Introduction
Pancreatic cancer (PC) is the seventh leading cause
of cancer-related death globally (1). Pancreatic ductal adenocarcinoma (PDAC)
accounts for >90% of PC cases and is the most typical type of
PC. Despite advances in treatment, PDAC has a low survival rate, as
the 5-year overall survival rate is <10% (2). Sarcomatoid (spindle cell) carcinoma
(SC) is an aggressive form of carcinoma composed of malignant
spindle cells, with or without a coexisting epithelial cell
component. Though it can occur in all organs of the body, it mainly
affects the respiratory tract, lungs, breasts and kidneys, and in
extremely rare cases, the pancreas (3). Undifferentiated SC of the pancreas
(SCP) is an aggressive malignant neoplasm originating in the
pancreas with a poor prognosis. Following the World Health
Organization (WHO) classification (Fifth edition, 2019), SCP
represents a histologically undifferentiated PDAC subtype,
accounting for 2–3% of all PDACs and its variants (4–6);
however, the histogenesis of this carcinoma type remains debatable.
Currently, the clinicopathological features, molecular landscape
and therapeutic strategies for SCP are poorly understood due to its
low incidence. The present review aimed to describe the
histogenesis, diagnosis, genetic characteristics, prognosis and
treatment of SCP, specifically focusing on the molecular
alterations to elucidate potential targets for precision therapy.
The eligible cases of SCP from the Affiliated Lihuili Hospital of
Ningbo University (Ningbo, China) were retrospectively collated to
summarize the single-center experience and a literature review was
performed by searching PubMed (https://pubmed.ncbi.nlm.nih.gov) and the China Science
Periodical Database (wanfangdata.com.cn) from January 1, 1990 up to
August 31, 2023 with a combination of the following keywords:
‘sarcomatoid carcinoma’; ‘undifferentiated carcinoma’; and
‘pancreatic cancer’. Studies that reported an explicit
histopathological diagnosis of SCP with follow-up data were
considered eligible for inclusion in the present review. The
present review excluded certain articles where terminologies such
as ‘carcinosarcoma’, ‘sarcoma-like’ or ‘carcinosarcomatous
histology’ were used, and collected and analyzed data from 38
patients with SCP (Table SI)
(7–28).
Histological ontogeny
Histologically, SCP predominantly comprises
mesenchymatoid spindle-shaped tumor cells originating from
pancreatic ducts and acinus, but it does not exhibit glandular
differentiation. The tumor displays a distinct biphasic component
of carcinoma and sarcoma. Carcinosarcoma of the pancreas (CSP) also
originates in the pancreas and has similar biphasic features
(29). Previously published studies
often use the terms ‘carcinosarcoma’ and ‘sarcomatoid carcinoma’
interchangeably and the definitions of these terms vary in the
literature (30,31). According to the fifth edition of the
WHO classification of exocrine pancreatic tumors from 2019, SCP and
CSP are classified as undifferentiated carcinoma of the pancreas
(UCP) (32). UCP, a subtype of
PDAC, represents a group of rare tumors that account for ~5% of PC
(30). The primary difference
between UCP and PDAC is that UCP is a hypercellular tumor with
minimal stroma and scant desmoplastic reaction, whereas
conventional PDAC has a considerable amount of desmoplastic stroma
with few neoplastic cells/glands. Based on the aforementioned WHO
classification of exocrine pancreatic tumors, SCP consists of
spindle-shaped cells that may contain allogenic components, such as
bone and cartilage. The microscopic description is a critical
indicator for differentiating SCP from CSP. SCP is defined as a
neoplasm composed of >80% atypical spindle cells, with or
without heterogenic differentiation. Pathologically, CSP is defined
as a UCP subtype composed of a combination of round epithelioid
cells and spindle sarcoma cells, with each component constituting
~30% of the tumor (Table I)
(32). In addition, there is a
transitional zone between the epithelioid and sarcomatoid cells in
SCs, whilst in carcinosarcomas, these two portions are separated
without such a transitional zone (Table
I) (16,31). The sarcomatous tissues of the SCs
exhibit biphasic expression of mesenchymal markers and epithelial
markers, and ultrastructures of epithelial cells (11,16).
Sarcomatoid components in carcinosarcomas do not have this feature;
they express only mesenchymal markers and are negative for
epithelial-derived markers (Table
I) (13,32). However, the pathogenesis of SC
remains unclear. Researchers have presented the following
hypotheses to explain the phenomenon that a particular tumor
exhibits both epithelial and sarcomatous traits biphasically: i)
Conversion: The sarcomatous components are transformed from
cancerous components by metaplasia; ii) collision: The sarcoma and
carcinoma grow independently adjacent to each other; and iii)
combination: Bidirectional differentiation of primitive totipotent
stem cells into epithelium and sarcoma tissue (33–36).
The genetic alterations in sarcoma and epithelial components have
been reported to be nearly identical in pancreatic mucinous cystic
neoplasms and sarcomatous stroma, which is consistent with the
‘combination theory’ that the two components of the neoplasms have
the same clonal origin and subsequently differentiate into the
epithelial and sarcomatous components of the carcinosarcomas
(37). Since the emergence of the
epithelial-mesenchymal transition (EMT) theory in cancerous tissues
in the 1990s, EMT has been reported by certain researchers to
explain the histogenesis mechanism of carcinosarcoma. They have
pointed out that the complete transformation of epithelial cells
into mesenchymal cells is a continuous process driven by the EMT
program, generating cells that exhibit a series of intermediate
phenotypic states. This process is regulated by contextual signals
and the intracellular gene circuitry of the cells. Therefore, cells
in intermediate states may exhibit the characteristics of
mesenchymal cells but can retain certain epithelial markers
(38). In certain instances,
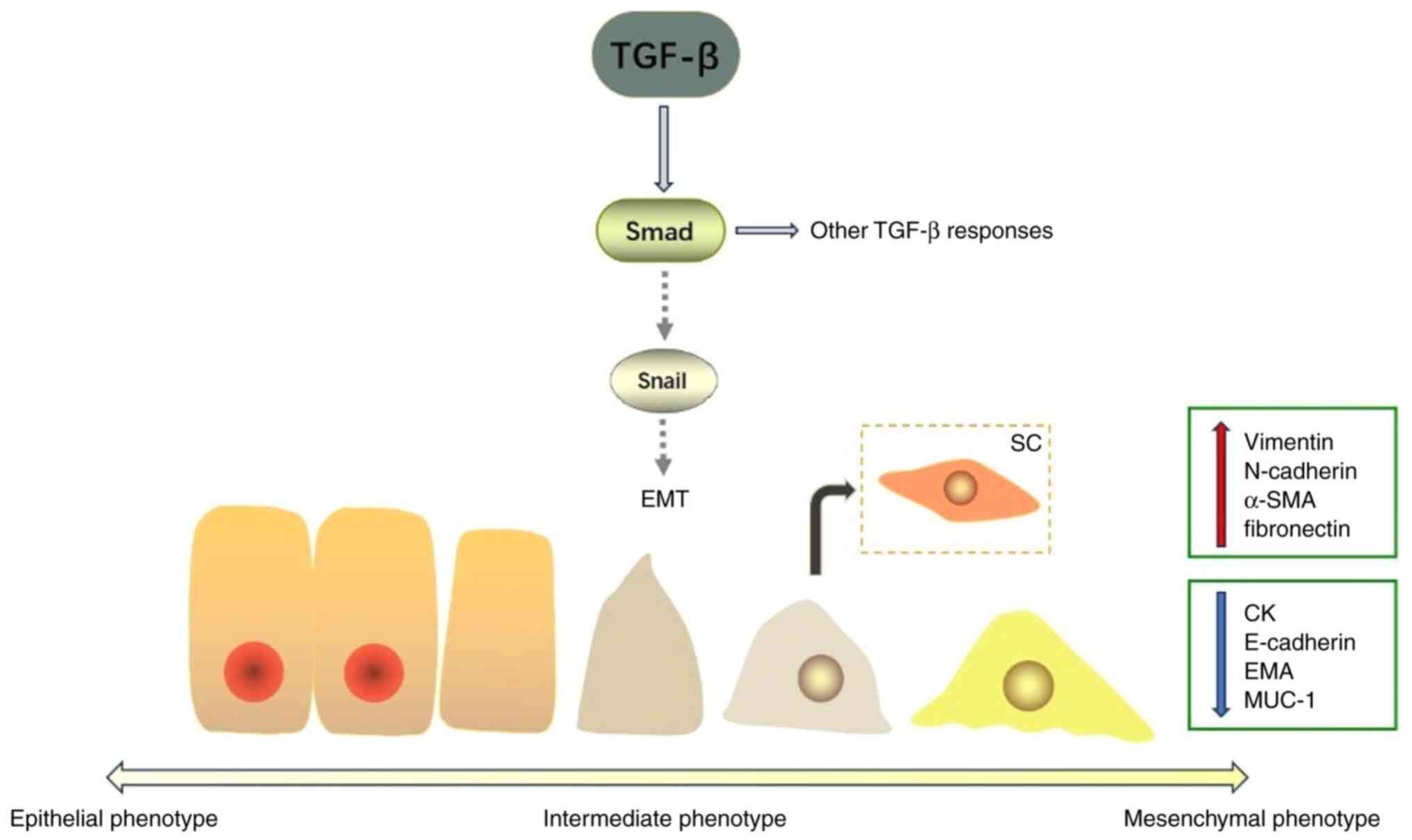
transforming growth factor-β (TGF-β) can act as an oncogene
to promote the proliferation of normal epithelial cells. Therefore,
TGF-β is considered to induce EMT in pancreatic cells and to
promote the formation of SCP (Fig.
1) (39). In other tumor types,
TGF-β is also a powerful tumor suppressor inhibiting the
multiplication of pre-malignant cells by triggering apoptosis. This
dual effect of TGF-β is mainly mediated by the Smad pathway
(40). Furthermore, the
pathological type, cellular context and specific environment
determine the tumor responsiveness to TGF-β (41–43).
Ren et al (10) reported
that the plasma interleukin (IL)-11 and TGF-β levels were
notably higher in patients with SCP compared with those in healthy
controls and patients with PDAC. IL-11 is a TGF-β target
gene and TGF-β induces IL-11 production in several cell
types. The Smad tumor suppressor pathway mediates the expression of
IL-11 and connective tissue growth factor via TGF-β
(44). Furthermore, TGF-β
may be a critical driver of sarcomatoid transdifferentiation in
renal clear cell carcinoma (45).
Kimura et al (26,27) assessed the expression of
fibronectin, Snail and phosphorylated (p)Smad2/3 in the sarcomatoid
tissues of 3 patients with SCP. Fibronectin is an extracellular
marker of spindle metaplasia during EMT (46). Snail is a zinc-finger transcription
factor that represses the transcription of E-cadherin, which
is involved in the regulation of EMT during embryonic development.
pSmad2 and pSmad3 are regarded as critical intracellular
transduction molecules that transmit TGF-β signals from the
cell surface into the nucleus (47,48).
The aforementioned studies reported that sarcomatoid components may
be converted from cancerous components via EMT mediated by TGF-β.
However, further studies are required to elucidate the molecular
mechanisms underlying the processes of cellular differentiation
leading to SCP.
 | Table I.Histopathological characteristics of
undifferentiated carcinoma of the pancreas. |
Table I.
Histopathological characteristics of
undifferentiated carcinoma of the pancreas.
| Characteristic | Sarcomatoid
carcinoma | Carcinosarcoma |
|---|
| Spindle cells,
% | >80 | >30 |
| Epithelioid cells,
% | <20 | >30 |
| Presence of
transitional zone | Yes | No |
| Markers
expressed | Mesenchymal and
epithelial | Only
mesenchymal |
Diagnosis
According to the aforementioned WHO guidelines, the
diagnosis of SCP is highly dependent on pathology and
immunohistochemistry (IHC); however, it is challenging to
distinguish SCP from other pancreatic tumors preoperatively.
Potential preoperative diagnosis includes PDAC, mucinous cystic
neoplasm, pseudocyst and solid pseudopapillary tumor (18). Several studies have also reported
certain specific imaging features of SCP, such as large irregular
cystic-solid masses, which were prone to invading adjacent organs
(9,12,14,17,26).
Furthermore, biopsy is crucial for the preoperative diagnosis of
SCP and fine needle aspiration guided by endoscopic ultrasound has
relatively high sensitivity and specificity for the diagnosis of
solid pancreatic masses. The final diagnosis requires histological
examination and immunohistochemical analysis. Due to the poor
prognosis and aggressive clinical behavior, the early diagnosis of
SCP is critical (15,20,29).
Clinical manifestation and laboratory
examination
Based on the 38 patients with SCP in the present
study (Table SI), the incidence of
SCP was higher in middle-aged and elderly individuals, with the age
of onset ranging between 48–88 years. The average age of onset was
65 years. SCP was more common in male patients than in female
patients (12:7). Patients with SCP typically presented with
abdominal symptoms (73.7%), weight loss (18.4%), loss of appetite,
fatigue, vomiting and emaciation. Jaundice (18.4%) occurred when
tumors involved the head of the pancreas blocking the biliary
tract, similar to the symptoms observed with PDAC; however, 2
patients mainly complained of back pain and 4 patients were
asymptomatic during the initial pre-operative visit but had tumors
that were found incidentally. Abdominal palpation revealed
tenderness in the upper abdomen and an abdominal mass was detected
in certain cases. The analysis of serum tumor markers in 18
patients revealed elevated levels of carbohydrate antigen (CA)19-9
(61.1%) and CA 12-5 (33.3%), whilst in the remaining cases, the
levels were within the normal range. Elevated liver enzyme or
bilirubin levels were observed when tumors were located in the
pancreatic head block bile ducts (7,8,14–16).
Imaging features
Most cases of SCP were reported based on
pathological examinations; however, in 28 cases, distinct imaging
features were reported. According to the data of the 28 cases
(Table SI), SCP primarily occurred
in the pancreatic head and pancreatic tail (head, n=13; body, n=6;
and tail, n=9). The tumors were quite large, ranging between 2.4–20
cm, with a mean maximum diameter of 6.47 cm. Generally, SCPs
exhibited the following features: i) Rapid growth with the presence
of non-uniformly enhanced large irregular cystic-solid and cystic
masses; ii) enhanced computed tomography (CT) revealed moderate
tumor enhancement, with the lowest CT value in the arterial phase
and the most significant in the portal phase; iii) SCPs were highly
aggressive and prone to invading adjacent organs; iv) the tail of
the pancreas did not atrophy in general, and SCP at the head of the
pancreas frequently pressed the pancreatic duct and bile duct,
causing dilation of the pancreatic duct and bile duct; and v)
necrosis was common, which may be related to the rapid growth and
insufficient vessels of the tumor, and the mixed cystic-solid
structure (49). Nevertheless,
distinguishing the undifferentiated sarcomatoid carcinoma (USC)
subtype from PDAC with cystic changes is challenging. Compared with
the latter, extrapancreatic vascular and perineural invasion,
peripheral organ infiltration and parenchymal atrophy of the
pancreas are more common in UCP (50).
Pathological features
Among the data we collected from the literature,
histological examination revealed that SCP is primarily
characterized by a dominance of spindle cells (15–28).
These cells are heteromorphic, active in terms of nuclear division
and arranged in a disorganized or interleaved pattern. They account
for >80% of the tumor cells (4,51) and
are occasionally accompanied by multinucleated giant cells
(52). The epithelial components,
accounting for <20%, can be adenocarcinomas or squamous cell
carcinomas. As demonstrated in Table
SI, adenocarcinoma was the most common epithelial component,
which was mainly poorly differentiated (n=8) and moderately
differentiated (n=5), whilst no highly differentiated
adenocarcinoma was found (n=0). A certain case exhibited an
epithelial component of a mucinous cystic neoplasm (18), and another exhibited two different
epithelial components, including adenocarcinoma and squamous
carcinoma (12). Furthermore, one
case exhibited an area of calcification/ossification with scattered
large, atypical cells adjacent to malignant polymorphic spindle
cell hyperplasia; the final diagnosis was SCP with heterologous
elements (osteosarcomatous differentiation) (20).
IHC serves an indispensable role in the pathological
diagnosis of SCP. The most notable feature of SCP is the expression
of both epithelial and mesenchymal markers by its sarcomatoid
components, as observed by immunohistochemical staining; however,
CSP does not express the former. Vimentin is one of the most
typically expressed mesenchymal markers with the highest positivity
rate (7). Notably, other
mesenchymal markers such as myogenic markers, including smooth
muscle actin, myoglobin and desmin, neurogenic markers and
osteogenic markers may be expressed in the corresponding
components. Conversely, commonly used epithelial markers include
cytokeratin (CK)7, CK19, CK (pan), CAM5.2 and epithelial membrane
antigen, which are expressed in the epithelial and sarcomatoid
areas (9,12–15).
In addition, the transmembrane glycoprotein mucin 1 (MUC1) has been
reported to be present in several adenocarcinomas. In one case, its
expression was observed in the sarcomatoid region of SCP (7). The expression of MUC1 is also
associated with tumor metastasis and recurrence (7). Furthermore, an additional two cases
exhibited the loss of membranous E-cadherin expression in tumor
cells of the sarcomatoid lesion (12,24).
The loss of E-cadherin is also a sign of EMT (48). According to data in the present
study (Table SI), the average
Ki-67 index of sarcomatoid cells in SCP was 38.5%, ranging between
11–90%.
Genetic features
At present, there are numerous reports of the
relationship between PDAC carcinogenesis and several gene
alterations, including mutations in genes such as KRAS, TP53,
SMAD4 and CDKN2A (29,53).
Due to the rarity of SCP, only a few individual cases have been
reported (20,54,55),
without any large prospective studies, and its genetic
characterization has not been fully described. As a subtype of
PDCA, SCP has a genetical landscape mimicking PDAC (29). Recently, Ding et al (54) assessed the clinicopathological and
genetic characterization of 71 patients with USC, which included
five USCs of the pancreas. This study reported the presence of
highly frequent TP53, RB1, TER0054 and KRAS
alterations. In particular, mutations in TP53 and
KRAS were identified in all cases and the KRAS
mutation was reported to be associated with a poor prognosis
(54). Furthermore, a
next-generation sequencing (NGS) analysis of 10 SCP samples
revealed that SCP was genetically similar to PDAC. It was also
reported that 100% of these samples exhibited KRAS
mutations, 90% exhibited TP53 mutations and 60% exhibited
CDKN2A mutations. However, SCP also exhibited several
critical genetic characteristics that were distinct from PDAC.
SMAD4, a tumor suppressor gene that is altered in 50–60% of
conventional PDAC cases (56,57),
was mutated in only 1 SCP case (10%). This type of mutation may
indicate early metastasis of the tumor (56). Furthermore, tumor cells in PDAC
rarely revealed recurrent KRAS amplification, whilst it was
present in 3/10 cases. Similarly, rhabdoid carcinoma of the
pancreas (RCP) is a subtype of UCP. A cohort study on RCP revealed
this amplification in 5/13 (38%) patients (58). Moreover, in at least a subset of
patients with PDAC, KRAS amplification may be a genetic
driver for the acquisition of undifferentiated morphology. Finally,
two potential molecular therapeutic targets such as the POLQ
mutation and MCL1 amplification that did not belong to the
typical PDAC molecular landscape were detected in 2 cases (55).
PDAC is occasionally associated with germline
BRCA mutations. The BRCA genes, including
BRCA1 and BRCA2, encode proteins involved in
repairing the broken DNA double strands via the homologous
recombination pathway (20,59). A previous large prospective study
reported that patients with pathogenic BRCA1/2 variants may
benefit from poly-ADP ribose polymerase (PARP) inhibitor treatment,
an emerging therapy targeting the genes involved in DNA maintenance
(59). A study reported mutations
in TP53 and KRAS, as well as BRCA2 (20). The molecular profiles of the SCP
cases we collected are summarized in Table II. Overall, these genomic profiling
results indicate encouraging outcomes for precise targeted therapy
in SCP.
| Table II.Summary of genomic alterations in the
cohort of sarcomatoid carcinoma of the pancreas. |
Table II.
Summary of genomic alterations in the
cohort of sarcomatoid carcinoma of the pancreas.
|
|
|
|
| Gene
alteration |
|
|---|
|
|
|
|
|
|
|
|---|
| First author/s,
year | Case | TMB,
muations/mb | MSI | Gene | Variation | Mutation type | Frequency | (Refs.) |
|---|
| Zhang et al,
2023 | 11 | 6.2 | No | KRAS | p.G12V | NA | 55 | (20) |
|
|
|
|
| NTRK3 | p.T261A | NA | 48 |
|
|
|
|
|
| BRCA2 | p.L698P | NA | 21 |
|
| Gkountakos | 1 | 7.6 | No | KRAS | p.G12V | Substitution:
Missense | 68 | (55) |
| et al,
2022 |
|
|
| TP53 | p.M237I | Substitution:
Missense | 29 |
|
|
|
|
|
| SMAD4 | p.Q28fs*17 | Deletion:
Frameshift | 36 |
|
|
| 2 | 8.6 | No | KRAS | p.G12V | Substitution:
Missense | 62 |
|
|
|
|
|
| TP53 | p.G245S | Substitution:
Missense | 41 |
|
|
| 3 | 6.7 | No | KRAS | p.Q61H | Substitution:
Missense | 40 |
|
|
|
|
|
| TP53 | p.Y220C | Substitution:
Missense | 37 |
|
|
| 4 | 5.4 | No | KRAS | p.Q61H | Substitution:
Missense | 57 |
|
|
|
|
|
| TP53 | p.C176Y | Substitution:
Missense | 26 |
|
|
| 5 | 5.4 | No | KRAS | p.G12D | Substitution:
Missense | 13 |
|
|
| 6 | 4.9 | No | KRAS | p.G12D | Substitution:
Missense | 51 |
|
|
|
|
|
| TP53 | p.R248Q | Substitution:
Missense | 38 |
|
|
|
|
|
| PHF6 | p.F19_G29del | Deletion: In
frame | 17 |
|
|
| 7 | 11.9 | No | KRAS | p.G12C | Substitution:
Missense | 25 |
|
|
|
|
|
| TP53 | p.P177_C182del | Deletion: In
frame | 20 |
|
|
| 8 | 10.8 | No | KRAS | p.G12D | Substitution:
Missense | 48 |
|
|
|
|
|
| TP53 | p.K101* | Substitution:
Nonsense | 35 |
|
|
|
|
|
| CDKN2 | p.H83Y | Substitution:
Missense | 37 |
|
|
| 9 | 7.0 | No | KRAS | p.G12D | Substitution:
Missense | 57 |
|
|
|
|
|
| TP53 | p.R175H | Substitution:
Missense | 92 |
|
|
|
|
|
| CDKN2 | p.P81L | Substitution:
Missense | 82 |
|
|
| 10 | 5.4 | No | KRAS | p.G12D | Substitution:
Missense | 77 |
|
|
|
|
|
| TP53 | p.R273C | Substitution:
Missense | 50 |
|
|
|
|
|
| POLQ | c.7389þ1G>A | Substitution:
Splice site | 44 |
|
| Present study | 12 | 4.0 | No | BRCA1 | p.I1824fs | Deletion:
Frameshift | 53 | - |
|
|
|
|
| Tp53 | p.Y234C | Substitution:
Missense | 14 |
|
|
|
|
|
| KRAS | p.Q61H | Substitution:
Missense | 9 |
|
At present, validated predictive biomarkers for
immunotherapy include programmed death ligand 1 (PD-L1), as well as
microsatellite instability and tumor mutation load. As presented in
Table II, all previously mentioned
cases exhibited low levels of tumor mutation burden with
microsatellite-stable states, indicating poor outcomes of
immunotherapy. Notably, Lehrke et al (60) reported that patients with UCP with
osteoclast-like giant cells (OCGCs) expressed PD-L1 significantly
more frequently than patients with PDAC (63 vs. 15%; P<0.01).
Another study retrospectively evaluated PD-L1 and Notch expression
in 6 cases of SCP (61). The
combined positive score (CPS) is an index that can be used to
evaluate PD-L1 expression in tumor's and is obtained by dividing
the number of PD-L1-stained cells, namely tumor cells, macrophages
and lymphocytes, by the total number of viable tumor cells
multiplied by 100 (61). A CPS ≥1
was typical in 5 cases of SCP (83%) and 3 of the subjects (50%) had
a CPS ≥50. This finding indicates an improved effectiveness in SCP
compared with conventional PDAC. However, high expression levels of
Notch1 and Notch3 were also reported in all cases. Further
immunofluorescence analysis revealed that, when the expression
levels of PD-L1 and Notch3 were upregulated within the cytoplasmic
or membranous compartments of the sarcomatoid cells, both proteins
were co-localized in the same cells, providing a rationale for
future research in anticipation of evaluating the potential
crosstalk between the PD-L1/programmed cell death protein 1 axis
and the Notch3 pathway (61).
Therefore, further studies on the significance of immunotherapy are
required.
The research into molecular classifications and
genetic signatures has long spurred the development of novel
therapeutic strategies, enabling medical practitioners to make
accurate and personalized decisions. Table III summarizes gene alterations in
SCP compared with in PDAC. Nevertheless, due to the low incidence
of SCP, there are limited genomic profiling data available. Thus,
further studies based on larger cohorts of patients with SCP are
warranted to explore the genetic features of SCP.
| Table III.Gene alterations of sarcomatoid
carcinoma of the pancreas. |
Table III.
Gene alterations of sarcomatoid
carcinoma of the pancreas.
| First author/s,
year | Key gene | Mutation rate | Alteration | (Refs.) |
|---|
| Gkountakos et
al, 2022 | SMAD4 | 1/10 | Downregulation | (55) |
|
| POLQ | 2/10 | Upregulation |
|
|
| KRAS
amplification | 3/10 | Upregulation |
|
|
| MCL1
amplification | 2/10 | Upregulation |
|
| Agaimy et
al, 2015 | KRAS
amplification | 5/13 | Upregulation | (58) |
| Silvestris et
al, 2021 | PD-L1 CPS
≥1 | 5/6 | Upregulation | (61) |
|
| Notch2 | 0/6 | Downregulation |
|
|
| Notch3 | 6/6 | Upregulation |
|
Prognosis and treatment
Due to its rarity, the surgical protocols,
postoperative adjuvant treatments and overall prognosis of SCP are
insufficiently described in the literature. Notably, no direct
comparisons between SCP and PDAC have been made; however, patients
with SCP tend to have worse survival rates than those with PDAC
(11,28). Generally, most patients present with
an advanced, unresectable state of the disease, with only ~12% of
the patients surviving >5 years (62). For patients presenting with
resectable disease (10–15%), surgery followed by adjuvant
chemotherapy is the standard therapeutic approach, with an
anticipated median overall survival of 54.4 months (53).
The poor prognosis of SCP makes identifying
effective treatments a top priority. Notably, total R0 surgical
extirpation is the sole opportunity for a radical cure (28,63)
and it has been reported that patients who did not undergo complete
R0 tumor extirpation had an early recurrence, leading to mortality
in <3 months. Surgical procedures mainly include
pancreaticoduodenectomy and distal pancreatectomy. Occasionally it
is necessary to remove the surrounding invaded organs to ensure the
complete removal of the tumor. In addition to surgery, patients can
benefit from postoperative adjuvant treatments, especially
chemotherapy (29). A study
analyzing the prognoses of 261 patients with UCP indicated that
surgery was the first choice for resectable UCP and that adjuvant
therapies needed to be introduced immediately (64). Generally, patients with UCP were
administered the same regimens as those with more common PDACs.
Albumin paclitaxel and gemcitabine, and fluorouracil, irinotecan,
leucovorin and oxaliplatin, the first-line chemotherapy regimens
for PDAC, are also the preferred choices of adjuvant therapy for
SCP (53,65). Furthermore, gemcitabine has been
reported to be effective in patients with tumor recurrence or
portal vein thrombosis (65). A
multicenter cohort study retrospectively analyzed the outcomes of
50 patients with unresectable UCP and assessed the efficacy of
several chemotherapies. It was reported that the median overall
survival of these patients was 4.08 months and a
paclitaxel-containing regimen was associated with a relatively
longer survival (65). Gkountakos
et al (29) also reported
that complete surgical resection followed by PDAC-standardized
adjuvant chemotherapy was the only tangible possibility for
long-term survival in patients with SCP. Another retrospective
study reported 8 patients with SCP in a single center. 2/8 cases
underwent R0 resection and received adjuvant therapy with the
tumors located in the body/tail of the pancreas, surviving >5
years. Furthermore, one of the aforementioned cases had a survival
of ~16 years in spite of lymph node metastasis, representing the
longest survival time of patients with SCP in the literature, to
the best of our knowledge (28).
Additionally, immune checkpoint inhibitors are increasingly being
administered in several types of cancer; however, PDAC has shown a
limited response to immunotherapy compared with other tumor types.
It has been reported that PD-L1 expression is more frequent in SCP
and UCP. Therefore, immunotherapy has become a promising treatment
option (29,59); however, its effectiveness in SCP
needs to be confirmed in large prospective studies.
Molecular alterations may serve as targets for
precise therapy. These abnormal genetic events can be detected by
NGS and can be used to find approaches to selectively kill tumor
cells (66). SCP is genetically
similar to PDAC (29,55). In general, the main stages of
tumorigenesis include oncogene activation and tumor suppressor
inactivation. Notably, numerous researchers are working to develop
strategies to target oncogenes such as KRAS; however, no
KRAS inhibitor has reached the clinical application stage at
present (67). Advances have been
made in clinical and preclinical trials of treatments targeting
TP53, CDKN2A and SMAD4, the three major tumor
suppressors of PDAC (53), and
further studies are warranted to assess whether the reactivators
clinically improve the prognosis of patients. Moreover, the genes
involved in chromatin stabilization and remodeling, such as
BRCA and KDM6A, have been reported to be deficient in
patients with PDAC and SCP (66).
It is encouraging that administering PARP inhibitors to block
base-excision repair leaves both double- and single-stranded DNA
breaks unrepaired, leading to death of the cells with BRCA
dysfunction (68). Furthermore, a
phase III trial reported that, among patients with germline
BRCA mutations and metastatic PC, the progression-free
survival was longer in patients with maintenance olaparib
administration than in those with placebo administration (69). Therefore, precise therapy based on
molecular alterations is a promising approach.
In PDAC, several histopathological factors have been
reported as prognostic factors, including tumor grade, R0 resection
margin, lymph node status and adjuvant therapy (28,70).
In SCP, a comparison analysis of these factors is not adequate.
Notably, evidence has suggested a possible association between
cellular senescence induced by TGF-β and long-term survival could
be interpreted as a promising finding. The study reported positive
staining for fibronectin, Snail and pSmad2/3 at the IHC level in
the tumor cells of 3 patients with SCP. γ-H2AX, p53 and p21,
typically used as markers of cellular senescence, were observed in
the sarcomatoid component of a case with long-term survival but not
in the others. Consistent with this finding, the Ki-67 labelling
index of the long-term survivor was the lowest compared with that
of the other 2 patients (26,27).
The Ki-67 labelling index is a strong prognostic factor in
pancreatic neuroendocrine tumor (71); however, its clinical significance in
PDAC has not been thoroughly evaluated. Therefore, TGF-β-mediated
senescence and a low Ki-67 labelling index may be critical in
reducing the proliferation and metastasis of sarcomatous cells.
Furthermore, OCGCs, the multinucleated giant cells with abundant
cytoplasm resembling giant cell tumors of the bone, have previously
been reported to protect against anaplastic carcinoma, with
long-term survival reported in ~50% of patients in a previous study
(30,72).
A single-center experience
We included cases with histological diagnosis of
PDAC and excluded cases with a previous history of malignant
tumors. Between August 2015 and August 2023, 603 cases of PDAC,
including 7 cases of SCP, were pathologically confirmed at the
Affiliated Lihuili Hospital of Ningbo University (Ningbo, China)
and the prevalence of SCP in all PDACs was 1.16%, which is lower
than that previously reported in the literature (1,2). Of
the 7 cases (Table SI), 3
exhibited abdominal pain, 3 exhibited jaundice and 1 was
asymptomatic. Serum bilirubin, mainly direct bilirubin, alanine
aminotransferase and aspartate aminotransferase increased in all 3
cases with jaundice. CA 19-9 was increased in 5 cases, and CA 12-5
was increased in 3 cases (Table
IV). All the patients underwent contrast-enhanced CT. CT
revealed that the pancreatic mass was cystic-solid or cystic, with
inhomogeneous or mild enhancement, and the boundary was mostly
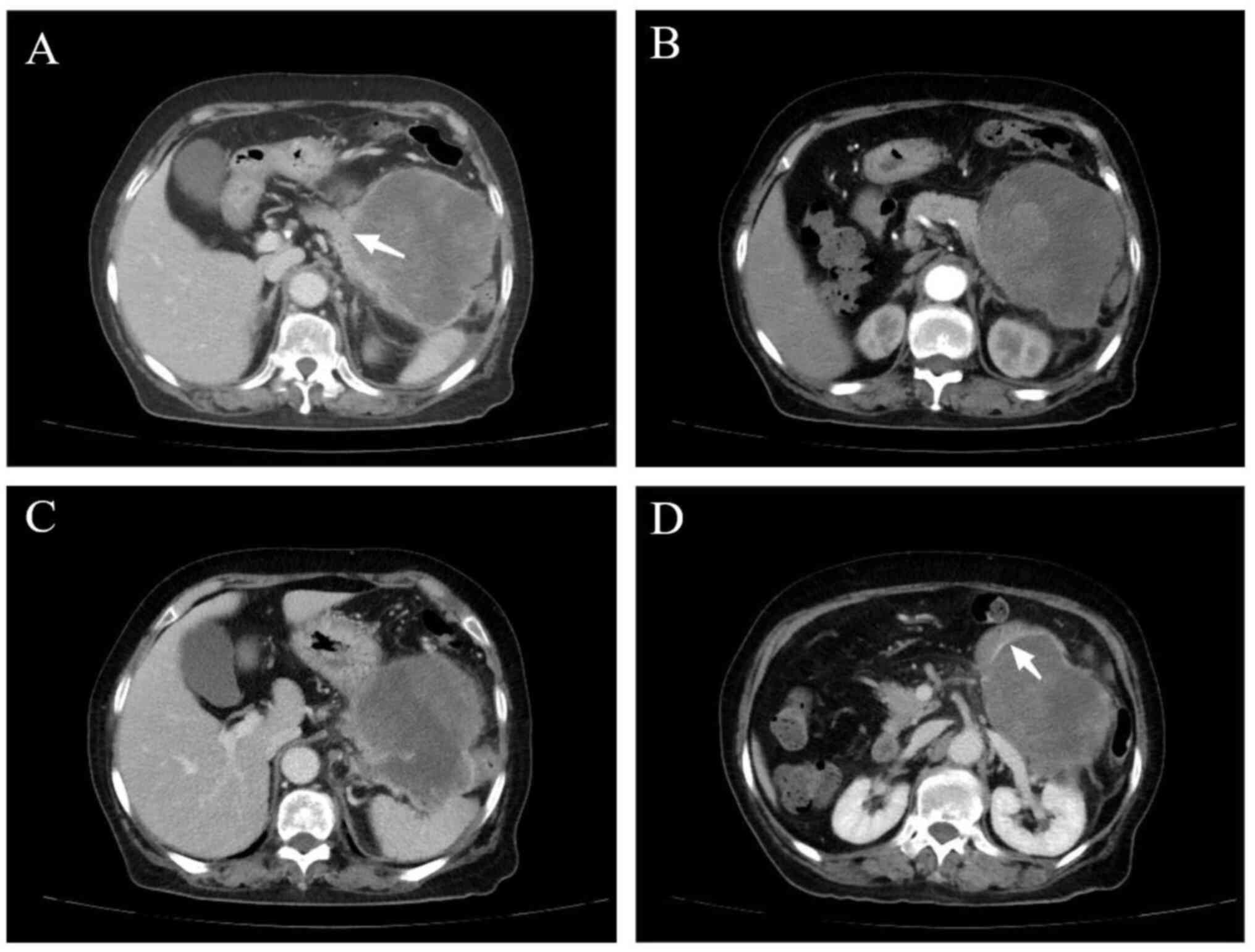
unclear (data not shown). Fig. 2
presents the imaging features of a typical case.
| Table IV.Serological alterations in cases of
sarcomatoid carcinoma of the pancreas at the Affiliated Lihuili
Hospital of Ningbo University (Ningbo, China). |
Table IV.
Serological alterations in cases of
sarcomatoid carcinoma of the pancreas at the Affiliated Lihuili
Hospital of Ningbo University (Ningbo, China).
|
| Case |
|
|---|
|
|
|
|
|---|
| Parameter | 1 | 2 | 3 | 4 | 5 | 6 | 7 | Normal range |
|---|
| TB, mmol/l | 9 | 8.5 | 149.6 | 192.6 | 13 | 17.2 | 135.3 | 0-23.0 |
| DB, mmol/l | 2.7 | 2.1 | 127.4 | 165.2 | 4.5 | 6.2 | 116.8 | 0-8.0 |
| ALT, U/l | 11 | 11 | 233 | 164 | 25 | 22 | 195 | 7-40 |
| AST, U/l | 19 | 14 | 116 | 81 | 26 | 28 | 133 | 13-35 |
| CA19-9, U/ml | 38 | 13.1 | 56.9 | 39.3 | 109.2 | 1986 | 22 | 0-37.0 |
| CA12-5, U/ml | 101.7 | 4.6 | 83.2 | 6.2 | 6.6 | 134.7 | 5.5 | 0-30.2 |
As indicated in Table
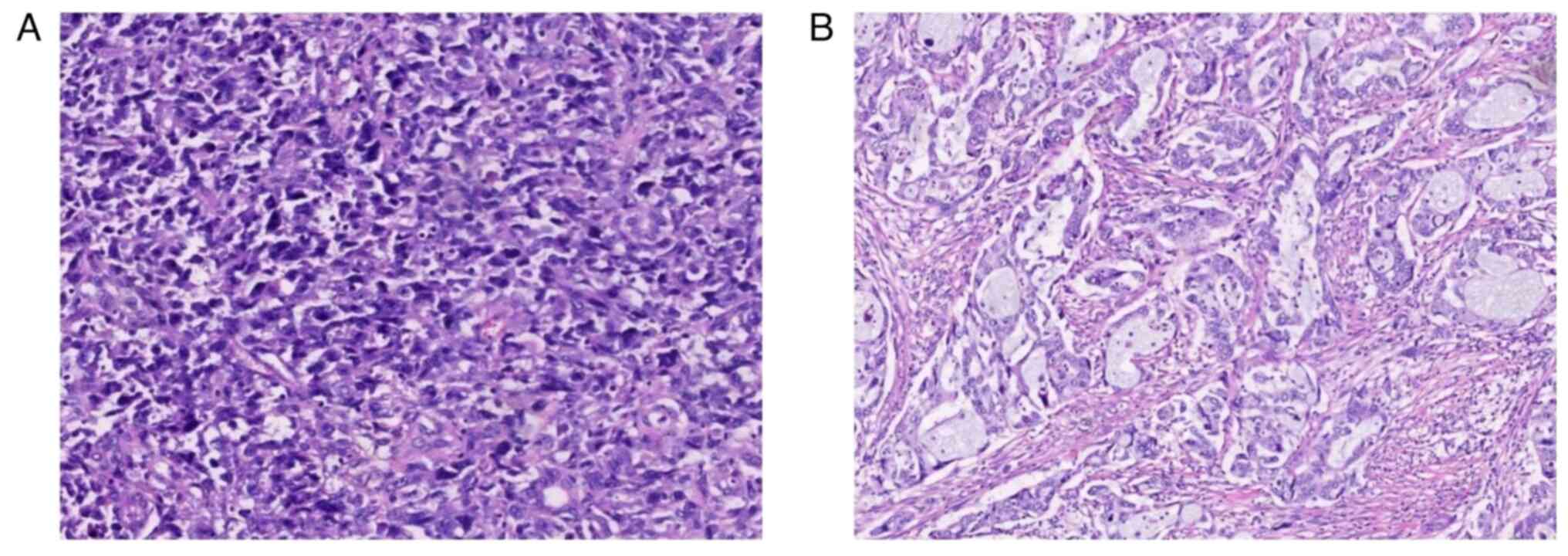
SI, 6 patients underwent radical surgery and histological
examination (Fig. 3), and all had
lymphovascular and perineural invasion. The tumor invaded adjacent
organs (duodenum, n=2; stomach, n=1; and colon, n=1) in 4 patients
(57.1%). A total of 2 patients (28.6%) had no lymph node
metastases, whereas the remaining patients had ≥1 positive lymph
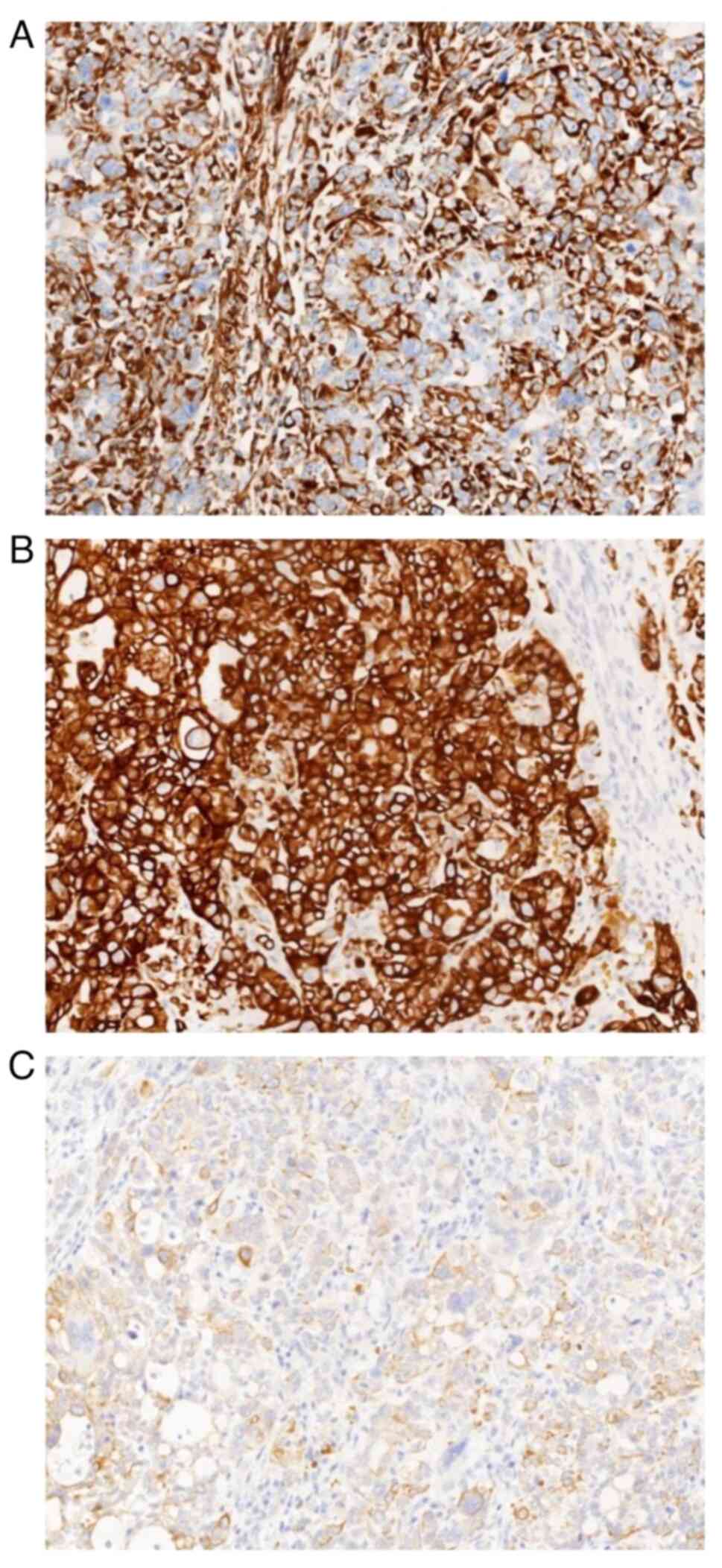
node metastasis. All samples examined by immunohistochemical
staining were positive for vimentin and CK (Fig. 4). The protocol was as follows:
Samples of all cases were fixed with 3.7% neutral formaldehyde
solution, dehydrated, embedded in paraffin and slices were
subjected to H&E staining according to routine procedures to
prepare slides observed under a light microscope. All paraffin
blocks containing tumors were stained with 34BE12+P540s double
labelling. Immunohistochemical staining for certain markers in the
cases was performed at the time of diagnosis. Immunohistochemical
analysis was performed at the clinical laboratory of our
institution using the Roche BenchMark automated system (Roche
Diagostics) with appropriate controls. The following primary
antibodies were applied: CK7 [cat. no. ZM-0071; 1:200 dilution;
Zhongshang Goldenbridge (ZSGB)-Bio], CK19 (cat. no. l1006;
pre-diluted antibody; Biolynx), CK20 (cat. no. ZA-0574; 1:100
dilution; ZSGB-Bio), CK8/18 (cat. no. ZM-0315; pre-diluted
antibody; ZSGB-Bio), MUC1 (cat. no. CMM-0251; pre-diluted antibody;
Celnovte), smooth muscle actin (cat. no. CAM-0191; pre-diluted
antibody; Celnovte), human melanoma black 45 (cat. no. ZM-0187; 1:1
dilution; ZSGB-Bio), vimentin (cat. no. ZM-0260; 1:200 dilution;
ZSGB-Bio) and Ki-67 (cat. no. ZM-0166; 1:800 dilution; ZSGB-Bio).
All antibodies were incubated for 30 min at room temperature. Next,
a conjugated secondary antibody was added (cat. no. DS0003;
pre-diluted antibody; ZSGB-Bio) and incubated at room temperature
for 30 min. Subsequently, visualization was performed by applying
0.1% 3,3′-di-aminobenzidine tetrahydrochloride solution for 5 min
at room temperature. The sections were finally counterstained with
Mayer's hematoxylin for 1 min at room temperature, dehydrated and
mounted with coverslips after being embedded in mounting medium.
The slides were stored at room temperature. The sections were
viewed under a light microscope by two independent pathologists
blinded to the patients' clinical data. Immunoreactivity was
evaluated in a semiquantitative manner to assess the percentage of
immunopositive tumor cells: Negative (−), 0%; focal, <25%;
moderate, 25–75%; and diffuse, >75%.
In all 3 surviving patients, the tumor was located
in the distal pancreas rather than in the pancreatic head, without
distant metastases. Among them, 1 patient underwent a gene test.
The NGS revealed TP53 and KRAS mutations, and a
pathogenic variant of the germline BRCA1 gene. Therefore,
the patient received a PARP inhibitor because of their poor
tolerance to chemotherapy; to the best of our knowledge, this is
the first report of this drug administration to a patient with SCP
in the literature. It was encouraging that a good result was
obtained after administering olaparib to the patient with a
germline BRCA mutation and this prompted continuation of the
genetic testing of patients with rare tumors.
Accordingly, as presented in Table SI, the 3-month and 1-year mortality
rates of the patients with SCP exceeded 23.3 and 46.7%,
respectively, despite aggressive surgical management, with many
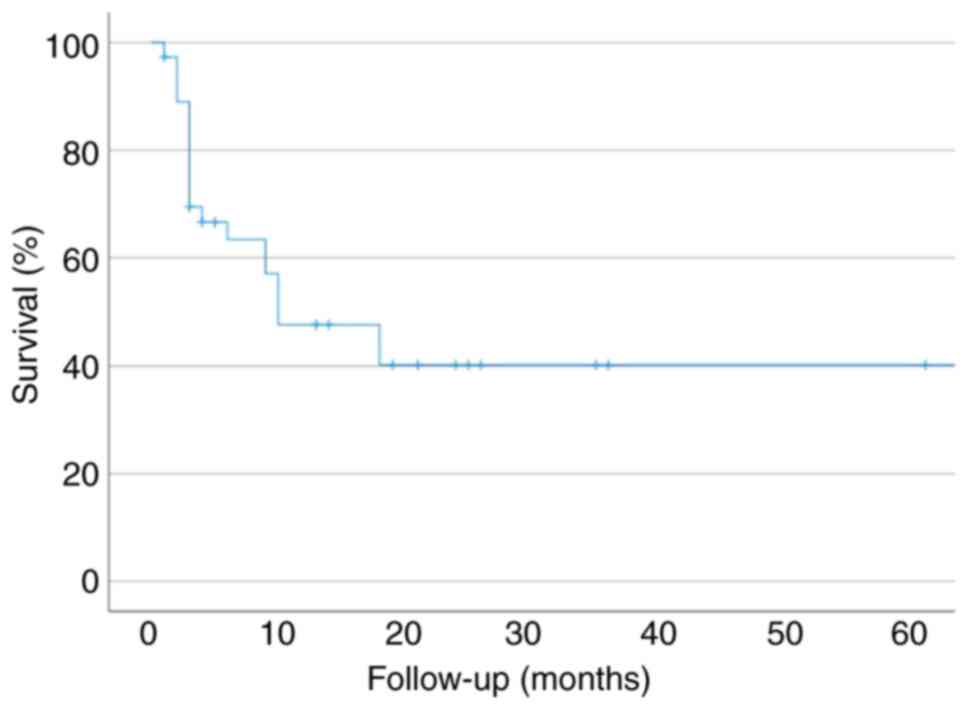
succumbing to early metastasis. Subsequently, Kaplan-Meier curves
of the survival outcomes of all patients with SCP were plotted
(Fig. 5), and the median overall
survival time was 10 months. Despite the small sample size and
incomplete follow-up data, the data indicates that SCP is
associated with a worse prognosis.
Conclusions
SCP is a rare subtype of PDAC and is generally
considered to be an aggressive neoplasm with a poor prognosis.
Nevertheless, the low incidence and the incomplete understanding of
its clinical course hinder the possibility of performing
large-scale studies on patients with SCP. At present, the treatment
strategy for SCP is empirical therapy based on medical research of
PDAC. Similarly, surgical resection followed by PDAC-standardized
adjuvant chemotherapy is the most likely treatment option for
achieving long-term survival. A considerable portion of patients
with SCP may benefit from emerging immunotherapy-based strategies
in the near future. Notably, patients with SCP frequently exhibit
TP53 and KRAS mutations, highlighting the hereditary
homogeneities with PDAC; however, there are also certain crucial
distinctions. Particularly, certain molecular alterations in SCP,
including BRCA mutation, MCL1 amplification and
POLQ mutation, uncover more genetic features and provide
novel therapeutic targets. For example, PARP inhibitors aim to
selectively kill carcinoma cells with BRCA mutation. Lately,
several clinical trials have confirmed the partial efficacy of
olaparib, prompting further investigation to achieve synthetic
lethality in PC. For this reason, there is an urgent need for
genomic and transcriptomic studies based on larger cohorts of
patients with SCP to explore its molecular profile in greater depth
and to identify its histogenesis.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present research was funded by Ningbo City Health Technical
Key Youth Program (grant. no. 2021QNJSGG-WK).
Availability of data and materials
The data generated in the present study may be found
in the CNGB Sequence Archive of China National GeneBank DataBase
under accession number CNP0005730 or the following URL: https://db.cngb.org/search/project/CNP0005730/. All
other data generated in the present study may be requested from the
corresponding author.
Authors' contributions
SM, CL, YM and KW conceived and designed the study.
HZ performed the pathological examination of the patients in our
center. YM, HZ and KW collected and analyzed the data from our
single center. YM, YY, YH, SW, HZ and JM performed the literature
searches and drafted the manuscript. All authors have read and
approved the final manuscript. YM and KW confirm the authenticity
of all the raw data.
Ethics approval and consent to
participate
The present study was approved by The Ethics
Committee of The Affiliated Lihuili Hospital of Ningbo University
(Ningbo, China; approval no. KY2021PJ263) and was performed in
accordance with the Declaration of Helsinki.
Patient consent for publication
Written informed consent for publication (case 32)
was obtained from the patient and their relative.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
SC
|
sarcomatoid carcinoma
|
|
SCP
|
sarcomatoid carcinoma of the
pancreas
|
|
PC
|
pancreatic cancer
|
|
CSP
|
carcinosarcoma of the pancreas
|
|
PDAC
|
pancreatic ductal adenocarcinoma
|
|
USC
|
undifferentiated SC
|
|
WHO
|
World Health Organization
|
|
UCP
|
undifferentiated carcinoma of the
pancreas
|
|
RCP
|
rhabdoid carcinoma of the pancreas
|
|
TGF-β
|
transforming growth factor-β
|
|
EMT
|
epithelial-mesenchymal transition
|
|
IL
|
interleukin
|
|
IHC
|
immunohistochemistry
|
|
PARP
|
poly ADP-ribose polymerase
|
|
OCGCS
|
osteoclast-like giant cells
|
|
CK
|
cytokeratin
|
|
MUC1
|
transmembrane glycoprotein mucin
1
|
|
NGS
|
next-generation sequencing
|
|
PD-L1
|
programmed death-ligand 1
|
|
CPS
|
combined positive score
|
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sarantis P, Koustas E, Papadimitropoulou
A, Papavassiliou AG and Karamouzis MV: Pancreatic ductal
adenocarcinoma: Treatment hurdles, tumor microenvironment and
immunotherapy. World J Gastrointest Oncol. 12:173–181. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Seok JY and Kim YB: Sarcomatoid
hepatocellular carcinoma. Korean J Hepatol. 16:89–94. 2010.(In
Korean). View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nagtegaal ID, Odze RD, Klimstra D, Paradis
V, Rugge M, Schirmacher P, Washington KM, Carneiro F and Cree IA;
WHO Classification of Tumours Editorial Board, : The 2019 WHO
classification of tumours of the digestive system. Histopathology.
76:182–188. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Luchini C, Capelli P and Scarpa A:
Pancreatic ductal adenocarcinoma and its variants. Surg Pathol
Clin. 9:547–560. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bazzichetto C, Luchini C, Conciatori F,
Vaccaro V, Di Cello I, Mattiolo P, Falcone I, Ferretti G, Scarpa A,
Cognetti F and Milella M: Morphologic and Molecular landscape of
pancreatic cancer variants as the basis of new therapeutic
strategies for precision oncology. Int J Mol Sci. 21:88412020.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Higashi M, Takao S and Sato E: Sarcomatoid
carcinoma of the pancreas: A case report with immunohistochemical
study. Pathol Int. 49:453–456. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
De la Riva S, Muñoz-Navas MA, Betés M,
Súbtil JC, Carretero C and Sola JJ: Sarcomatoid carcinoma of the
pancreas and congenital choledochal cyst. Gastrointest Endosc.
64:1005–1006. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yao J, Qian JJ, Zhu CR, Bai DS and Miao Y:
Laparoscopic left pancreatectomy for pancreatic sarcomatoid
carcinoma: A case report and review of the literature. Oncol Lett.
6:568–570. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ren CL, Jin P, Han CX, Xiao Q, Wang DR,
Shi L, Wang DX and Chen H: Unusual early-stage pancreatic
sarcomatoid carcinoma. World J Gastroenterol. 19:7820–7824. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kane JR, Laskin WB, Matkowskyj KA, Villa C
and Yeldandi AV: Sarcomatoid (spindle cell) carcinoma of the
pancreas: A case report and review of the literature. Oncol Lett.
7:245–249. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lu BC, Wang C, Yu JH, Shen ZH and Yang JH:
A huge adenosquamous carcinoma of the pancreas with sarcomatoid
change: An unusual case report. World J Gastroenterol.
20:16381–16386. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yepuri N, Pruekprasert N and Naous R:
High-grade malignant pancreatic neoplasm with sarcomatoid features.
AME Case Rep. 2:392018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xie Y, Xiang Y, Zhang D, Yao X, Sheng J,
Yang Y and Zhang X: Sarcomatoid carcinoma of the pancreas: A case
report and review of the literature. Mol Med Rep. 18:4716–4724.
2018.PubMed/NCBI
|
|
15
|
Bukhari N and Joudeh A: Early stage
anaplastic sarcomatoid carcinoma of the pancreas, a case report. Am
J Case Rep. 20:597–601. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhou DK, Gao BQ, Zhang W, Qian XH, Ying LX
and Wang WL: Sarcomatoid carcinoma of the pancreas: A case report.
World J Clin Cases. 7:236–241. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Toledo PF, Berger Z, Carreño L, Cardenas
G, Castillo J and Orellana O: Sarcomatoid carcinoma of the
pancreas-a rare tumor with an uncommon presentation and course: A
case report and review of literature. World J Clin Cases.
9:3716–3725. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lim HJ, Kang HS, Lee JE, Min JH, Shin KS,
You SK and Kim KH: Sarcomatoid carcinoma of the
pancreas-multimodality imaging findings with serial imaging
follow-up: A case report and review of literature. World J Clin
Cases. 9:3102–3113. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harada Y, Sakai A, Ikegawa T, Shiomi H,
Masuda A and Kodama Y: Pancreatic sarcomatoid carcinoma with
intraductal growth protruding from the papilla of vater. Am J
Gastroenterol. 117:19002022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang ML, Sabatino ME, Castillo CFD,
Mattia AR and Chebib I: Cytologic, histologic, and molecular
features of pancreatic sarcomatoid undifferentiated carcinoma with
heterologous osteosarcomatous transformation. Diagn Cytopathol.
51:E164–E169. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen LL, Chen J, Bu P, Chen CW and Xiang
XX: Two cases of sarcomatoid pancreatic carcinoma. Chin J
Gastroenterol Hepatol. 23:837–838. 2014.
|
|
22
|
Hu QL, Li HQ and Xia TY: A case of
sarcomatoid carcinoma of the pancreas. Shijie Huaren Xiaohua Zazhi.
23:707–710. 2015.
|
|
23
|
Zhao ZB, Ji Y and Kong HF: Pancreatic
sarcomatoidcarcinoma with liver metastasis: A case report. Chin J
Geriatr. 37:225–226. 2018.(In Chinese).
|
|
24
|
Jiang YS, Tao LY, Yang MW, He RZ, Shen Y
and Sun YW: A case of sarcomatoid carcinoma of the pancreas. Chin J
Pract Surg. 39:879–880. 2019.(In Chinese).
|
|
25
|
Ren PT: Two cases of sarcomatoid carcinoma
of the pancreas. Chin J Gen Srug. 34:366–367. 2019.(In
Chinese).
|
|
26
|
Kimura T, Fujimoto D, Togawa T, Ishida M,
Iida A, Sato Y and Goi T: Sarcomatoid carcinoma of the pancreas
with rare long-term survival: A case report. World J Surg Oncol.
18:1052020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kimura T, Togawa T, Iida A, Noriki S, Sato
Y and Goi T: Does cellular senescence play an important role in the
prognosis of sarcomatoid carcinoma of the pancreas? World J Surg
Oncol. 19:792021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Blair AB, Burkhart RA, Griffin JF, Miller
JA, Weiss MJ, Cameron JL, Wolfgang CL and He J: Long-term survival
after resection of sarcomatoid carcinoma of the pancreas: An
updated experience. J Surg Res. 219:238–243. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gkountakos A, Simbolo M, Bariani E, Scarpa
A and Luchini C: Undifferentiated sarcomatoid carcinoma of the
pancreas: From histology and molecular pathology to precision
oncology. Int J Mol Sci. 23:12832022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Clark CJ, Graham RP, Arun JS, Harmsen WS
and Reid-Lombardo KM: Clinical outcomes for anaplastic pancreatic
cancer: A population-based study. J Am Coll Surg. 215:627–634.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guarino M, Tricomi P, Giordano F and
Cristofori E: Sarcomatoid carcinomas: Pathological and
histopathogenetic considerations. Pathology. 28:298–305. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
WHO Classification of Tumours Editorial
Board, . Digestive System Tumours. WHO classification of tumours.
5th edition. International Agency for Research on Cancer; Lyon:
2019
|
|
33
|
Li J, Wei T, Zhang J, Wei S, Chen Q, Chen
BW, Zhou Y, Wen L, Qin H, Bai X and Liang T: Carcinosarcoma of the
pancreas: Comprehensive clinicopathological and molecular
characterization. HPB (Oxford). 22:1590–1595. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ruess DA, Kayser C, Neubauer J,
Fichtner-Feigl S, Hopt UT and Wittel UA: Carcinosarcoma of the
pancreas: Case report with comprehensive literature review.
Pancreas. 46:1225–1233. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Thompson L, Chang B and Barsky SH:
Monoclonal origins of malignant mixed tumors (carcinosarcomas).
Evidence for a divergent histogenesis. Am J Surg Pathol.
20:277–285. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cantrell LA, Blank SV and Duska LR:
Uterine carcinosarcoma: A review of the literature. Gynecol Oncol.
137:581–588. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
van den Berg W, Tascilar M, Offerhaus GJ,
Albores-Saavedra J, Wenig BM, Hruban RH and Gabrielson E:
Pancreatic mucinous cystic neoplasms with sarcomatous stroma:
Molecular evidence for monoclonal origin with subsequent divergence
of the epithelial and sarcomatous components. Mod Pathol. 13:86–91.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang Y and Weinberg RA:
Epithelial-to-mesenchymal transition in cancer: Complexity and
opportunities. Front Med. 12:361–373. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cantelli G, Crosas-Molist E, Georgouli M
and Sanz-Moreno V: TGFBeta-induced transcription in cancer. Semin
Cancer Biol. 42:60–69. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Massagué J: TGFbeta signalling in context.
Nat Rev Mol Cell Biol. 13:616–630. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Arteaga CL: Inhibition of TGFbeta
signaling in cancer therapy. Curr Opin Genet Dev. 16:30–37. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Massagué J: TGFbeta in cancer. Cell.
134:215–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kang Y, He W, Tulley S, Gupta GP,
Serganova I, Chen CR, Manova-Todorova K, Blasberg R, Gerald WL and
Massagué J: Breast cancer bone metastasis mediated by the Smad
tumor suppressor pathway. Proc Natl Acad Sci USA. 102:13909–13914.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Boström AK, Möller C, Nilsson E, Elfving
P, Axelson H and Johansson ME: Sarcomatoid conversion of clear cell
renal cell carcinoma in relation to epithelial-to-mesenchymal
transition. Hum Pathol. 43:708–719. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lien HC, Lee YH, Juang YL and Lu YT:
Fibrillin-1, a novel TGF-beta-induced factor, is preferentially
expressed in metaplastic carcinoma with spindle sarcomatous
metaplasia. Pathology. 51:375–383. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kawarada Y, Inoue Y, Kawasaki F, Fukuura
K, Sato K, Tanaka T, Itoh Y and Hayashi H: TGF-β induces p53/Smads
complex formation in the PAI-1 promoter to activate transcription.
Sci Rep. 6:354832016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Saitoh M: Epithelial-mesenchymal
transition is regulated at post-transcriptional levels by
transforming growth factor-β signaling during tumor progression.
Cancer Sci. 106:481–488. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Husain AN, Colby TV, Ordóñez NG, Krausz T,
Borczuk A, Cagle PT, Chirieac LR, Churg A, Galateau-Salle F, Gibbs
AR, et al: Guidelines for pathologic diagnosis of malignant
mesothelioma: A consensus statement from the international
mesothelioma interest group. Arch Pathol Lab Med. 133:1317–1331.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Shi HY, Xie J and Miao F: Pancreatic
carcinosarcoma: First literature report on computed tomography
imaging. World J Gastroenterol. 21:1357–1361. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Watanabe M, Miura H, Inoue H, Uzuki M,
Noda Y, Fujita N, Yamazaki T and Sawai T: Mixed
osteoclastic/pleomorphic-type giant cell tumor of the pancreas with
ductal adenocarcinoma: Histochemical and immunohistochemical study
with review of the literature. Pancreas. 15:201–208. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Mugaanyi J and Lu C, Huang J and Lu C:
Undifferentiated pancreatic carcinomas, clinical features and
therapeutic options: What we know. Cancers (Basel). 14:61022022.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Park W, Chawla A and O'Reilly EM:
Pancreatic cancer: A review. JAMA. 326:851–862. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ding Y, Shao Y, Na C, Yin JC, Hua H, Tao
R, Jiang Y, Hu R, He X, Miao C, et al: Genetic characterisation of
sarcomatoid carcinomas reveals multiple novel actionable mutations
and identifies KRAS mutation as a biomarker of poor prognosis. J
Med Genet. 59:10–17. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gkountakos A, Mafficini A, Lou E, Malleo
G, Salvia R, Calicchia M, Silvestris N, Racila E, Amin K, Veronese
N, et al: Genomic characterization of undifferentiated sarcomatoid
carcinoma of the pancreas. Hum Pathol. 128:124–133. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Iacobuzio-Donahue CA, Fu B, Yachida S, Luo
M, Abe H, Henderson CM, Vilardell F, Wang Z, Keller JW, Banerjee P,
et al: DPC4 gene status of the primary carcinoma correlates with
patterns of failure in patients with pancreatic cancer. J Clin
Oncol. 27:1806–1813. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Blackford A, Serrano OK, Wolfgang CL,
Parmigiani G, Jones S, Zhang X, Parsons DW, Lin JCH, Leary RJ,
Eshleman JR, et al: SMAD4 gene mutations are associated with poor
prognosis in pancreatic cancer. Clin Cancer Res. 15:4674–4679.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Agaimy A, Haller F, Frohnauer J, Schaefer
IM, Ströbel P, Hartmann A, Stoehr R and Klöppel G: Pancreatic
undifferentiated rhabdoid carcinoma: KRAS alterations and SMARCB1
expression status define two subtypes. Mod Pathol. 28:248–260.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Waddell N, Pajic M, Patch AM, Chang DK,
Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al:
Whole genomes redefine the mutational landscape of pancreatic
cancer. Nature. 518:495–501. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lehrke HD, Graham RP, Mcwilliams RR,
Lam-Himlin DM, Smyrk TC, Jenkins S, Dong H and Zhang L:
Undifferentiated pancreatic carcinomas display enrichment for
frequency and extent of PD-L1 expression by tumor cells. Am J Clin
Pathol. 148:441–449. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Silvestris N, Argentiero A, Brunetti O,
Sonnessa M, Colonna F, Delcuratolo S, Luchini C, Scarpa A, Lonardi
S, Nappo F, et al: PD-L1 and Notch as novel biomarkers in
pancreatic sarcomatoid carcinoma: A pilot study. Expert Opin Ther
Targets. 25:1007–1016. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Halbrook CJ, Lyssiotis CA, Pasca di
Magliano M and Maitra A: Pancreatic cancer: Advances and
challenges. Cell. 186:1729–1754. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Hermanek P and Wittekind C: Residual tumor
(R) classification and prognosis. Semin Surg Oncol. 10:12–20. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Shiihara M, Higuchi R, Izumo W, Furukawa T
and Yamamoto M: A comparison of the pathological types of
undifferentiated carcinoma of the pancreas. Pancreas. 49:230–235.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Imaoka H, Ikeda M, Maehara K, Umemoto K,
Ozaka M, Kobayashi S, Terashima T, Inoue H, Sakaguchi C, Tsuji K,
et al: Clinical outcomes of chemotherapy in patients with
undifferentiated carcinoma of the pancreas: A retrospective
multicenter cohort study. BMC Cancer. 20:9462020. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Qian Y, Gong Y, Fan Z, Luo G, Huang Q,
Deng S, Cheng H, Jin K, Ni Q, Yu X and Liu C: Molecular alterations
and targeted therapy in pancreatic ductal adenocarcinoma. J Hematol
Oncol. 13:1302020. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Zorde Khvalevsky E, Gabai R, Rachmut IH,
Horwitz E, Brunschwig Z, Orbach A, Shemi A, Golan T, Domb AJ, Yavin
E, et al: Mutant KRAS is a druggable target for pancreatic cancer.
Proc Natl Acad Sci USA. 110:20723–20728. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zhu H, Wei M, Xu J, Hua J, Liang C, Meng
Q, Zhang Y, Liu J, Zhang B, Yu X and Shi S: PARP inhibitors in
pancreatic cancer: Molecular mechanisms and clinical applications.
Mol Cancer. 19:492020. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Ashworth A: A synthetic lethal therapeutic
approach: Poly(ADP) ribose polymerase inhibitors for the treatment
of cancers deficient in DNA double-strand break repair. J Clin
Oncol. 26:3785–3790. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Serrano PE, Cleary SP, Dhani N, Kim PT,
Greig PD, Leung K, Moulton CA, Gallinger S and Wei AC: Improved
long-term outcomes after resection of pancreatic adenocarcinoma: A
comparison between two time periods. Ann Surg Oncol. 22:1160–1167.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Philips P, Kooby DA, Maithel S, Merchant
NB, Weber SM, Winslow ER, Ahmad S, Kim HJ, Scoggins CR, McMasters
KM and Martin RCG II: Grading using Ki-67 index and mitotic rate
increases the prognostic accuracy of pancreatic neuroendocrine
tumors. Pancreas. 47:326–331. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Strobel O, Hartwig W, Bergmann F, Hinz U,
Hackert T, Grenacher L, Schneider L, Fritz S, Gaida MM, Büchler MW
and Werner J: Anaplastic pancreatic cancer: Presentation, surgical
management, and outcome. Surgery. 149:200–208. 2011. View Article : Google Scholar : PubMed/NCBI
|