Introduction
Atractyloside, a diterpenoid glycoside, is a
naturally occurring active component common in medicines and animal
feed, particularly in plants (Asteraceae and Atractylis)
native to east Asia. Atractyloside has been used in traditional
Chinese medicine to treat nasal congestion and allergic rhinitis
(1). Numerous studies (2–4) have
demonstrated the biological functions of atractyloside, indicating
that it is a strong candidate for the treatment of illnesses,
including digestive disorders, liver injury and diabetes. According
to a pharmacological clinical study in humans (1), atractyloside has been demonstrated to
inhibit the mitochondrial ATP translocase, also known as the
adenine nucleotide translocator, and to effectively reduce fat
accumulation in the liver (steatosis) and improve insulin
sensitivity, which protects the liver. Cho et al (5) revealed that atractyloside is a modest
hypoglycemic agent in splenocytes, thus suggesting that it may be
used to treat diabetes. Previous research has demonstrated that
atractyloside inhibits mitochondrial ATP transporters, leading to
cancer cell death (6).
The incidence and mortality rates of cancer are
rapidly increasing worldwide. In both sexes, lung cancer is the
most commonly diagnosed type of cancer (11.6% of total cases) and
the leading cause of cancer-related deaths (18.4% of total cancer
deaths) worldwide (7). Lung cancer
is histologically classified as small-cell lung cancer (SCLC) and
non-SCLC (NSCLC), and ~85% of patients have NSCLC. Of these
patients, lung squamous cell carcinoma and lung adenocarcinoma are
the most common subtypes, accounting for 40 and 20–25% of global
cases, respectively (8,9). Notably, the application of biomarkers
for NS CLC is clinically beneficial. Epidermal growth factor
receptor (EGFR) is a well-known biomarker for NSCLC management
(10). Although small-molecule
tyrosine kinase inhibitors (TKIs), such as gefitinib, have curative
effects, relapse caused by EGFR mutations usually lead to patients
succumbing to the disease 2 years after the first diagnosis
(11). First-generation EGFR
inhibitors, such as gefitinib and erlotinib, have significantly
improved the survival of patients with NSCLC; however, the
secondary EGFR-T790M mutation leads to clinical resistance to
first-generation EGFR-TKIs (11–13).
New biomarkers may improve the diagnosis and treatment of
NSCLC.
Brother of the regulator of imprinted sites
(BORIS, also known as CTCFL)which is a paralog of
CCCTC-binding factor, is commonly expressed in most types of
cancer, whereas it is not expressed in the corresponding normal
tissues; therefore, it is considered a potential therapeutic target
for lung cancer (14–16). In our previous study, it was
revealed that BORIS suppressed apoptosis and enhanced
5-fluorouracil resistance in colorectal cancer (17), and BORIS has also been
reported to increase resistance to cisplatin treatment in NSCLC
(12). Debruyne et al
(18) reported that BORIS
may be associated with various tumor occurrences, including brain
cancer and cervical cancer, drug resistance and the prognosis of
patients with cancer. Based on the ubiquitous expression of
BORIS and the high incidence of EGFR resistance in NSCLC, it
is worth studying whether BORIS influences targeted
therapies for lung cancer.
In the present study, the association between
BORIS and TKI-resistant NSCLC was assessed. In addition,
atractyloside was used to mimic BORIS knockdown to study the
therapeutic function of BORIS on the prevention of TKI resistance.
The results revealed that atractyloside could facilitate TKIs to
suppress NSCLC cell proliferation.
Materials and methods
Cell culture
NSCLC cancer cell lines H1299, PC-9, PC-9-IR and
H1975, and the colorectal cancer cell line Caco2 were purchased
from The Cell Bank of Type Culture Collection of The Chinese
Academy of Sciences. All cells were cultured in RPMI 1640 medium
(Nanjing BioChannel Biotechnology Co., Ltd.) containing 10%
heat-inactivated fetal bovine serum (GeminiBio) at 37°C in an
incubator containing 5% CO2.
Cell transfection and treatment
Lipofectamine® RNAiMAX reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) was used to transfect
small interfering RNA (siRNA) into cells. According to the
manufacturer's protocol, H1975, PC-9 and PC-9IR cells at 70%
confluence were used for transfection. Briefly, 1 pmol siRNA/well
was used for the transfection of cells on a 96-well plate, and 30
pmol siRNA/well was used for the transfection of cells on a 6-well
plate. After a 5-min incubation at room temperature, the
RNAiMAX-siRNA mixture was added to either 6-well or 96-well plates.
The cells were incubated for 48 h without media replacement, after
which subsequent experiments were performed. The 96-well plates
were used for MTT and cell proliferation assays. The 6-well plates
were used for the analysis of transcript or protein expression
levels. The siRNA sequences used in the present study are listed in
Table I. Negative control siRNA and
siBORIS were synthesized by Xiangyin Biotechnology Co., Ltd.
The cells were then incubated at 37°C in an incubator containing 5%
CO2. After 48 h, the cells were used for subsequent
experiments. Following transfection, cells were treated with their
respective drug treatments, with the control group receiving an
equal volume of DMSO. Atractyloside (cat. no. HY-N1462), the TKI
inhibitor gefitinib (cat. no. HY-50895) and erlotinib (cat. no.
HY-50896) were purchased from MedChemExpress. Cells were treated
with gefitinib (50 µM) 4 h post-transfection at room temperature.
Cells were subjected to experiments after 48 or 96 h of gefitinib
treatment. In addition, cells were treated with gefitinib (1 µM),
erlotinib (5 µM), or atractyloside (1, 2.5 or 5 µM) for 48 h at
37°C prior to performing Cell Counting Kit (CCK)-8 assays, western
blotting and reverse transcription-quantitative PCR (RT-qPCR).
 | Table I.siRNA sequences using for
BORIS knockdown. |
Table I.
siRNA sequences using for
BORIS knockdown.
| siRNA | Forward, 5′-3′ | Reverse, 5′-3′ |
|---|
| Negative control
siRNA |
UUCUCCGAACGUGUCACGUdTdT |
ACGUGACACGUUCGGAGAAdTdT |
| BORIS
siRNA |
GGAAAUACCACGAUGCAAATT |
UUUGCAUCGUGGUAUUUCCtt |
Cell viability analysis
A total of 3,000 cells/well were seeded in a 96-well
plate for transfection or drug treatment. Subsequently, MTT (500
µg/ml; cat. no. M2128; Sigma-Aldrich; Merck KGaA) was added to the
cells and incubated for 4 h at 37°C, and 100 µl dimethyl sulfoxide
was added for 15 min at room temperature. Signals were recorded
using a BioTek Synergy 2 plate reader at a wavelength of 490 nm
(BioTek; Agilent Technologies, Inc.).
CCK-8
A total of 3,000 cells/well were seeded in a 96-well
plate and underwent drug treatment. After treatment with drugs
(gefitinib, 1 µM; erlotinib, 5 µM; atractyloside, 5 µM) for 48 h at
37°C, the cell culture medium was discarded, and 100 µl medium
containing 10 µl CCK-8 (cat. no. K1018; APeXBIO Technology LLC)
reagent was added. The cells were then incubated for 1 h at 37°C
and signals were recorded using a BioTek Synergy 2 plate reader at
a wavelength of 450 nm.
Western blotting
H1299, Caco2, PC-9 and H1975 cells were cultured in
a 6-well plate and were lysed using RIPA buffer (cat. no. 20188;
MilliporeSigma) containing PMSF (1:100; cat. no. ST506; Beyotime
Institute of Biotechnology) and Roche cOmplete™ Protease Inhibitor
Cocktail (1:25; cat. no. 04693116001; Sigma-Aldrich; Merck KGaA).
After centrifugation at 12,000 × g for 30 min at 4°C, the
supernatants were collected, and the total protein was quantified
using a detergent-compatible Bradford protein assay kit (cat. no.
P0006C; Beyotime Institute of Biotechnology). Samples (30 µg/lane)
were separated by SDS-PAGE on a 10% gel and were transferred onto a
PVDF membrane (cat. no. ISEQ00010-PVDF; MilliporeSigma). The
membrane was blocked with a protein-free rapid blocking buffer
(cat. no. PS108P; New Cell & Molecular Biotech Co., Ltd.) for
15 min at room temperature and then incubated at 4°C overnight with
the following antibodies: Mouse anti-GAPDH (1:500,000; cat. no.
60004-1-Ig; Proteintech Group, Inc.), rabbit anti-XRCC4 (1:1,000;
cat. no. 15817-1-AP; Proteintech Group, Inc.), mouse anti-BORIS
(1:1,000; cat. no. sc-377085; Santa Cruz Biotechnology, Inc),
rabbit anti-AKT (1:1,000; cat. no. 9272; Cell Signaling Technology,
Inc.) and mouse anti-phosphorylated (p)-AKT (1:1,000; cat. no.
66444-1-Ig; Proteintech Group, Inc.). After washing with TBS-1%
Tween (TBST) three times (10 min/wash), the membrane was incubated
with HRP-conjugated secondary antibodies (anti-rabbit and
anti-mouse; 1:5,000; cat. nos. DW-GAR007 and DW0990-100; Hangzhou
Dawen Biological Co., Ltd.) for 2 h at room temperature. Signals
were detected after washing with TBST three times (10 min/wash)
using the Ultrasensitive ECL Kit (cat no. P2300; New Cell &
Molecular Biotech Co., Ltd)and ChemiDoc XRS+ system (Bio-Rad
Laboratories, Inc.). The relative expression of the protein bands
was semi-quantified using ImageJ version 1.53 software (National
Institutes of Health).
RT-qPCR
The RNA of treated cells was extracted using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) through ethanol precipitation. cDNA was reverse transcribed
using the Hifair® II 1st Strand cDNA Synthesis Kit (gDNA
digester plus) (cat. no. 11121ES60; Shanghai Yeasen Biotechnology
Co., Ltd.), and was used for qPCR analysis. For RT, the temperature
settings were as follows: 25°C for 5 min, 42°C for 30 min and 85°C
for 5 min. qPCR was performed using the 2X T5 Fast qPCR Mix (SYBR
Green; cat. no. 11201ES08; Shanghai Yeasen Biotechnology Co., Ltd.)
and a CFX connect real-time PCR detection system (Bio-Rad
Laboratories, Inc.). According to the manufacturer's protocol, the
thermocycling conditions were as follows: Initial denaturation at
95°C for 5 min, followed by 40 cycles of denaturation at 95°C for
15 sec and annealing/extension at 60°C for 30 sec. GAPDH was used
as an internal reference for normalization. The primers used for
qPCR are listed in Table II. The
qPCR results were analyzed using the 2−ΔΔCq method
(19).
| Table II.Primer sequences used for reverse
transcription-quantitative PCR. |
Table II.
Primer sequences used for reverse
transcription-quantitative PCR.
| Gene name | Forward, 5′-3′ | Reverse, 5′-3′ |
|---|
| BORIS
(CTCFL) |
CAGGCCCTACAAGTGTAACGACTGCAA |
GCATTCGTAAGGCTTCTCACCTGAGTG |
| GAPDH |
CCCACTCCTCCACCTTTGAC |
TGTTGCTGTAGCCAAATTCGT |
| XRCC4 |
ATGGCTCCTCAGGAGAATCAGC |
GAGGTCTTCTGGGCTGCTGTTT |
| MSH6 |
CCAAGGCGAAGAACCTCAAC |
ACCAGGGGTAACCCTCCATC |
|
BRCA-1 |
ACTCTGAGGACAAAGCAGCG |
CATCCCTGGTTCCTTGAGGG |
| c-myc |
AAGCCAAGGACTGTCTGAACG |
GGGACGAGTAATTCTTTCCCCT |
Bioinformatics analysis
BORIS expression in NSCLC was determined
using the R2 Genomics Analysis and Visualization Platform
(http://r2.amc.nl). The Gene Expression Omnibus
(https://www.ncbi.nlm.nih.gov/geo/)
datasets GSE19188 (20) and
GSE63074 (21) were utilized for
bioinformatics analysis.
Statistical analysis
GraphPad Prism 8 software (Dotmatics) was used for
all statistical analyses. All experiments were performed in
triplicate. Data are presented as the mean ± standard deviation.
Statistical differences were calculated using one-way or two-way
ANOVA followed by Tukey's multiple comparisons test, paired
Student's t-test or unpaired Student's t-test. P<0.05 was
considered to indicate a statistically significant difference.
Results
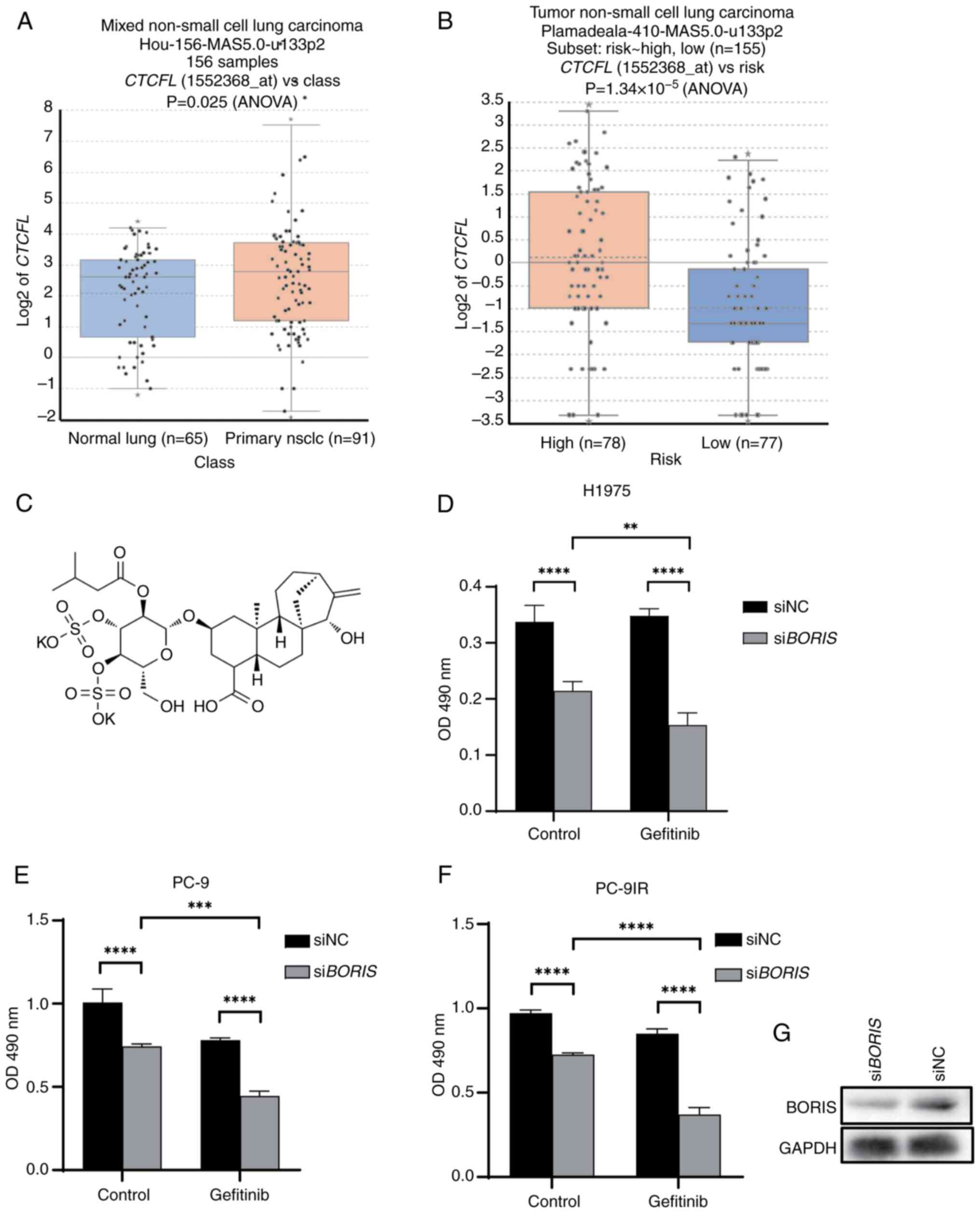
Bioinformatics analysis
Bioinformatics analysis was performed using the R2
Genomics Analysis and Visualization Platform. Based on the GSE19188
dataset, BORIS expression was elevated in primary NSCLC
tissues compared with in normal lung tissues (Fig. 1A). From the GSE63074 dataset,
survival rate data were used to perform a risk stratification
analysis, categorizing patients into high-risk and low-risk groups
according to a previously described method (22); the analysis indicated a significant
upregulation of BORIS expression in the high-risk NSCLC group
(Fig. 1B). Given that EGFR
upregulation or mutation was prevalent in high-risk NSCLC cases,
further investigation into the potential association between BORIS
and EGFR expression/mutation is warranted.
BORIS knockdown inhibits H1975 cell
viability
The H1975 cell line comprises NSCLC cells resistant
to gefitinib due to the T790M mutation. The present study knocked
down BORIS in H1975 cells and the results demonstrated that
the cell viability was significantly decreased in response to
successful transfection with siBORIS (Fig. 1D). This finding indicated that the
presence of BORIS may maintain the stability of H1975 cells
and that its knockdown could be beneficial for treating
drug-resistant lung cancer.
BORIS knockdown, alongside gefitinib
treatment, inhibits NSCLC cell viability
To further explore the function of BORIS in
TKI resistance, PC-9, PC-9IR and H1975 cells underwent BORIS
knockdown and gefitinib treatment. PC-9 is an EGFR wild-type NSCLC
cell line, whereas PC-9IR and H1975 are EGFR-mutant cells that are
resistant to TKIs.
siBORIS effectively reduced the viability of
NSCLC cells (Fig. 1D-F). All cell
transfections were successful. When siBORIS was combined
with gefitinib treatment, cell viability was significantly reduced
compared with gefitinib treatment alone, with a more obvious effect
observed on the drug-resistant cell lines H1975 and PC9-IR
(Fig. 1D and 1F). Verification of
the knockdown efficiency of siBORIS is presented in Fig. 1G. These findings suggested that the
knockdown of BORIS may be beneficial for lung cancer
resistance. Western blot analysis confirmed that the expression of
BORIS was decreased in response to siBORIS.
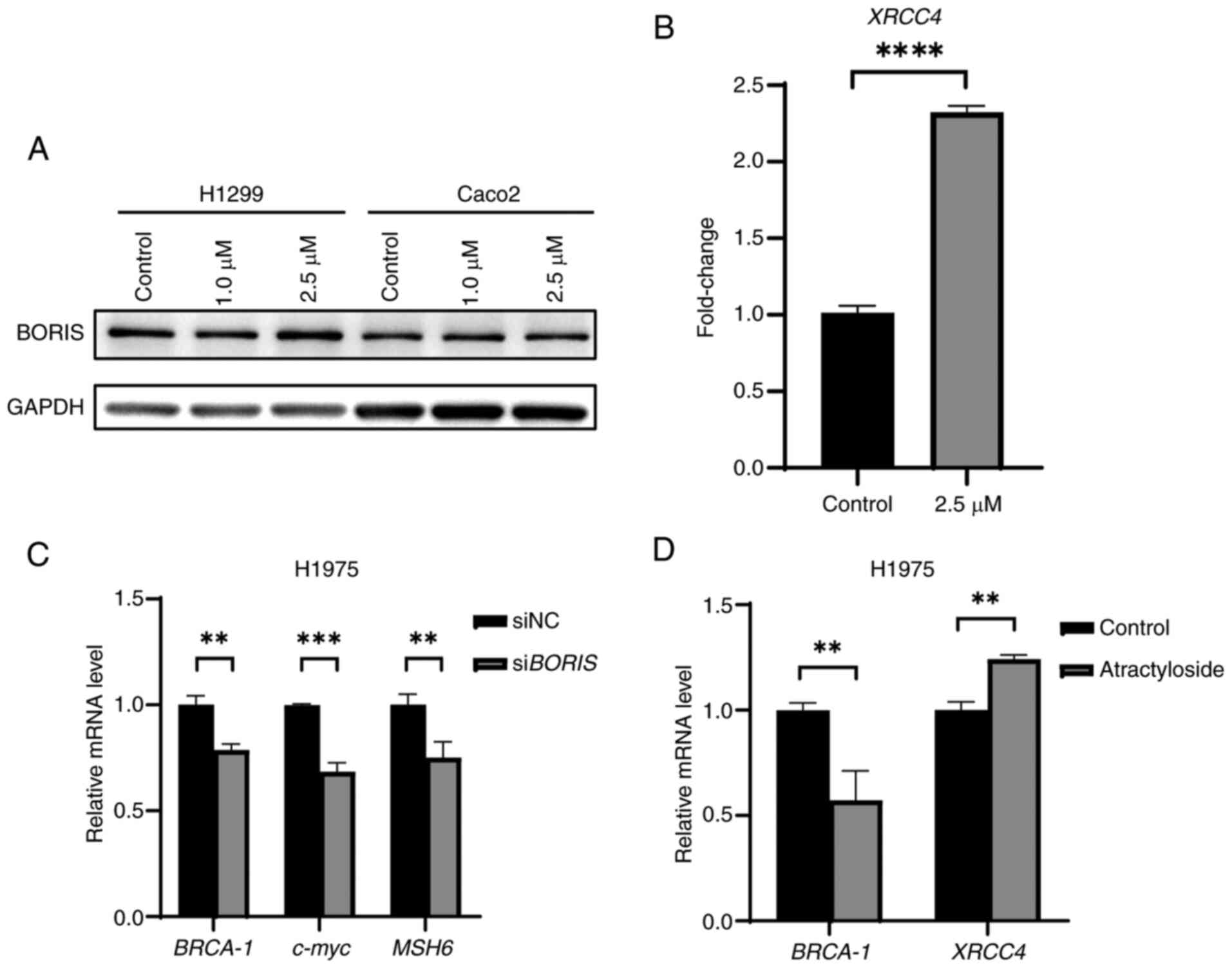
Atractyloside mimics BORIS knockdown
to suppress NSCLC viability
In a previous study, a drug that could mimic
BORIS knockdown was identified (23). To identify bioactive drugs that
might mimic the effects of BORIS knockdown, genes regulated
in BORIS-silenced Caco2 cells were analyzed using microarray
and a connectivity map database was screened for associated drugs
in our previous study (23). Based
on gene expression patterns and drug correlation analysis,
metronidazole and atractyloside were identified as promising
candidates for further study (23).
These previous findings using the Caco2 cell line demonstrated that
atractyloside (Fig. 1C) inhibited
cell viability (23).
The H1299 (wild-type EGFR) and H1975 (EGFR mutation)
NSCLC cells were used for assessing the response of siBORIS or
atractyloside treatment. The results demonstrated that siBORIS
treatment downregulated the expression of DNA repair-related genes,
including BRCA-1, MSH6 and c-myc (Fig.
2B). Atractyloside treatment, on the other hand, increased
XRCC4 expression while downregulating BRCA-1 (Fig. 2D), which is consistent with our
previous observations (23). In
H1299 and H1975 cells, atractyloside treatment resulted in DNA
damage and upregulation of XRCC4 expression (Fig. 2B and D). These results indicated
that atractyloside could mimic the effects of siBORIS to regulate
the downstream genes (Fig. 2C and
D); however, atractyloside treatment did not influence BORIS
expression (Fig. 2A). In the
present study atractyloside was used instead of siBORIS in
subsequent experiments to regulate BORIS-related downstream
genes.
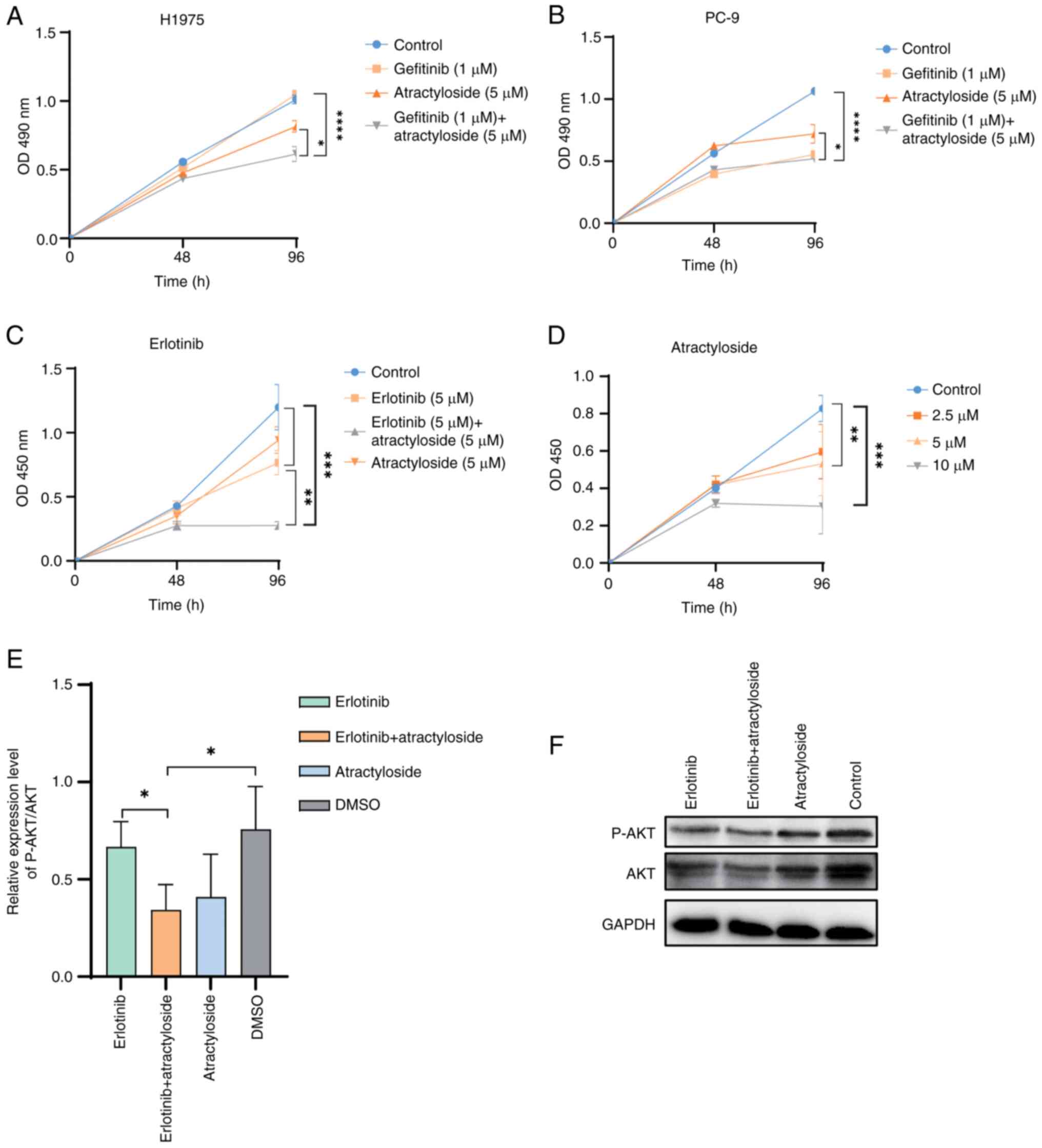
Combination of atractyloside and
gefitinib treatment reduces the proliferation of NSCLC cells
To avoid using a high concentration of
atractyloside, which would induce mitochondrial permeability
transition and cause apoptosis (4),
5 µM atractyloside was selected for application in two lung cancer
cell lines, the wild-type cell line PC-9 and the T790M mutant
drug-resistant cell line H1975.
In PC-9 cells, it was observed that, after 2 days of
administration, gefitinib inhibited cell proliferation, and after 4
days administration, the proliferation of PC-9 cells was
significantly inhibited by gefitinib (Fig. 3A). Atractyloside was shown to
suppress PC-9 cell proliferation, but was less effective than
gefitinib. In addition, the effect of the two-drug combination on
cell proliferation was not significant, thus indicating that
atractyloside had little effect on wild-type lung cancer cells. In
H1975 cells, gefitinib at a concentration of 1 µM did not affect
cell proliferation (Fig. 3B).
However, when used in combination with atractyloside, cell
proliferation was significantly decreased, indicating that the
combined administration of atractyloside and gefitinib may affect
the proliferation of TKI-resistant cells.
To further verify the inhibitory effect of
combination therapy on EGFR-mutant cells, the 2nd-generation
EGFR-TKI inhibitor erlotinib was used. After treatment with the
drugs for 96 h, a combination of erlotinib and atractyloside
suppressed H1975 cell proliferation better than erlotinib alone
(Fig. 3C). Atractyloside
demonstrated cytotoxic effects on cancer cells, with increasing
concentrations inhibiting H1975 cell proliferation (Fig. 3D). When combined with gefitinib, the
effect of atractyloside on drug-resistant lung cancer cells was
stronger than that on wild-type cells, indicating that the
BORIS pathway may be associated with lung cancer resistance.
Western blot analysis indicated that atractyloside may suppress
NSCLC cell proliferation by inhibiting AKT phosphorylation
(Fig. 3E and F). This finding
aligns with the results of a previous study demonstrating that AKT
phosphorylation promotes lung cancer cell proliferation (24). AKT is a downstream factor of EGFR
and can be used to examine the severity of cancer; therefore, the
inhibition of AKT phosphorylation indicated that EGFR-related
signaling was suppressed by atractyloside.
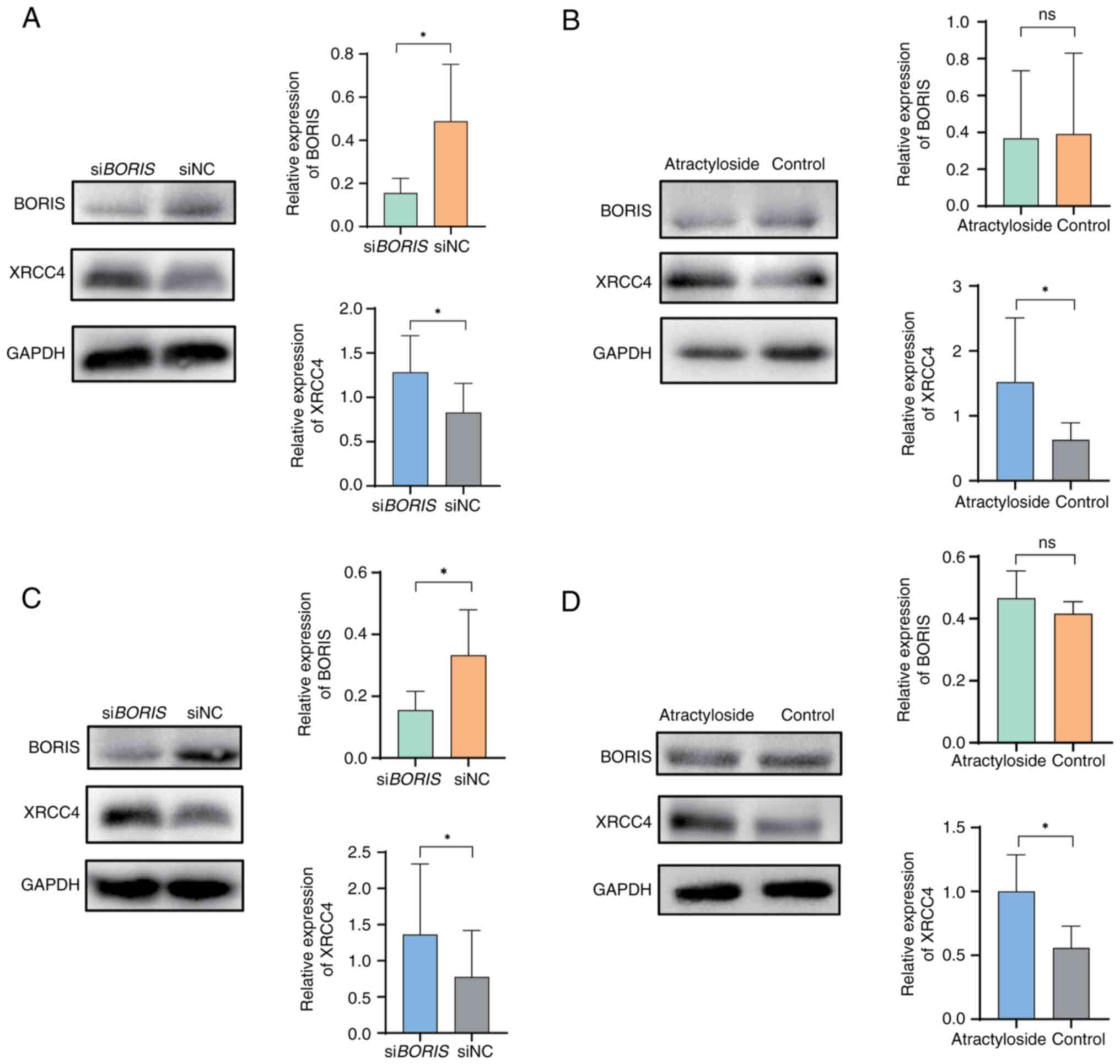
Atractyloside induces DNA damage in
NSCLC
After knockdown of the expression of BORIS in
the H1975 cell line, a decrease in the expression levels of the
homologous recombination-related genes c-myc, BRCA-1 and the
mismatch repair gene MSH6 (25), was detected (Fig. 2C). These findings were consistent
with our previous results (12) and
indicated the existence of BORIS-stabilized cell DNA. To
further verify the effects of atractyloside on H1975 and PC-9
cells, XRCC4 protein expression was detected. The data demonstrated
a consistent trend of XRCC4 upregulation, observed in response to
both BORIS knockdown and atractyloside treatment (Fig. 4A-D). In PC-9 cells, BORIS
knockdown elevated the expression of XRCC4 (Fig. 4A), as did atractyloside (Fig. 4B). In addition, in H1975 cells,
BORIS knockdown elevated the expression of XRCC4 (Fig. 4C), as did atractyloside (Fig. 4D). In summary, atractyloside may
disrupt DNA stability in H1975 cells and PC-9 cells.
Discussion
Gefitinib is a small-molecule EGFR-TKI that blocks
the intracellular receptor binding site of adenosine triphosphate
(ATP); this blocks downstream signal transduction, inhibits tumor
cell proliferation and promotes apoptosis, all of which have a
significant effect on the treatment of advanced NSCLC (13,26).
However, the median survival time for patients from 61 centers
across 11 European and Asia-Pacific countries with advanced NSCLC
was revealed to be only 7–9 months, and drug resistance, frequently
arising from secondary mutations, presents a significant obstacle
to effective treatment (13). Among
mutations, EGFR-T790M is considered the main cause of acquired drug
resistance. This mutation competitively reduces binding with
EGFR-TKIs to confer drug resistance by increasing the affinity
between EGFR and ATP (27).
Therefore, attention is required to identify novel ways to deal
with this acquired drug resistance.
Through the analysis of BORIS expression in
NSCLC in the present study, it was revealed that BORIS
expression was increased in tissues from patients with high-risk
NSCLC. As EGFR mutations are usually related with high-risk NSCLC,
it may be hypothesized that BORIS is associated with EGFR
mutations. In addition, BORIS knockdown or atractyloside
treatment promoted TKI resistance in H1975 NSCLC cells with T790M
mutation. These findings suggested that inhibiting BORIS could be a
promising strategy for combination therapy with first-generation
EGFR inhibitors in NSCLC. The prognosis of a number of patients
with NSCLC is poor because of secondary drug-resistance gene
mutations (27). However, the
notable effect of BORIS knockdown provides a novel
opportunity for treatment. In addition, atractyloside, a
BORIS knockdown mimetic, holds promise as a means for
treatment of NSCLC. However, the lack of in vivo experiments
is a limitation of the present study. In future studies, we plan to
conduct in-depth research on atractyloside and verify its medicinal
value in xenograft models, since animal experiments may better
reflect the occurrence and development of NSCLC. Future research
may also construct stable drug-resistant H1975 and PC-9IR NSCLC
cell lines with BORIS overexpression, and may assess
treatment of drug resistance in lung carcinoma in situ and
in a brain metastasis model of lung cancer.
In the present study, the knockdown of BORIS
in H1975 cells or the administration of atractyloside decreased the
expression of homologous recombination-related genes, such as
BRCA-1. In addition, BORIS can influence DNA repair
pathways, such as non-homologous end recombination, were
compensatively upregulated, indicating that BORIS could
stabilize the DNA of NSCLC cells. The H1299 cell line, expressing
wild-type EGFR, was used to confirm that atractyloside treatment
could induce XRCC4. Notably, the expression of DNA damage
repair genes, including BRCA-1, c-myc and MSH6, was
detected only in H1795 cells, which harbor an EGFR mutation. EGFR
mutations or amplifications in NSCLC cells cause resistance to TKIs
and induce downstream constitutive AKT phosphorylation, whereas
inhibition of AKT reverses resistance to TKIs. In a study on
neuroblastoma, ALK-mutated neuroblastoma cells were resistant to
the ALK inhibitor TAE684 (18).
Resistant cells exhibited upregulation of BORIS, which could
lead to wide-ranging changes in chromatin interactions and
transcriptional reprogramming. A 10-fold gain in genome-wide
occupancy by BORIS was observed in resistant cells (22,891
vs. 2,211 in the sensitive cells) (18). We observed increased BORIS
expression in TKI-resistant NSCLC cells and identified AKT as a
potential downstream target of BORIS activation in these cells. The
present study observed that atractyloside may inhibit AKT activity
and suppress NSCLC cell proliferation, but did not affect the
expression of BORIS. Inhibition of AKT by atractyloside
suggests crosstalk between BORIS and factors downstream of
EGFR.
In summary, BORIS expression was increased in
patients with high-risk lung cancer, as determined by comparing
groups with different survival rates. Notably, atractyloside is an
inhibitor of the BORIS pathway and may be a potential
therapeutic drug against TKI resistance.
Acknowledgements
Not applicable.
Funding
This study was supported by grants from the Zhejiang Provincial
Natural Science Foundation of China (grant no. HDMY22H318024), the
Medical and Health Science and Technology Project of Zhejiang
Province (grant no. 2022RC128), and the Foundation of the Zhejiang
Academy of Medical Sciences to Yanmei Zhang.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
Conceptualization of the study was directed by YQ
and YZ. The experiments were performed by WY, CL, NZ and YZ. The
original draft was written by CL, WY and YZ. The review, editing
and revisions were completed by YQ and YZ. The visualization of the
data and the generation of the figures were performed by CL and NZ.
The funding was provided by YQ and YZ. All authors read and
approved the final version of the manuscript. WY and CL confirmed
the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
NSCLC
|
non-small cell lung cancer
|
|
BORIS
|
brother of the regulator of imprinted
sites
|
|
ATP
|
adenosine triphosphate
|
|
TKI
|
tyrosine kinase inhibitor
|
References
|
1
|
Chen LY, Hu A and Chang CJ: The
degradation mechanism of toxic atractyloside in herbal medicines by
decoction. Molecules. 18:2018–2028. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xu J, Liu C, Shi K, Sun X, Song C, Xu K
and Liu Y: Atractyloside-A ameliorates spleen deficiency diarrhea
by interfering with TLR4/MyD88/NF-κB signaling activation and
regulating intestinal flora homeostasis. Int Immunopharmacol.
107:1086792022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang P, Cheng X, Sun H, Li Y, Mei W and
Zeng C: Atractyloside protect mice against liver steatosis by
sctivation of autophagy via ANT-AMPK-mTORC1 Signaling Pathway.
Front Pharmacol. 12:7366552021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li H, Shi X, Jiang H, Kang J, Yu M, Li Q,
Yu K, Chen Z, Pan H and Chen W: CMap analysis identifies
Atractyloside as a potential drug candidate for type 2 diabetes
based on integration of metabolomics and transcriptomics. J Cell
Mol Med. 24:7417–7426. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cho J, Zhang Y, Park SY, Joseph AM, Han C,
Park HJ, Kalavalapalli S, Chun SK, Morgan D, Kim JS, et al:
Mitochondrial ATP transporter depletion protects mice against liver
steatosis and insulin resistance. Nat Commun. 8:144772017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ling X, Zhou Y, Li SW, Yan B and Wen L:
Modulation of mitochondrial permeability transition pore affects
multidrug resistance in human hepatocellular carcinoma cells. Int J
Biol Sci. 6:773–783. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu WJ, Du Y, Wen R, Yang M and Xu J: Drug
resistance to targeted therapeutic strategies in non-small cell
lung cancer. Pharmacol Ther. 206:1074382020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Herbst RS, Morgensztern D and Boshoff C:
The biology and management of non-small cell lung cancer. Nature.
553:446–454. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Aran V and Omerovic J: Current approaches
in NSCLC targeting K-RAS and EGFR. Int J Mol Sci. 20:57012019.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lu X, Yu L, Zhang Z, Ren X, Smaill JB and
Ding K: Targeting EGFRL858R/T790M and
EGFRL858R/T790M/C797S resistance mutations in NSCLC:
Current developments in medicinal chemistry. Med Res Rev.
38:1550–1581. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang Y, Song Y, Li C, Ren J, Fang M, Fang
J and Wang X: Brother of regulator of imprinted sites inhibits
cisplatin-induced DNA damage in non-small cell lung cancer. Oncol
Lett. 20:2512020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mok TSK, Kim SW, Wu YL, Nakagawa K, Yang
JJ, Ahn MJ, Wang J, Yang JC, Lu Y, Atagi S, et al: Gefitinib plus
chemotherapy versus chemotherapy in epidermal growth factor
receptor mutation-positive non-small-cell lung cancer resistant to
first-line gefitinib (IMPRESS): Overall survival and biomarker
analyses. J Clin Oncol. 35:4027–4034. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Soltanian S and Dehghani H: BORIS: A key
regulator of cancer stemness. Cancer Cell Int. 18:1542018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Loukinov D: Targeting CTCFL/BORIS for the
immunotherapy of cancer. Cancer Immunol Immunother. 67:1955–1965.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Asano T, Hirohashi Y, Torigoe T, Mariya T,
Horibe R, Kuroda T, Tabuchi Y, Saijo H, Yasuda K, Mizuuchi M, et
al: Brother of the regulator of the imprinted site (BORIS) variant
subfamily 6 is involved in cervical cancer stemness and can be a
target of immunotherapy. Oncotarget. 7:11223–11237. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang Y, Fang M, Song Y, Ren J, Fang J and
Wang X: Brother of regulator of imprinted sites (BORIS) suppresses
apoptosis in colorectal cancer. Sci Rep. 7:407862017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Debruyne DN, Dries R, Sengupta S, Seruggia
D, Gao Y, Sharma B, Huang H, Moreau L, McLane M, Day DS, et al:
BORIS promotes chromatin regulatory interactions in
treatment-resistant cancer cells. Nature. 572:676–680. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hou J, Aerts J, den Hamer B, van Ijcken W,
den Bakker M, Riegman P, van der Leest C, van der Spek P, Foekens
JA, Hoogsteden HC, et al: Gene expression-based classification of
non-small cell lung carcinomas and survival prediction. PLoS One.
5:e103122010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang S, Reitze NJ, Ewing AL, McCreary S,
Uihlein AH, Brower SL, Wang D, Wang T, Gabrin MJ, Keating KE, et
al: Analytical Performance of a 15-Gene Prognostic Assay for
Early-Stage Non-Small-Cell Lung Carcinoma Using RNA-Stabilized
Tissue. J Mol Diagn. 17:438–445. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu CQ, Ding K, Strumpf D, Weir BA,
Meyerson M, Pennell N, Thomas RK, Naoki K, Ladd-Acosta C, Liu N, et
al: Prognostic and predictive gene signature for adjuvant
chemotherapy in resected non-small-cell lung cancer. J Clin Oncol.
28:4417–4424. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fang M, Song Y, Ren J, Yuan H, Fang J, Yan
D, Zhang Y and Wang X: Atractyloside mimics BORIS knockdown
to induce DNA damage in colorectal cancer cells. Int J Clin Exp
Pathol. 11:3286–3293. 2018.PubMed/NCBI
|
|
24
|
Wang R, Wang S, Li Z, Luo Y, Zhao Y, Han
Q, Rong XZ, Guo YX and Liu Y: PLEKHH2 binds β-arrestin1 through its
FERM domain, activates FAK/PI3K/AKT phosphorylation, and promotes
the malignant phenotype of non-small cell lung cancer. Cell Death
Dis. 13:8582022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Luoto KR, Meng AX, Wasylishen AR, Zhao H,
Coackley CL, Penn LZ and Bristow RG: Tumor cell kill by c-MYC
depletion: Role of MYC-regulated genes that control DNA
double-strand break repair. Cancer Res. 70:8748–8759. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Arteaga CL: ErbB-targeted therapeutic
approaches in human cancer. Exp Cell Res. 284:122–130. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jin X, Wang J, Shen H, Ran R, Xu K, Zhang
W, Tong X and Feng L: Curcumin co-treatment ameliorates resistance
to gefitinib in drug- resistant NCI-H1975 lung cancer cells. J
Tradit Chin Med. 37:355–360. 2017. View Article : Google Scholar : PubMed/NCBI
|