P=7.08×10−7, (15)]. To
generate insights into the role of BTK in the pathogenesis
of CML, BTK expression in treatment-naïve and
imatinib-resistant cell lines was examined on mRNA and protein
level. BTK was significantly downregulated on mRNA (0.5 µM:
−33%; 2 µM: −87%, both P<0.001) and protein level in 2 µM
imatinib-resistant cells confirming the BTK downregulation in
imatinib resistance (Fig. 1A and
B). To investigate, whether BTK downregulation occurs in
response to imatinib treatment, BTK mRNA expression was
measured after short-term exposure to imatinib of 3–24 h. Hence,
imatinib treatment led to a significant reduction in BTK
mRNA levels by 33 to 62% (3 h: P=0.002, 6 h: P=0.005, 12 h: P=0.03,
24 h: P=0.001, Fig. 1C).
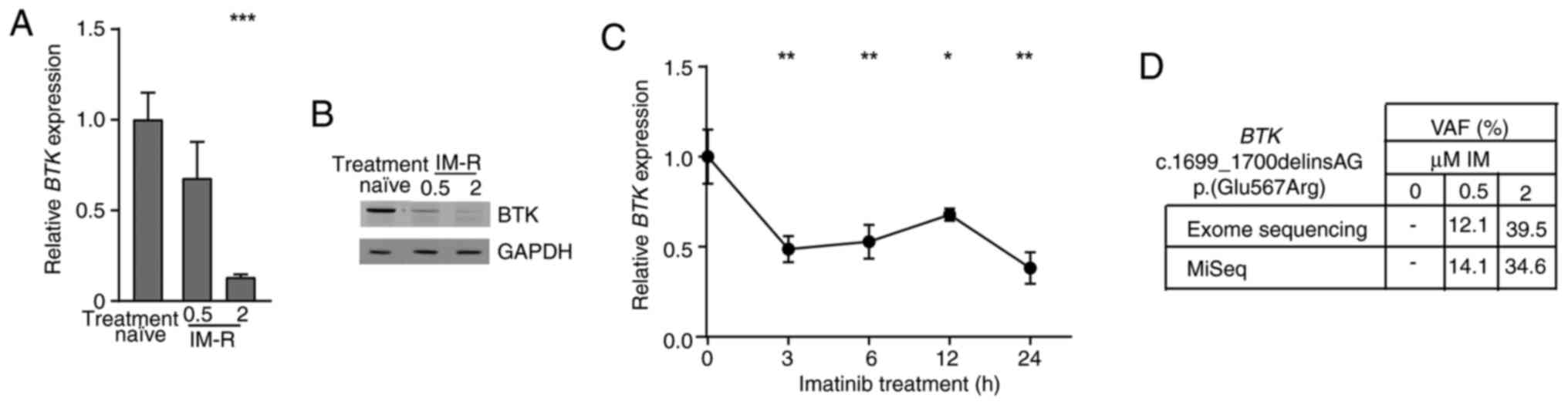 | Figure 1.BTK downregulation and gain of
BTK variant p.(Glu567Arg)/E567R in imatinib resistance. (A)
BTK mRNA expression in imatinib resistance analyzed by
RT-qPCR and normalized to GAPDH, TBP and HPRT1 as
housekeeping genes and treatment-naïve K-562 cells. n=3. (B)
Immunoblotting of BTK protein levels in imatinib-resistant
cells compared with GAPDH. n=3. (C) BTK mRNA expression in
response to treatment with 2 µM imatinib for 0 to 24 h analyzed by
RT-qPCR and normalized to GAPDH, TBP and HPRT1 and 0
h. n=3. (D) VAF of the BTK dinucleotide variant
c.1699_1700delinsAG p.(Glu567Arg)/E567R in imatinib-resistant cell
lines (0.5 and 2 µM imatinib) compared with treatment naïve K-562
cells obtained from exome and in-depth sequencing. Error bars
indicate standard deviation. *P<0.05, **P<0.01, ***P<0.001
compared with (A) treatment-naïve and (C) 0 h. IM, imatinib; IM-R,
imatinib resistance; VAF, Variant allele frequencies; BTK,
Bruton's tyrosine kinase; RT-qPCR, reverse
transcription-quantitative PCR; HPRT1, Hypoxanthine-guanine
phosphoribosyltransferase. |
As previously reported, the novel BTK
dinucleotide variant [NM_000061.3, c.1699_1700delinsAG
p.(Glu567Arg)] was detected in imatinib-resistant cells [PRJEB60564
(16)]. This variant leads to an
amino acid exchange from glutamic acid to arginine in the BTK
kinase domain, which is likely to have a detrimental effect on
protein function according to prediction tools (CADD: 25.2). In
depth-sequencing confirmed the presence of this variant in 0.5
(14%) and 2 µM imatinib-resistant cells (34%, considering the K-562
triploidy, Fig. 1D). Overall, our
findings show a downregulation of BTK expression and a gain
of a novel dinucleotide BTK variant in imatinib
resistance.
Loss of BTK activity or its expression
promotes imatinib resistance
Since a putative pathogenic BTK dinucleotide
variant p.(Glu567Arg) was detected in imatinib resistance, the
question arose whether deterioration of the kinase function through
inhibition could also influence the imatinib susceptibility of CML
cells. Thus, treatment-naïve cells were exposed to the BTK-specific
inhibitor ibrutinib and compared to single treatment with imatinib.
Treatment with imatinib significantly decreased cell viability of
treatment-naïve, but not imatinib-resistant K-562 cells by 73 to
89% (300–2,000 nM: P<0.001), while ibrutinib did not affect the
cells (Fig. 2A and B). However, in
a combined treatment with low dose (100 nM) imatinib, the presence
of ibrutinib dose-dependently increased cell viability by 11 to 37%
(ibrutinib (nM): 100: P=0.05, 500: P=0.02, 1,000: P=0.002, Fig. 2C), while this was not observed in
combination with high dose (2 µM) imatinib (Fig. 2D).
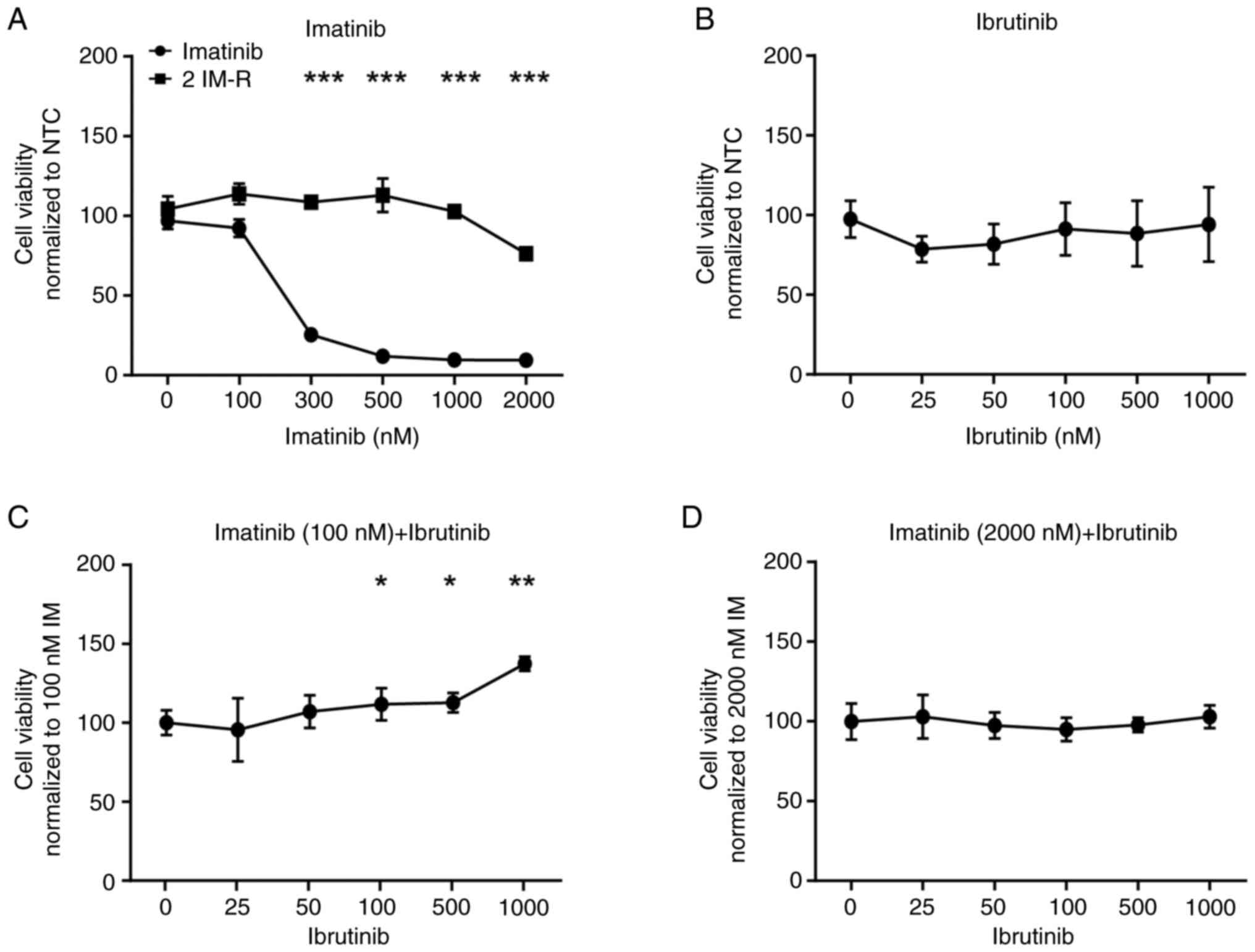 | Figure 2.Inhibition of BTK by ibrutinib
hampers susceptibility to low dose imatinib. Cell viability of
K-562 cells after exposure to (A) imatinib (0–2,000 nM), (B)
ibrutinib (0–1,000 nM), as well as to (C) 100 or (D) 2,000 nM
imatinib, respectively, in a dose-dependent combination with
ibrutinib for 48 h. Data were normalized to NTC. n=3. Error bars
indicate standard deviation. *P<0.05, **P<0.01, ***P<0.001
compared with the respective cell line at 0 nM. IM, imatinib; IM-R,
imatinib resistance; BTK, Bruton's tyrosine kinase; NTC, no
treatment controls. |
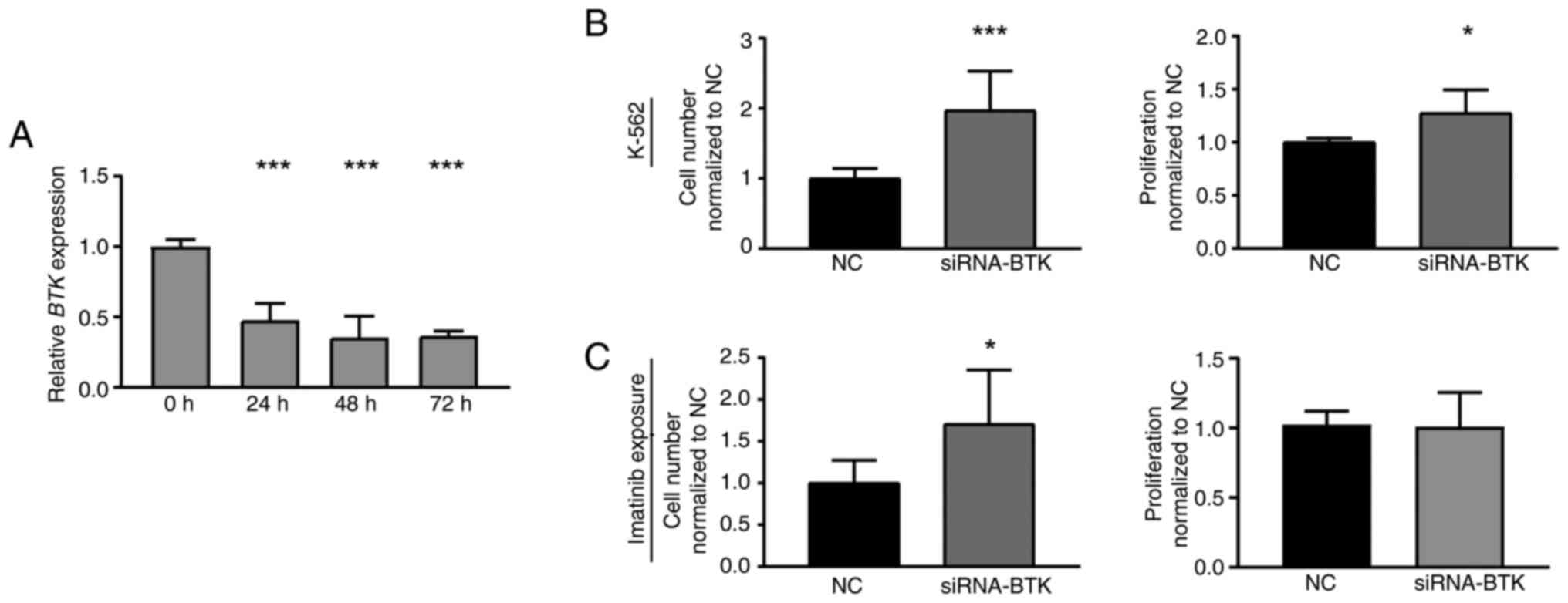
To investigate whether loss of BTK expression
also affects K-562 cells, a siRNA-mediated BTK knockdown was
performed in treatment-naïve CML cells. Successful BTK
downregulation (P<0.001, Fig.
3A) led to a significant increase in cell number (97.2%,
P<0.001) and proliferation rate of CML cells compared to
negative control-transfected cells (27.5%, P=0.01, Fig. 2B). After subsequent treatment with
imatinib, BTK knockdown cells also showed increased cell
numbers (70.6%, P=0.04), while proliferation was not significantly
altered (Fig. 2C). Overall, these
findings indicate that both, inhibition and the loss of BTK
expression, are beneficial for CML cells and reduce imatinib
susceptibility.
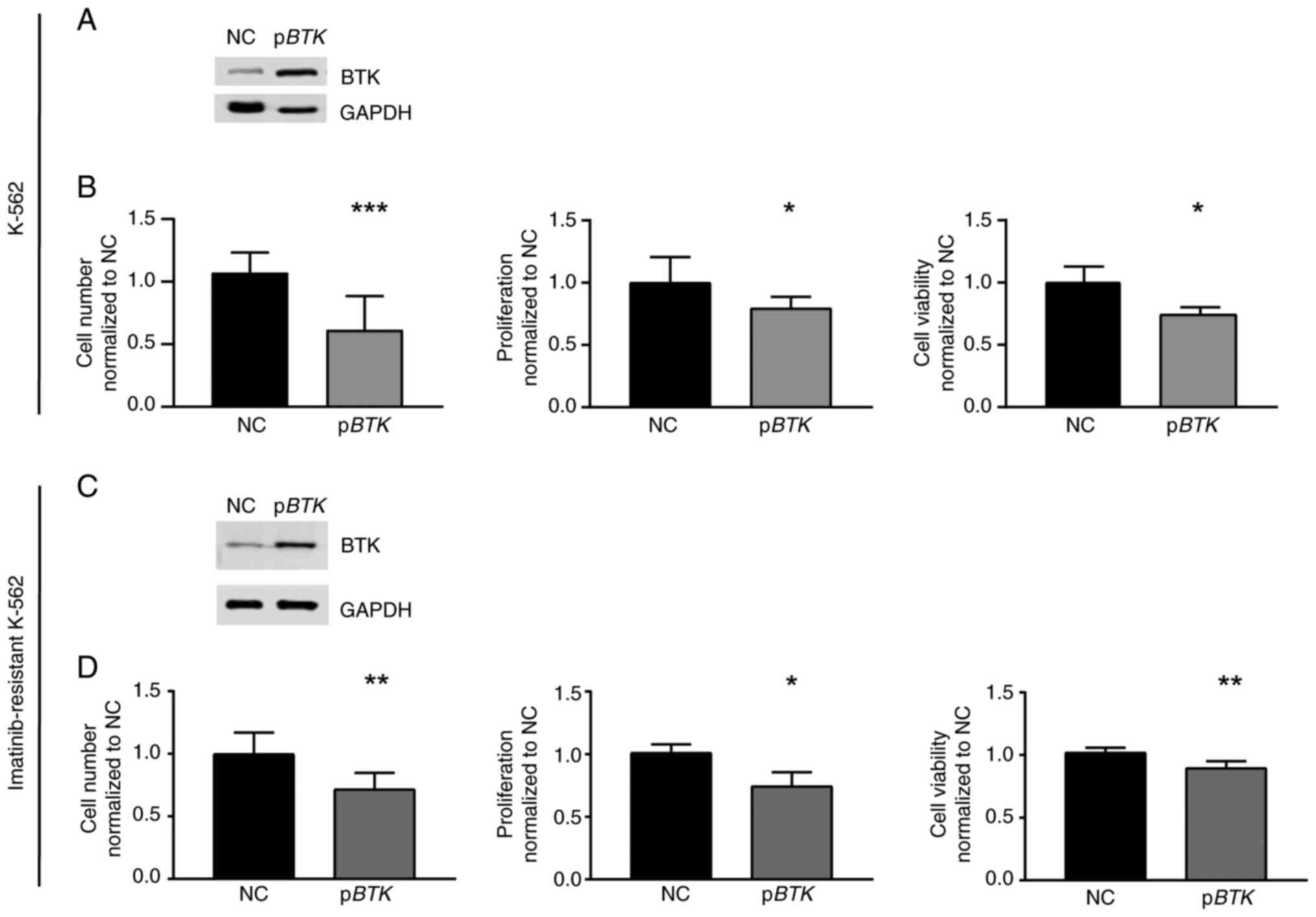
BTK overexpression and its rescue
reinstate imatinib susceptibility
Vice versa, to analyze whether high BTK
expression is detrimental for CML cells under imatinib exposure,
treatment-naïve cells were stably transfected with a
BTK-encoding plasmid to induce BTK overexpression
(Fig. 4A) and challenged with 2 µM
imatinib. In response to imatinib, the presence of BTK resulted in
a reduction in cell number (−25.9%, P=0.03), proliferation (−20.5%,
P=0.04), and cell viability compared to negative
control-transfected cells (−25.7%, P<0.001, Fig. 4B). These data indicate that the
presence of BTK augments the response to imatinib treatment.
Since BTK was significantly downregulated in
imatinib-resistant cells, restoration of BTK expression in
these cells was performed by transfection experiments followed by
imatinib exposure. After successful restoration of BTK
expression, cell number (−28.4%, P=0.002), proliferation (−26.7%,
P=0.04) and cell viability (−12.4%, P=0.003) were decreased
compared to negative control-transfected cells indicating an
improved response to imatinib (Fig. 4C
and D). Therefore, our findings demonstrate that the
restoration of BTK in imatinib-resistant cells reinstates
imatinib susceptibility.
Influence of BTK variants on imatinib
susceptibility
As the BTK variant p.(Glu567Arg) was acquired
in imatinib resistance, the question arose whether this mutation
affects CML cells and could be involved in the development of
resistance. Stable transfection of BTK wild-type and the
p.(Glu567Arg) variant in treatment-naïve K-562 cells was performed.
For comparison, the pathogenic kinase-dead BTK p.(Glu567Lys)
(33) and benign p.(Glu567Gly)
variants were also transfected. BTK mRNA expression was
significantly increased in all stable transfected cell lines
indicating successful overexpression (P<0.001, Fig. 5A). On protein level, however, in
p.(Glu567Arg) and p.(Glu567Lys)-expressing cells, a reduction in
BTK protein level was observed, while p.(Glu567Gly) had the highest
BTK levels pointing to decreased protein stability in the presence
of lysine or arginine at this residue (Fig. 5A). To investigate if the cells
harboring BTK variants are still able to respond to ibrutinib
treatment, the cells were exposed to ibrutinib (Figs. 2B, S1). The presence of 100 nM ibrutinib
resulted in increased cell viability for p.(Glu567Arg) (22.6%,
P=0.008), similarly to p.(Glu567Lys) (45.6%, P=0.006), but not for
p.(Glu567Gly) compared to cells overexpressing BTK wild-type
(Fig. 5B). After exposure to 2 µM
imatinib, the presence of the p.(Glu567Arg) variant led to an
increase in cell number (57.2%, P=0.003), proliferation (36.5%,
P<0.001) and cell viability (60.1%, P<0.001) during imatinib
treatment compared to wild-type BTK (Fig. 5C). A similar effect was also
observed for BTK p.(Glu567Lys), as increased cell numbers
(35.4%, P=0.04), proliferation (61.2%, P<0.001) and cell
viability were observed under imatinib exposure (29.5%, P<0.001,
Fig. 5B). The benign variant
p.(Glu567Gly), however, did not affect the response to imatinib
treatment. These findings show that p.(Glu567Arg), as well as
p.(Glu567Lys), result in diminished imatinib susceptibility.
Overall, our data show that the acquired BTK p.(Glu567Arg)
variant appears to be relevant for the development of imatinib
resistance.
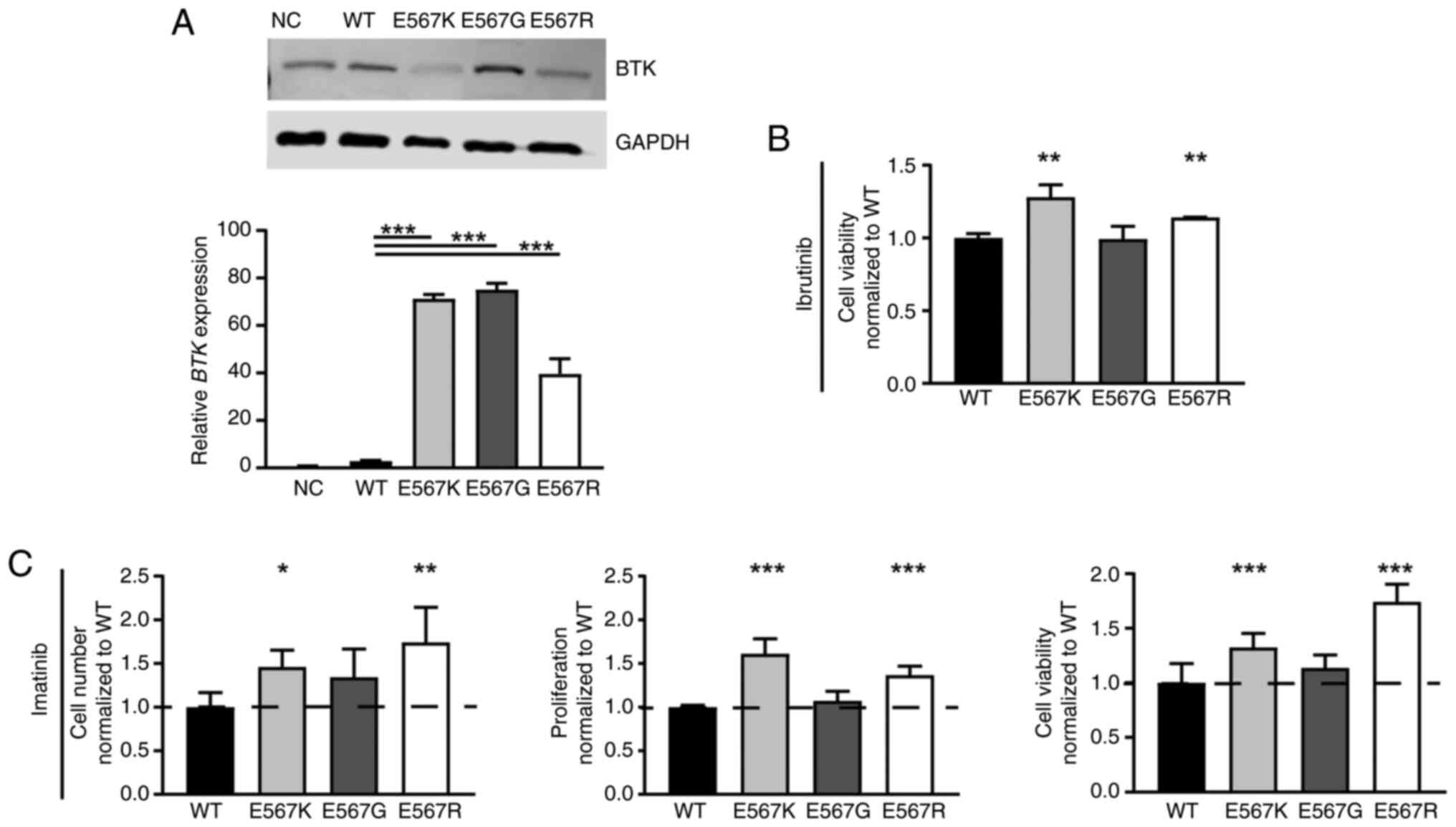 | Figure 5.Presence of BTK
p.(Glu567Lys)/E567K and p.(Glu567Arg)/E567R, but not
p.(Glu567Gly)/E567G promotes imatinib resistance. (A) BTK
mRNA and protein levels after stable transfection of BTK WT,
the variants p.(Glu567Lys)/E567K, p.(Glu567Gly)/E567G and
p.(Glu567Arg)/E567R and the NC into treatment naïve K-562 cells
analyzed by immunoblotting compared to GAPDH (top) and by RT-qPCR
normalized to GAPDH, TBP and HPRT1 (bottom). n=3.
Presence of the variants in the stable transfected cell lines was
confirmed using Sanger sequencing. (B) Cell fitness after
overexpression of BTK WT or the variants E567K, E567G or
E567R after exposure to 100 nM ibrutinib. n=3. Data were normalized
to WT. (C) Cell number, proliferation and cell viability of
BTK variant cell lines after treatment with 2 µM imatinib
compared to WT cells. n=3. Data were normalized to WT. Error bars
indicate standard deviation. *P<0.05, **P<0.01, ***P<0.001
compared with WT. WT, wild-type; BTK, Bruton's tyrosine
kinase; NC, empty vector-control; RT-qPCR, reverse
transcription-quantitative PCR; HPRT1, Hypoxanthine-guanine
phosphoribosyltransferase; NC, negative control. |
Discussion
The Bruton's tyrosine kinase (BTK) was initially
associated with X-linked agammaglobulinemia (XLA), a primary
immunodeficiency disorder characterized by a severe blockade of B
cell development in the bone marrow resulting in a lack of B cells
and serum antibodies due to (18).
Since its discovery, BTK was found to be crucial in oncogenic
signaling being important for B cell development and mature B cell
function. Accordingly, BTK is deregulated in several B cell
malignancies, including CLL, small lymphocytic leukemia (SLL), MCL
and follicular lymphoma, as well as in various other tumors, such
as pancreatic, lung, breast, ovarian, prostate and colorectal
cancer (17,34). In addition, BTK has been shown to be
an essential player in chronic graft-vs.-host disease or
Waldenström macroglobulinemia (17,35).
In the present study, we analyzed the role of BTK in the
myeloproliferative disease CML. A downregulation of BTK
expression, as well as the acquired BTK p.(Glu567Arg)
variant were detected in imatinib resistance. Subsequent
transfection experiments demonstrated that the presence of
BTK affects the response to imatinib in CML cells. Further,
imatinib susceptibility was restored after rescue of BTK
expression in imatinib-resistant cells. In addition, the BTK
p.(Glu567Arg) variant seems to be pathogenic to the protein
function resulting in a loss-of-function promoting imatinib
resistance.
BTK is a 77 kDa protein composed of 659 amino acids
and five domains: A plectrin homology (PH), a Tec homology (TH), an
SH3 and SH2 domain, and the C-terminal kinase domain (36). The SH2 domain mutation p.(Thr316Ala)
and the kinase domain mutations p.(Thr474Ile/Ser),
p.(Cys481Ser/Ala/Phe/Gly/Arg/Tyr) and p.(Leu528Trp) with
p.(Cys481Ser) are the main BTK mutations, associated with
BTKI resistance in B cell malignancies (36). Clinical trials of secondary drug
resistant CLL have shown the presence of the BTK
p.(Cys481Ser) mutation in approximately 80% of patients resulting
in a loss of drug binding while maintaining kinase activity
(36,37). This mutation has also been detected
in drug resistant or relapsed MCL and diffuse large B cell lymphoma
(DLBCL) leading to a sustained BTK signaling and,
consequently, to tumor progression (38). In the present study, the BTK
p.(Glu567Arg) variant, which is located at the C-terminal end of
the kinase domain, was acquired in imatinib resistance (16,39).
This variant showed similar effects on imatinib susceptibility as
the published p.(Glu567Lys) variant, which was shown to suppress
BTK-mediated NLRP3 inflammasome activation in brain ischemia
(33). Loss-of-function variants at
this amino residue, such as the exchange to glutamine
p.(Glu567Gln), have previously been associated with XLA (40). The p.Glu567 residue was found to
form an ionic bond to Arg641, which stabilizes the kinase structure
(41). Therefore, mutations at this
residue are likely to affect BTK stability and may also explain the
reduction in BTK expression in the presence of p.(Glu567Arg)
in imatinib resistance observed here. Further, stable transfection
of p.(Glu567Arg) was shown to result in increased levels of
BTK mRNA, but not protein indicating increased protein
turnover or reduced translation. Interestingly, both, BTK
p.(Glu567Arg) and p.(Glu567Lys) variants also resulted in an
increased cell fitness in the presence of ibrutinib, which could be
a results of decreased binding of ibrutinib to the variant
proteins.
BCR::ABL1 TKIs are tremendously successful in CML
treatment. However, therapy resistance remains a clinical problem
with a need for TKI alternatives for 20–25% of CML patients within
five years after therapy onset (4).
In B cell malignancies, the TKI ibrutinib also shows significant
clinical impact with overall response rates ranging from 90 and
97%, e.g., in CLL (42,43). Ibrutinib is a covalent kinase
inhibitor, which irreversibly binds to the BTK cysteine 481 in the
catalytic site preventing autophosphorylation at tyrosine 223 and
thereby, downstream signaling (39). Interestingly, it was shown that BTK
is also a target of the second generation BCR::ABL1 inhibitor
dasatinib (44). In the present
study, we found that inhibiting BTK by ibrutinib or knocking down
its expression prevented CML cells from imatinib resulting in
increased cell proliferation rates. These findings contradict a
study on primary CML stem cells, where BTK inhibition with
ibrutinib was demonstrated as a potential therapy approach in
combination with BCR::ABL1 TKI therapy to eradicate leukemic stem
cells mediated by inhibition of the Fc gamma receptor IIb (FcγRIIb,
CD32B) and a case report from concomitant use of imatinib and
ibrutinib in a patient with combined CML and CLL (45,46).
However, a study on Ba/F3 cells expressing BCR::ABL1 wild-type or
p.(Thr315Ile) mutation found that BTK depletion did not
affect viability or proliferation indicating that BTK is not
essential for leukemogenesis (47).
Consistent with our findings of a BTK downregulation at 0.5
and 2 µM imatinib, concentrations that reflect the observed
imatinib plasma levels in patients undergoing therapy (48), decreased BTK levels were also
shown in imatinib-resistant CML patients (49). In general, BTK appears to be
relevant for TKI resistance and CML progression only in cases of a
persistent CML, as present in our analyzed K-562 cell line model,
but not in the development of the disease. Thus, the BTK
downregulation in the imatinib-resistant cell line as presented
here, may result in a deregulation of BTK-dependent signaling
pathways that are likely involved in the development of imatinib
resistance, such as Ras-Map-signaling, to circumvent the
imatinib-mediated BCR::ABL1 inhibition. However, the BTK
deregulation, but also the acquisition of BTK mutations in CML
patients should be further analyzed in a clinical study to
understand the influence on resistance.
Due to its function as a kinase and its role as an
oncogene, it is generally assumed that BTK would be
upregulated or gain-of-function mutations would be present in drug
resistance. Accordingly, in previous studies on CLL and MCL, an
upregulated BTK expression relative to non-malignant cells
have been observed (20,50). However, we found that BTK
downregulation is associated with CML progression and imatinib
resistance. Our results contradict the common assumption that
upregulated kinases, such as BTK, enhance tumor progression
and their downregulation suppresses it. However, this discrepancy
is consistent with the description of BTK as a
context-dependent oncogene or tumor suppressor gene capable to
either increase proliferation and cell survival or induce apoptosis
and senescence, particularly by affecting p53-signaling (51). Therefore, BTK should be
considered as a pleiotropic gene with opposing effects in cancer,
acting as an oncogene especially in B cell malignancies, but also
as a tumor suppressor in other neoplasms.
In conclusion, the present data show that BTK
is involved in the development of imatinib resistance and
progression of chronic myeloid leukemia in an in
vitro-model. Further, the gain of the BTK
c.1699_1700delinsAG p.(Glu567Arg) variant in imatinib-resistant
cells further highlights the significance of BTK in imatinib
resistance. These findings demonstrate that BTK is not only
relevant in B cell malignancies, but may also be involved in other
hematopoietic disorders, either as an oncogene or tumor suppressor
gene, as well as a factor modulating treatment efficacy. Clinical
investigations should confirm whether BTK inhibitors should be
considered prior to their usage in imatinib-resistant chronic
myeloid leukemia.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Ms. Irina Naujoks,
Ms. Kerstin Viertmann and Ms. Anna Jürgensen (Institute of
Experimental and Clinical Pharmacology, Kiel, Germany) and Ms.
Claudia Becher (Institute of Human Genetics, Kiel, Germany), for
their technical assistance. The authors would like to also thank
Dr. D. Langfeldt, Ms. Manuela Pendziwiat and Dr. B. Löscher
(Institute of Clinical Molecular Biology, Kiel Germany) for their
technical support. The authors would like to acknowledge the
Institute of Clinical Molecular Biology in Kiel (Kiel, Germany) for
providing Sanger sequencing as supported in part by the DFG
Clusters of Excellence ‘Precision Medicine in Chronic Inflammation’
and ‘ROOTS’.
Funding
This study was funded by the Medical Faculty of the University
of Kiel.
Availability of data and materials
The data generated in the present study may be
requested re from the corresponding author. The exome sequencing
and gene expression data in the present study may be found in the
European Nucleotide Archive (ENA) repository under the accession
number (PRJEB60565; http://www.ebi.ac.uk/ena/) and the GEO dataset
accession number (GSE227347; http://www.ncbi.nlm.nih.gov/gds).
Authors' contributions
MK and IN conceptualized the study. MK designed the
research. LS performed the experiments. LS, MK and IV analyzed the
data. LS, IC, MK interpreted the data. LS and MK wrote the original
draft. All authors read and approved the final version of the
manuscript. MK and LS confirmed the authenticity of all the raw
data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing
interests.
Authors' information
Dr Inga Nagel, https://orcid.org/0000-0001-5174-4454; Professor
Ingolf Cascorbi, https://orcid.org/0000-0002-2182-9534; Dr Meike
Kaehler, https://orcid.org/0000-0002-2401-6037.
References
|
1
|
Druker BJ, Tamura S, Buchdunger E, Ohno S,
Segal GM, Fanning S, Zimmermann J and Lydon NB: Effects of a
selective inhibitor of the Abl tyrosine kinase on the growth of
Bcr-Abl positive cells. Nat Med. 2:561–566. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Deininger MW, Goldman JM and Melo JV: The
molecular biology of chronic myeloid leukemia. Blood. 96:3343–3356.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Quintás-Cardama A and Cortes J: Molecular
biology of bcr-abl1-positive chronic myeloid leukemia. Blood.
113:1619–1630. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Milojkovic D and Apperley J: Mechanisms of
resistance to imatinib and second-generation tyrosine inhibitors in
chronic myeloid leukemia. Clin Cancer Res. 15:7519–7527. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hochhaus A, Baccarani M, Silver RT,
Schiffer C, Apperley JF, Cervantes F, Clark RE, Cortes JE,
Deininger MW, Guilhot F, et al: European LeukemiaNet 2020
recommendations for treating chronic myeloid leukemia. Leukemia.
34:966–984. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cortes J, Jabbour E, Kantarjian H, Yin CC,
Shan J, O'Brien S, Garcia-Manero G, Giles F, Breeden M, Reeves N,
et al: Dynamics of BCR-ABL kinase domain mutations in chronic
myeloid leukemia after sequential treatment with multiple tyrosine
kinase inhibitors. Blood. 110:4005–4011. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gorre ME, Mohammed M, Ellwood K, Hsu N,
Paquette R, Rao PN and Sawyers CL: Clinical resistance to STI-571
cancer therapy caused by BCR-ABL gene mutation or amplification.
Science. 293:876–880. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jabbour E, Parikh SA, Kantarjian H and
Cortes J: Chronic myeloid leukemia: Mechanisms of resistance and
treatment. Hematol Oncol Clin North Am. 25:981–995. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Baccarani M, Deininger MW, Rosti G,
Hochhaus A, Soverini S, Apperley JF, Cervantes F, Clark RE, Cortes
JE, Guilhot F, et al: European LeukemiaNet recommendations for the
management of chronic myeloid leukemia: 2013. Blood. 122:872–884.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
O'Hare T, Zabriskie MS, Eiring AM and
Deininger MW: Pushing the limits of targeted therapy in chronic
myeloid leukaemia. Nat Rev Cancer. 12:513–526. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Braun TP, Eide CA and Druker BJ: Response
and resistance to BCR-ABL1-targeted therapies. Cancer Cell.
37:530–542. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kaehler M and Cascorbi I: Pharmacogenomics
of impaired tyrosine kinase inhibitor response: Lessons learned
from chronic myelogenous leukemia. Front Pharmacol. 12:6969602021.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
de Lavallade H and Kizilors A: The
Importance of mutational analyses in chronic myeloid leukaemia for
treatment choice. Eur Med J Oncol. 4:86–95. 2016.
|
|
14
|
Cilloni D and Saglio G: Molecular
pathways: BCR-ABL. Clin Cancer Res. 18:930–937. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kaehler M, Litterst M, Kolarova J, Böhm R,
Bruckmueller H, Ammerpohl O, Cascorbi I and Nagel I: Genome-wide
expression and methylation analyses reveal aberrant cell adhesion
signaling in tyrosine kinase inhibitor-resistant CML cells. Oncol
Rep. 48:1442022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kaehler M, Osteresch P, Künstner A, Vieth
S, Esser D, Möller M, Busch H, Vater I, Spielmann M, Cascorbi I and
Nagel I: Clonal evolution in tyrosine kinase inhibitor-resistance:
Lessons from in vitro-models. Front Oncol. 13:12008972023.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wen T, Wang J, Shi Y, Qian H and Liu P:
Inhibitors targeting Bruton's tyrosine kinase in cancers: Drug
development advances. Leukemia. 35:312–332. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pal Singh S, Dammeijer F and Hendriks RW:
Role of Bruton's tyrosine kinase in B cells and malignancies. Mol
Cancer. 17:572018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mattsson PT, Vihinen M and Smith CI:
X-linked agammaglobulinemia (XLA): A genetic tyrosine kinase (Btk)
disease. Bioessays. 18:825–834. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hendriks RW, Yuvaraj S and Kil LP:
Targeting Bruton's tyrosine kinase in B cell malignancies. Nat Rev
Cancer. 14:219–232. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ntanasis-Stathopoulos I, Gavriatopoulou M,
Fotiou D and Dimopoulos MA: Current and novel BTK inhibitors in
Waldenstrom's macroglobulinemia. Ther Adv Hematol.
12:20406207219895862021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Palma M, Mulder TA and Österborg A: BTK
inhibitors in chronic lymphocytic leukemia: Biological activity and
immune effects. Front Immunol. 12:6867682021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bond DA, Alinari L and Maddocks K: Bruton
tyrosine kinase inhibitors for the treatment of mantle cell
lymphoma: Review of current evidence and future directions. Clin
Adv Hematol Oncol. 17:223–233. 2019.PubMed/NCBI
|
|
24
|
Byrd JC, Hillmen P, Ghia P, Kater AP,
Chanan-Khan A, Furman RR, O'Brien S, Yenerel MN, Illés A, Kay N, et
al: Acalabrutinib versus ibrutinib in previously treated chronic
lymphocytic leukemia: Results of the first randomized phase III
trial. J Clin Oncol. 39:3441–3452. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hillmen P, Eichhorst B, Brown JR, Lamanna
N, O'Brien SM, Tam CS, Qiu L, Kazmierczak M, Zhou K, Šimkovič M, et
al: Zanubrutinib versus ibrutinib in relapsed/refractory chronic
lymphocytic leukemia and small lymphocytic lymphoma: Interim
analysis of a randomized phase III trial. J Clin Oncol.
41:1035–1045. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lozzio CB and Lozzio BB: Human chronic
myelogenous leukemia cell-line with positive Philadelphia
chromosome. Blood. 45:321–334. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kaehler M, Dworschak M, Rodin JP,
Ruemenapp J, Vater I, Penas EMM, Liu C, Cascorbi I and Nagel I:
ZFP36L1 plays an ambiguous role in the regulation of cell expansion
and negatively regulates CDKN1A in chronic myeloid leukemia cells.
Exp Hematol. 99:54–64.e7. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kaehler M, Ruemenapp J, Gonnermann D,
Nagel I, Bruhn O, Haenisch S, Ammerpohl O, Wesch D, Cascorbi I and
Bruckmueller H: MicroRNA-212/ABCG2-axis contributes to development
of imatinib-resistance in leukemic cells. Oncotarget.
8:92018–92031. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Waetzig V, Haeusgen W, Andres C, Frehse S,
Reinecke K, Bruckmueller H, Boehm R, Herdegen T and Cascorbi I:
Retinoic acid-induced survival effects in SH-SY5Y neuroblastoma
cells. J Cell Biochem. 120:5974–5986. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bruhn O, Lindsay M, Wiebel F, Kaehler M,
Nagel I, Böhm R, Röder C and Cascorbi I: Alternative
polyadenylation of ABC transporters of the C-family (ABCC1, ABCC2,
ABCC3) and implications on posttranscriptional Micro-RNA
regulation. Mol Pharmacol. 97:112–122. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rentzsch P, Schubach M, Shendure J and
Kircher M: CADD-Splice-improving genome-wide variant effect
prediction using deep learning-derived splice scores. Genome Med.
13:312021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ito M, Shichita T, Okada M, Komine R,
Noguchi Y, Yoshimura A and Morita R: Bruton's tyrosine kinase is
essential for NLRP3 inflammasome activation and contributes to
ischaemic brain injury. Nat Commun. 6:73602015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Uckun FM and Venkatachalam T: Targeting
solid tumors with BTK inhibitors. Front Cell Dev Biol.
9:6504142021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Treon SP, Xu L, Guerrera ML, Jimenez C,
Hunter ZR, Liu X, Demos M, Gustine J, Chan G, Munshi M, et al:
Genomic landscape of waldenstrom macroglobulinemia and its impact
on treatment strategies. J Clin Oncol. 38:1198–1208. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Smith CIE and Burger JA: Resistance
mutations to BTK inhibitors originate from the NF-κB but not from
the PI3K-RAS-MAPK Arm of the B cell receptor signaling pathway.
Front Immunol. 12:6894722021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ahn IE and Brown JR: Targeting Bruton's
tyrosine kinase in CLL. Front Immunol. 12:6874582021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chiron D, Di Liberto M, Martin P, Huang X,
Sharman J, Blecua P, Mathew S, Vijay P, Eng K, Ali S, et al:
Cell-cycle reprogramming for PI3K inhibition overrides a
relapse-specific C481S BTK mutation revealed by longitudinal
functional genomics in mantle cell lymphoma. Cancer Discov.
4:1022–1035. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gu D, Tang H, Wu J, Li J and Miao Y:
Targeting Bruton tyrosine kinase using non-covalent inhibitors in B
cell malignancies. J Hematol Oncol. 14:402021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Väliaho J, Faisal I, Ortutay C, Smith CI
and Vihinen M: Characterization of all possible single-nucleotide
change caused amino acid substitutions in the kinase domain of
Bruton tyrosine kinase. Hum Mutat. 36:638–647. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Speletas M, Kanariou M,
Kanakoudi-Tsakalidou F, Papadopoulou Alataki E, Arvanitidis K,
Pardali E, Constantopoulos A, Kartalis G, Vihinen M, Sideras P and
Ritis K: Analysis of Btk mutations in patients with X-linked
agammaglobulinaemia (XLA) and determination of carrier status in
normal female relatives: A nationwide study of Btk deficiency in
Greece. Scand J Immunol. 54:321–327. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Burger JA, Barr PM, Robak T, Owen C, Ghia
P, Tedeschi A, Bairey O, Hillmen P, Coutre SE, Devereux S, et al:
Long-term efficacy and safety of first-line ibrutinib treatment for
patients with CLL/SLL: 5 years of follow-up from the phase 3
RESONATE-2 study. Leukemia. 34:787–798. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Broccoli A, Argnani L, Morigi A, Nanni L,
Casadei B, Pellegrini C, Stefoni V and Zinzani PL: Long-term
efficacy and safety of ibrutinib in the treatment of CLL patients:
A real life experience. J Clin Med. 10:58452021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hantschel O, Rix U, Schmidt U,
Bürckstümmer T, Kneidinger M, Schütze G, Colinge J, Bennett KL,
Ellmeier W, Valent P and Superti-Furga G: The Btk tyrosine kinase
is a major target of the Bcr-Abl inhibitor dasatinib. Proc Natl
Acad Sci USA. 104:13283–13288. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Parting O, Langer S, Kuepper MK, Wessling
C, Li S, Braunschweig T, Chatain N, Maié T, Costa IG, Crysandt M,
et al: Therapeutic inhibition of FcγRIIb signaling targets leukemic
stem cells in chronic myeloid leukemia. Leukemia. 34:2635–2647.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shea LK, Mikhail FM, Forero-Torres A and
Davis RS: Concomitant imatinib and ibrutinib in a patient with
chronic myelogenous leukemia and chronic lymphocytic leukemia. Clin
Case Rep. 5:899–901. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
MacPartlin M, Smith AM, Druker BJ,
Honigberg LA and Deininger MW: Bruton's tyrosine kinase is not
essential for Bcr-Abl-mediated transformation of lymphoid or
myeloid cells. Leukemia. 22:1354–1360. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Peng B, Lloyd P and Schran H: Clinical
pharmacokinetics of imatinib. Clinl Pharmacokinet. 44:879–894.
2005. View Article : Google Scholar
|
|
49
|
Villuendas R, Steegmann JL, Pollán M,
Tracey L, Granda A, Fernández-Ruiz E, Casado LF, Martínez J,
Martínez P, Lombardía L, et al: Identification of genes involved in
imatinib resistance in CML: A gene-expression profiling approach.
Leukemia. 20:1047–1054. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhu S, Gokhale S, Jung J, Spirollari E,
Tsai J, Arceo J, Wu BW, Victor E and Xie P: Multifaceted
immunomodulatory effects of the BTK inhibitors ibrutinib and
acalabrutinib on different immune cell subsets-beyond B
lymphocytes. Front Cell Dev Biol. 9:7275312021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Rada M, Barlev N and Macip S: BTK: A
two-faced effector in cancer and tumour suppression. Cell Death
Dis. 9:10642018. View Article : Google Scholar : PubMed/NCBI
|