Introduction
The International Agency for Research on Cancer of
the World Health Organization ranked cervical cancer (CC) fourth in
terms of global incidence and third in terms of cancer-associated
deaths among women in 2022, with 662,301 new cases and 348,874
associated deaths reported. Of these cases and deaths, 90% were
reported from low- to middle-income countries (1).
There are numerous factors involved in the emergence
and progression of CC. The most notable is persistent infection
with the human papillomavirus (HPV). Additional factors include a
compromised immune system, spontaneous mutations and
lifestyle-related factors, such as smoking, alcohol consumption,
hormonal therapies, malnutrition and a sedentary lifestyle, as well
as vaginal dysbiosis associated with Sneathia sp.,
Fusobacterium sp., Pseudomonas sp., Streptococcus
sp., human immunodeficiency virus and Chlamydia sp.
(2), and mutations in cancer-driver
genes, which can initiate carcinogenesis and promote disease
progression by acting directly or influencing the activity of other
genes (3). The IntOgen database,
updated to 2023, provides valuable information on cancer driver
genes and their pathways. Through the analysis of 266 cohorts
comprising 33,019 samples across 73 types of cancer, the IntOgen
database has classified 619 cancer driver genes. These findings
highlight the broad impact and diversity of cancer driver genes in
cancer research (4). The IntOGen
database has analyzed four cohorts of patients with cervical
squamous cell carcinoma, comprising 467 samples in total. The CC
cohorts present with 9,749,109 mutations and 44 mutated cancer
driver genes, including PIK3-catalytic subunit a (PI3KCA), FAT
atypical cadherin (FAT1), SMAD4, F-Box And WD repeat domain
containing 7 (FBXW7), kelch-like ECH-associated protein 1 (KEAP1),
E1A-associated cellular p300 transcriptional co-activator protein
(EP300), AKT1 and nuclear factor erythroid 2-related factor 2
(NRF2). Cellular-MYC (c-MYC) and cAMP response element-binding
binding protein (CREBBP), are not reported as driver genes in CC,
but are in other types of cancer. Examples for c-MYC include
Burkitt's lymphoma, malignant lymphoma and acute myeloid leukemia,
while examples for CREBBP include bladder urothelial carcinoma,
non-small cell lung cancer, ovarian epithelial tumor and
hepatocellular carcinoma (4).
Yes-associated protein 1 (YAP1) and TEA domain family member 2
(TEAD2) have not been reported as driver genes.
The NRF2 pathway is a detoxifying pathway activated
by high levels of ROS. The NRF2 and KEAP1 genes form a complex
where KEAP1 anchors to the cytoskeleton. When the KEAP1-NRF2 link
is lost, NRF2 can relocate to the nucleus and activate the
transcription of genes necessary to eliminate toxins within the
cell. The NRF2 pathway inhibits the development of multiple
diseases, including cancer and neurodegenerative diseases (5). However, in addition to the protective
role of NRF2 in normal cells, its role in cancer cells has also
been highlighted, where aberrant signal transduction in the NRF2
pathway leads to the constitutive activation of NFE2L2 and thereby
the positive regulation of its target genes, promoting cell
survival (6). Dual activity of
NFE2L2 has been observed in various cancer types, where its
overactivation induces the expression of genes that allow tumor
cells to acquire altered survival capabilities through mechanisms
such as self-renewal capacity and repression of apoptosis (6).
The Notch pathway is involved in cellular
differentiation and embryonic development, but has also been
identified as hyperactive in cancer (7). c-Myc, the most studied proto-oncogene
of the Myc family, is altered in various solid tumors, leukemias
and lymphomas, and is involved in functions such as cell
differentiation, cell adhesion, migration and angiogenesis, among
others (8).
Finally, the Hippo signaling pathway is a kinase
cascade that recognizes FAT1 as the primary initiator of the
pathway. Under normal conditions, its leading role is to promote
cell death and differentiation, and inhibit proliferation.
Therefore, this pathway regulates cell fate, differentiation and
organ growth (9).
The present study aimed to analyze the
aforementioned genes in the different stages of CC, which are
classified according to the cervical epithelial abnormalities
caused by the presence of HPV in cervical intraepithelial neoplasia
(CIN) 1, the earliest form of precancerous lesions. CIN 2 and CIN 3
are grouped as high-grade squamous intraepithelial lesions. The
last group is the cervical cancerous stages known as in situ
and invasive CC (10).
All the genes analyzed in the present study are
involved in signaling pathways closely related to cancer
development and are mostly cancer-driver genes (11). The identification of driver genes
and understanding their roles in cancer development is crucial for
the development of targeted therapies and personalized treatment
strategies for patients with cancer.
Materials and methods
Ethics statement
The present study used formalin-fixed,
paraffin-embedded (FFPE) cervical tissue, as approved by the
Research Ethics Committee of the General Hospital Zacatecas ‘Luz
Cosío González’, (Mexico City, Mexico; approval no. 0131/2018). The
FFPE cervical tissue samples used were ≥10 years old from the
Department of Pathology hospital archive; therefore, the
requirement for patient consent was waived. All protocols were
designed and performed according to the principles of the
Declaration of Helsinki.
FFPE cervical tissue
Patients were previously diagnosed at the Department
of Pathology, General Hospital Zacatecas ‘Luz González Cosío’, and
their samples divided into the following groups: Non-precancerous
or non-cancerous tissue (control), CIN 1, CIN 2, CIN 3, in
situ CC or invasive CC. For the reverse
transcription-quantitative PCR (RT-qPCR) assays, a total of 81
samples were analyzed, which consisted of 12 control cervical
tissue (non-neoplastic), 16 CIN 1 samples, 14 CIN 2 samples, 12 CIN
3 samples, 18 samples of in situ CC and 9 samples of
invasive CC. For the immunohistochemistry (IHC) analyses in tissue
microarrays (TMAs), 159 independent samples were examined,
including 24 samples of non-neoplastic cervical tissue, 42 CIN 1
samples, 33 CIN 2 samples, 16 CIN 3 samples, 32 in situ CC
samples and 12 samples of invasive CC. The inclusion criteria for
both patient sample cohorts were: i) Sufficient quantity of fixed
tissue; and ii) sufficient RNA, in the case of the RT-qPCR assays.
The sample exclusion criteria were: i) Insufficient quantity of
malignant tissue; and ii) insufficient RNA, in the case of the
RT-qPCR assays.
Gene expression analysis
RNA extraction from FFPE tissue samples
From each FFPE cervical tissue sample, 10 sections
of 5 µm in thickness were obtained using a microtome. Tissue
sections were deparaffinized at 37°C through washes with xylene for
1 h and 96 and 70% ethyl alcohol for 5 min for each wash. RNA
extraction was carried out using TRIzol™ (cat. no.
15596018; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol for RNA extraction in mammalian cells, with
minor modifications. A total of 500 µl proteinase K buffer [50 mM
Tris (pH 8.0), 400 mM NaCl, 2 mM EDTA and 4% SDS] and 50 µl of 20
mg/ml proteinase K were added to the deparaffinized samples and
incubated overnight, with 400-rpm agitation at 50°C. The next day,
samples were incubated for 1 h at 90°C without agitation to break
the cross-linkage between nucleic acids and proteins, to release
the nucleic acids (12). Following
this, the manufacturer's protocol for RNA extraction in mammalian
cells was continued. The concentration and purity of the extracted
RNA were determined using a NanoDrop™ One
Spectrophotometer (Thermo Fisher Scientific, Inc.). The extracted
RNA was subsequently used for the synthesis of cDNA.
Synthesis of cDNA
DNA decontamination was performed on the extracted
RNA through digestion with the DNase I, RNase-free kit (1 U/µl)
(cat. no. EN0521; Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions. The concentration and purity of the
decontaminated DNA were measured using a NanoDrop One
Spectrophotometer (Thermo Fisher Scientific, Inc.). cDNA synthesis
was performed using the SuperScript™ IV Reverse
Transcriptase kit (cat. no. 18090200; Thermo Fisher Scientific,
Inc.), following the manufacturer's instructions, with some
modifications. Random hexamers (50 ng/µl; cat. no. C118A; Promega
Corporation) were added and incubated at 70°C for 10 min, followed
by incubation at 45°C for 5 min. Next, the RT SuperScript 5X
Buffer, DTT (100 nM), dNTPs (10 mM), RNase inhibitor RiboLock (40
U/µl) and RT SuperScript enzyme (200 U/µl) were added, followed by
overnight incubation at 45°C (13).
The next day, samples were incubated for 10 min at 70°C. The newly
synthesized cDNA was stored at −70°C until use.
qPCR
The qPCR mix contained cDNA from each sample,
primers (10 pmol of each primer), water and Maxima™
SYBR® Green/ROX qPCR Master Mix (cat. no. K0222; Thermo
Fisher Scientific, Inc.). The qPCR was performed using the
QuantStudio 1™ Real-Time PCR system (cat. no. A40426;
Thermo Fisher Scientific, Inc.). The following thermocycling
conditions were used for PCR: 50°C for 2 min, 95°C for 10 min, and
40 cycles of 95°C for 15 sec and 57.5°C for 1 min. Primer sequences
were designed using the mRNA sequence for each gene as reported in
the National Center for Biotechnology Information database
(https://www.ncbi.nlm.nih.gov/nucleotide/) (Table I). The relative expression of each
gene was determined using the 2−ΔΔCq method (14), normalized to GAPDH gene expression.
Samples with a Cq value >39 were discarded.
 | Table I.Sequences of primers used for reverse
transcription-quantitative PCR. |
Table I.
Sequences of primers used for reverse
transcription-quantitative PCR.
| Gene | NCBI accession
no. | Sequence
(5′-3′) |
|---|
| FAT1 | NM_005245.4 | F:
TTCAAAATAGGTGAAGAGACAGGTG |
|
|
| R:
TTGTGATGAGACCTGTTTTAGGATG |
| YAP1 | NM_001195045.2 | F:
GCCCACTCGGGATGTAACTT |
|
|
| R:
AACCCTTTGGTCTCCGACAG |
| TEAD2 | NM_001256658.2 | F:
GTAGAGTTCTCAGCCTTCGTG |
|
|
| R:
ATTTGTCGTAGATCTGCCGG |
| SMAD4 | NM_005359.6 | F:
GAGGTTATGGTTCTGGGTGG |
|
|
| R:
GAAAGGCACTGGACAAACATG |
| NRF2 | NM_006164.5 | F:
ATAGCTGAGCCCAGTATC |
|
|
| R:
CATGCACGTGAGTGCTCT |
| KEAP1 | NM_203500.2 | F:
CCGGGAGTACATCTACATGC |
|
|
| R:
TGATGCAGGCGTGGAAG |
| PIK3CA | NM_006218.4 | F:
TGGTTAAAGATCCAGAAGTACAGG |
|
|
| R:
TTTGGCAATTCTGGTGAAGATTC |
| AKT 1 | NM_005163.2 | F:
GCTGAAGAGATGGAGGTGTC |
|
|
| R:
AGGATCACCTTGCCGAAAG |
| MYC | NM_002467.6 | F:
AATGAAAAGGCCCCCAAGGTAGTTATCC |
|
|
| R:
GTCGTTTCCGCAACAAGTCCTCTTC |
| CREBBP | NM_004380.3 | F:
TCAAACCCCAGTTCAGCC |
|
|
| R:
AGGGACTCTGTTATCAATGCTG |
| EP300 | NM_001429.4 | F:
GTGAACTCTCCTATAATGCCTCC |
|
|
| R:
ATGAAGAGCTGGTTGAGGAAG |
| FBXW7 | NM_033632.3 | F:
AAAGAGTTGTTAGCGGTTCTCG |
|
|
| R:
CCACATGGATACCATCAAACTG |
| GAPDH | NM_002046.7 | F:
CACCATGGAGAAGGCTGG |
|
|
| R:
TGCTGATGATCTTGAGGCTG |
Protein expression analysis
TMA construction
A total of 159 FFPE samples from various stages of
CC were selected, including non-lesioned, precancerous and
cancerous samples, and analyzed for TMA construction. Each tissue
sample had been previously examined using hematoxylin and eosin
staining to define the diagnostic areas. The selected tissues were
punched with a 1-mm needle and transferred onto a recipient
paraffin block using a Chemicon Advanced Tissue Arrayer ATA100
(Chemicon International; Thermo Fisher Scientific, Inc.). Tissue
sections of 5 µm in thickness were cut from each TMA and placed
onto positively charged slides (VWR Superfrost Plus; VWR
International; Avantor, Inc.).
IHC analysis on TMA
Protein expression analysis was performed using IHC
on the TMA. The following primary antibodies were used at a 1:50
dilution: NRF2 (cat. no. Y414975), KEAP1 (cat. no. Y400489), PIK3CA
(cat. no. Y011508), AKT (cat. no. Y409091), SMAD4 (cat. no.
Y401229), CMYC (cat. no. Y080045) and CREBBP (cat. no. Y401909)
(all Applied Biological Materials, Inc.), FAT1 (cat. no.
NBP1-84565; Novus Biologicals, Ltd.; Bio-Techne) and TEAD2 (cat.
no. AB273017; Abcam). Protein expression levels were detected using
the DAB HRP Brown IHC detection system from Bio SB, Inc., following
the manufacturer's protocol. The slides were imaged using the
Aperio LV1-Real-time Digital Pathology System (Leica Biosystems),
and tissue images were analyzed using the Aperio ImageScope
software (version 12.3.3.5048; Leica Biosystems).
Image analysis
The expression levels of each protein in the tumor
fraction of the samples within the microarray were assessed using a
semi-quantitative method involving visual analysis. A numerical
score was assigned to: i) The intensity of staining (intensity);
and ii) the proportion of positive cells showing expression of the
protein of interest (positivity). A visual appraisal was conducted,
in which three independent expert evaluators, in a blinded
analysis, assigned scores from 0 to 3 as follows: No staining, 0;
weak staining, 1; moderate staining, 2; and intense staining, 3,
which reflected the percentage of expression from 0–100%. Scores
from 1 to 4 were also assigned as follows: 0–25% Tissue expression,
1; 26–50% expression, 2; 51–75% expression, 3; and 76–100%
expression, 4, reflecting the percentage of positivity from 0–100%
(Fig. 1). The proteins of interest
were assessed in each of the tumors from cervical cancer patients,
analyzing their expression intensity categorized as high, medium,
low or absent, reflected on a scale from 0 to 3.
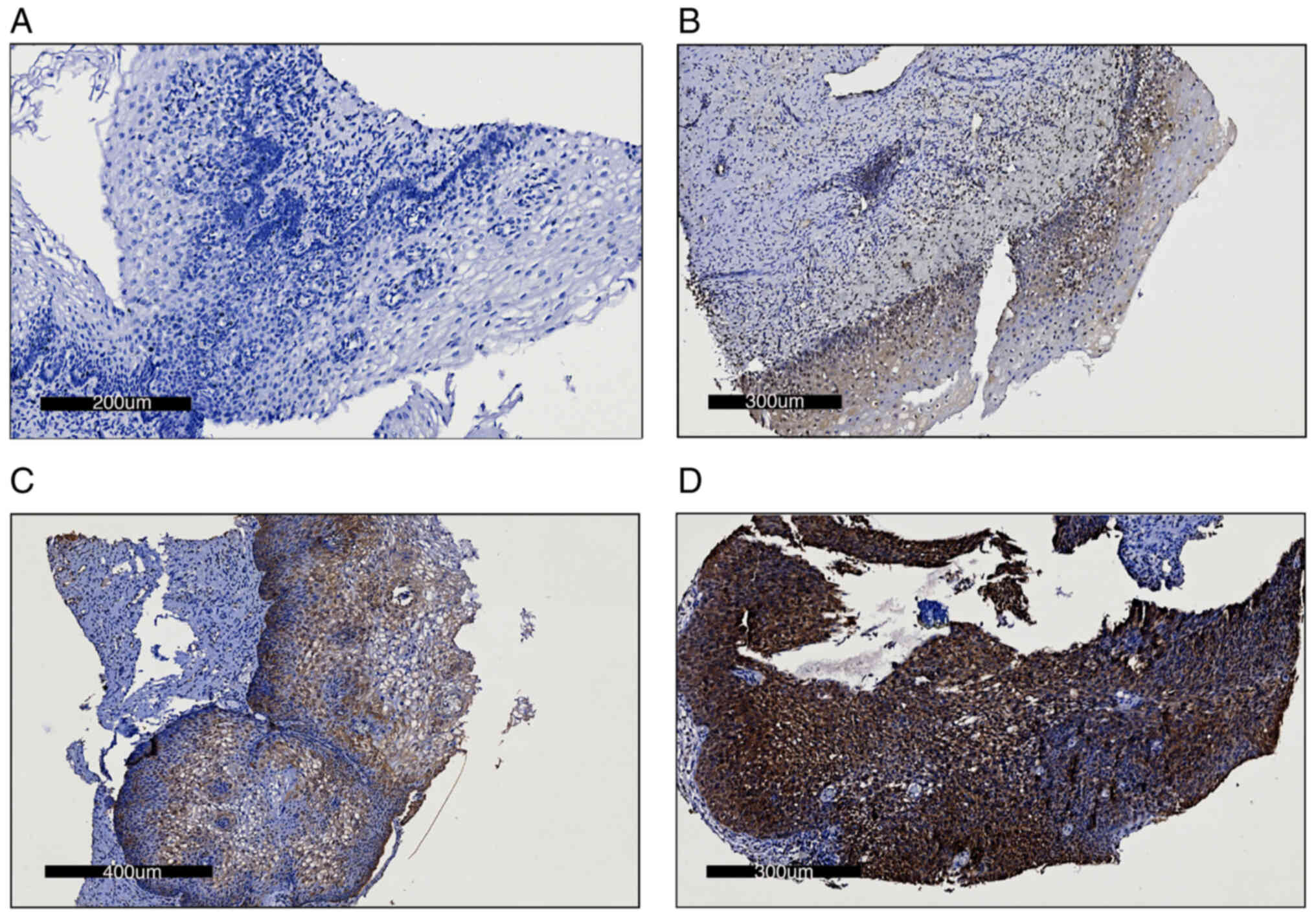 | Figure 1.Immunohistochemistry images of the
staining intensity of cervical cancer tumor samples. Representative
images of (A) no staining, assigned 0 points (scale bar, 200 nm),
(B) weak staining, assigned 1 point (scale bar, 300 nm), (C)
moderate staining, assigned 2 points (scale bar, 400 nm) and (D)
intense staining, assigned 4 points (scale bar, 300 nm). |
Statistical analysis
The data generated were analyzed for normality using
the Shapiro-Wilk test. Subsequently, a comparative analysis of
relative expression levels among the different groups was conducted
using the Kruskal-Wallis test, followed by the Dunn multiple
comparisons test. P<0.05 was considered to indicate a
statistically significant difference. P<0.05 was used to
indiciate a statistically significant difference. Statistical
analysis was performed using GraphPad Prism software (version 9.0;
Dotmatics).
Bioinformatic analysis
Kaplan-Meier plots
Analysis was conducted using data retrieved from the
Kaplan-Meier plotter platform (https://www.kmplot.com/analysis/) (15), with the following parameters:
Pan-Cancer RNA-seq generated database, patients stratified by
median age, automatic selection of the best cut-off value, overall
survival and no restriction by cancer stage or cellular
content.
Search Tool for Retrieval of
Interacting Genes/Proteins (STRING)
The analysis of protein-protein interaction networks
and functional enrichment analysis was performed using a STRING
Core Data Resource version 12.0 (https://string-db.org) with the following parameters:
Full STRING network; type of interaction, evidence; and interaction
source, experiments, database, co-occurrence and co-expression
(16). The cellular processes were
analyzed within which the aforementioned selected genes were
involved (Table II).
| Table II.Inference of potentially dysregulated
cellular processesa. |
Table II.
Inference of potentially dysregulated
cellular processesa.
| Term
description | Term ID, GO
no. | Observed gene
count, n | Background gene
count, n |
Strengthb | False discovery
ratec | Matching proteins
in the network |
|---|
| 0009628 | Response to abiotic
stimulus | 5 | 1,107 | 1.25 | 0.0038 | CREBBP, PIK3CA,
NRF2, AKT1 and c-MYC |
| 0002682 | Regulation of
immune system process | 5 | 1,438 | 1.14 | 0.0073 | CREBBP, PIK3CA,
NRF2, AKT1 and c-MYC |
| 0042592 | Homeostatic
process | 5 | 1,406 | 1.15 | 0.0073 | CREBBP, PIK3CA,
NRF2, AKT1 and c-MYC |
| 0009966 | Regulation of
signal transduction | 5 | 2,978 | 0.82 | 0.0314 | CREBBP, PIK3CA,
NRF2, AKT1 and c-MYC |
| 0031325 | Positive regulation
of cellular metabolic process | 5 | 3,114 | 0.8 | 0.0361 | CREBBP, PIK3CA,
NRF2, AKT1 and c-MYC |
| 0051173 | Positive regulation
of nitrogen compound metabolic process | 5 | 3,166 | 0.79 | 0.0373 | CREBBP, PIK3CA,
NRF2, AKT1 and c-MYC |
| 0006950 | Response to
stress | 5 | 3,358 | 0.77 | 0.0428 | CREBBP, PIK3CA,
NRF2, AKT1 and c-MYC |
| 0010604 | Positive regulation
of macromolecule metabolic process | 5 | 3,533 | 0.75 | 0.0442 | CREBBP, PIK3CA,
NRF2, AKT1 and c-MYC |
| 0036293 | Response to
decreased oxygen levels | 4 | 291 | 1.73 | 0.0038 | CREBBP, NRF2, AKT1
and c-MYC |
| 0080135 | Regulation of
cellular response to stress | 4 | 712 | 1.35 | 0.012 | CREBBP, NRF2, AKT1
and c-MYC |
| 0043066 | Negative regulation
of apoptotic process | 4 | 891 | 1.25 | 0.0218 | PIK3CA, NRF2, AKT1
and c-MYC |
| 0045944 | Positive regulation
of transcription by RNA polymerase II | 4 | 1,250 | 1.1 | 0.0314 | CREBBP, NRF2, AKT1
and c-MYC |
| 0051247 | Positive regulation
of protein metabolic process | 4 | 1,512 | 1.02 | 0.0428 | PIK3CA, NRF2, AKT1
and c-MYC |
| 0033554 | Cellular response
to stress | 4 | 1,572 | 1 | 0.0442 | PIK3CA, NRF2, AKT1
and c-MYC |
| 0036294 | Cellular response
to decreased oxygen levels | 3 | 136 | 1.94 | 0.0089 | NRF2, AKT1and
c-MYC |
| 0009411 | Response to UV | 3 | 150 | 1.9 | 0.0089 | CREBBP, AKT1 and
c-MYC |
| 2001242 | Regulation of
intrinsic apoptotic signaling pathway | 3 | 169 | 1.84 | 0.0102 | NRF2, AKT1 and
c-MYC |
| 0071375 | Cellular response
to peptide hormone stimulus | 3 | 240 | 1.69 | 0.0102 | PIK3CA, NRF2 and
AKT1 |
| 0062197 | Cellular response
to chemical stress | 3 | 272 | 1.64 | 0.0218 | PIK3CA, NRF2 and
AKT1 |
| 0001666 | Response to
hypoxia | 3 | 278 | 1.63 | 0.0229 | CREBBP, NRF2 and
c-MYC |
| 0043276 | Anoikis | 2 | 22 | 2.82 | 0.0232 | PIK3CA and
AKT1 |
| 1902176 | Negative regulation
of oxidative stress-induced intrinsic apoptotic signaling
pathway | 2 | 26 | 2.64 | 0.0102 | NRF2 and AKT1 |
| 0044030 | Regulation of DNA
methylation | 2 | 25 | 2.5 | 0.0128 | PIK3CA and
c-MYC |
| 0034405 | Response to fluid
shear stress | 2 | 34 | 2.97 | 0.0218 | NRF2 and AKT1 |
| 0016242 | Negative regulation
of macroautophagy | 2 | 35 | 2.35 | 0.0254 | PIK3CA and
AKT1 |
| 0046326 | Positive regulation
of glucose import | 2 | 36 | 2.34 | 0.0255 | NRF2 and AKT1 |
| 0031295 | T cell
co-stimulation | 2 | 42 | 2.27 | 0.0272 | PIK3CA and
AKT1 |
| 0035924 | Cellular response
to vascular endothelial growth factor stimulus | 2 | 42 | 2.27 | 0.0272 | PIK3CA and
AKT1 |
| 0043491 |
Phosphatidylinositol 3-kinase/protein
kinase B signal transduction | 2 | 49 | 2.24 | 0.0272 | PIK3CA and
AKT1 |
| 0007173 | Epidermal growth
factor receptor signaling pathway | 2 | 49 | 2.21 | 0.0235 | PIK3CA and
AKT1 |
| 0043491 |
Phosphatidylinositol 3-kinase/protein
kinase B signal transduction | 2 | 53 | 2.17 | 0.0814 | PIK3CA, AKT1 |
| 0043536 | Positive regulation
of blood vessel endothelial cell migration | 2 | 53 | 2.17 | 0.0814 | NRF2 and AKT1 |
| 0008286 | Insulin receptor
signaling pathway | 2 | 64 | 2.09 | 0.0979 | PIK3CA and
AKT1 |
| 0006112 | Energy reserve
metabolic process | 2 | 66 | 2.08 | 0.0873 | AKT1 and c-MYC |
| 0043542 | Endothelial cell
migration | 2 | 69 | 2.06 | 0.0994 | PIK3CA and
AKT1 |
| 0048661 | Positive regulation
of smooth muscle cell proliferation | 2 | 85 | 1.97 | 0.0412 | PIK3CA and
AKT1 |
| 0036294 | Cellular response
to decreased oxygen levels | 3 | 135 | 1.94 | 0.0089 | NRF2, AKT1 and
c-MYC |
| 0034644 | Cellular response
to UV | 2 | 30 | 1.94 | 0.0459 | CREBBP and
c-MYC |
| 2000777 | Positive regulation
of proteasomal ubiquitin-dependent protein catabolic process
involved in cellular response to hypoxia | 2 | 50 | 1.91 | 0.0412 | NRF2 and AKT1 |
| 0009411 | Response to UV | 3 | 150 | 2.0 | 0.0102 | CREBBP, AKT1 and
c-MYC |
Results
Protein expression and protein
interactions
The present study analyzed the expression of
transcripts and proteins of 12 genes related to the Notch, Hippo
and NRF2 signal transduction pathways. Fig. 1 summarizes the cellular positivity
and intensity in the expression found in precancerous lesions,
cancerous lesions and control cervical tissue through
immunohistochemistry assays for the proteins analyzed.
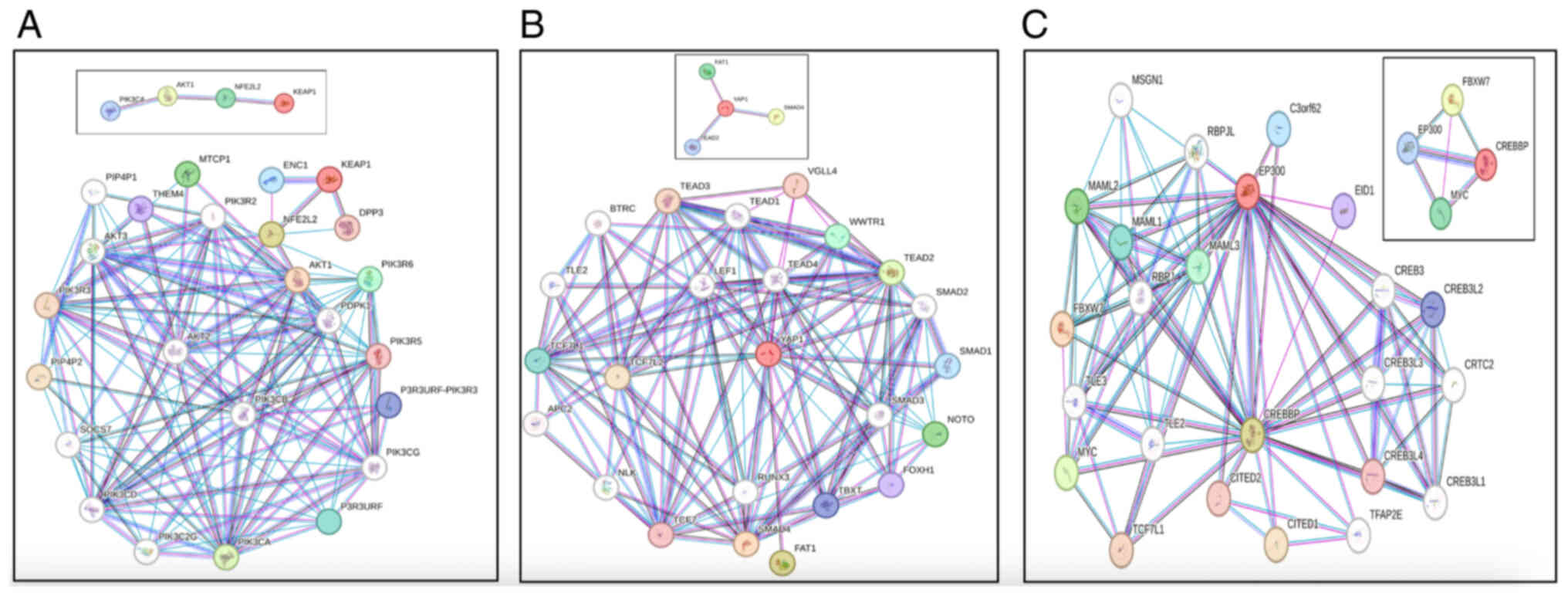
To investigate the relationship between gene
expression and CC, the protein-protein interactions of three
critical molecular nodes, namely the Hippo, Notch and NRF2
pathways, were analyzed (Fig.
2).
The STRING analysis showed that the Hippo pathway
had a close association with genes within the SMAD and TEAD
families (Fig. 2A). It was
demonstrated that the SMAD family is linked to the Notch pathway
through TGF-β, a crucial pathway in cancer development (17).
Through gene interaction analysis using the STRING
platform, it was demonstrated that Notch pathway associations
belonged to MYC pathway genes, such as EP300, FBXW7, CREBBP, TL2,
TL3, TCF7L1 and MAML3 (Fig. 2B). By
contrast, CREBBP mainly networked with EP300, CREB3L4, CREB3,
CREB3L2, CITED2 and CITED1. Additionally, FBXW7 interacted with
CREBBP, EP300, MAML2, RBPJ and MAML3. Moreover, EP300 was
associated with CREB3, CREB3L2, RBPJL, MAML1, MAML1, TLE3, TL2 and
CITED2. In summary, a large majority of the genes resulting from
the present interaction analyses were associated with
carcinogenesis and cell proliferation (18).
The NRF2/KEAP1 axis establishes crosstalk with the
PI3K/AKT1 axis in cellular detoxification through reactive oxygen
species (ROS) (19). These axes
converged through the direct interaction of AKT and NRF2 (Fig. 2C). In this context, the interactions
of genes that belong to the selected nodes of interest with other
proteins were recreated from known significative interactions, such
as PIK3CA with PDPK1, AKT1 and AKT3, known for their oncogenic
activity and promotion of tumor growth (18). AKT, by contrast, serves a vital role
in cancer development and progression through interactions with
NRF2, PIK3CA, PDPK1 and AKT2. NRF2 directly interacted with KEAP1,
AKT1, AKT2, AKT3, critical genes that regulate tumorigenesis and
cancer progression (17).
Furthermore, KEAP1 interacted with DPP3, ENC1 and NRF2.
Hippo pathway in CC
To investigate the relevance of these signaling
nodes, survival rates among patients with CC associated with the
expression levels of each aforementioned gene were explored. The
mRNA and protein expression levels were assessed in tissue samples
obtained from patients diagnosed with CIN 1, 2 and 3, in
situ CC and invasive CC, using cervical tissue samples without
neoplasia as a control for expression levels (Fig. 3, Fig.
4, Fig. 5).
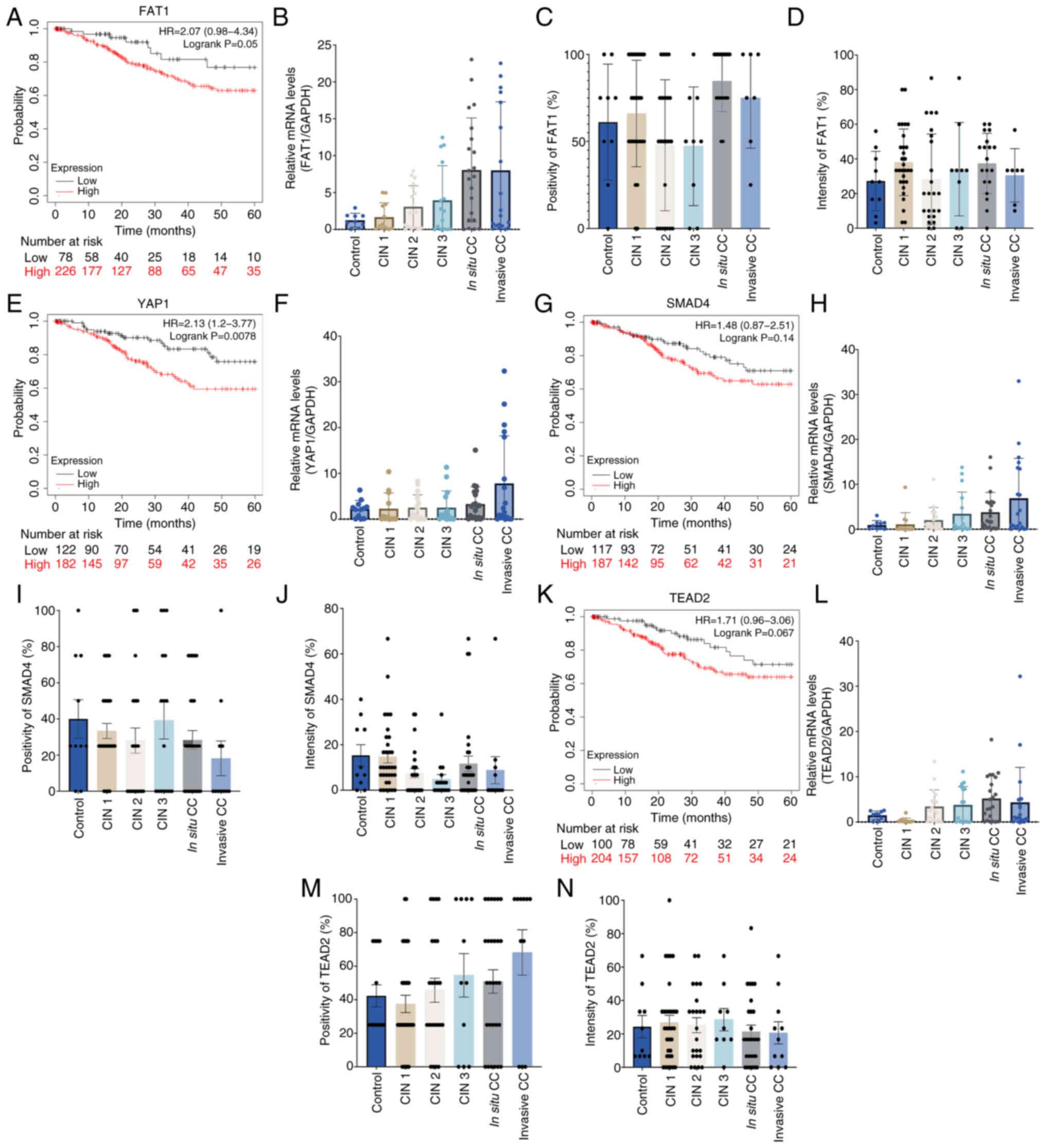
The Hippo pathway is a cascade of serine/threonine
kinases that serves a role in organ growth, survival and
differentiation (9). A recent study
showed that dysregulation of the Hippo pathway was associated with
the onset of certain types of cancer (20). Analysis was conducted of four
molecules of the Hippo pathway, namely FAT1, YAP1, SMAD4 and TEAD2,
in samples from patients with CC.
Kaplan-Meier curves demonstrated positive hazard
ratio (HR) values for the high expression groups of the four genes,
YAP1 (HR, 2.13), FAT1 (HR, 2.07), TEAD2 (HR, 1.7), SMAD 4 (HR,
1.48) and which indicated lower survival rates within the first 60
months from diagnosis (Fig. 3A, E, G
and K). This suggested that a lower survival rate was
associated with high mRNA expression levels of the genes involved
in the Hippo pathway. However, no significant differences in the
mRNA expression levels of FAT1, YAP1, SMAD4 and TEAD2 were
identified, potentially due to the dispersion of values in these
Hippo pathway genes, although a notable increase in expression
levels in invasive CC samples was observed compared with those in
the control (Fig. 3B, F, H and L).
At the protein level, there was no significant difference in the
percentage of cells expressing the Hippo pathway proteins nor in
the intensity in CC samples when compared with the control sample
group or between different stages of CC (Fig. 3C, D, I, J, M and N).
Notch pathway in CC
The Notch pathway is a highly conserved cell
signaling system in a large proportion of multicellular organisms
(21); it is crucial in various
cellular processes, including cell proliferation, differentiation
and apoptosis. Dysregulation of the Notch pathway has been
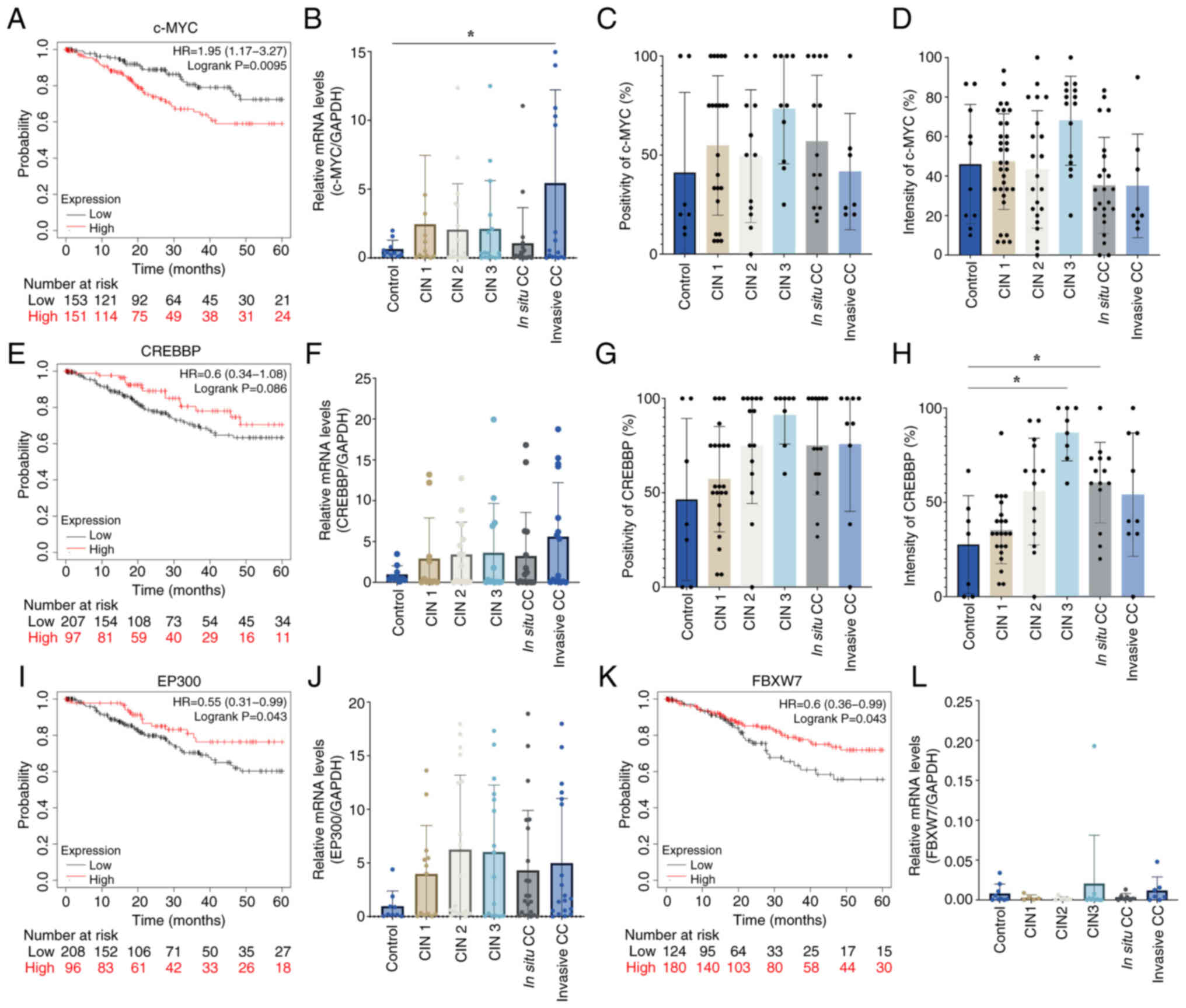
implicated in numerous human diseases, including cancer (22). Analysis of four proteins from the
Notch pathway was conducted, namely c-MYC, CREBBP, EP300 and FBXW7,
in tissue samples from patients with different stages of CC. The
analysis demonstrated an HR of 1.95 for c-MYC, which suggested that
patients with high expression levels of c-MYC have a lower survival
rate up to 60 months (Fig. 4).
However, EP300, CREBBP and FBXW7 showed HR values of 0.55, 0.6 and
0.6 respectively, which suggested that patients with CC with high
levels of these proteins have a slight increase in survival rate at
60 months (Fig. 4A, E, I and
K).
Analysis of the mRNA expression levels of c-MYC
showed a significant increase in expressions levels in the invasive
stage of CC compared with the control group and with the other
genes analyzed in this pathway (Fig.
4B, F, J and L). At the protein level, no significant
difference in either the percentage of positivity or intensity
staining was detected (Fig. 4C and
D); however, a significant increase in the protein intensity of
CREBBP in the CIN 3 and in situ stages of CC was
demonstrated (Fig. 4F-H).
NRF2 pathway in CC
NRF2 is a protein encoded by the NFE2L2 gene. NRF2
is a crucial regulator of the body's antioxidant response, as it is
involved in cellular defense mechanisms against oxidative stress
and inflammation (23,24). However, NRF2 signaling disruption
has been implicated in a number of diseases, including cancer
(25). In the present study,
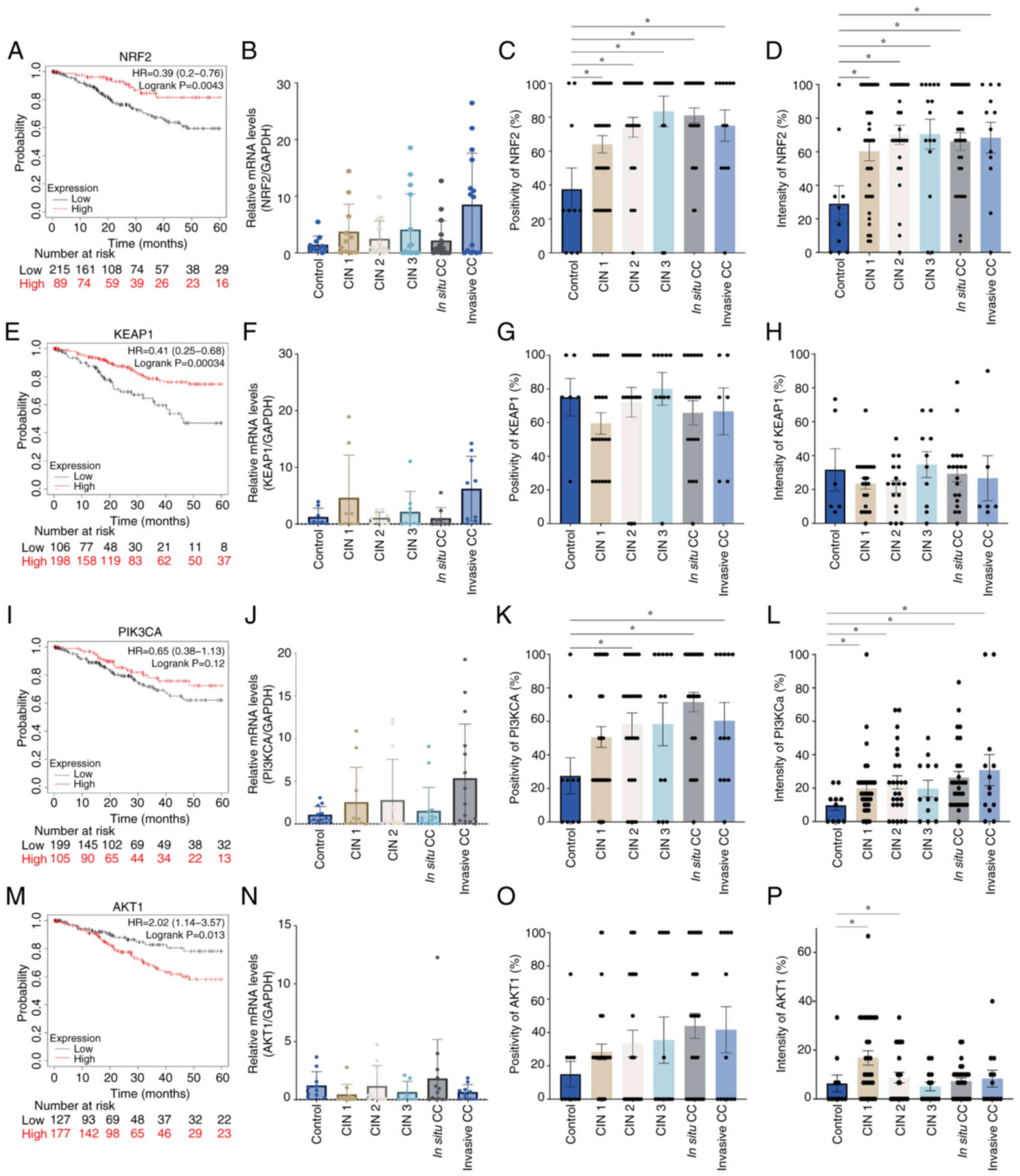
analysis of PI3K, AKT, NRF2 and KEAP1 as NRF2 pathway components in
patients with CC was conducted.
Survival analyses indicated that a number of
proteins associated with the NRF2 pathway exhibited a HR <1,
including NRF2 (HR, 0.39; Fig. 5A),
KEAP1 (HR, 0.41; Fig. 5E) and PI3K
(HR, 0.65; Fig. 5I). This indicated
that patients with elevated mRNA expression levels of these
proteins showed an increased survival rate. By contrast, the AKT1
protein survival curve exhibited an HR of 2.02, which suggested
that patients with high AKT1 expression levels had a lower survival
probability at 60 months (Fig. 5A).
However, analysis of mRNA expression levels of all four proteins
demonstrated no significant differences when comparing CC with the
control groups. Analysis of protein expression levels, however,
demonstrated increased positivity of PI3KCA in CIN 2, in
situ and invasive CC, while shown increased intensity in CIN 1,
CIN 2, in situ and invasive CC. Significantly increased AKT1
protein expression intensity was found in the CIN 1 and CIN 2
stages of CC. Furthermore, NRF2 exhibited protein upregulation in
all tumor samples, regardless of stage.
A summary analysis of the cellular processes
potentially affected by the proteins c-Myc, CREBBP, PI3K, AKT1 and
NRF2, which are dysregulated in patients with CC, was conducted
(Fig. 6A). c-MYC from the Notch
pathway showed an HR of 2.00 on Kaplan-Meier analysis. AKT1 from
the NRF2 pathway showed an HR of 1.70, while NRF2 and KEAP1 from
the same pathway exhibited the lowest HR of 0.39 and 0.41,
respectively (Fig. 6B). Analysis of
the mRNA and protein expression levels of the 12 proteins showed
that NRF2 protein expression levels measured by IHC were increased
in all cancer groups, regardless of stage. AKT1 was upregulated in
the CIN 1 and CIN 2 stages, and PI3K in the CIN 2, in situ
and invasive stages of CC. Moreover, mRNA expression levels of
c-MYC from the Notch pathway were also increased at the invasive
stage, while CREBBP protein was downregulated in the CIN 3 and
in situ stages of CC. Bioinformatic analysis indicated the
cellular processes in which these five dysregulated proteins may be
involved, among which responses to abiotic stimuli, regulation of
the immune system and homeostasis maintenance were identified.
Through analysis of the modified processes according to their
strength, which according to STRING, is a measure Log10
(observed/expected) that describes how large the enrichment effect
is. The present results showed that even without involvement of the
aforementioned five genes, the main processes involved were
‘anoikis’ with the participation of two proteins (PIK3CA and AKT1),
and ‘negative regulation of oxidative stress-induced intrinsic
apoptotic signaling pathway’ (NRF2 and AKT1) (Table II).
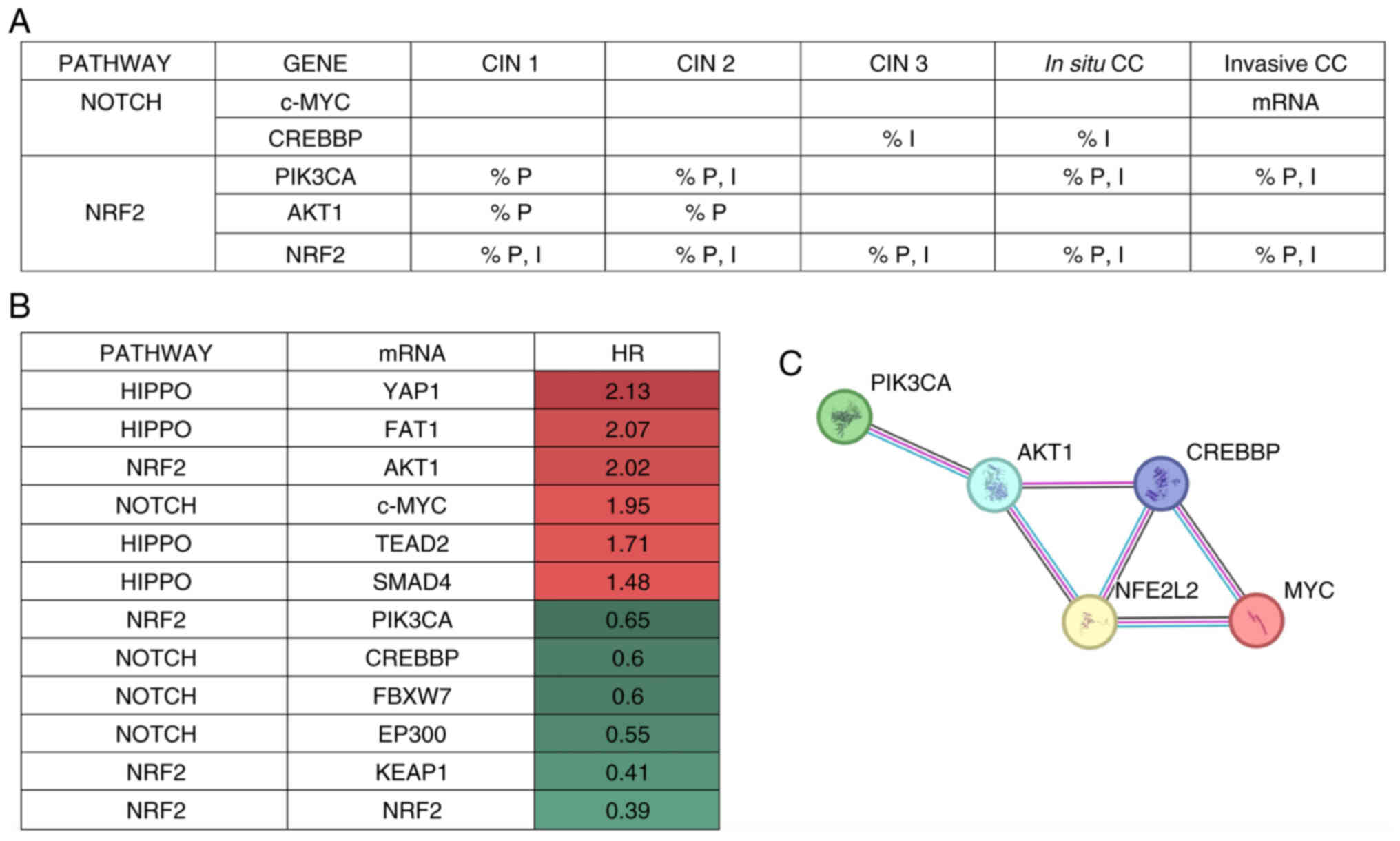 | Figure 6.Integrated analysis of results of the
present study. (A) Summary of HR values obtained from Kaplan-Meier
curve analyses. (B) Notch and NRF2 pathway proteins are
differentially expressed in CC tissue samples. (C) Interactions
among differentially expressed proteins in CC tissue samples. The
colored lines represent the interaction source as follows:
Experiments, pink; database, aqua blue; co-occurrence, navy blue;
and co-expression, black. Created in STRING function network
(version 12.0). An HR value of 1 means there is no difference
between the groups. An HR value of 2 means there is double the risk
of death. An HR value of 0.5 means that there is half the risk of
patient death. Red indicates a higher risk level, while green means
less risk. HR, hazard ratio; CIN, cervical intraepithelial
neoplasia; PI3KCA; PIK3-catalytic subunit a; FAT1, FAT atypical
cadherin; FBXW7, F-box and WD repeat domain containing 7; KEAP1,
kelch-like ECH-associated protein 1; EP300, E1A-associated cellular
p300 transcriptional co-activator protein; NRF2, nuclear factor
erythroid 2-related factor 2; c-MYC, cellular-MYC; CREBBP, cAMP
response element-binding binding protein; YAP1, yes-associated
protein 1; TEAD2, TEA domain family member 2. |
Discussion
Cancer arises from genetic alterations, which can be
either hereditary or induced by an individual's lifestyle or
environmental factors. These genetic alterations confer selective
advantages to the emerging cancer cell clones, and enable them to
resist apoptosis, achieve replicative immortality, induce
angiogenesis, activate invasion and metastasis, reprogram cellular
metabolism, and evade growth-suppressing signals and the immune
response (26). Additionally, the
association of cancer cells with the tumor microenvironment
promotes disease progression, tumor growth, angiogenesis and
metastasis (27).
The genes analyzed in the present study are involved
in the NRF2, Notch and Hippo pathways, which are a part of a
cluster of pathways related to cancer development and progression
that serve crucial roles in cellular processes, which makes them
potential targets for the study of novel treatments and
technological advances in diagnosis.
Kaplan-Meier survival curve analyses suggested that
the genes that provided more valuable information associated with
survival were c-MYC from the Notch pathway, and NRF2, KEAP1 and
AKT1 from the NRF2 pathway. This data demonstrated that patients
with CC with high gene expression levels of c-MYC or AKT1; or low
gene expression levels of NRF2 and KEAP1 have a decreased
probability of survival.
Analysis of mRNA expression levels of the genes that
form the three signal transduction pathways demonstrated that c-MYC
mRNA provided relevant information regarding invasive CC samples.
c-MYC is a widely studied proto-oncogene in solid tumors (28). Alterations in this gene have been
demonstrated in patients who develop HPV-positive CC. Furthermore,
the integration of HPV sequences near the c-MYC locus has been
demonstrated in some cervical cell lines and genital tumors, which
suggests a synergistic role of HPV and the c-MYC proto-oncogene in
CC development (29).
By contrast, CREBBP protein expression levels (Notch
pathway) were increased in CIN 3 and in situ stages of CC in
the present study. Limited information is available regarding the
status of CREBBP in CC. However, this gene is known to be altered
in at least 5% of cancer cases, with an increased prevalence of
CREBBP mutations observed in breast, bladder, lung and colon cancer
types (30,31). CREBBP contributes to tumor
progression and to tumor microenvironment development (32); its role in transcriptional
regulation involves serving as a scaffold between specific DNA
regions and the transcription machinery, as well as in histone
acetylation and interactions with transcription factors (33). CREBBP has a dual function; it binds
to transcription factors and associates them with larger molecular
complexes. Additionally, as an acetyltransferase, the protein
possesses transformative capabilities, primarily on histones, which
promote chromatin accessibility (33). Further research is needed to
understand CREBBP functions in CC.
Protein level analysis through measurement of the
percentage of positivity and intensity of protein expression levels
of the tissue samples demonstrated that proteins from the NRF2
pathway were upregulated in all tumor samples compared with
non-cancerous tissue samples. It has been well documented that the
primary role of the NRF2 pathway is to detoxify cells by inducing
the expression of antioxidant enzymes such as heme oxygenase,
catalase, glutathione peroxidase and superoxide dismutase. These
enzymes can neutralize ROS in the early stages of cancer
development, which protects cells from damage by oxidative stress
(25). However, the NRF2 pathway is
also known for its dual action. In the tumor microenvironment, the
NRF2/KEAP1 complex aids disease progression by the inactivation of
chemotherapy drugs through ROS elimination, which protects cancer
cells from drug-induced cell death (23,24).
Based on the results of the present study, it could be suggested
that this pair of genes are potential markers for the onset of
cervical malignant neoplasia, as upregulation of NRF2 and
inhibition of KEAP1 were observed even in the earliest stages of
CC.
The NRF2/KEAP1 axis establishes crosstalk with the
PI3K/AKT1 axis in cellular detoxification through ROS. Increased
ROS levels activate this pathway through the inhibition of
phosphatases, such as PTEN, or the activation of oncogenes, such as
AKT (16,19). AKT1 biological functions have been
extensively studied and are linked to survival, proliferation,
metabolism and cellular growth, all of which serve critical
functions in carcinogenesis (34).
In the present study, AKT1 protein expression levels were increased
in the CIN 1 and CIN 2 stages of CC, suggesting that the early
stages of CC may be supported by the AKT1 pathway. In addition,
previous studies have demonstrated that the viral oncoproteins HPV
E6 and E7 can induce activation of the AKT/PI3K pathway (35). PI3KCA serves a crucial role in
cellular functions, such as growth, proliferation, differentiation,
motility, survival and intracellular trafficking, and is often
hyperactive in cancer, being attributed to certain roles in cancer
development (36). In patients with
CC, it was reported that an amplification of PI3KCA 3q26.3, the
most common mutation of PI3KCA in CC, is implicated in the
progression of cervical dysplastic cells towards invasive cancer
(37). Consistently, the present
study showed an increase in the PI3KCA expression levels in the
in situ and invasive CC samples.
The expression levels of 12 proteins associated with
the Hippo, Notch and NRF2 pathways that serve essential roles and
are dysregulated in certain types of cancer, were analyzed in CC.
The results demonstrated that in CC, the Hippo pathway did not show
changes between tumor and normal samples. At the mRNA level,
increased expression levels of c-MYC (Notch pathway) and AKT1 (NRF2
pathway), and decreased levels of NRF2, showed an association with
a reduced survival rate through Kaplan-Meier analysis. In the
invasive stage of CC, high expression levels of c-MYC mRNA were
demonstrated. NRF2 protein expression levels showed a significant
change in all precancerous and cancerous stages samples compared
with those in normal samples. The invasive stage samples also
exhibited increased protein expression levels of PI3KCA, another
protein from the NRF2/KEAP1 pathway family. CREBBP (Notch pathway)
and PI3K (NRF2 pathway) were upregulated in the in situ CC
samples. During earlier stages of CC, the AKT1 protein was
upregulated. The present study indicates that the proteins of the
Notch pathway (c-MYC and CREBBP) and the NRF2 pathway (NRF2/KEAP1
and AKT1) are essential and are dysregulated in CC.
Acknowledgements
The authors would like to thank Dr. Nayeli Gabiño
(Clinical Applications Development, National Institute of Genomic
Medicine, Mexico City, Mexico) for their pathology and
immunohistochemistry services.
Funding
This study was supported by the National Council of Humanities,
Science and Technologies (México) through the Frontier Science 2019
call (grant no. 6368). The authors would also like to thank INMEGEN
for their support to publish this article.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
GMA and VM were responsible for study
conceptualization and experimental design. Experimental procedures
and data analysis were performed by JELG, PAE, KIVS, RCO, FL, VM,
JMZ, PPS and GMA. VM, JMZ and GMA prepared the manuscript draft.
JELG, PAE, KIVS, RCO, FL and GMA were responsible for
conceptualization and design of tables and figures. JELG, PAE,
KIVS, RCO, FL, VM, JMZ, PPS and GMA participated in the writing,
review and editing of the manuscript. JELG and GMA confirm the
authenticity of all the raw data. GMA was responsible for funding
acquisition. All authors have read and approved the final version
of the manuscript.
Ethics approval and consent to
participate
The study was conducted in accordance with the
Declaration of Helsinki and approved by the Ethics Committee of
Hospital General Zacatecas ‘Luz Cosío González’ (Mexico City,
Mexico; approval no. 0131/2018; March 14, 2018) for the FFPE
cervical tissue study. The FFPE tissues were obtained from hospital
files ≥10 years of age. Therefore, according to the Hospital
General Zacatecas policies, signed consent from patients was not
necessary for the use of archived material.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Singh D, Vignat J, Lorenzoni V, Eslahi M,
Ginsburg O, Lauby-Secretan B, Arbyn M, Basu P, Bray F and
Vaccarella S: Global estimates of incidence and mortality of
cervical cancer in 2020: A baseline analysis of the WHO global
cervical cancer elimination initiative. Lancet Glob Health.
11:e197–e206. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Morales-Figueroa GG, Bravo-Parra M,
Olivas-Matas KM, Esparza-Romero J, Valenzuela-Zamorano M,
Olivas-López OM and Quihui-Cota L: Associated factors with human
papillomavirus infection in adult women from northwest Mexico.
Biotecnia. 25:133–139. 2022. View Article : Google Scholar
|
|
3
|
Ostroverkhova D, Przytycka TM and
Panchenko AR: Cancer driver mutations: Predictions and reality.
Trends Mol Med. 29:554–566. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Martínez-Jiménez F, Muiños F, Sentís I,
Deu-Pons J, Reyes-Salazar I, Arnedo-Pac C, Mularoni L, Pich O,
Bonet J, Kranas H, et al: A compendium of mutational cancer driver
genes. Nat Rev Cancer. 20:555–572. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Uruno A and Yamamoto M: The KEAP1-NRF2
system and neurodegenerative diseases. Antioxid Redox Signal.
38:974–988. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fox DB, Garcia NMG, McKinney BJ, Lupo R,
Noteware LC, Newcomb R, Liu J, Locasale JW, Hirschey MD and Alvarez
JV: NRF2 activation promotes the recurrence of dormant tumour cells
through regulation of redox and nucleotide metabolism. Nat Metab.
2:318–334. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guenter R, Patel Z and Chen H: Notch
signaling in thyroid cancer. Notch Signaling in Embryology and
Cancer. Vol. 1287. Reichrath J and Reichrath S: Springer
International Publishing; Cham: pp. 155–168. 2021, View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Llombart V and Mansour MR: Therapeutic
targeting of ‘undruggable’ MYC. EBioMedicine. 75:1037562022.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fu M, Hu Y, Lan T, Guan KL, Luo T and Luo
M: The Hippo signalling pathway and its implications in human
health and diseases. Signal Transduct Target Ther. 7:3762022.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mendoza-Almanza G, Ortíz-Sánchez E,
Rocha-Zavaleta L, Rivas-Santiago C, Esparza-Ibarra E and Olmos J:
Cervical cancer stem cells and other leading factors associated
with cervical cancer development. Oncol Lett. 18:3423–3432.
2019.PubMed/NCBI
|
|
11
|
Sanchez-Vega F, Mina M, Armenia J, Chatila
WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia
S, et al: Oncogenic signaling pathways in the cancer genome atlas.
Cell. 173:321–337.e10. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Belder N, Coskun Ö, Doganay Erdogan B, Ilk
O, Savas B, Ensari A and Özdağ H: From RNA isolation to microarray
analysis: Comparison of methods in FFPE tissues. Pathol Res Pract.
212:678–685. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Farragher SM, Tanney A, Kennedy RD and
Paul Harkin D: RNA expression analysis from formalin fixed paraffin
embedded tissues. Histochem Cell Biol. 130:435–445. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Győrffy B: Discovery and ranking of the
most robust prognostic biomarkers in serous ovarian cancer.
Geroscience. 45:1889–1898. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res.
43((Database Issue)): D447–D452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Blokzijl A, Dahlqvist C, Reissmann E, Falk
A, Moliner A, Lendahl U and Ibáñez CF: Cross-talk between the Notch
and TGF-beta signaling pathways mediated by interaction of the
Notch intracellular domain with Smad3. J Cell Biol. 163:723–728.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Papatheodorou I, Moreno P, Manning J,
Muñoz-Pomer Fuentes A, George N, Fexova S, Fonseca NA, Füllgrabe A,
Green M, Huang N, et al: Expression Atlas update: from tissues to
single cells. Nucleic Acids Res. 48:D77–D83. 2020.PubMed/NCBI
|
|
19
|
Kma L and Baruah TJ: The interplay of ROS
and the PI3K/Akt pathway in autophagy regulation. Biotechnol Appl
Biochem. 69:248–264. 2022. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mokhtari RB, Ashayeri N, Baghaie L, Sambi
M, Satari K, Baluch N, Bosykh DA, Szewczuk MR and Chakraborty S:
The Hippo pathway effectors YAP/TAZ-TEAD oncoproteins as emerging
therapeutic targets in the tumor microenvironment. Cancers (Basel).
15:34682023. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li X, Yan X, Wang Y, Kaur B, Han H and Yu
J: The Notch signaling pathway: A potential target for cancer
immunotherapy. J Hematol Oncol. 16:452023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kar R, Jha SK, Ojha S, Sharma A, Dholpuria
S, Raju VSR, Prasher P, Chellappan DK, Gupta G, Singh SK, et al:
The FBXW7-NOTCH interactome: A ubiquitin proteasomal system-induced
crosstalk modulating oncogenic transformation in human tissues.
Cancer Rep (Hoboken). 4:e13692021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Panda H, Wen H, Suzuki M and Yamamoto M:
Multifaceted roles of the KEAP1-NRF2 system in cancer and
inflammatory disease milieu. Antioxidants (Basel). 11:5382022.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang Y, Yang W, Yang L, Wang T, Li C, Yu
J, Zhang P, Yin Y, Li R and Tao K: Nrf2 inhibition increases
sensitivity to chemotherapy of colorectal cancer by promoting
ferroptosis and pyroptosis. Sci Rep. 13:143592023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Aranda-Rivera AK, Cruz-Gregorio A,
Arancibia-Hernández YL, Hernández-Cruz EY and Pedraza-Chaverri J:
RONS and oxidative stress: An overview of basic concepts. Oxygen.
2:437–478. 2022. View Article : Google Scholar
|
|
26
|
Hanahan D: Hallmarks of cancer: New
dimensions. Cancer Discov. 12:31–46. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao Y, Shen M, Wu L, Yang H, Yao Y, Yang
Q, Du J, Liu L, Li Y and Bai Y: Stromal cells in the tumor
microenvironment: Accomplices of tumor progression? Cell Death Dis.
14:5872023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Medda A, Compagnoni M, Spini G, Citro S,
Croci O, Campaner S, Tagliabue M, Ansarin M and Chiocca S:
c-MYC-dependent transcriptional inhibition of autophagy is
implicated in cisplatin sensitivity in HPV-positive head and neck
cancer. Cell Death Dis. 14:7192023. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Haręża DA, Wilczyński JR and Paradowska E:
Human papillomaviruses as infectious agents in gynecological
cancers. Oncogenic properties of viral proteins. Int J Mol Sci.
23:18182022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Peck B, Bland P, Mavrommati I, Muirhead G,
Cottom H, Wai PT, Maguire SL, Barker HE, Morrison E, Kriplani D, et
al: 3D functional genomics screens identify CREBBP as a targetable
driver in aggressive triple-negative breast cancer. Cancer Res.
81:847–859. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jiang Y, Guo X, Liu L, Rode S, Wang R, Liu
H and Yang ZQ: Metagenomic characterization of lysine
acetyltransferases in human cancer and their association with
clinicopathologic features. Cancer Sci. 111:1829–1839. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhu Y, Wang Z, Li Y, Peng H, Liu J, Zhang
J and Xiao X: The role of CREBBP/EP300 and its therapeutic
implications in hematological malignancies. Cancers (Basel).
15:12192023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vannam R, Sayilgan J, Ojeda S,
Karakyriakou B, Hu E, Kreuzer J, Morris R, Herrera Lopez XI, Rai S,
Haas W, et al: Targeted degradation of the enhancer lysine
acetyltransferases CBP and p300. Cell Chem Biol. 28:503–514.e12.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hua H, Zhang H, Chen J, Wang J, Liu J and
Jiang Y: Targeting Akt in cancer for precision therapy. J Hematol
Oncol. 14:1282021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang HM, Lu YJ, He L, Gu NJ, Wang SY, Qiu
XS, Wang EH and Wu GP: HPV16 E6/E7 promote the translocation and
glucose uptake of GLUT1 by PI3K/AKT pathway via relieving miR-451
inhibitory effect on CAB39 in lung cancer cells. Ther Adv Chronic
Dis. 11:20406223209571432020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tewari D, Patni P and Bishayee A, Sah AN
and Bishayee A: Natural products targeting the PI3K-Akt-mTOR
signaling pathway in cancer: A novel therapeutic strategy. Semin
Cancer Biol. 80:1–17. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Voutsadakis IA: 3q26 amplifications in
cervical squamous carcinomas. Curr Oncol. 28:2868–2880. 2021.
View Article : Google Scholar : PubMed/NCBI
|