Introduction
Classical Hodgkin lymphoma (CHL), a malignancy of
the lymphatic system, is one of the most prevalent types of
lymphoma, exhibiting an incidence of 2–3 cases per 100,000
individuals annually in developed countries (1). It is generally regarded as a highly
curable disease, especially with the use of standard first-line
chemotherapy and, in some cases, radiotherapy (2). Peripheral T-cell lymphoma (PTCL)
represents a group of aggressive non-Hodgkin lymphomas (NHLs)
(3), among which, PTCL, not
otherwise specified (PTCL-NOS), constitutes approximately one-third
of all PTCL cases (4). PTCL-NOS is
the most frequently encountered subtype of PTCL in North America
and Europe, excluding Native Americans, accounting for ~30% of all
PTCL diagnoses (5). These
malignancies involve a heterogeneous collection of mature T-cell
neoplasms, typically characterized by a complex clinicopathological
presentation and an aggressive clinical course, leading to a poor
prognosis (5,6). The coexistence of two or more types of
lymphoma within the same or different organs is described as
composite lymphoma or discordant lymphoma. The histological
patterns are well-defined and clearly demarcated, often comprising
two or three types of NHL or a combination of Hodgkin lymphoma (HL)
with NHL (7). CHL originates from
germinal center B lymphocytes (8)
and PTCL-NOS originates from post-thymic mature T lymphocytes
(5), and both are typically
associated with a complex tumor microenvironment. The simultaneous
or sequential occurrence of HL and NHL in the same patient is rare
(9,10), and the association between such
discordant lymphomas and immune system disorders or HL treatment
remains unclear. The present study describes the case of a patient
who was initially diagnosed with nodular sclerosing-type CHL, which
later evolved into peripheral T-cell lymphoma, not otherwise
specified (PTCL-NOS), after 3 years. The clinicopathological
features and potential mechanisms underlying this transformation
were further explored through a review of the literature.
Case report
A 73-year-old man was diagnosed with CHL at Yantai
Yuhuangding Hospital (Yantai, China) in December 2012. Fresh ~4-cm
lymph node specimens were obtained by surgical resection from the
left neck, and were homogenized and suspended in RPMI medium
(Gibco; Thermo Fisher Scientific, Inc.) for flow cytometry.
Histological examination revealed features indicative of nodular
sclerosing-type CHL. After obtaining appropriate written informed
consent, including an explanation of the risks and benefits, left
posterior iliac crest bone marrow aspiration and biopsy were
performed. Flow cytometry revealed no monotypic population and
CD25-positive cells within normal limits (data not shown). A
complete blood count performed in December 2012 showed unremarkable
white blood cell morphology and an elevated eosinophil count of 31%
(normal count, 0.4–8%), but with no immature eosinophils. Red blood
cell indices were essentially normal, with minimal anisocytosis and
poikilocytosis, while platelets exhibited normal granularity. The
left cervical lymph node was excised, and complete destruction of
the lymph node structure was revealed under low magnification in a
light microscope field (Olympus BX53M; Olympus Corporation). The
fibrotic lymph node capsule divided the lymph node into nodules of
varying sizes (Fig. 1A). Large
mononuclear cells, occasionally binucleated or multinucleated, with
prominent nuclei and nucleoli were observed amidst numerous
neutrophils, eosinophils and lymphocytes within the nodules
(Fig. 1B).
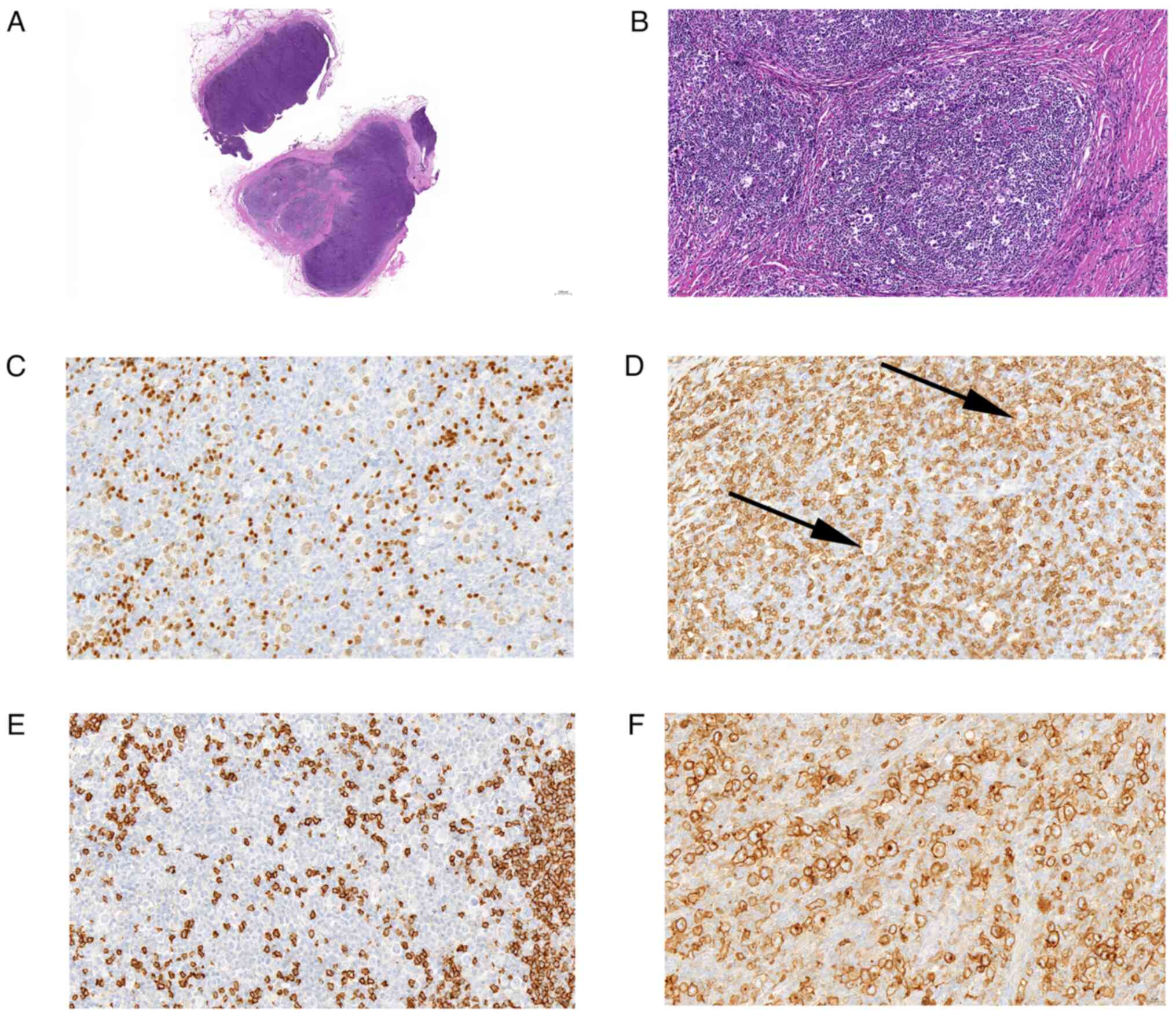 | Figure 1.H&E and partial
immunohistochemical analysis were used to diagnose classical
Hodgkin lymphoma (nodular sclerosis type). (A) Under low power, the
lymph node capsule was fibrotic and divided by the thick fiber
bundle into nodules of different sizes (H&E; magnification,
×9). (B) Large cells were mainly mononuclear, occasionally
binucleated or multinucleated, with large nuclei and nucleoli
(H&E; magnification, ×200). Immunohistochemically, the cell
surface of B cells was (C) positive for PAX5 (magnification, ×194)
and (D) negative for CD20 (black arrows indicate the negative
cells; magnification, ×400). Cell surface of other cells was (E)
negative for CD3 and (F) positive for CD30 (magnification, ×400).
H&E, hematoxylin and eosin. |
For immunohistochemistry, the specimen was fixed in
10% neutral formalin for 12 h at room temperature, embedded in
paraffin and sectioned into 4-µm continuous slices.
Immunohistochemical staining was performed using the BenchMark
ULTRA immunohistochemistry staining system (Roche Diagnostics). The
specific steps are as follows: i) Paraffin-embedded sections were
dewaxed and hydrated at 72°C, followed by rinsing with PBS for 4
min; ii) according to the requirements of the primary antibodies
(Table I), antigen retrieval was
performed at 95°C for 30 min; iii) sections were rinsed with PBS,
and then incubated with an endogenous peroxidase inhibitor (3%
H2O2) at 37°C for 4 min; iv) sections were
rinsed again with PBS and then incubated with primary antibodies at
37°C for 32 min; v) the peroxidase-conjugated secondary antibody
(Table I; 1:20) was added to the
sections at 37°C for 8 min; vi) freshly prepared DAB color
development reagent was added to the sections at 37°C for 8 min to
visualize staining; vii) sections were rinsed with running water to
terminate the color development and were counterstained with
hematoxylin for 8 min at 37°C; viii) the blue color was restored
with PBS for 8 min, and the sections were dehydrated with gradient
ethanol, cleared with xylene and sealed with neutral balsam. Known
positive tissue (normal lymph nodes from the same patient) was used
as a positive control and PBS was used instead of the primary
antibody as a negative control. The stained sections were scanned
using a 3D Pannoramic SCAN digital slide scanner [Bio-One
Scientific Instrument (Beijing) Co., Ltd.]. Immunohistochemical
staining demonstrated positivity for PAX5 (Fig. 1C), and negativity for CD20 (Fig. 1D), CD10, immunoglobulin (Ig)κ and λ
chains, and IgD on the surface of B cells (data not shown). T cells
were negative for CD3 (Fig. 1E),
PD1 and CD57 (data not shown), whereas other cells were positive
for CD30 (Fig. 1F), CD15 and Bcl-6,
and negative for Bcl-2 and epithelial membrane antigen (data not
shown). CD21 and CD23 staining highlighted the follicular dendritic
cell meshwork (data not shown), while Epstein-Barr virus
(EBV)-encoded small RNA (EBER) staining was negative, as determined
by in situ hybridization (11) (data not shown). In January 2013, the
patient received four cycles of doxorubicin, bleomycin, vinblastine
and dacarbazine chemotherapy (ABVD regimen; doxorubicin 25
mg/m2, on the 1st and 15th day; bleomycin 10
mg/m2, on the 1st and 15th day; vincristine 6
mg/m2, on the 1st and 15th day; dacarbazine 375
mg/m2, on the 1st and 15th day; each cycle is 28 days),
resulting in complete remission.
 | Table I.Antibody details. |
Table I.
Antibody details.
| Antibody | Clone no. | Cat. no. |
|---|
| Primary
antibodies |
|
|
|
CD1a | EP80 | ZA-0544 |
|
CD2 | UMAB6 | ZM-0278 |
|
CD3 | EP41 | ZA-0503 |
|
CD4 | EP204 | ZA-0519 |
|
CD5 | EP77 | ZA-0510 |
|
CD8 | SP16 | ZA-0508 |
|
CD10 | UMAB235 | ZM-0283 |
|
CD15 | MMA+BY87 | ZM-0037 |
|
CD20 | L26 | ZM-0039 |
|
CD21 | EP64 | ZA-0525 |
|
CD23 | EP75 | ZA-0516 |
|
CD30 | EP154 | ZA-0591 |
|
CD45 |
2B11&PD7/26 | ZM-0183 |
|
CD57 | NK1 | ZM-0058 |
|
CD68 | PG-M1 | ZM-0464 |
|
Bcl2 | EP36 | ZA-0536 |
|
Bcl6 | OTIR1D9 | ZA-0583 |
|
EMA | GP14 | ZM-0095 |
|
EBER | CS1-4 | ZM-0105 |
|
Igκ | CH15 | ZM-0160 |
|
Igλ | SHL53 | ZA-0544 |
|
IgD | / | ZA-0443 |
|
PAX5 | EP156 | ZA-0566 |
|
Pd1 | UMAB199 | ZM-0381 |
|
TdT | EP285 | ZA-0625 |
| Secondary
antibodies |
|
|
| Goat
Anti-Rabbit | / | A0545 |
| Goat
Anti-Mouse | / | A4416 |
A total of 3 years later, the patient presented with
enlarged lymph nodes in the left groin. A fresh left groin lymph
node specimen, obtained in October 2015, consisted of a
~2.0×1.5×1.0 cm lymph node, with an attached ellipse of
healthy-looking skin and benign adipose tissue measuring 2.5×1.0
cm. The patient underwent a karyotype test in October 2015,
exhibiting a karyotype of 46, XY. Histological examination using a
light microscope revealed a disrupted lymph node structure with
features suggestive of dermatopathic lymphadenitis with nodular
T-zone hyperplasia under low-power magnification (Fig. 2A) and small atypical T cells amidst
abundant blood vessels under high-power magnification (Fig. 2B). Immunohistochemical staining
identified notable CD3 expression (Fig.
2C) and a partial CD8 T-cell population(data not shown).
Immunohistochemically, the Ki67 proliferation rate was ~98%
(Fig. 2D). Consistent with a
diagnosis of PTCL, strong clonal T-cell receptor (TCR)-γ
(TRG) gene rearrangement was detected by molecular analysis
(PCR) (12) (Fig. 2E) and flow cytometry (Fig. 3A-C) with increased proliferation.
Minimal superficial perivascular lymphocytic inflammation with
scattered eosinophils was observed in the skin section (data not
shown), and no evident T-cell lymphoma was identified.
Immunohistochemical staining of B cells revealed negativity for
CD20 and PAX5, and positivity for CD2, CD3, CD5 and CD8, while T
cells were negative for CD4 and PD1, and other cell types were
negative for CD45, CD30, CD15, CD1a, CD68, terminal
deoxyribonucleotidyl transferase and EBER. Peripheral blood and
bone marrow aspirates, and a biopsy from the iliac crest exhibited
hypercellular bone marrow (60% cellularity) with panhyperplasia,
including ~20% eosinophils in both peripheral blood and bone marrow
(Fig. 2F). The marrow had an
estimated myeloid:erythroid ratio of 4:1, with no lymphoid
aggregates. Flow cytometry (13)
was positive for T-cell lymphoma, and the significance of the
positive TCR gene rearrangement was unclear in the context
of a normal complete blood count and the absence of morphologic
lymphoid aggregates. The molecular findings may have reflected
differences in sampling or may have been positive due to the
presence of some clonal cells in the peripheral blood. No HL was
detected. The patient subsequently received six cycles of
cyclophosphamide, doxorubicin, vincristine and prednisolone (CHOP
regimen; cyclophosphamide 75 mg/m2, intravenous
injection, on the 1st day; doxorubicin 50 mg/m2;
vincristine 1.4 mg/m2, up to a maximum of 2 mg,
intravenous bolus injection, on the 1st day; prednisone 60
mg/m2, oral administration, from the 1st to 5th day; 21
days per cycle) but was lost to follow-up in October 2022, having
achieved complete remission prior to this time.
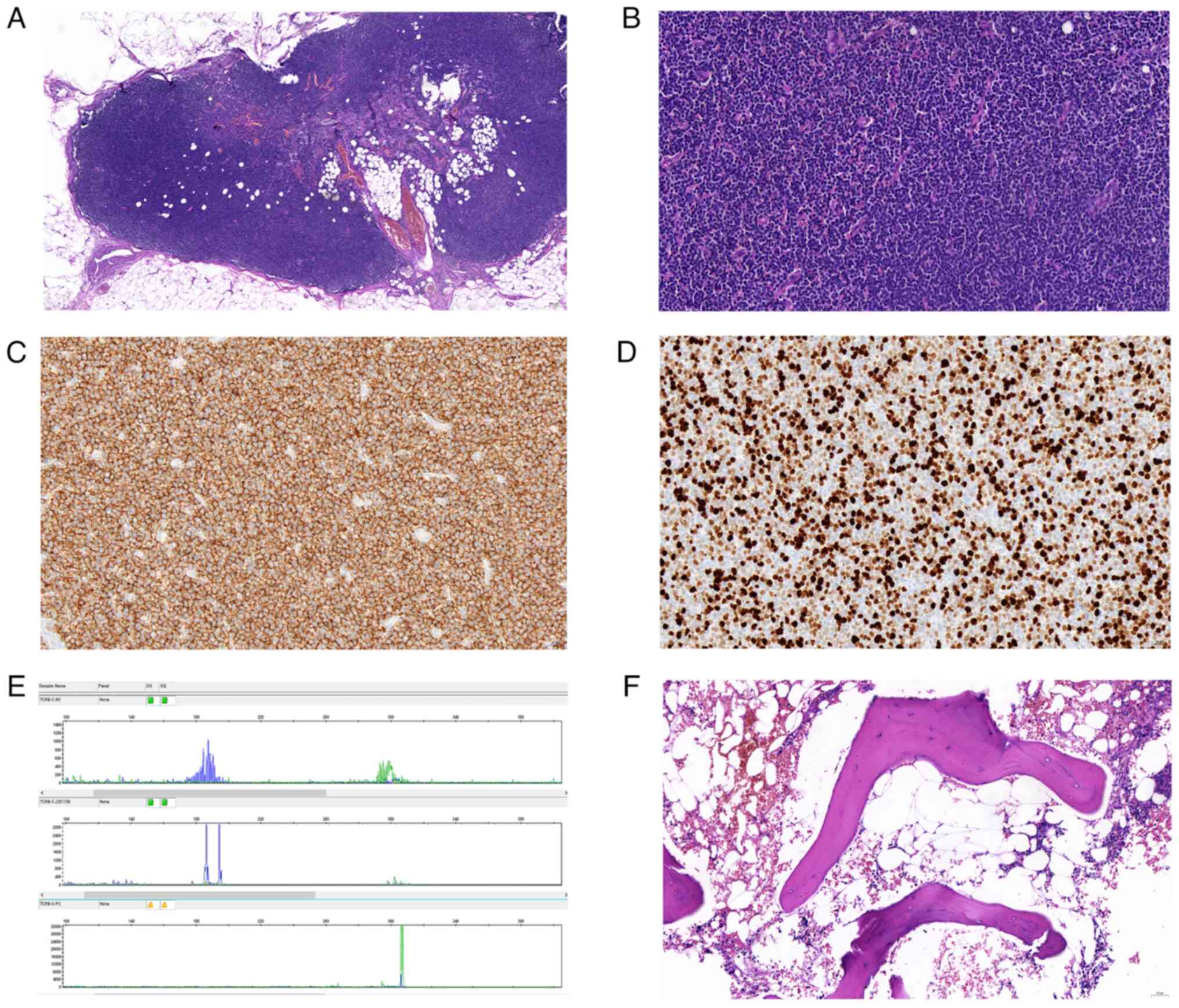 | Figure 2.H&E, partial immunohistochemistry
and T-cell receptor-γ rearrangement analysis for the diagnosis of
peripheral T-cell lymphoma. (A) Under low power, no lymphoid
aggregates were identified in T-cell immunostaining (H&E;
magnification, ×47). (B) Under high power, dermatopathic
lymphadenitis with nodular T-zone hyperplasia of small atypical T
cells, histiocytes, Langerhans cells and dendritic cells was
detectde (H&E; magnification, ×400). Immunohistochemistry
showed (C) positive CD3 staining (magnification, ×400); (D) a Ki67
proliferation rate of ~98% Ki67 (magnification, ×400). (E)
Molecular study (PCR) showed strong clonal T-cell receptor-γ gene
rearrangement. (F) Bone marrow biopsy was hypercellular (60%) with
an estimated myeloid:erythroid ratio of 4:1 (magnification, ×200).
H&E, hematoxylin and eosin. |
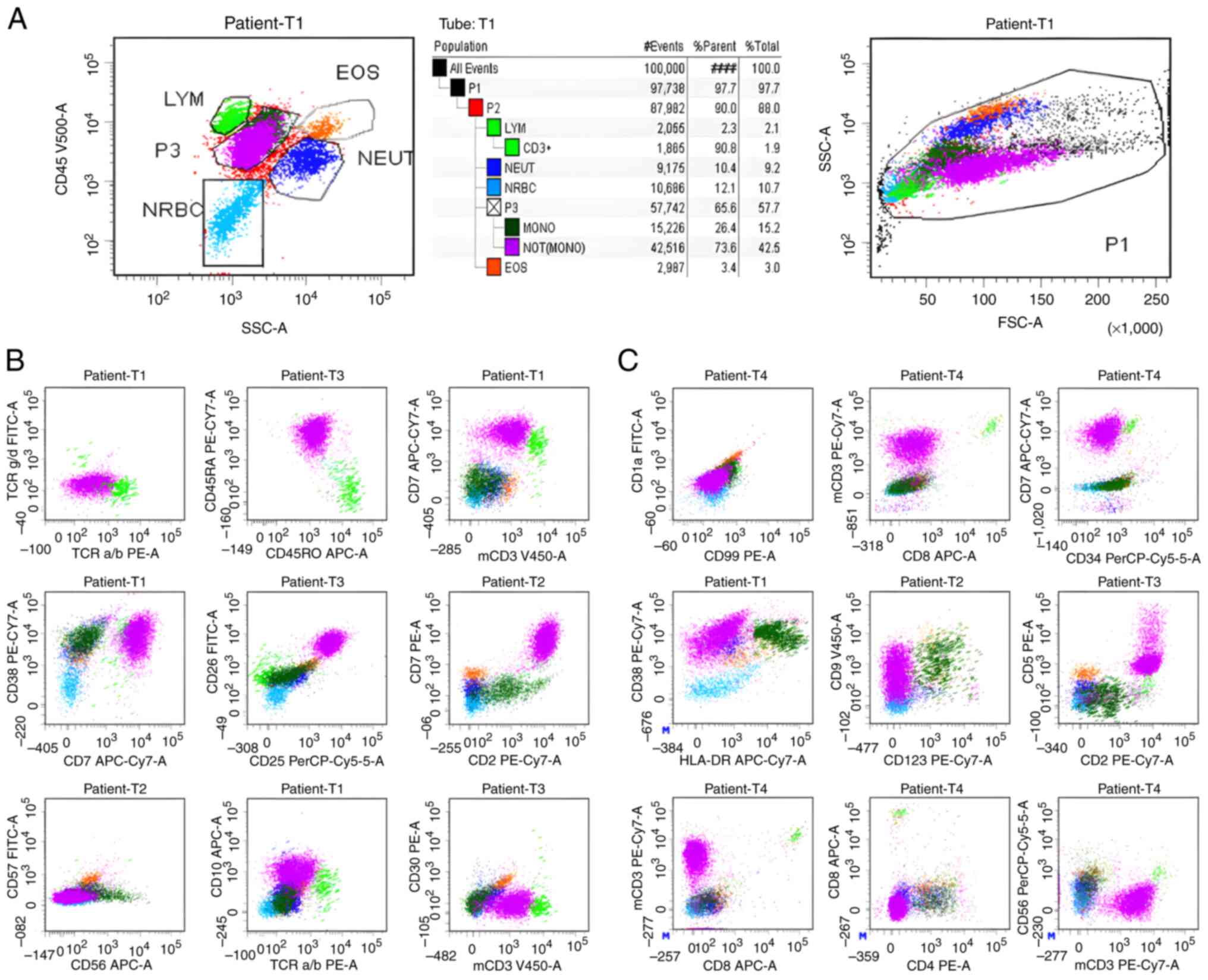 | Figure 3.(A-C) Bone marrow aspiration biopsy
diagnosis was peripheral T-cell lymphoma. (A) In the bone marrow
sample, abnormal expression of T-cell antigens was observed, with
abnormal T cells accounting for 42.5% of nucleated cells. (B) In
the CD45/SSC primitive cell region, a cluster of abnormal cells
with a larger FSC (red) was detected, which expressed CD7, CD2,
CD45RA, CD38, CD25 and CD26, and weakly expressed CD10 and mCD3.
(C) Part of the aforementioned red cell cluster expressed CD9,
weakly expressed CD5, lacked expression of CD4, CD8 and the
primitive cell marker CD34. In general, the cells in this cluster
expressed TCRab (dim), CD25, CD26, mCD3 (dim), CD7str/+, CD5dim/+,
CD45RA, CD2, CD38; and did not express CD57, CD8, CD45RO, CD16,
CD56, TCRgd, CD4, CD30, CD10, CD99, CD1a, CD34, HLA-DR. FSC,
forward scatter; dim, diminished; str, strong. |
Discussion
CHL is typically classified as a germinal center
B-cell neoplasm (4), with T-cell
lineage accounting for <5% of all cases (14). This observation is consistent with
the updated World Health Organization classification of
hematolymphoid neoplasms in 2017 (13–17).
In addition, T-cell NHL is uncommon, constituting only 7–12% of all
NHL cases (10,18). The co-occurrence of NHL and HL is
rare, particularly when the NHL component originates from the
T-cell lineage (19–21). Various explanations have been
proposed to account for the development of HL followed by T-cell
lymphoma, including therapy-induced effects,
immunodeficiency-related factors and tumor biological interactions
(7,22,23).
A search of the PubMed (https://pubmed.ncbi.nlm.nih.gov/) database identified
10 cases, including the present case, in which CHL transformed into
PTCL-NOS following treatment. A summary of the clinicopathological
features of these cases is provided in Table II. The male-to-female ratio was 9:2
(males 81.82%) and the ages ranged between 18 and 76 years (mean
age, 64 years), with most patients being middle-aged or elderly
(>45 years old; 9/11; 81.82%), although a few patients were
younger. The clinical presentations varied among cases but lymph
node enlargement and weight loss were commonly reported. Most
biopsies used for CHL diagnosis were obtained from enlarged lymph
nodes. Regarding CHL staging, cases 2 (7), 5 (24), 6 (25), 7 (26) and 8 (27) (Table
II) were specifically staged, but staging of the other cases
was unclear and was inferred from available data. Most diagnoses
occurred at an advanced stage.
| Table II.Clinical data of patients with CHL
transformed into PTCL after treatment. |
Table II.
Clinical data of patients with CHL
transformed into PTCL after treatment.
| First author,
year | Case no./Age,
years/Sex | Clinical
presentation | Biopsy site for CHL
diagnosis | CHL subtype | CHL stage | EBV infection | Treatment for
CHL | Time from diagnosis
of CHL to PTCL-NOS | Biopsy site for
PTCL-NOS diagnosis | Treatment for
PTCL-NOS | Prognosis | Mechanisms of
transformation | (Refs.) |
|---|
| Huettl, 2019 | 1/18/M | Enlarged
retroperitoneal LNs, splenomegaly, trephine multiple bone specimen
lesions and B-symptoms | Bone marrow | NA | NA |
EBER+ | 6BEACOPP
escalated | 28 months | Liver biopsy | NA | D | T-cell
clonality | (26) |
| Brown, 2004 | 2/32/M | Cervical
lymphadenopathy, prolonged bronchitis | Right posterior
cervical LN | NA | IIIA |
EBER+ | 6ABVD | 2 years | Left submandibular
lymphadenopathy | Salvage
chemotherapy followed by autologous stem cell transplantation | NA |
Immunodeficiency | (7) |
| Nakazaki, 2022 | 3/47/M | Scaly erythematous
patches on the upper and lower limbs with marked pigmentation,
extensive squamous erythema and obvious pigmentation of the right
armpit | Right axillary
LN | NS | NA |
EBV-LMP-1− | 6AAVD | 2 years | Left thigh | NA | NA | Blocking of IL-13
and IL-4 pathways | (9) |
| Mohrmann, 2000 | 4/47/M | Right axillary LN
enlargement, mild fatigue, weight loss and flu-like symptom | Right axillary
LN | NS | NA |
EBV-LMP-1− | 6ABVD | 5 years | Posterior cervical
LN | NA | Remission | Immunodeficiency,
chemotherapy | (29) |
| Wlodarska,
1993 | 5/54/M | Weight loss, fever
and symptoms of arthritis | Cervical LN | NS | IIIA | NA | 8MOPP/ABVD | 2 years | Cervical LN | NA | NA |
Immunodeficiency | (24) |
| Chang, 2015 | 6/64/F | Abrupt weight loss,
abdominal pain and bloating | Left
supraclavicular LN | NS | IIIB, or possibly
stage IV/BE |
EBER− | 3ABVD | 2 years | Right inguinal
LN | 3 cycles
etoposide-containing regimen | D | Immunodef
iciency | (25) |
| Huettl, 2019 | 7/65/M | Enlargement of LNs
in left armpit and neck, weight loss | Cervical LN | NS | IB |
EBER− | 4ABVD, 30 Gy
radiotherapy | 10 months | Left-sided inguinal
lymphadenopathy and urosepsis | Antibiotics | NA | T-cell
clonality | (26) |
| Niedobitek,
2000 | 8/65/M | Weight loss, fever
and general malaise | Left axillary
LN | NS | IVB |
EBER+ | COPP/ABVD | 4 years | Cervical LN | CEVD | D | EBV infection | (27) |
| Zhu, 2016 | 9/72/M | Weight loss, poor
mental health, emaciation, occasional fever | Cervical LN | MC | NA |
EBER+ | 6ABVD | 3 years | Cervical LN | NA | D | Chemotherapy | (28) |
| Oka, 2000 | 10/76/F | A palm sized, ill-
defined and elastic hard tumor on the right forearm and multiple
subcutaneous tumors on the back; B symptoms, including fever and
general signs | Submandibular
LN | NA | NA |
EBER+ | ABVD | 9 years | Skin tumor | CHOP | D | EBV infection | (30) |
| Song, 2024 | Present
case/73 | LN enlargement | Left cervical
LN | NS | NA |
EBER− | 4ABVD | 3 years | Left inguinal
LN | CHOP | NA | Chemotherapy and
immunodeficiency | NA |
Regarding EBV infection, except for case 5 (18) in whom the association was not
clearly indicated, cases 1 (26), 2
(7), 8 (27) and 9 (28) were reported to be EBV-positive at
the initial diagnosis of CHL, whereas cases 3 (9), 4 (29), 6 (25), 7 (26) and the current case were
EBV-negative. In terms of the mechanism underlying transformation,
Huettl et al (26) reported
the case of two patients in whom lymphoma transformation was
suggested to be related to T-cell clonality, whereas Nakazaki et
al (9) reported one patient in
whom lymphoma transformation could be associated with the
interleukin (IL)-13 and IL-4 pathways. Additionally, lymphoma
transformation might have been related to immunodeficiency in cases
4 (29), 5 (24), 6 (25) and the current case, while there was
a possible association with chemotherapy in cases 4 (29), 6 (25), 9 (28) and the present case, and a potential
link to EBV infection in cases 8 (27) and 10 (30). Some researchers (30) have suggested EBV infection as a
possible factor in the early onset of T-cell lymphoma, whereas
others (9) have proposed different
mechanisms. Nakazaki et al (9) reported a case of HL in a patient
treated with dupilumab for 1 year, who was diagnosed with a rare
combination of discordant HL and PTCL. This previous study
emphasized the need for vigilance regarding the potential
development of lymphoma associated with the IL-13 and IL-4 pathways
in patients with unresponsive atopic dermatitis treated with
dupilumab, and suggested the need to consider the possibility of
complex or discordant lymphomas in the diagnosis and treatment of
lymphoma (9). Other researchers
have argued that most cases of PTCL following treatment for HL
could result from therapy-induced immunodeficiency, rather than
from clonal progression (7). Clonal
rearrangement of TCR genes, as an accepted diagnostic
feature of T-cell lymphoma, was observed in most cases according to
a previous study (31). There have
also been reports of Ig heavy-chain gene rearrangements in
Reed-Sternberg cells (31,32), but further studies are needed to
clarify this phenomenon. A previous study (31) proposed that TCR and
IGH clonal rearrangements indicated that the tumor cells
were not clonally related, but may occur simultaneously and inhabit
the same immune microenvironment. Brown et al (7) described four patients with composite
lymphomas, one of whom showed TCR rearrangement in
Reed-Sternberg cells, raising the possibility that HL may evolve
from basal T-cell NHL. The present study describes the case of an
elderly male patient who developed PTCL-NOS 3 years after
chemotherapy for an initial diagnosis of nodular-sclerosing CHL.
Mohrmann and Arber (29) reported a
similar case of composite lymphoma at presentation, although their
patient was diagnosed with PTCL-NOS after 2 years of intermittent
chemotherapy; however, the existence of composite lymphoma at the
time of CHL diagnosis remains uncertain, because only a cervical
lymph node was obtained and confirmed as HL. The occurrence of
PTCL-NOS in this previously described case may thus be related to
chemotherapy.
Following diagnosis, nine patients, including the
current patient, underwent ABVD chemotherapy, whereas case 1
(26) received bleomycin,
etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine
and prednisone (BEACOPP regimen) and radiotherapy, respectively,
but all progressed to PTCL-NOS at varying intervals, with the
longest duration being 9 years in case 10 (30) and the shortest being 10 months in
case 7 (26). Regarding the
treatment plans for patients diagnosed with PTCL-NOS, the treatment
regimen was not specified in cases 1 (26), 3 (9), 4 (29)
and 5 (24), while the other
patients received different treatment modalities. Brown et
al (7) reported on one patient
(case 2) who underwent autologous stem cell transplantation
post-chemotherapy, whereas Chang et al (25) documented a patient (case 6) who was
treated with an etoposide regimen for three cycles. Huettl et
al (26) reported a patient
(case 7) who was treated with antibiotic therapy, and Niedobitek
et al (27) reported on one
patient (case 8) treated with cyclophosphamide, etoposide,
vindesine and dexamethasone (CEVD regimen). Additionally, Zhu et
al (28) reported on a patient
(case 9) who did not receive any treatment, whereas Oka et
al (30) reported on a patient
(case 10) who was treated with the CHOP regimen. Concerning the
prognosis, cases 2 (7), 3 (9), 5 (24)
and 7 (26) had unspecified
outcomes. The current patient and case 4 (29) achieved remission after treatment;
however, the remaining patients succumbed during follow-up.
Notably, all CHL subtypes presented with nodular sclerosis, except
for cases with unspecified subtypes [cases 1 (26), 2 (7), 10 (30)] and one patient [case 9 (28)] with mixed-cell type. Cases 3
(9), 4 (29), 5 (24), 7 (26), 8 (27), 9 (28) and the present case predominantly
involved middle-aged and elderly men initially diagnosed with CHL
that subsequently transformed into PTCL-NOS post-treatment. Cases 6
(25) and 10 (30) involved elderly women, while one
patient (case 10) (30) was
initially diagnosed with mixed-cell type of HL, which transformed
into PTCL-NOS after treatment. These cases suggest that increased
attention should be paid to the treatment of middle-aged and
elderly patients with nodular sclerosing-type CHL in clinical
practice, given the higher likelihood of transformation into
PTCL-NOS post-treatment in these patients. Once PTCL-NOS develops,
the prognosis is typically poor. No apparent immunodeficiency was
noted in the patient described in the present study prior to CHL
diagnosis, suggesting that PTCL-NOS may have been primarily caused
by the chemotherapy. The transformation of CHL into PTCL-NOS
post-treatment is rare, but the mechanism underlying this
transformation may be associated with the complex tumor
microenvironment of CHL. Although the present study conducted a
retrospective review of the relevant literature, the lack of
practical clinical data and experience may have led to different
conclusions, recommendations and actual situations, representing a
significant limitation of this study. The precise mechanisms thus
remain unclear. Notably, a full understanding of rare and/or
complex diseases often requires the accumulation of data over a
long period of time, and despite the thorough exploration of
existing research materials, the scarcity of cases means that some
pathophysiological processes may have been missed. The conclusions
are also limited by the lack of detailed analysis of different
patient groups. Rare or complex diseases may manifest differently
and have distinct mechanisms in diverse populations, influenced by
factors such as age, sex, genetic background and lifestyle;
however, these factors were not fully considered because of
constraints in terms of research resources and time, potentially
leading to a less understanding of specific patient groups.
In conclusion, the transformation from CHL to
PTCL-NOS following treatment is rare, with a predilection towards
men. Nodular sclerosing-type CHL is the predominant subtype prone
to transformation into PTCL-NOS. Patients undergoing this
transformation typically exhibit a poor prognosis, with potential
mechanisms linked to the intricate tumor microenvironment
characteristic of CHL. Future research should thus place greater
emphasis on integration with clinical practice, to collect and
analyze clinical data to validate and refine the theoretical
models. In summary, although this case report and literature review
may improve understanding of the relevant mechanisms underlying the
transformation to PTCL-NOS following treatment for CHL, there
remain a number of limitations and challenges. Further research and
analysis are needed to clarify the relevant mechanisms and provide
stronger support for the accurate diagnosis and effective treatment
of patients.
Acknowledgements
Not applicable.
Funding
This project was supported by the Shandong Natural Science
Foundation (grant no. ZR2022MH297) and the project Yantai Science
and Technology Plan (grant no. 2021MSGY043).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
LS, YaY and GY designed the project. LS wrote the
draft, and WW, NZ and GY revised the manuscript. NZ, YP and LS
completed the revision of the article, including image
modification, data management, data analysis and figure generation.
WW performed flow cytometry. YiY, JW and YG prepared the figures,
collected data and prepared tissue slides. SW and YW collected the
literature and analyzed the data. PY and XS performed the molecular
test and pathological diagnosis. GY and YaY confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Based on the ethical rules for biomedical research
related to humans issued by the national health and family planning
commission, and The Declaration of Helsinki, the Ethical Review
Board discussed the study protocol and informed consent, and voted
anonymously on June 21, 2020. The Institutional Ethical Review
Board decided that the main participants qualified for clinical
study, the study design was eligible, practical and scientific, and
the rights and interests of the patients were fully protected. The
present study was approved by the Institutional Ethical Review
Board of Yantai Yuhuangding Hospital (approval no. [2020-43]) and
this was valid for 1 year from the date of approval.
Patient consent for publication
Written informed consent was obtained from the
patient prior to the enrollment into this case report.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yung L and Linch D: Hodgkin's lymphoma.
Lancet. 361:943–951. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brice P, de Kerviler E and Friedberg JW:
Classical Hodgkin lymphoma. Lancet. 398:1518–1527. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vose J, Armitage J and Weisenburger D;
International T-Cell Lymphoma Project, : International peripheral
T-cell and natural killer/T-cell lymphoma study: Pathology findings
and clinical outcomes. J Clin Oncol. 26:4124–4130. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Swerdlow SH, Campo E, Harris NL, Jaffe ES,
Pileri SA, Stein H and Thiele J: WHO Classification of Tumours of
Haematopoietic and Lymphoid Tissues. 4th Edition. Vol 2.
International Agency for Cancer Research; Lyon: 2017
|
|
5
|
Weiss J, Reneau J and Wilcox RA: PTCL,
NOS: An update on classification, risk-stratification, and
treatment. Front Oncol. 13:11014412023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moskowitz AJ, Stuver RN and Horwitz SM:
Current and upcoming treatment approaches to common subtypes of
PTCL (PTCL NOS, ALCL, TFHs). Blood blood. Feb 2–2024.(Epub ahead of
print). View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brown JR, Weng AP and Freedman AS: Hodgkin
disease associated with T-cell non-Hodgkin lymphomas: Case reports
and review of the literature. Am J Clin Pathol. 121:701–708. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weniger MA and Küppers R: Molecular
biology of Hodgkin lymphoma. Leukemia. 35:968–981. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nakazaki K, Yoshida M, Masamoto Y,
Shinozaki-Ushiku A, Ikemura M, Hisamoto T, Yasunaga M, Sato S and
Kurokawa M: Discordant lymphomas of classic Hodgkin lymphoma and
peripheral T-cell lymphoma following dupilumab treatment for atopic
dermatitis. Int J Hematol. 116:446–452. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thirumala S, Esposito M and Fuchs A: An
unusual variant of composite lymphoma: A short case report and
review of the literature. Arch Pathol Lab Med. 124:1376–1378. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hamilton-Dutoit SJ, Raphael M, Audouin J,
Diebold J, Lisse I, Pedersen C, Oksenhendler E, Marelle L and
Pallesen G: In situ demonstration of Epstein-Barr virus small RNAs
(EBER 1) in acquired immunodeficiency syndrome-related lymphomas:
Correlation with tumor morphology and primary site. Blood.
82:619–624. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gong Q, Wang C, Zhang W, Iqbal J, Hu Y,
Greiner TC, Cornish A, Kim JH, Rabadan R, Abate F, et al:
Assessment of T-cell receptor repertoire and clonal expansion in
peripheral T-cell lymphoma using RNA-seq data. Sci Rep.
7:113012017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vermeer MH, Moins-Teisserenc H, Bagot M,
Quaglino P and Whittaker S: Flow cytometry for the assessment of
blood tumour burden in cutaneous T-cell lymphoma: Towards a
standardized approach. Br J Dermatol. 187:21–28. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Müschen M, Rajewsky K, Bräuninger A, Baur
AS, Oudejans JJ, Roers A, Hansmann ML and Küppers R: Rare
occurrence of classical Hodgkin's disease as a T cell lymphoma. J
Exp Med. 191:387–394. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Seitz V, Hummel M, Marafioti T,
Anagnostopoulos I, Assaf C and Stein H: Detection of clonal T-cell
receptor gamma-chain gene rearrangements in Reed-Sternberg cells of
classic Hodgkin disease. Blood. 95:3020–3024. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Krenacs L, Wellmann A, Sorbara L,
Himmelmann AW, Bagdi E, Jaffe ES and Raffeld M: Cytotoxic cell
antigen expression in anaplastic large cell lymphomas of T- and
null-cell type and Hodgkin's disease: Evidence for distinct
cellular origin. Blood. 89:980–989. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Oudejans JJ, Kummer JA, Jiwa M, van der
Valk P, Ossenkoppele GJ, Kluin PM, Kluin-Nelemans JC and Meijer CJ:
Granzyme B expression in Reed-Sternberg cells of Hodgkin's disease.
Am J Pathol. 148:233–240. 1996.PubMed/NCBI
|
|
18
|
Steinhoff M, Hummel M, Assaf C,
Anagnostopoulos I, Treudler R, Geilen CC, Stein H and Orfanos CE:
Cutaneous T cell lymphoma and classic Hodgkin lymphoma of the B
cell type within a single lymph node: composite lymphoma. J Clin
Pathol. 57:329–331. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Delabie J, Greiner TC, Chan WC and
Weisenburger DD: Concurrent lymphocyte predominance Hodgkin's
disease and T-cell lymphoma. A report of three cases. Am J Surg
Pathol. 20:355–362. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bräuninger A, Hansmann ML, Strickler JG,
Dummer R, Burg G, Rajewsky K and Küppers R: Identification of
common germinal-center B-cell precursors in two patients with both
Hodgkin's disease and non-Hodgkin's lymphoma. N Engl J Med.
340:1239–1247. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang Q, Wilczynski SP, Chang KL and Weiss
LM: Composite recurrent hodgkin lymphoma and diffuse large B-cell
lymphoma: One clone, two faces. Am J Clin Pathol. 126:222–229.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Amini RM, Enblad G, Sundström C and
Glimelius B: Patients suffering from both Hodgkin's disease and
non-Hodgkin's lymphoma: A clinico-pathological and
immuno-histochemical population-based study of 32 patients. Int J
Cancer. 71:510–516. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Steinhoff M, Assaf C, Anagnostopoulos I,
Geilen CC, Stein H and Hummel M: Three coexisting lymphomas in one
patient: Genetically related or only a coincidence? J Clin Pathol.
59:1312–1315. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wlodarska I, Delabie J, De Wolf-Peeters C,
Mecucci C, Stul M, Verhoef G, Cassiman JJ and Van den Berghe H:
T-cell lymphoma developing in Hodgkin's disease: Evidence for two
clones. J Pathol. 170:239–248. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chang SH and Lee HR: Peripheral T Cell
Non-Hodgkin's Lymphoma following Treatment of Hodgkin's Lymphoma.
Case Rep Oncol Med. 2015:4383852015.PubMed/NCBI
|
|
26
|
Huettl KS, Staiger AM, Stehle A, Bonzheim
I, Horn H, Borgmann V, Ott M, Fend F and Ott G: Peripheral T-cell
lymphoma NOS arising in patients with classical Hodgkin lymphoma of
cytotoxic phenotype. Leuk Lymphoma. 60:3561–3564. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Niedobitek G, Baumann I, Brabletz T,
Lisner R, Winkelmann C, Helm G and Kirchner T: Hodgkin's disease
and peripheral T-cell lymphoma: Composite lymphoma with evidence of
Epstein-Barr virus infection. J Pathol. 191:394–399. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhu X, Wu MJ, Yin WJ and Sun WY:
Peripheral T-cell lymphoma secondary to EBV positive classical
Hodgkin's lymphoma: A case report and literature review. Chin J
Cancer Prev Treat. 1:56–59. 2016.(In Chinese).
|
|
29
|
Mohrmann RL and Arber DA: CD20-Positive
peripheral T-cell lymphoma: Report of a case after nodular
sclerosis Hodgkin's disease and review of the literature. Mod
Pathol. 13:1244–1252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Oka K, Nagayama R, Iijima S, Yonekawa N,
Hirosawa K, Yatabe Y and Mori N: Epstein-Barr virus-associated
lymphoproliferative disorder presenting with classical Hodgkin
lymphoma and developing as peripheral T-cell lymphoma 9 years
later: A case report of composite lymphoma. Pathol Int. 61:752–755.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gualco G, Chioato L, Van Den Berg A, Weiss
LM and Bacchi CE: Composite lymphoma: EBV-positive classic Hodgkin
lymphoma and peripheral T-cell lymphoma: A case report. Appl
Immunohistochem Mol Morphol. 17:72–76. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Stein H, Diehl V, Marafioti T, Jox A, Wolf
J and Hummel M: The nature of Reed-Sternberg cells, lymphocytic and
histiocytic cells and their molecular biology in Hodgkin's disease.
Lippincott Williams &Wilkins; Philadelphia, PA, USA: pp.
121–138. 1999
|














