Introduction
Primary liver cancer is among the six most
widespread malignancies worldwide, and has the third highest
mortality rate globally (1–3). Liver cancer is closely associated with
chronic liver disease in >90% of cases, and causes of cirrhosis
are important risk factors for liver cancer. Alcohol consumption,
diabetes, obesity-induced non-alcoholic steatohepatitis and
hepatitis B and V viruses are all critical risk elements for liver
cancer, in addition to biliary cirrhosis and hemochromatosis
(4,5). Currently, the primary treatment
options for liver cancer are radical resection or liver
transplantation. However, for patients with advanced, recurrent
liver cancer or those who are not suitable for surgery, the
prognosis remains unsatisfactory. Despite some advances in the
diagnosis, treatment and management of liver cancer, its overall
survival remains poor due to the high rates of relapse, vascular
invasion or distant metastasis (6).
Therefore, it is urgently necessary to explore effective and
representative biomarkers and new predictive tools.
Circulating tumor cells (CTCs) are tumor cells that
have been shed from a primary or metastatic lesion into the
bloodstream, which are rare in healthy individuals (7–9). CTCs
exist as single cells or multicellular aggregates known as
circulating tumour microemboli (CTMs) (10). Studies in mouse models have
confirmed that CTMs are more metastatic than individual CTCs, with
results suggesting that the injection of clusters of aggregated
cancer cells significantly increases the formation of tumours
compared to the injection of the same number of individual cancer
cells into mice (11–13). Heterotopic CTMs contain many helper
cells, such as red blood cells, fibroblasts and immune cells, which
contribute to the metastatic survival of CTMs, rather than just an
aggregation of individual cancer cells (14). As an essential component of liquid
biopsy technology, CTCs play an essential role in the diagnosis and
treatment of cancer, carrying heterogeneous information about the
primary tumor and serving as an effective biomarker and modeling
tool. Researchers have found that CTCs serve a key role in the
metastatic process of tumors. Therefore, the isolation and
identification of CTCs with non-invasive biopsy can be widely
applied for the early diagnosis, real-time efficacy monitoring and
prognosis evaluation of tumors (12,15–18).
In general, it has been shown that higher levels of CTCs are
associated with a worse outcome in patients with tumors. For
example, in two studies of patients with liver cancer, the duration
of survival was significantly shorter and associated with poor
clinical features in the CTC-positive cohort (19,20).
Similarly, Sun et al (21)
found that the risk of tumor recurrence increased in patients with
liver cancer when the preoperative CTC count was ≥2/7.5 ml,
particularly at a-fetoprotein levels of ≤400 ng/ml. With advances
in technology, and the genomic, transcriptomic and proteomic
analysis of CTCs at the single-cell level, as well as the
refinement of CTC in vitro models, our understanding of the
critical role of CTCs in cancer has been improved (22,23).
Nevertheless, the biological functions of CTCs in tumors at the
molecular level have not been fully elucidated. Therefore, the
present study aimed to identify the CTC/CTM-related genes (CRGs) in
liver cancer and explore their clinical significance.
In the present study, a comprehensive analysis of
the transcriptomic data and clinical information of liver cancer in
The Cancer Genome Atlas (TCGA) and the International Cancer Genome
Consortium (ICGC) databases was performed. Analysis of these data
in combination with mRNA data associated with liver cancer from the
GSE117623 dataset led to the identification of 258 CRGs.
Subsequently, a prognostic model and risk subgroups for patients
with liver cancer were constructed based on five CRGs, and the
associations between different subgroups of patients and immune
markers such as immune infiltration, immune checkpoints and tumor
mutation burden (TMB) were analyzed. Finally, the detection
efficacy and clinical value of the model were evaluated, and
chemotherapeutic agents with potential therapeutic value were
screened.
Materials and methods
Data sources
The transcriptome profiles and the corresponding
clinicopathological data of patients with liver cancer were
obtained from TCGA database (https://portal.gdc.cancer.gov/) as the training
cohort. In addition, RNA-sequencing (RNA-seq) data and clinical
trait information from patients with liver cancer were downloaded
from the ICGC database (LIRI-JP dataset; http://icgc.org/) and Gene Expression Omnibus (GEO;
http://www.ncbi.nlm.nih.gov/) for
validation. Specifically, 12,518 CRGs in the GSE117623 dataset were
downloaded from the GEO database (24). Transcriptomic and matched clinical
data from the IMvigor210 cohort of patients treated with anti-PD-L1
were collected (research-pub.gene.com/IMvigor210CoreBiologies) to
explore the value of model genes in assessing response to
immunotherapy (25).
Identification of candidate genes
To acquire the differentially expressed genes (DEGs)
associated with CTCs/CTMs, the limma R package (version 2.7,
bioinf.wehi.edu.au/limma) was used to process the RNA-seq data
using a false discovery rate (FDR) <0.05 and |log2 (fold
change)|>2 as the cutoff criteria. A Venn diagram was then
constructed using a Venn webtool (http://bioinformatics.psb.ugent.be/webtools/Venn/) to
illustrate the intersection among TCGA-DEGs, ICGC-DEGs and genes
from the GSE117623 dataset. These intersected genes were considered
to be the CRGs.
Pathway enrichment and protein-protein
interaction (PPI) network analysis
Gene Ontology (GO) and Kyoto Encyclopedia of Genes
and Genomes (KEGG) enrichment analyses were conducted to explore
the functional roles and pathways associated with the CRGs using
the clusterProfiler R package (version 3.19) (26). The cut-offs were set as P<0.05
and FDR <0.05. Gene set enrichment analysis (GSEA) was performed
to investigate the common biological pathways (27) using cp.kegg.v7.1.symbols.gmt as a
reference gene set with a threshold of P<0.05, to screen for key
enriched pathways in different risk groups. In addition,
interactions among the CRGs were illustrated by the construction of
a PPI network using the STRING database (https://string-db.org/), with an interaction score
>0.7 being considered significant. Moreover, Cytoscape software
was utilized to visually represent the PPI network. Specifically,
the Cytoscape plug-in Molecular Complex Detection (MCODE) (version
2.0.3) was utilized to identify the highly interconnected modules
of the PPI network with the following criteria: Degree cut-off, 2;
node score cut-off, 0.2; k-core, 2; and max. depth, 100 (28). In addition, another Cytoscape
plug-in, cytoHubba (version 0.1), was used to rank the nodes in the
network according to their network functionality (29). The gene set variation analysis
(GSVA) package (version 3.19) was used to explore the signaling
pathways between high- and low-risk groups (30).
Construction and validation of the
risk prognostic model
Univariate Cox regression analysis was performed to
determine the prognostic CRGs and the CRGs associated with survival
time, with P<0.01 considered to be statistically significant.
Then, least absolute shrinkage and selection operator (LASSO)
penalized Cox regression analysis was performed to further filter
prognostic CRGs associated with the overall survival (OS) of
patients with liver cancer (31).
Subsequently, a risk signature was developed via stepwise
multivariate Cox proportional hazards regression analysis.
Prognostic gene signatures were constructed based on linear
combinations of regression coefficients derived by multiplying the
LASSO Cox regression model coefficients by their mRNA expression
levels (32): Risk score=∑ (βmRNA ×
mRNA)n, where β represents the regression coefficient for the mRNA,
mRNA represents the expression level of the mRNA, and n represents
the specific gene. Receiver operating characteristic (ROC) curves
and Kaplan-Meier curves were constructed to evaluate the predictive
performance of the prognostic model in TCGA cohort. pheatmap R
package (version 1.0.12;
cran.r-project.org/web/packages/pheatmap/index.html) to plot images
describing gene expression heatmaps, risk scores and OS for high
and low risk groups. Data from the ICGC and GEO databases were used
as external validation data to test the predictive capability of
the model.
Identification of liver cancer
subtypes
A non-negative matrix factorization (NMF) clustering
algorithm was utilized to analyze the five signature genes in the
risk score model, and determine the subtypes of CRGs in liver
cancer using the NMF R package (version 0.27) (33). Using conformal, scatter and
silhouette features, the optimal number of clusters with n=2 was
determined.
Establishing the predictive
nomogram
Nomograms are widely used as a tools for the
prognostic analysis of patients with tumors (34). A simplified liver cancer nomogram
was constructed for each dataset based on the CRG model and its
predictive performance was evaluated by plotting calibration
curves.
Bioinformatics analysis of the
prognostic signature
The association between the low- and high-risk
groups and clinical characteristics were explored using Chi-square
tests, and the results were displayed as a heatmap. In addition,
the associations between the signature genes and immune cell
infiltration were analyzed. Six algorithms, namely CIBERSORT-ABS
(35), TIMER (36) (https://cistrome.shinyapps.io/timer/), QUANTISEQ
(37), MCPCOUNTER (38), XCELL (39) and EPIC (40,41),
were used to evaluate the differences in the immune
microenvironment between the two risk groups. Tumor-associated
immune comprehensive score was assessed via ImmunoPhenoScore in R
package IOBR (42) (version 0.99.9,
http://github.com/IOBR/IOBR). Waterfall
plots for the two risk groups were produced using the maftools
(github.com/PoisonAlien/maftools) R package (version 3.19).
Differences in the expression of major histocompatibility complex
(MHC) molecules, human leukocyte antigen (HLA) signature,
chemokines and potential immune checkpoints were also compared
between the two groups. To investigate the association between
signature genes and immune subtypes, ‘Subtypes’ module of the
TISIDB database (http://cis.hku.hk/TISIDB/index.php). Pearson
correlation coefficients of the signature genes expression with the
immune checkpoints (PD-1, PD-L1 and CTLA4) were calculated using R
language to assess the correlation. In this study, the OCLR
algorithm and the Primary Cell Biology Consortium (PCBC, http://progenitorcells.org/) stemness score model were
used to calculate the mRNAsi of cells in the TCGA-LIHC dataset and
to assess the correlation between the stemness index and the risk
score (43). Pearson correlation
coefficients of the signature genes expression with the immune
checkpoints were calculated using the R language to assess the
correlation.
Screening potential therapeutic small
molecule drugs for liver cancer
To identify small molecule compounds that may be
suitable for the treatment of liver cancer, the pRRophetic
(genemed.uchicago.edu/~pgeeleher/pRRophetic/) R package (version
3.19) was used to calculate the half-maximal inhibitory
concentration (IC50) based on data from the Genomics of
Drug Sensitivity in Cancer database (44).
Cell culture and transfection
HepG2 and MHCC97H human liver cancer cells (cat.
nos. CTCC-001-0014 and CTCC-400-0192, respectively) were obtained
from the Meisen Chinese Tissue Culture Collections. The cell lines
were authenticated by short tandem repeat testing. Both cell lines
were cultivated in high-glucose Dulbecco's modified Eagle's medium
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.) at 37°C in 5%
CO2. Two small interfering RNA (siRNAs) targeting tumor
protein p53 inducible protein 3 (TP53I3), namely si-TP53I3-1 and
si-TP53I3-2, and an siRNA negative control were synthesized by and
purchased from Sangon Biotech Co., Ltd. The sequences of siRNAs are
listed in Table SI. Cell
transfection was conducted in 6-well plates when cell confluence
was 60–70%, with a final siRNA concentration of 50 nM per well.
Transfection of the liver cancer cells was performed using
Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) following the manufacturer's instructions.
Transfection was performed for 6–8 h at 37°C in 5% CO2.
The cells were harvested at 24 h post-transfection for reverse
transcription-quantitative PCR (RT-qPCR) analysis and at 48 h
post-transfection for western blot and in vitro functional
assessment.
Western blot analysis
Cells were lysed on ice with RIPA buffer (Wuhan
Boster Biological Technology, Ltd.) containing protease inhibitor
cocktail (MedChemExpress) for 20 min. The protein contents of the
cell lysates were quantified using a BCA protein assay kit
(Beyotime Institute of Biotechnology). Then, 30 µg protein/lane was
separated by 10% SDS-PAGE (Boster Biological Technology) and
transferred to PVDF membranes (EMD Millipore). The membranes were
blocked with 5% defatted milk at room temperature for 2 h, then
incubated with anti-TP53I3 (#14828-1-AP; 1:1,000; Proteintech
Group, Inc.) and anti-ACTB (#AC006; 1:3,000; ABclonal Biotech Co.,
Ltd.). primary antibodies at 4°C for 12–16 h, followed by
HRP-conjugated Affinipure goat anti-rabbit IgG (H+L) (SA00001-2;
1:5,000; Proteintech) secondary antibodies at room temperature for
2 h, and the signal was detected using Pierce® ECL
Western Blotting Substrate (Thermo Fisher Scientific, Inc.).
Finally, the bands were detected and analyzed using
ChemiDoc™ XRS+ with Image Lab™ software
(version 6.0, Bio-Rad Laboratories, Inc.).
RT-qPCR
Total RNA was extracted from cells using FreeZol
reagent (Vazyme Biotech Co., Ltd.) and synthesized into cDNA using
PrimeScript™ RT Master Mix (Takara Bio, Inc.), according
to the manufacturer's instructions. qPCR was then carried out using
the CFX96 Real-Time PCR System (Bio-Rad Laboratories, Inc.) with
the SYBR Green PCR kit (Thermo Fisher Scientific, Inc.) according
to the standard protocol. The thermocycling conditions used were as
follows: 95°C for 30 sec pre-cycling, and then 40 cycles of 95°C
for 10 sec and 60°C for 30 sec. The primer pairs were synthesized
by Sangon Biotech Co., Ltd. and their sequences are presented in
Table SII. The relative expression
of TP53I3 was calculated using the formula 2−ΔΔCq with
GAPDH as the reference gene (34).
Cell proliferation assay
Cell Counting Kit 8 (CCK-8) assay (ABclonal Biotech
Co., Ltd.) was utilized to assess the proliferation ability of the
cells. Cells (3,000/well) were plated in a 96-well plate and
incubated overnight at 37°C to allow adhesion. At 24, 48 and 74 h,
100 µl 10% CCK-8 solution was added to each well and the cells were
cultured in a cell incubator for 2 h, after which absorbance was
measured at 450 nm using a microplate reader (Thermo Fisher
Scientific, Inc.).
Colony formation assay
Cells were seeded into 6-well plates at a
concentration of 1,000 cells/well. The cells were cultured at 37°C
with 5% CO2 in fresh medium and allowed to grow for 14
days. The colonies were then fixed for 15 min at room temperature
in 4% paraformaldehyde (Wuhan Servicebio Technology Co., Ltd.), and
stained with crystal violet (0.5% wt./vol.) at room temperature for
15 min. Finally photographs of the plates were taken and the
colonies were quantified using an inverted microscope (Guangzhou
Micro-shot Technology Co., Ltd.). The number of colonies was
counted manually. Each independently counted colony refers to a
cell cluster of ≥50 cells. The experiment was repeated three
times.
5-Ethynyl-2′-deoxyuridine (EdU)
detection
The BeyoClick™ EdU-555 Cell Proliferation
Kit (Beyotime Institute of Biotechnology) was employed to
investigate the proliferation rate of the human liver cancer cells
according to the manufacturer's protocols. Briefly, after
incubation with 1X EdU (10 µM) solution for 2 h at 37°C, cells were
fixed with paraformaldehyde (4%) for 30 min at room temperature,
then permeabilized with 0.3% Triton X-100 for 15 min and finally
stained with Hoechst 33342 and 4′,6-diamidino-2-phenylindole in the
absence of light for 30 min at room temperature. Finally, the cells
were imaged by fluorescence microscopy.
Statistical analysis
Bioinformatics analysis and mapping were
accomplished using R software. Survival rates were compared using
Kaplan-Meier analysis with the calculation of P-values using
log-rank tests, or the 2-stage test in the plot with late-stage
crossover (cran.r-project.org/web/packages/TSHRC/TSHRC.pdf). In
addition, the Chi-square test was used for comparisons between
categorical variables, and unpaired Student's t-test was utilized
to evaluate the discrepancies between the two risk groups.
Correlations between variables were assessed using Spearman's
correlation test. The cell groups were compared by one-way ANOVA
followed by Dunnett's post hoc tests. For each statistical
analysis, P<0.05 was considered to indicate a statistically
significant result.
Results
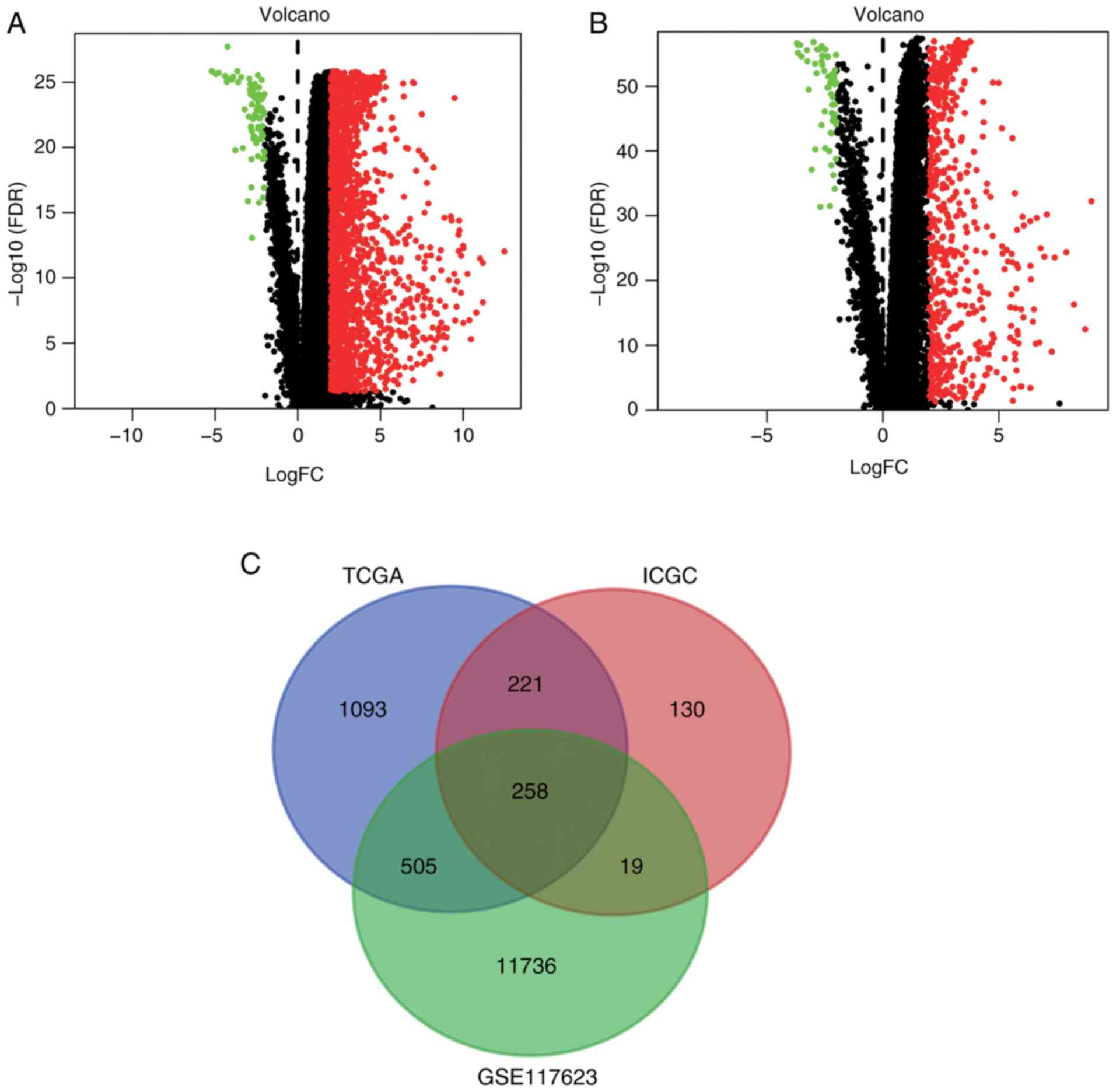
Differentially expressed CRGs
The liver cancer (liver hepatocellular carcinoma)
gene expression profiles were downloaded from TCGA and ICGC portals
and 1,622 and 628 DEGs, respectively, were screened out using the
limma R package. The DEGs from TCGA and ICGC databases are shown as
volcano plots in Fig. 1A and B,
respectively. A Venn diagram was then constructed to filter out the
differentially expressed CRGs (Fig.
1C). The intersection of the DEGs from TCGA and ICGC databases
with the 12,518 CRGs from GSE117623 yielded a total of 258
differentially expressed CRGs (Table
SIII).
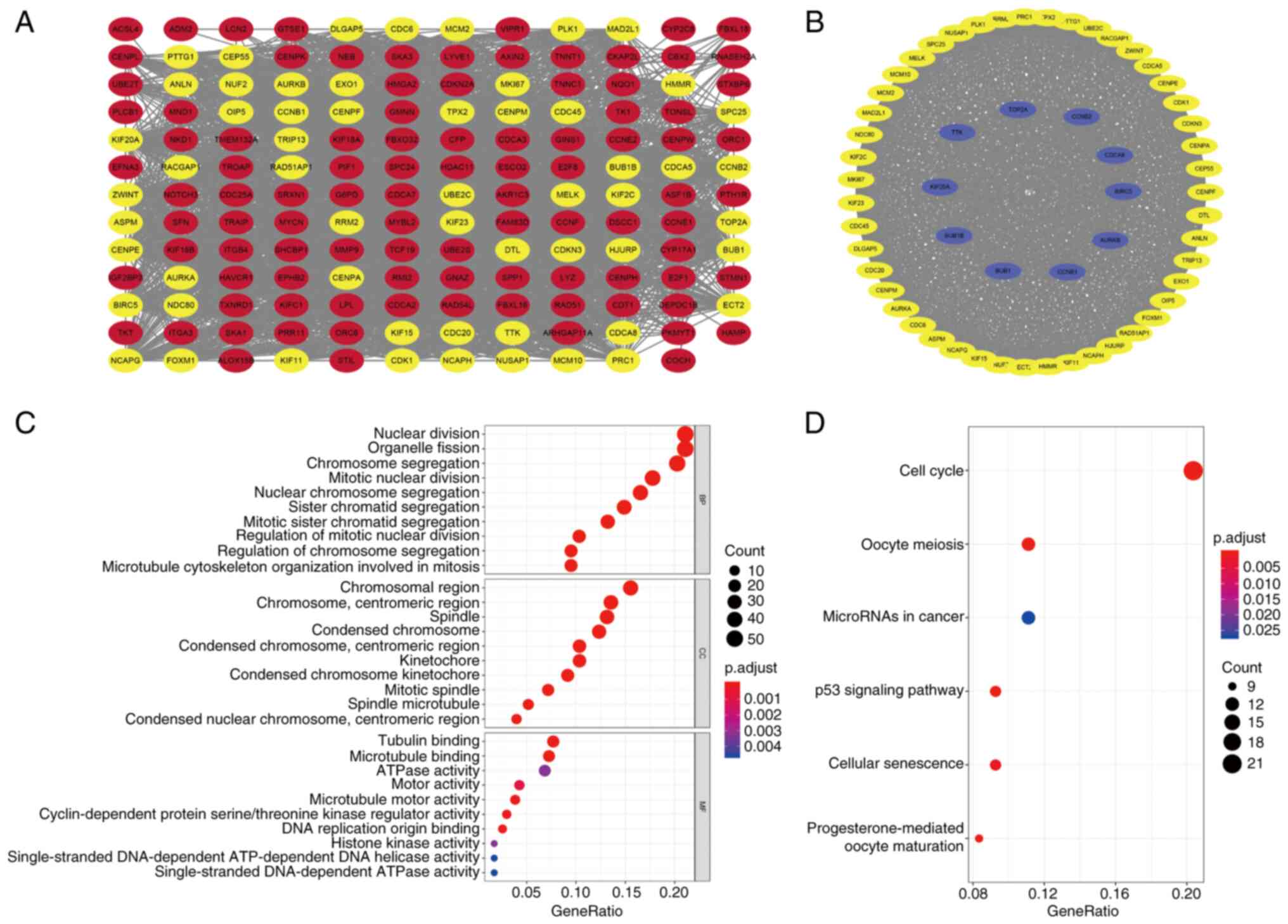
PPI network construction
To investigate the interrelationship of the
differentially expressed CRGs and identify hub genes, a PPI network
was constructed and module analysis performed to determine
co-expression networks. Firstly, the 258 differentially expressed
CRGs were uploaded to the STRING database, and the minimum required
interaction score was set to 0.7, which indicates a strong
interaction between the CRGs. The STRING interactions were then
analyzed using Cytoscape and the resulting co-expression network,
which contained 155 nodes and 2,731 edges, is shown in Fig. 2A. In addition, modules with >50
genes were identified using the MCODE plug-in and 10 hub genes in
that module, namely topoisomerase IIa, cyclin B2, cell division
cycle associated 8 (CDCA8), BIRC5, aurora kinase B, cyclin B1, BUB1
mitotic checkpoint serine/threonine kinase (BUB1), BUB1B, kinesin
family member 20A and TTK protein kinase, were characterized using
the cytoHubba plug-in (Fig. 2B).
This included 57 nodes and 1,497 edges. These potential hub genes
may be instrumental in the biological progression of liver
cancer.
GO and KEGG enrichment analyses
To explore the biological categories and biological
processes associated with the differentially expressed CRGs, GO and
KEGG enrichment analyses were conducted using R software, and the
enrichment results are shown in bubble charts (Fig. 2C and D). The GO enrichment analysis
revealed that the differentially expressed CRGs were principally
concentrated in the biological process terms ‘nuclear division’,
‘organelle fission’, ‘chromosome segregation’ and ‘mitotic nuclear
division’. In addition, the main cellular component terms included
‘chromosomal region’, ‘chromosome, centromeric region’, ‘spindle’
and ‘kinetochore’. Moreover, the molecular function terms
associated with the CRGs were ‘tubulin binding’,
‘microtubule-binding’, ‘ATPase activity’ and ‘motor activity’
(Fig. 2C). Regarding the KEGG
analysis, the primary terms are shown in Fig. 2D, which reveals that the
differentially expressed CRGs were particularly enriched in ‘cell
cycle’, ‘microRNAs in cancer’, ‘p53 signaling pathway’ and
‘cellular senescence’.
Construction of a prognostic model and
validation of the model in the ICGC cohort
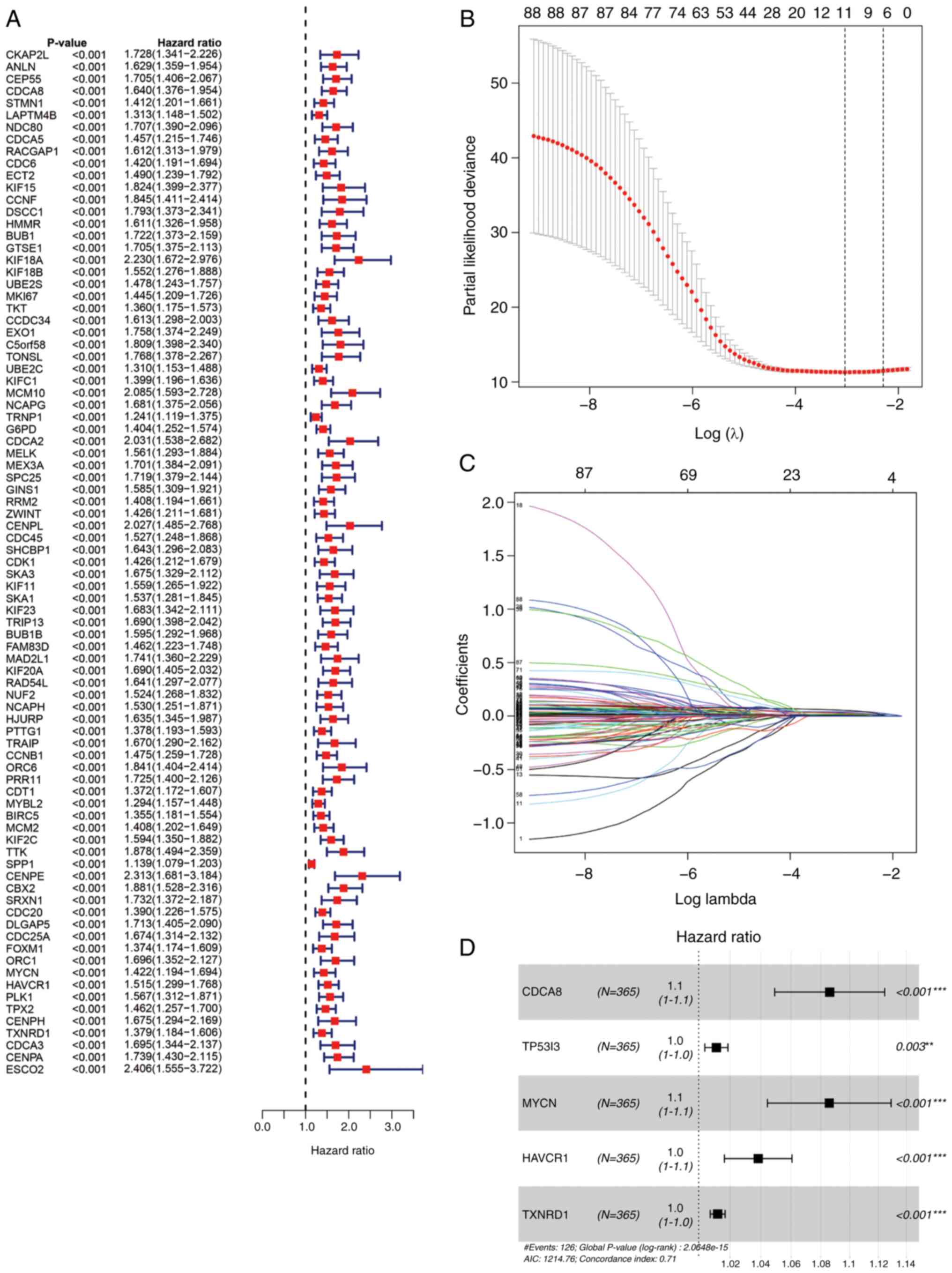
Univariate Cox regression analysis demonstrated that
88 CRGs were strongly associated with survival in patients with
liver cancer (P<0.01), all of which were prognostic risk factors
(Fig. 3A). Then, the 88 CRGs were
regression penalized using LASSO Cox regression to exclude
relatively insignificant parameters (Fig. 3B and C). Stepwise multivariate Cox
regression was subsequently employed to construct a predictive
signature for patients with liver cancer in TCGA cohort (Fig. 3D). The five genes in the signature
were CDCA8, TP53I3, hepatitis A virus cellular receptor 1 (HAVCR1),
MYCN proto-oncogene (MYCN) and thioredoxin reductase 1 (TXNRD1).
The formula for risk score calculation was as follows: Risk
score=(0.0826 × expression level of CDCA8) + (0.0112 × expression
level of TP53I3) + (0.0824 × expression level of MYCN) + (0.0376 ×
expression level of HAVCR1) + (0.0120 × expression level of TXNRD1)
(Table SIV). Patients in TCGA
cohort were classified into high- and low-risk groups using the
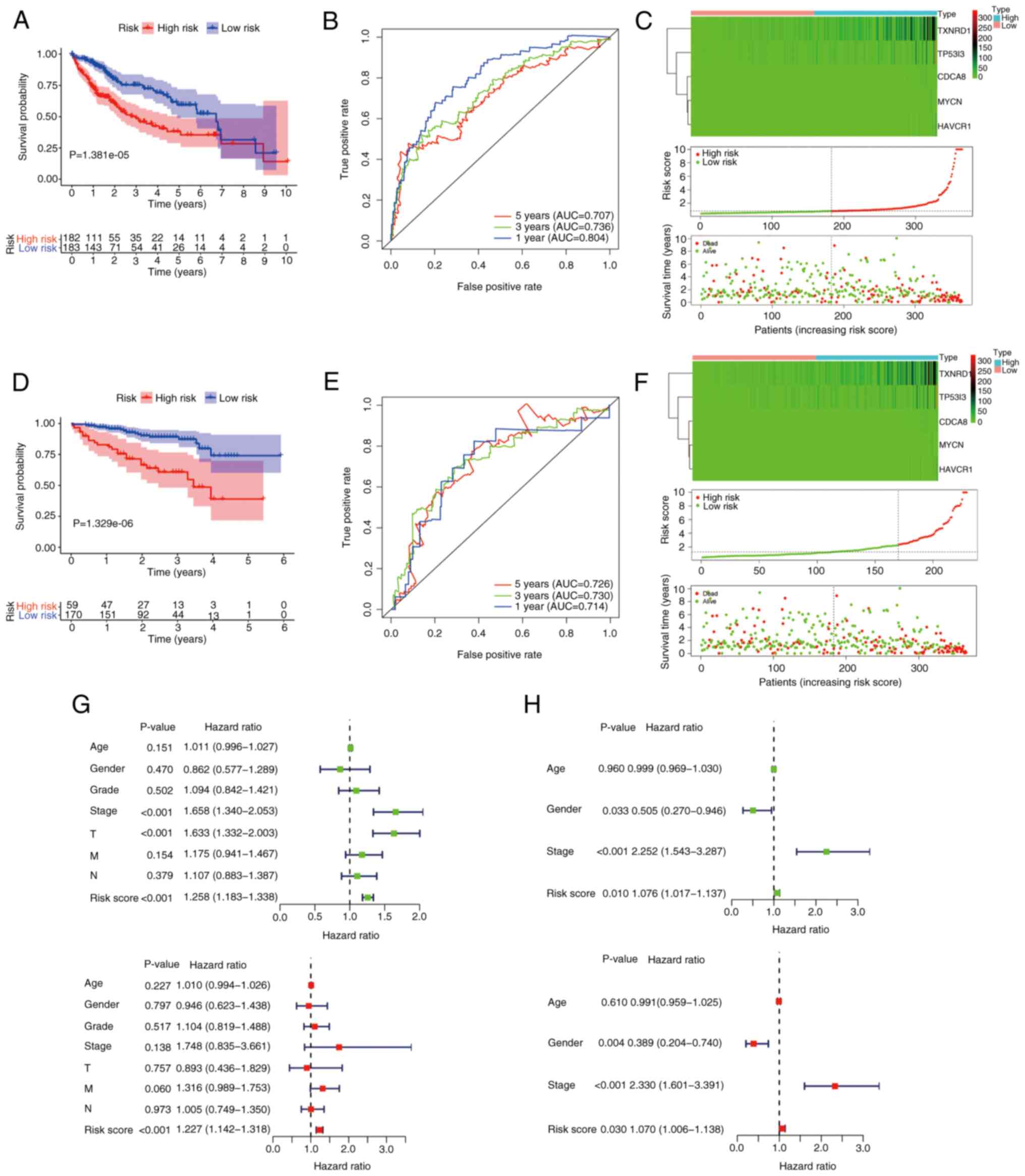
median predictive index as the cut-off point. As Fig. 4A shows, the low-risk group was
significantly associated with improved survival (P<0.05). To
evaluate the predictive ability of this prognostic signature, an
ROC analysis of the risk score was conducted. The area under the
curve (AUC) values predicted from the ROC curves for 1-, 3- and
5-year OS were 0.804, 0.736 and 0.707, respectively (Fig. 4B). In Fig. 4C, the upper panel shows the
expression heat map of the five prognostic model genes in the high-
and low subgroups, the middle panel reveals that the risk of
patients with liver cancer increases as risk score increases, and
the lower panel demonstrates the poor OS of the patients in the
high-risk group compared with those in the low-risk group. To
validate the predictive power of the signature, the same formula
was used to analyze the risk score of each patient in the ICGC
dataset, for independent external validation. The Kaplan-Meier
curves also displayed a poor prognosis of patients in the high-risk
group in this dataset (P<0.05; Fig.
4D). In addition, the ROC curve showed the strong predictive
ability of the risk-score signature for prognosis, with AUCs for
the prediction of 1-, 3- and 5-year OS of 0.714, 0.730 and 0.726,
respectively (Fig. 4E). Also, the
expression of the five CRGs and the mortality of the patients
increased as the risk scores increased (Fig. 4F).
Independent prognostic role of the
gene signature
To investigate whether the CTC/CTM-associated 5-gene
signature could be an independent prognostic factor for patients
with liver cancer, the prognostic value of this signature was
compared with that of several clinicopathological factors,
including age, sex, grade and American Joint Committee on Cancer
(AJCC) stage in both cohorts using univariate and multivariate Cox
regression analyses. For TCGA cohort, 365 valid patients were
included, 182 in the high-risk group and 183 in the low-risk group.
Univariate Cox analysis indicated that risk score [P<0.001;
hazard ratio (HR), 1.258; 95% confidence interval (95% CI),
1.183–1.338)], AJCC stage (P<0.001; HR, 1.658; 95% CI,
1.340–2.053) and T status (P<0.001; HR, 1.633; 95% CI,
1.332–2.003) were candidate factors. Further multivariate Cox
regression analysis emphasized that risk score was an independent
risk factor for patients with liver cancer (P<0.001; HR, 1.227;
95%CI, 1.142–1.318) (Fig. 4G). For
the ICGC cohort, 229 patients were included, 59 in the high-risk
group and 170 in the low-risk group. Univariate Cox regression
analysis demonstrated that sex (P=0.033; HR, 0.505; 95% CI,
0.270–0.946), risk score (P=0.010; HR, 1.076; 95% CI, 1.017–1.137)
and tumor stage (P<0.001; HR, 2.252; 95% CI, 1.543–3.287) were
potential risk factors. Multivariate Cox regression analysis also
indicated that sex (P=0.004; HR, 0.389; 95% CI, 0.204–0.740), risk
score (P=0.030; HR, 1.070; 95% CI, 1.006–1.138 and tumor stage
(P<0.001; HR, 2.330; 95% CI, 1.601–3.391) were independent
predictors for patients in the ICGC cohort (Fig. 4H). In conclusion, these findings
indicated that the 5-CRG risk signature was closely associated with
the clinical characteristics of patients with liver cancer, had a
fine predictive capacity and has potential as a prognostic
indicator for these patients.
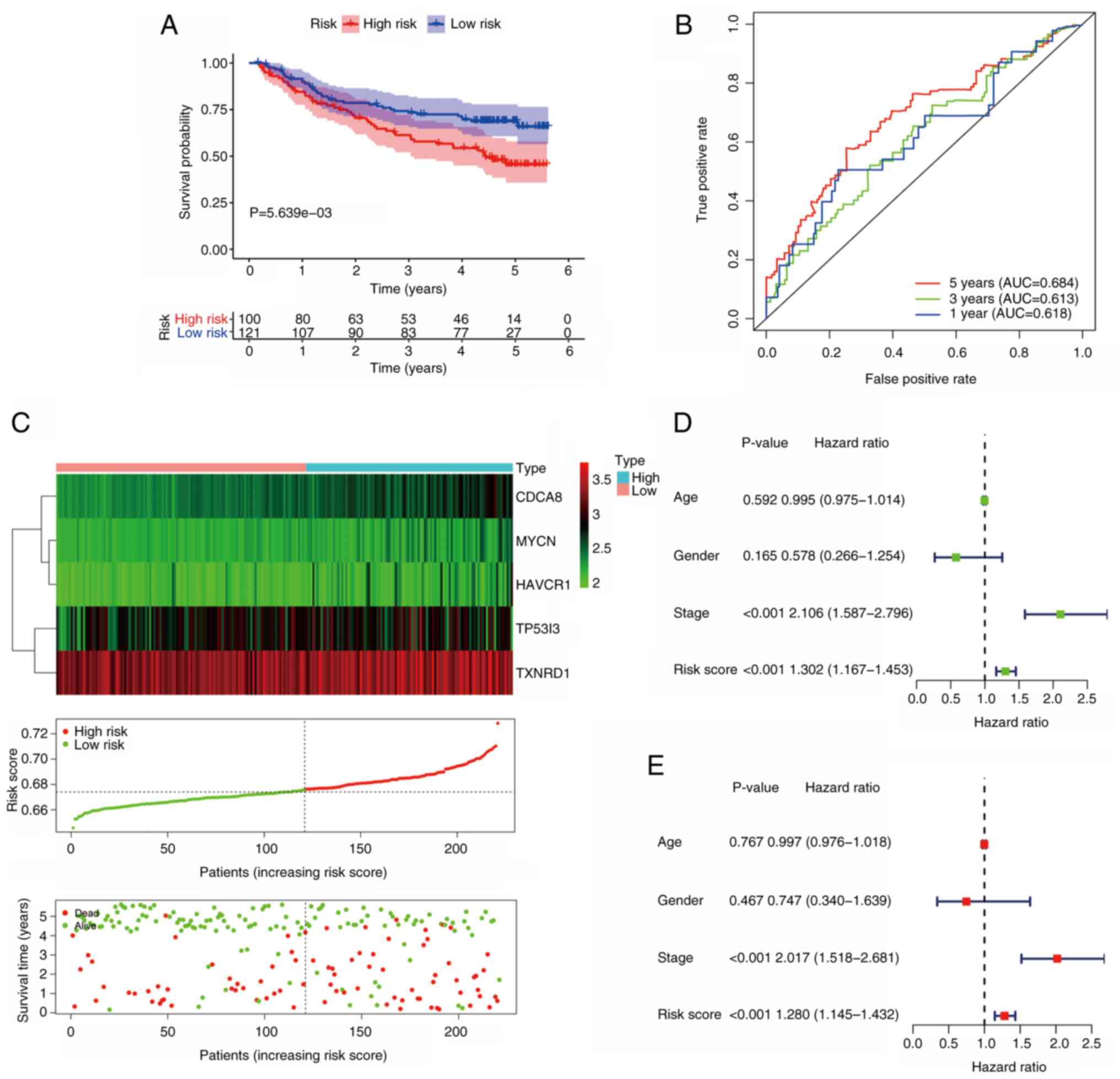
Validation of the signature in the GEO
cohort
To further verify the predictive power of the
prognostic signature, the GSE14520 dataset was analyzed. In this
dataset, 221 valid patients were included, with 100 in the
high-risk group and 121 in the low-risk group. As the results in
Fig. 5A illustrate, patients in the
low-risk group had improved survival outcomes compared with those
in the high-risk group (P<0.05). In the GEO cohort, the AUC for
5-year OS was 0.684 (Fig. 5B). The
expression of model genes, risk score distribution and survival
status for each patient in this validation cohort are shown in
Fig. 5C. Following univariate Cox
regression analysis (Fig. 5D), the
results of independent prognostic analysis revealed that risk score
(P<0.001; HR=1.280; 95% CI, 1.145–1.432), as well as AJCC stage
(P<0.001; HR, 2.017; 95% CI, 1.518–2.681) (Fig. 5E) were independent risk factors in
this cohort. These findings indicate that the prognostic model is
promising as a predictive signature.
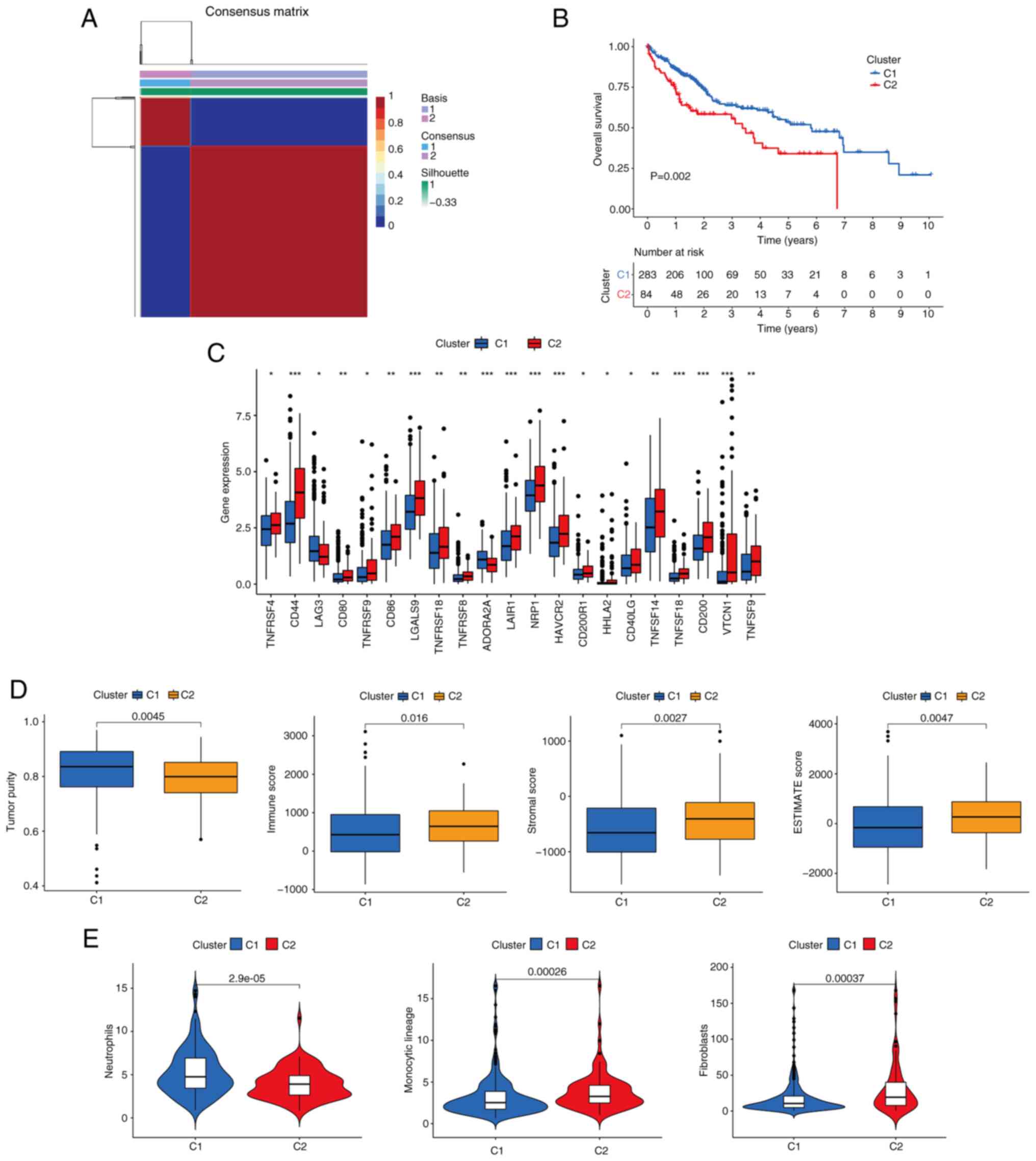
Identification of CTC/CTM-related
molecular subtypes
Patients were clustered into different subtypes
based on the expression levels of the five prognostic signature
genes using the NMF algorithm. To ensure the robustness of the
clustering results, the coefficient of correlation was used to
determine the optimal number of clusters, and when the number of
clusters was 2, clear boundaries were observed for both subtypes.
This indicated the stable and reliable clustering of the liver
cancer samples (Fig. 6A). The OS of
patients in cluster 1 (C1) was significantly improved compared with
that of C2 (P=0.002; Fig. 6B). Most
immune checkpoints were upregulated in the C2 group compared with
the C1 group (Fig. 6C). In
addition, it was also found that the level of immune infiltration
in the tumor microenvironment was also distinct in the two groups,
with immune score, stromal score and ESTIMATE score of the C1 group
being significantly lower than those of the C2 group (P<0.05;
Fig. 6D). This suggests that C1
molecular subtype tends to present ‘cold tumors’, whereas the C2
molecular subtype tends to present ‘hot tumors’. It was also noted
that the level of neutrophil infiltration was higher in the C1
group than in the C2 group, whereas the levels of monocytic lineage
and fibroblast infiltration were lower in the C1 group than in the
C2 group (P<0.05; Fig. 6E).
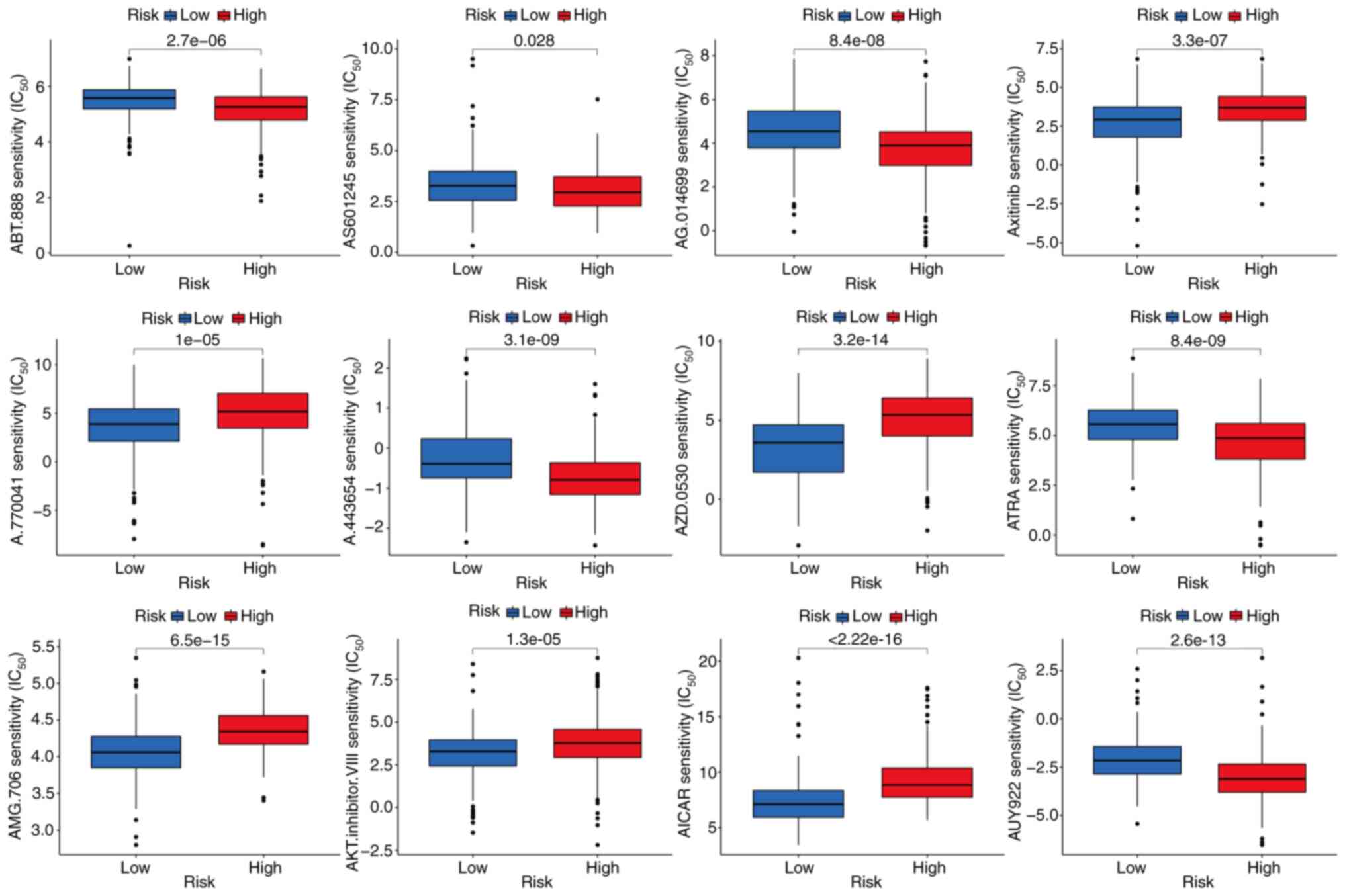
Identification of potentially
therapeutic small molecule drugs
The sensitivity of the high- and low-risk groups to
various chemotherapeutic agents was compared to evaluate drugs for
potential use in liver cancer. The findings indicate that the
low-risk group was associated with a higher IC50 for
chemotherapeutic compounds including ABT.888 (veliparib), AS601245
(an ATP-competitive JNK inhibitor), AG.014699 (rucaparib), A.443654
(a pan-Akt inhibitor), ATRA (tretinoin) and AUY922 (luminespib). By
contrast, axitinib, A.770041 (an LCK inhibitor), AZD.0530
(saracatinib), AMG.706 (motesanib), AKT.inhibitor.VIII and AICAR
(acadesine) had a higher IC50 in the high-risk group,
indicating that patients in the low-risk group may benefit more
from treatment with these compounds (P<0.05; Fig. 7). The sensitivity of the two liver
cancer subtypes to various chemotherapeutic drugs was also
evaluated. The results suggested that patients with the C1 subtype
might be more sensitive to metformin, lapatinib, elesclomol,
docetaxel, camptothecin, bosutinib, axitinib and vinblastine, while
patients in group C2 would likely benefit by treatment with
cisplatin, bortezomib, bleomycin, bicalutamide, mitomycin C,
imatinib, etoposide and gemcitabine (Fig. S1).
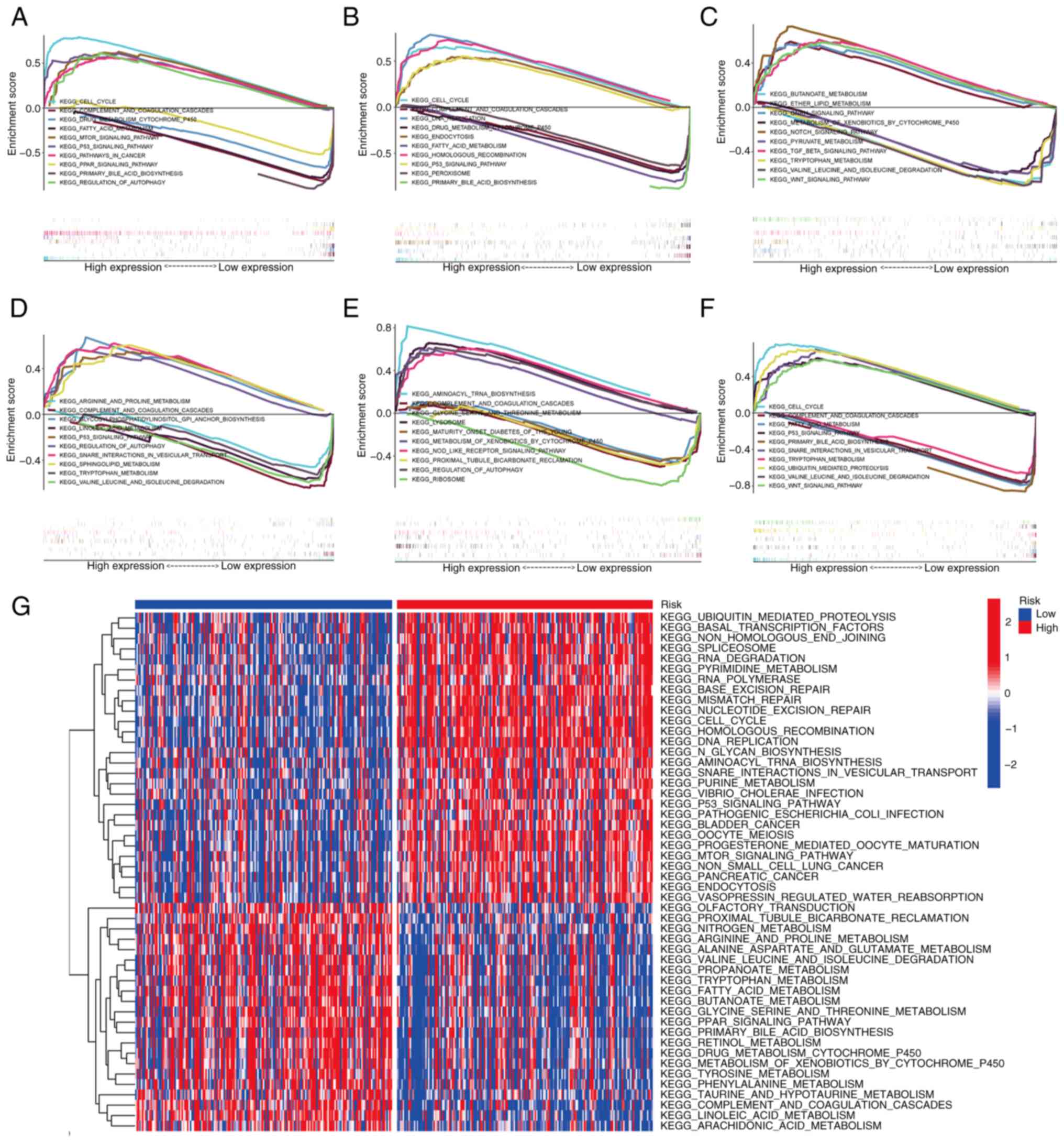
Pathway analysis by GSEA and GSVA
To further explore the molecular mechanism
associated with the signature genes and the prognostic module, GSEA
was performed in TCGA liver cancer cohort. Fig. 8A-E reveals the KEGG pathways of the
five signature genes, namely CDCA8, HAVCR1, MYCN, TP53I3 and
TXNRD1, showing the five most upregulated and downregulated
pathways for each gene. The signature genes are mainly concentrated
in KEGG pathways including ‘cell cycle’, ‘p53 signaling pathway’,
‘complement and coagulation cascades’ and ‘drug metabolism
cytochrome p450’. In addition, GSEA was used to compare the high-
and low-risk groups based on the risk scores. The KEGG pathways
enriched in the high and low risk groups are shown in Fig. 8F.
GSVA was also utilized to analyze the differences in
biological behavior between the high- and low-risk groups. The
results demonstrated that pathways associated with tumor
progression, such as ‘cell cycle’, ‘DNA replication’, ‘RNA
degradation’, ‘mTOR signaling pathway’ and ‘P53 signaling pathway’,
were mainly concentrated in the high-risk group. By contrast,
metabolism-related pathways, including ‘fatty acid metabolism’,
‘propanoate metabolism’, ‘butanoate metabolism’ and ‘tyrosine
metabolism’, were mainly present in the low-risk group of patients
(Fig. 8G).
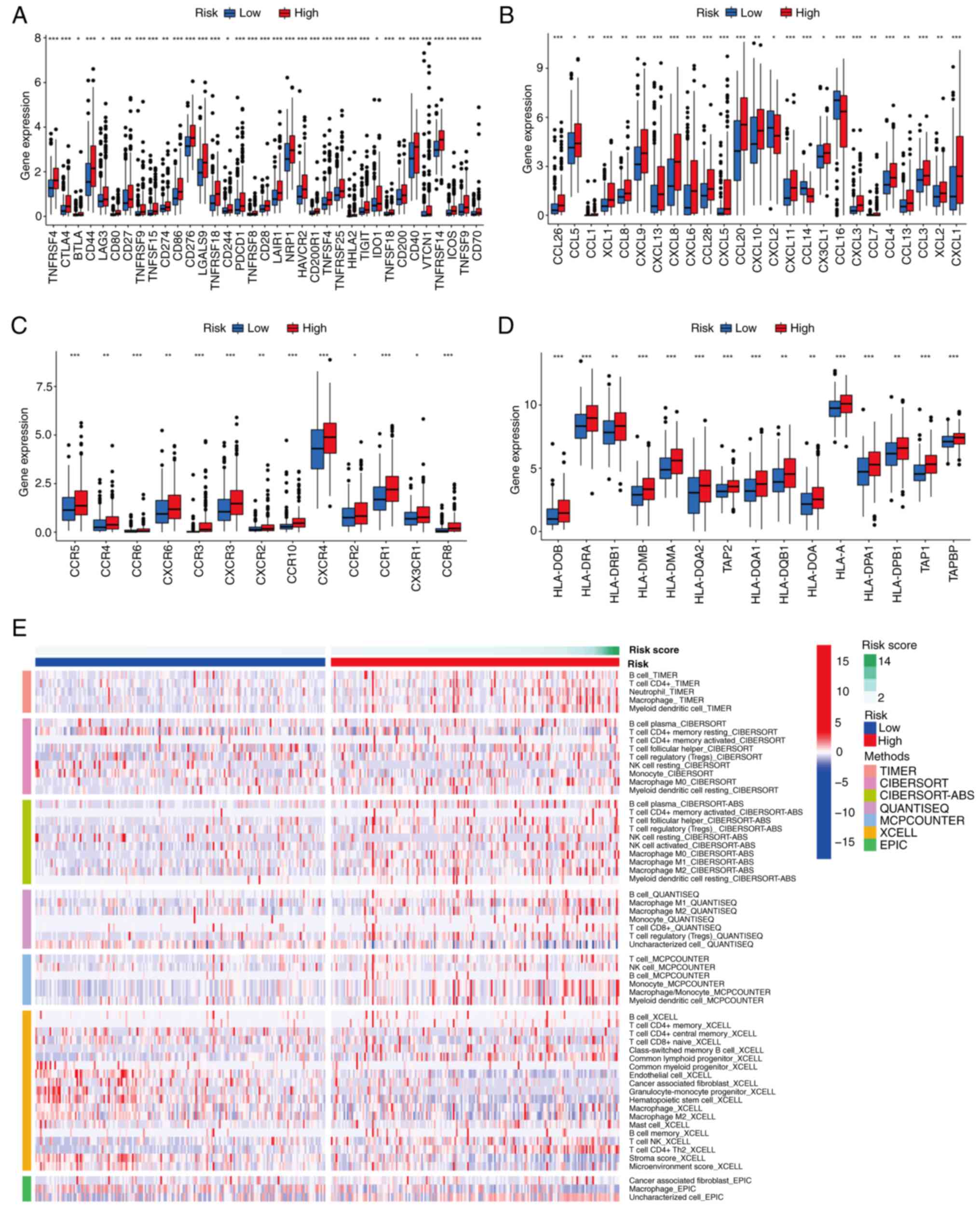
Differentiation of immune infiltration
between the two risk subgroups
In view of the important role of immune checkpoints
in tumor immunotherapy, the differential expression of immune
checkpoint genes was analyzed between risk subgroups. The results
revealed that common immune checkpoint genes, including cytotoxic
T-lymphocyte associated protein 4 (CTLA4), CD274, programmed cell
death 1, and T-cell immunoreceptor with Ig and ITIM domains were
upregulated in the high-risk group compared with the low-risk group
(P<0.05; Fig. 9A). This suggests
that the poor prognosis of high-risk patients with liver cancer may
at least partially be attributed to an immunosuppressive
microenvironment. Chemokines and their receptors are necessary for
the targeted migration of immune cells and the initiation and
execution of the immune response (45,46).
Therefore, the differential expression of chemokines and their
receptors was analyzed in the two risk subgroups, which revealed
higher levels of expression for the majority of these chemokines
and receptors in patients in the high-risk group (P<0.05;
Fig. 9B and C). An association
between risk score and HLA-associated gene expression was also
observed. As shown in Fig. 9D, the
abundance of HLA-related genes was higher in patients at high risk
than those in the low-risk group (P<0.05). The results of
algorithms were visualized using heat maps, including assessment of
immune cell infiltration in the two risk subgroups, and the results
suggest that the high-risk group has more abundant immune cell
infiltration (Fig. 9E). In
addition, further exploration of the association between risk score
and immune pathway activity revealed that cytolytic activity, type
I IFN response and type II IFN response scores were higher in the
low-risk group, and conversely, the MHC class I score was higher in
the high-risk group (P<0.05; Fig.
10F). These results demonstrate that patients in the high-risk
group are more likely to benefit from immunotherapy.
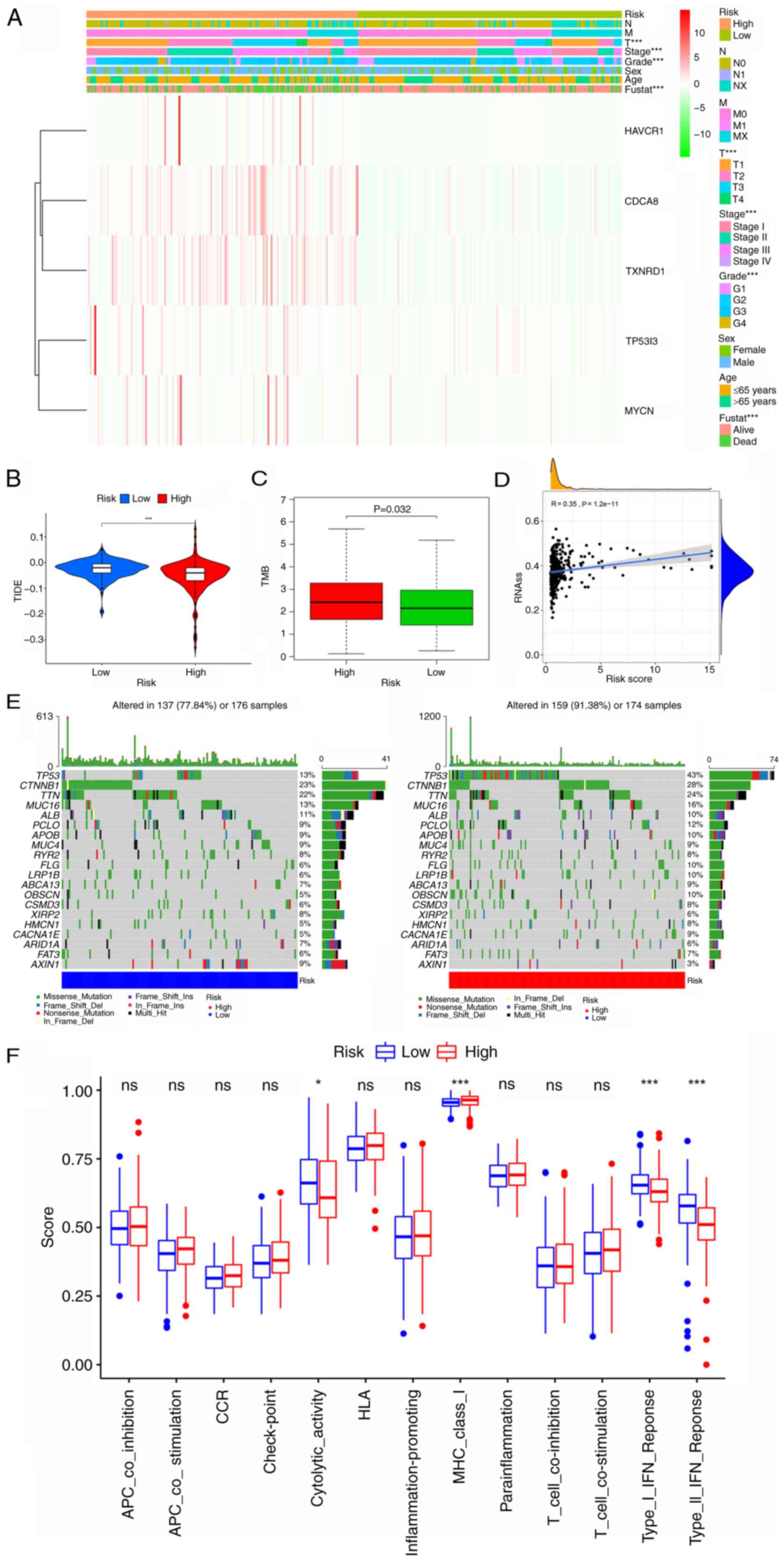 | Figure 10.Analysis of the clinical utility of
the CRG signature and comparison of TIDE, TMB, stem cell content,
the frequency of mutations and immune signaling pathways between
the high- and low-risk groups. (A) Heatmap showing the correlation
of the prognostic signature with clinicopathological
characteristics and five signature genes. (B and C) Boxplots
showing the difference in (B) TIDE and (C) TMB between the low- and
high-risk CRG groups. (D) Correlation between RNAss and the risk
score. (E) Mutation rate analysis of the two risk groups. (F)
Comparison of scores for immune-related pathways between the high-
and low-risk groups. *P<0.05 and ***P<0.001. CRG, circulating
tumor cell/circulating tumor microemboli-related gene; TIDE, tumor
immune dysfunction and exclusion; TMB, tumor mutation burden;
RNAss, RNA stemness score; fustat, follow-up status; HAVCR1,
hepatitis A virus cellular receptor 1; CDCA8, cell division cycle
associated 8; TXNRD1, thioredoxin reductase 1; TP53I3, tumor
protein p53 inducible protein 3; MYCN, MYCN proto-oncogene; ns, not
significant. |
Clinicopathological parameter
correlation analysis
To investigate the prognostic value of the CRG
signature in patients with different clinical features, a heat map
was drawn to reveal whether there was a potential association with
clinicopathological features in the high- and low-risk subgroups
(Fig. 10A). The expression levels
of CDCA8 and TXNRD1 were higher in the high-risk group than in the
low-risk group. In addition, the results revealed that the
high-risk score was closely associated with a higher T stage
(P<0.001), higher grade (P<0.001), higher tumor stage
(P<0.001) and poor patient survival status (P<0.001).
Analysis of the immunological value of
the CRG signature
Since TMB and tumor immune dysfunction and exclusion
(TIDE) are good indicators of the response to immunotherapy, sample
scores were calculated for each patient with liver cancer and
variability between the high- and low-risk subgroups was assessed.
The results revealed that the high-risk group had a higher TMB and
lower TIDE index, which further demonstrates that patients in the
high-risk group should be more responsive to immunotherapy
(P<0.05; Fig. 10B and C). The
mRNA expression-based stemness score revealed a positive
correlation between liver cancer tumor stemness and the risk score,
indicating that tumors in the high-risk group are more likely to
undergo malignant progression and thus lose their differentiated
phenotype (P<0.05; Fig. 10D).
In addition, the maftools R package was used to visualize the
differences in somatic mutation distribution between the high- and
low-risk groups. The results demonstrated that the high-risk group
had a higher mutation frequency compared with the low-risk group
(91.38 vs. 77.84%, respectively). The most mutated gene in the
low-risk group was catenin b1 (23%) and the most mutated gene in
the high-risk group was TP53 (43%) (Fig. 10E).
Correlation analysis of risk signature
genes and immune checkpoints
Immune checkpoints have an important role in immune
regulation, and immune checkpoint inhibitors are used in cancer
therapy. Therefore, the associations between the signature genes
and the expression of immune checkpoint genes, namely programmed
cell death protein 1 (PD-1), programmed death-ligand 1 (PD-L1) and
CTLA4, were investigated. The results in Fig. 11A indicate that the expression of
CDCA8 was positively correlated with that of the three immune
checkpoints, PD-1 (R=0.3; P=4.9×10−9), PD-L1 (R=0.32;
P=3.8×10−10) and CTLA4 (R=0.32; P=3.9×10−10).
In addition, the expression of HAVCR1 was positively correlated
with CTLA4 expression (R=0.32; P=1.3×10−10). TISIDB
portal was used to analyze the expression of signature genes in
different immune subtypes, specifically: C1, wound healing; C2,
IFN-g dominant; C3, inflammatory; C4, lymphocyte depleted; C5,
immunologically quiet; and C6, TGF-b dominant (47). The results indicated that the roles
of these five genes differ among the different immune subtypes,
with CACA8, MYCN and TXNRD1 being differentially expressed among
the immune subtypes. Specifically, the TISIDB analysis revealed
that CDCA8 was highly expressed in the C1 and C2 types, MYCN was
highly expressed in the C1 type, and TXNRD1 was mainly expressed in
the C2 and C4 types (Fig. 11B). In
addition, the IMvigor dataset was used to predict the
responsiveness of the five signature genes to atelelizumab
treatment. Notably, consistent with the previous findings, the
analysis suggested that patients with high expression of CDCA8 and
TXNRD1 may obtain improved treatment outcomes (Fig. 11C). The correlations between tumor
immune infiltration by CD4+ T cells, CD8+ T
cells, B cells, neutrophils, macrophages and dendritic cells, and
the expression of the five signature genes were also investigated
(Fig. S2). In this analysis,
correlation coefficients >0.3 and P<0.05 were considered as
distinctive; partial.cor denotes partial correlation, indicating
the correlation of gene expression with immune cell infiltration in
the TIMER database. The results show that CDCA8 expression is
positively correlated with the infiltration of six types of immune
cells: B cells (partial.cor, 0.441; P=9.08×10−18),
CD8+ T cells (partial.cor, 0.303;
P=1.03×10−8), CD4+ T cells (partial.cor,
0.359; P=6.74×10−12), macrophages (partial.cor, 0.439;
P=1.70×10−17), neutrophils (partial.cor, 0.368;
P=1.63×10−12) and dendritic cells (partial.cor, 0.465;
P=1.22×10−19). Similarly, HAVCR1 expression was found to
be positively correlated with the infiltration of B cells
(partial.cor, 0.302; P=1.14×10−8), macrophages
(partial.cor, 0.302; P=1.34×10−8), neutrophils
(partial.cor, 0.392; P=4.00×10−14) and dendritic cells
(partial.cor, 0.317; P=2.18×10−9), and TXNRD1 expression
was positively associated with neutrophil infiltration
(partial.cor, 0.322; P=8.67×10−10).
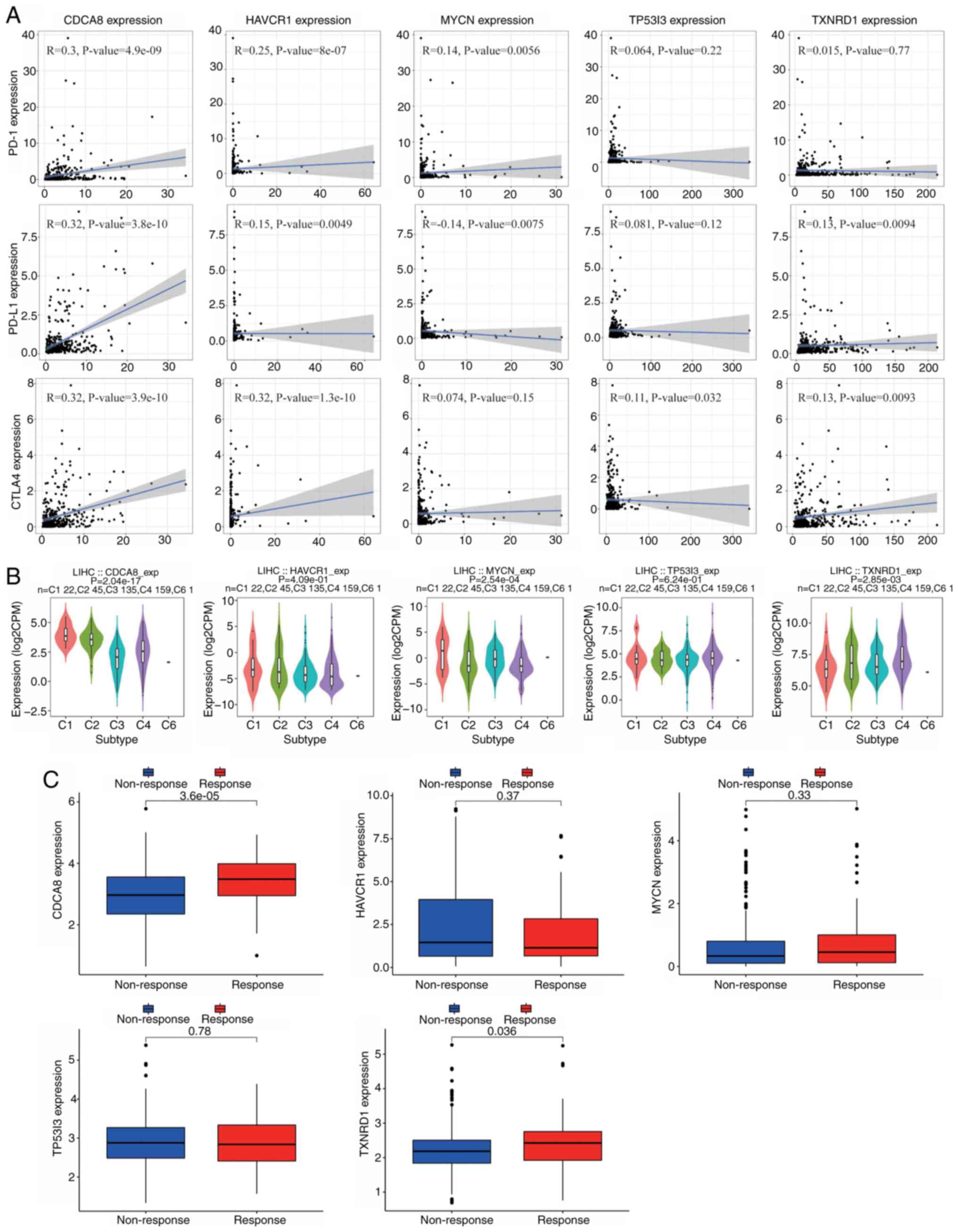 | Figure 11.Correlation analysis of five CRGs and
immunity markers. (A) Correlation between CRGs and the immune
checkpoints PD-1, PD-L1 and CTLA4. (B) Analysis of the role of the
five CRGs in different immune subtypes. (C) Expression levels of
the five CRGs in the IMvigor210 cohort. CRGs, circulating tumor
cell/circulating tumor microemboli-related genes; PD-1, programmed
cell death protein 1; PD-L1, programmed death-ligand 1; CTLA4,
cytotoxic T-lymphocyte associated protein 4; CDCA8, cell division
cycle associated 8; HAVCR1, hepatitis A virus cellular receptor 1;
MYCN, MYCN proto-oncogene; TP53I3, tumor protein p53 inducible
protein 3; TXNRD1, thioredoxin reductase 1; LIHC, liver
hepatocellular carcinoma; exp, expression; CPM, counts per million
reads. |
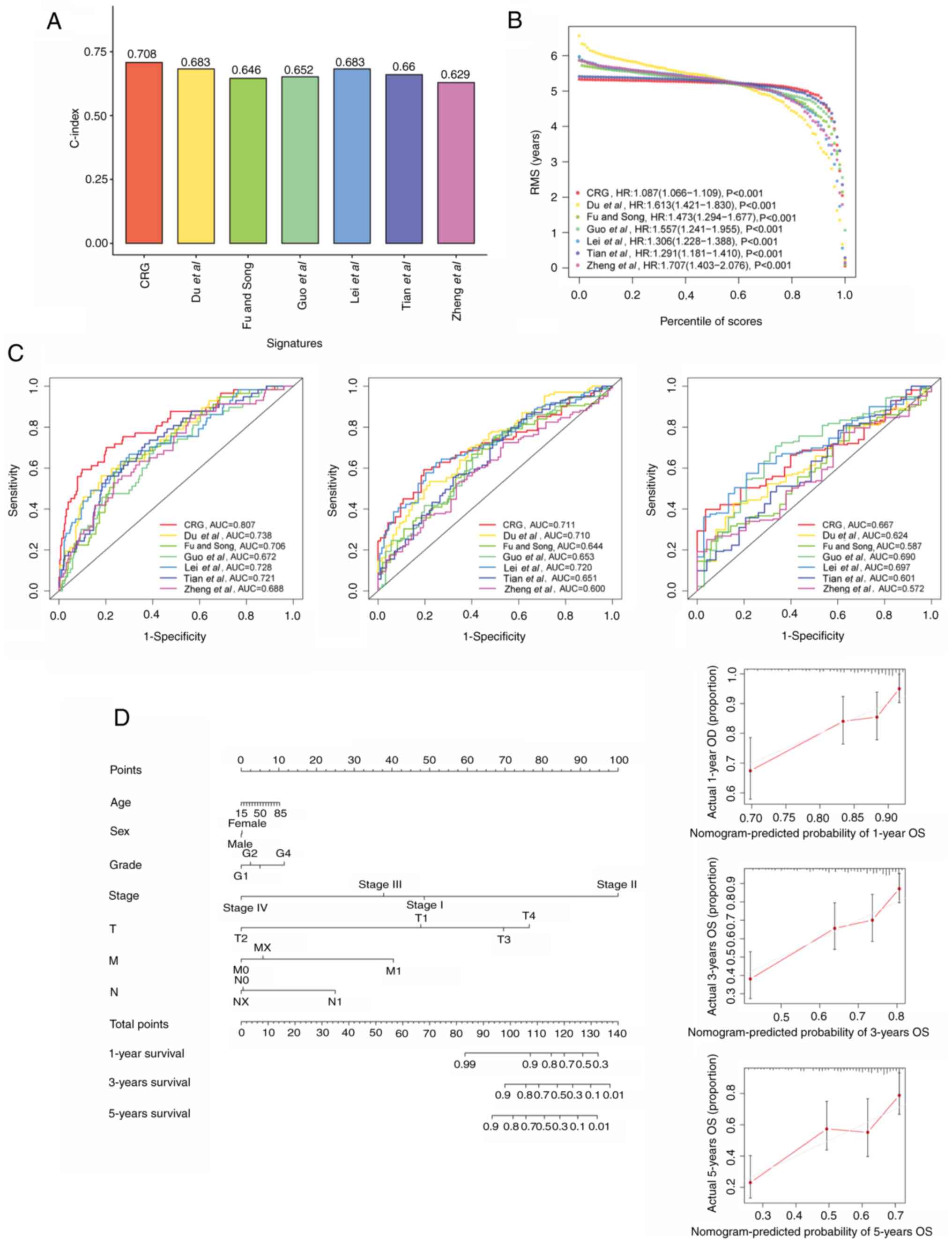
Comparison of the CRG signature with
external prognostic models
To better assess the predictive efficacy of the CRG
prognostic model, the risk signature was compared with six
published liver cancer prognostic models. The signature of Du et
al (48) was a m6A-based gene
signature; the signature of Fu and Song (49) was a pyroptosis-related gene
signature; the signature of Guo et al (50) was a signature containing nine genes;
Lei et al (51) devised a
starvation-based nine-mRNA signature; Tian et al (52) proposed a five-gene prognostic
signature for liver cancer; and the signature of Zheng et al
(53) comprised five
pyroptosis-related genes. When the accuracy of these models and the
current model were compared, it was found that the C-index and
restricted mean survival of the CRG signature were higher than
those of the other six models, which indicates that the present
model is optimal (Fig. 12A and B).
Additionally, the AUCs of the CRG model for 1-, 3- and 5-year OS
were 0.807, 0.711 and 0.667, respectively, which were higher than
those of the other signatures, which validates the previous results
(Fig. 12C).
Establishment and validation of a
predictive nomogram
To forecast the survivability of patients with liver
cancer, a nomogram including factors such as age, sex, stage and
risk score was created to predict probability of OS at 1, 3, and 5
years in the TCGA cohort. In addition, calibration plots were
constructed to evaluate the predictive power of the nomogram
(Fig. 12D). Similarly, two
nomograms were also constructed for the ICGC and GSE14520 cohorts
(Fig. S3). These all indicate the
good predictive power of the model.
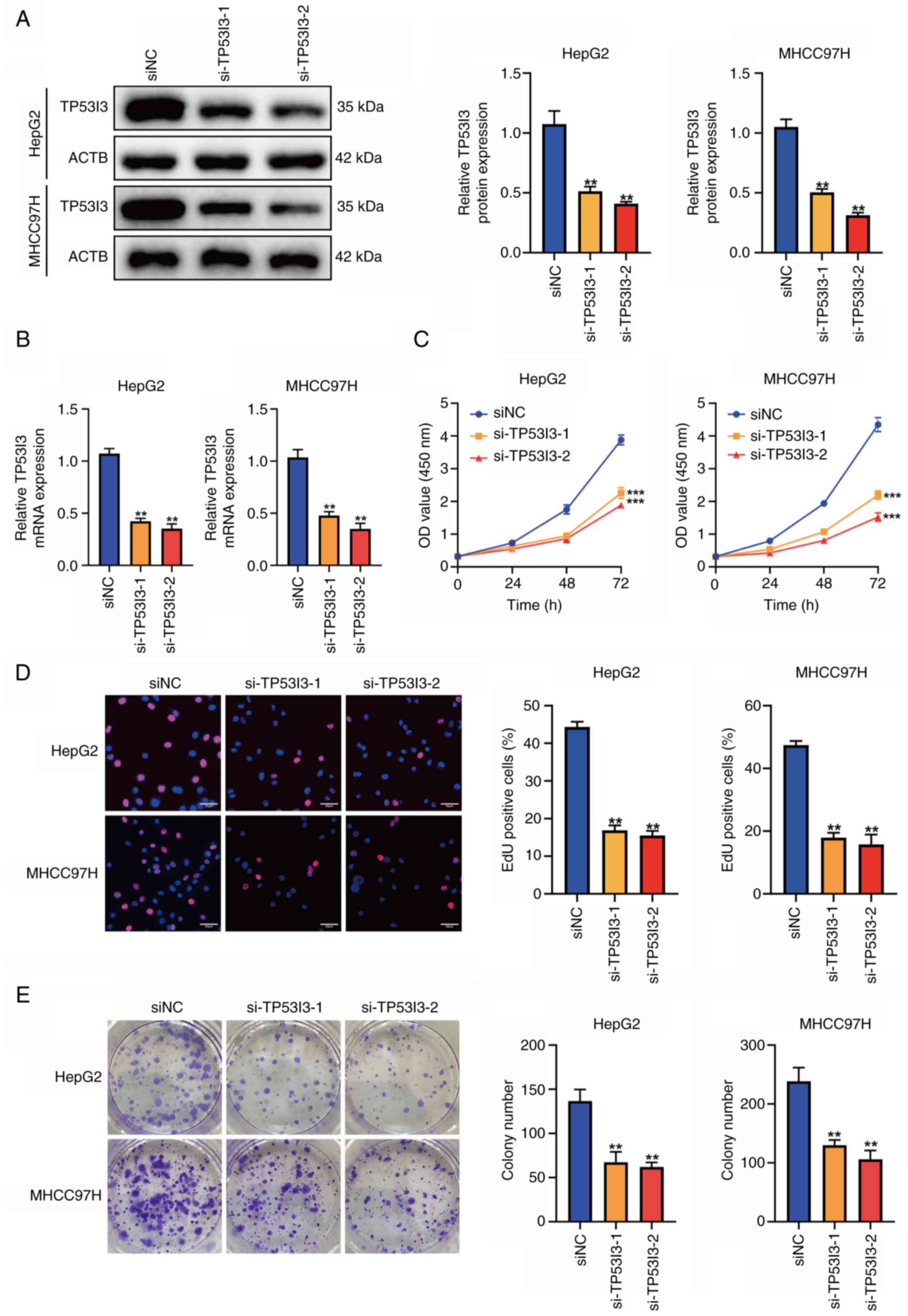
Downregulation of TP53I3 inhibits
liver cancer cell proliferation
Among the five signature genes, CDCA8, MYCN, HAVCR1
and TXNRD1 have previously been demonstrated to have a biological
regulatory function in liver cancer (vide infra), but TP53I3
has been poorly studied in liver cancer. Therefore, the role of
TP53I3 in liver cancer cells was evaluated using cellular
experiments. TP53I3 was knocked down in HepG2 and MHCC97H cells
using siRNA, and the transfection efficiency was verified by
western blot analysis and RT-qPCR (Fig. 13A and B). To explore the impact of
TP53I3 on the proliferation of liver cancer cells in vitro,
CCK-8, EdU and colony formation analyses were performed. The
results showed that the proliferation ability and colony formation
of the liver cancer cells was significantly suppressed after TP53I3
depletion (P<0.05; Fig. 13C-E),
which indicates that TP53I3 promotes the proliferation of liver
cancer cells.
Discussion
Liver cancer remains a significant challenge to
human health, with high rates of incidence and recurrence, even
after surgical resection. Numerous studies have demonstrated that
CTCs are tightly associated with the metastasis,
epithelial-mesenchymal transition and recurrence of malignant
tumors, including liver cancer (54–58).
Therefore, it is critical to screen molecules associated with CTCs
to identify biomarkers for the prediction of liver cancer. In the
present study, a reliable prognostic signature based on CRGs was
constructed and its clinical application in patients with liver
cancer was explored. The results showed that the CRG prognostic
model accurately predicted the prognosis and immunotherapy
sensitivity of patients with liver cancer.
In the present study, 258 CRGs were identified by
systematically analyzing the DEGs in TCGA and ICGC databases and
the CTC expression profiles of liver cancer. These genes were then
screened to construct a five-CRG signature in the TCGA cohort.
Kaplan-Meier survival and ROC analyses were performed to confirm
the prognostic value of the signature, and the results were
validated in ICGC and GEO cohorts. Univariate and multifactorial
Cox analyses further confirmed that the risk signature was able to
serve as an independent prognostic factor. In addition, nomograms
for all three cohorts showed the good predictive power of the
model. The genes in the prognostic signature were CDCA8, HAVCR1,
TP53I3, MYCN and TXNRD1, all of which have the potential to be used
as liver cancer prognostic risk genes. Previous studies have
demonstrated the ability of CDCA8 to promote cancer cell
proliferation and migration in several tumors, including esophageal
squamous cell carcinoma (59),
thyroid cancer (60), malignant
glioma and cutaneous melanoma (61). In addition, Jeon et al
(62) demonstrated that silencing
CDCA8 effectively suppressed liver cancer growth and stemness,
implying that CDCA8 may be a CTC-related gene. HAVCR1 is highly
expressed in a variety of tumors, including colorectal cancer,
non-small-cell lung cancer, clear cell renal cell carcinoma and
liver cancer, and is an independent prognostic factor (63–66).
Moreover, Ye et al (66)
found that T-cell immunoglobulin mucin-1+
(HAVCR1+) regulatory B cell infiltration was
significantly higher in liver tumor tissues compared with
paraneoplastic tissues in patients with liver cancer and promoted
the immune escape of liver cancer cells, implying that it could be
used as an immune therapeutic target. TP53I3, also known as
p53-inducible gene 3, is involved in the apoptosis process and DNA
damage response. Previous studies have revealed that TP53I3
promotes the invasion and metastasis of lung cancer cells and that
silencing TP53I3 increases the chemosensitivity of non-small cell
lung cancer cells to docetaxel (67,68).
Notably, the present study also demonstrated that the knockdown of
TP53I3 inhibited the proliferation ability of liver cancer cells in
cellular experiments. These findings may indicate a novel strategy
for the treatment of liver cancer. Qin et al (69,70)
highlighted that MYCN, a member of the MYC proto-oncogene family,
may be a stem cell-like marker for liver cancer and is potentially
a therapeutic target of acyclic retinoid for liver cancer. TXNRD1
is an antioxidant enzyme that has been reported to be overexpressed
in liver cancer. Lee et al (71) observed that the inhibition of TXNRD1
suppressed liver cancer cell proliferation, promoted apoptosis and
induced oxidative stress, suggesting that it could be used as a
therapeutic target for liver cancer. In conclusion, these previous
studies suggest that the five signature genes have an important
role in the development of liver cancer and may have potential as
therapeutic targets.
As indicated by KEGG analysis, CRGs may promote the
development, metastasis and recurrence of liver cancer via the cell
cycle and p53 signaling pathway. GSEA analysis of the five
signature genes and the high-risk group in the prognostic model
identified various oncogenesis-associated features, including the
terms ‘cell cycle’, ‘p53 signaling pathway’, ‘WNT signaling
pathway’ and ‘DNA replication’. In addition, GSVA results showed
that tumor progression-related pathways, such as ‘cell cycle’, ‘DNA
replication’, ‘mTOR signaling pathway’ and ‘P53 signaling pathway’,
were mainly concentrated in the high-risk group, which was
generally consistent with the GSEA results. On the basis of this, a
number of potential therapeutic agents were also evaluated, with
veliparib (72), ATRA (73,74)
and AUY922 (75) exhibiting high
drug sensitivity in the high-risk group, suggesting that these
agents are likely to be therapeutic candidates.
Immunotherapy is playing an increasingly important
role in liver cancer. Therefore, the relevance of the present model
to immune infiltration and immunotherapy was also analyzed in the
present study. Immune cell infiltration analysis demonstrated that
CDCA8 and HAVCR1 correlated with the infiltration abundance of
several immune cells, including B cells, CD8+ T cells,
macrophages, neutrophils and dendritic cells. In addition, immune
checkpoint expression, TMB scores and immune cell infiltration
levels were strongly associated with patients in the high-risk
subgroup. The analysis of somatic mutation rates also indicated
that patients in the high-risk group had an elevated frequency of
mutations and greater occurrence of TP53 mutations. It has been
proposed that TIDE scores may be used by oncologists to assist in
the selection of suitable patients for immune checkpoint inhibition
therapy (76). Consistent with
this, the present study found that patients in the high-risk group
had lower TIDE scores, while those in the low-risk group had higher
TIDE scores, indicating that the high-risk patients may benefit
more from immunotherapy. All these findings confirm that the
present model has good risk stratification capabilities and is
suitable for selecting the patients who may benefit from
immunotherapy.
Notably, this five-risk gene signature was also used
to identify liver cancer subgroups C1 and C2, of which C2 as a
high-risk subgroup showed a worse prognosis. Compared with group
C1, group C2 had a higher immune checkpoint expression and higher
stromal, immune and ESTIMATE scores for each sample, which also
suggested that patients in group C2 were more suitable for
immunotherapy. More importantly, several chemotherapeutic agents to
which C2 patients should be sensitive were also identified. These
were cisplatin (77), bortezomib
(78), bleomycin, bicalutamide,
mitomycin C, imatinib, etoposide and gemcitabine (79), which could improve the prognosis of
patients in the C2 group. In conclusion, the findings of this
analysis are helpful, but future studies are necessary to verify
this.
However, the study has some limitations. For
example, the regulatory role of these five CRGs in liver cancer
were not further investigated experimentally. Other external
validation of the model is lacking and must to be conducted in
clinical samples in the future. In addition, chemotherapy were not
analyzed. Therefore, additional studies and more evidence are
required to refine the present model in the future.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
Funding: Not applicable.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
LX was responsible for conceptualization,
methodology, software and writing the original draft of the
manuscript. QW performed validation, and reviewed and edited the
manuscript. KZ, XL and WY performed data analysis and
interpretation. XL and WY confirm the authenticity of all the raw
data. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CTC
|
circulating tumor cells
|
|
CTMs
|
circulating tumor microemboli
|
|
CRGs
|
CTCs/CTM-related genes
|
|
DEGs
|
differentially expressed genes
|
|
GO
|
Gene Ontology
|
|
GSEA
|
gene set enrichment analysis
|
|
GSVA
|
gene set variation analysis
|
|
ICGC
|
International Cancer Genome
Consortium
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
LASSO
|
least absolute shrinkage and
selection operator
|
|
NMF
|
non-negative matrix factorization
|
|
PPI
|
protein-protein interaction
|
|
ROC
|
receiver operating characteristic
|
|
TCGA
|
The Cancer Genome Atlas
|
|
TMB
|
tumor mutation burden
|
|
TIDE
|
tumor immune dysfunction and
exclusion
|
References
|
1
|
Global Burden of Disease Cancer
Collaboration, . Fitzmaurice C, Allen C, Barber RM, Barregard L,
Bhutta ZA, Brenner H, Dicker DJ, Chimed-Orchir O, Dandona R, et al:
Global, regional, and national cancer incidence, mortality, years
of life lost, years lived with disability, and disability-adjusted
life-years for 32 cancer groups, 1990 to 2015: A systematic
analysis for the global burden of disease study. JAMA Oncol.
3:524–548. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Villanueva A: Hepatocellular carcinoma. N
Engl J Med. 380:1450–1462. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Llovet JM, Kelley RK, Villanueva A, Singal
AG, Pikarsky E, Roayaie S, Lencioni R, Koike K, Zucman-Rossi J and
Finn RS: Hepatocellular carcinoma. Nat Rev Dis Primers. 7:62021.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Marrero JA, Kulik LM, Sirlin CB, Zhu AX,
Finn RS, Abecassis MM, Roberts LR and Heimbach JK:
Diagnosisstaging, and management of hepatocellular carcinoma: 2018
Practice guidance by the american association for the study of
liver diseases. Hepatology. 68:723–750. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kluger MD, Salceda JA, Laurent A, Tayar C,
Duvoux C, Decaens T, Luciani A, Van Nhieu JT, Azoulay D and Cherqui
D: Liver resection for hepatocellular carcinoma in 313 western
patients: Tumor biology and underlying liver rather than tumor size
drive prognosis. J Hepatol. 62:1131–1140. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao L, Wu X, Li T, Luo J and Dong D:
ctcRbase: The gene expression database of circulating tumor cells
and microemboli. Database (Oxford). 2020:baaa0202020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Alix-Panabières C and Pantel K:
Circulating tumor cells: Liquid biopsy of cancer. Clin Chem.
59:110–118. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Allard WJ, Matera J, Miller MC, Repollet
M, Connelly MC, Rao C, Tibbe AG, Uhr JW and Terstappen LW: Tumor
cells circulate in the peripheral blood of all major carcinomas but
not in healthy subjects or patients with nonmalignant diseases.
Clin Cancer Res. 10:6897–6904. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Plaks V, Koopman CD and Werb Z: Cancer.
Circulating tumor cells. Science. 341:1186–1188. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Szczerba BM, Castro-Giner F, Vetter M,
Krol I, Gkountela S, Landin J, Scheidmann MC, Donato C, Scherrer R,
Singer J, et al: Neutrophils escort circulating tumour cells to
enable cell cycle progression. Nature. 566:553–557. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Aceto N, Bardia A, Miyamoto DT, Donaldson
MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H, et al:
Circulating tumor cell clusters are oligoclonal precursors of
breast cancer metastasis. Cell. 158:1110–1122. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lo HC, Xu Z, Kim IS, Pingel B, Aguirre S,
Kodali S, Liu J, Zhang W, Muscarella AM, Hein SM, et al: Resistance
to natural killer cell immunosurveillance confers a selective
advantage to polyclonal metastasis. Nat Cancer. 1:709–722. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pereira-Veiga T, Schneegans S, Pantel K
and Wikman H: Circulating tumor cell-blood cell crosstalk: Biology
and clinical relevance. Cell Rep. 40:1112982022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cristofanilli M, Budd GT, Ellis MJ,
Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ,
Terstappen LW and Hayes DF: Circulating tumor cells, disease
progression, and survival in metastatic breast cancer. N Engl J
Med. 351:781–791. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hou JM, Krebs MG, Lancashire L, Sloane R,
Backen A, Swain RK, Priest LJ, Greystoke A, Zhou C, Morris K, et
al: Clinical significance and molecular characteristics of
circulating tumor cells and circulating tumor microemboli in
patients with small-cell lung cancer. J Clin Oncol. 30:525–532.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ye Q, Ling S, Zheng S and Xu X: Liquid
biopsy in hepatocellular carcinoma: Circulating tumor cells and
circulating tumor DNA. Mol Cancer. 18:1142019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
De Rubis G, Rajeev Krishnan S and Bebawy
M: Liquid biopsies in cancer diagnosis, monitoring, and prognosis.
Trends Pharmacol Sci. 40:172–186. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yu JJ, Xiao W, Dong SL, Liang HF, Zhang
ZW, Zhang BX, Huang ZY, Chen YF, Zhang WG, Luo HP, et al: Effect of
surgical liver resection on circulating tumor cells in patients
with hepatocellular carcinoma. BMC Cancer. 18:8352018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kelley RK, Magbanua MJ, Butler TM,
Collisson EA, Hwang J, Sidiropoulos N, Evason K, McWhirter RM,
Hameed B, Wayne EM, et al: Circulating tumor cells in
hepatocellular carcinoma: A pilot study of detection, enumeration,
and next-generation sequencing in cases and controls. BMC Cancer.
15:2062015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sun YF, Xu Y, Yang XR, Guo W, Zhang X, Qiu
SJ, Shi RY, Hu B, Zhou J and Fan J: Circulating stem cell-like
epithelial cell adhesion molecule-positive tumor cells indicate
poor prognosis of hepatocellular carcinoma after curative
resection. Hepatology. 57:1458–1468. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang Y and Navin NE: Advances and
applications of single-cell sequencing technologies. Mol Cell.
58:598–609. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Abouleila Y, Onidani K, Ali A, Shoji H,
Kawai T, Lim CT, Kumar V, Okaya S, Kato K, Hiyama E, et al: Live
single cell mass spectrometry reveals cancer-specific metabolic
profiles of circulating tumor cells. Cancer Sci. 110:697–706. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bhan I, Mosesso K, Goyal L, Philipp J,
Kalinich M, Franses JW, Choz M, Oklu R, Toner M, Maheswaran S, et
al: Detection and analysis of circulating epithelial cells in
liquid biopsies from patients with liver disease. Gastroenterology.
155:2016–2018.e11. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mariathasan S, Turley SJ, Nickles D,
Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita
JL, Cubas R, et al: TGFβ attenuates tumour response to PD-L1
blockade by contributing to exclusion of T cells. Nature.
554:544–548. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bader GD and Hogue CW: An automated method
for finding molecular complexes in large protein interaction
networks. BMC Bioinformatics. 4:22003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chin CH, Chen SH, Wu HH, Ho CW, Ko MT and
Lin CY: cytoHubba: Identifying hub objects and sub-networks from
complex interactome. BMC Syst Biol. 8 (Suppl 4):S112014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hänzelmann S, Castelo R and Guinney J:
GSVA: Gene set variation analysis for microarray and RNA-seq data.
BMC Bioinformatics. 14:72013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tibshirani R: The lasso method for
variable selection in the Cox model. Stat Med. 16:385–395. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lossos IS, Czerwinski DK, Alizadeh AA,
Wechser MA, Tibshirani R, Botstein D and Levy R: Prediction of
survival in diffuse large-B-cell lymphoma based on the expression
of six genes. N Engl J Med. 350:1828–1837. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gaujoux R and Seoighe C: A flexible R
package for nonnegative matrix factorization. BMC Bioinformatics.
11:3672010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Iasonos A, Schrag D, Raj GV and Panageas
KS: How to build and interpret a nomogram for cancer prognosis. J
Clin Oncol. 26:1364–1370. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Newman AM, Liu CL, Green MR, Gentles AJ,
Feng W, Xu Y, Hoang CD, Diehn M and Alizadeh AA: Robust enumeration
of cell subsets from tissue expression profiles. Nat Methods.
12:453–457. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li T, Fan J, Wang B, Traugh N, Chen Q, Liu
JS, Li B and Liu XS: TIMER: A web server for comprehensive analysis
of tumor-infiltrating immune cells. Cancer Res. 77:e108–e110. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Finotello F, Mayer C, Plattner C,
Laschober G, Rieder D, Hackl H, Krogsdam A, Loncova Z, Posch W,
Wilflingseder D, et al: Molecular and pharmacological modulators of
the tumor immune contexture revealed by deconvolution of RNA-seq
data. Genome Med. 11:342019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Becht E, Giraldo NA, Lacroix L, Buttard B,
Elarouci N, Petitprez F, Selves J, Laurent-Puig P, Sautès-Fridman
C, Fridman WH and de Reyniès A: Estimating the population abundance
of tissue-infiltrating immune and stromal cell populations using
gene expression. Genome Biol. 17:2182016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Aran D, Hu Z and Butte AJ: xCell:
Digitally portraying the tissue cellular heterogeneity landscape.
Genome Biol. 18:2202017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sturm G, Finotello F, Petitprez F, Zhang
JD, Baumbach J, Fridman WH, List M and Aneichyk T: Comprehensive
evaluation of transcriptome-based cell-type quantification methods
for immuno-oncology. Bioinformatics. 35:i436–i445. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Racle J and Gfeller D: EPIC: A tool to
estimate the proportions of different cell types from bulk gene
expression data. Methods Mol Biol. 2120:233–248. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zeng D, Ye Z, Shen R, Yu G, Wu J, Xiong Y,
Zhou R, Qiu W, Huang N, Sun L, et al: IOBR: Multi-omics
immuno-oncology biological research to decode tumor
microenvironment and signatures. Front Immunol. 12:6879752021.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Malta TM, Sokolov A, Gentles AJ,
Burzykowski T, Poisson L, Weinstein JN, Kamińska B, Huelsken J,
Omberg L, Gevaert O, et al: Machine learning identifies stemness
features associated with oncogenic dedifferentiation. Cell.
173:338–354.e15. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yang W, Soares J, Greninger P, Edelman EJ,
Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR, et
al: Genomics of drug sensitivity in cancer (GDSC): A resource for
therapeutic biomarker discovery in cancer cells. Nucleic Acids Res.
41:D955–D961. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ozga AJ, Chow MT and Luster AD: Chemokines
and the immune response to cancer. Immunity. 54:859–874. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Vilgelm AE and Richmond A: Chemokines
modulate immune surveillance in tumorigenesis, metastasis, and
response to immunotherapy. Front Immunol. 10:3332019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ru B, Wong CN, Tong Y, Zhong JY, Zhong
SSW, Wu WC, Chu KC, Wong CY, Lau CY, Chen I, et al: TISIDB: An
integrated repository portal for tumor-immune system interactions.
Bioinformatics. 35:4200–4202. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Du Y, Ma Y, Zhu Q, Liu T, Jiao Y, Yuan P
and Wang X: An m6A-related prognostic biomarker associated with the
hepatocellular carcinoma immune microenvironment. Front Pharmacol.
12:7079302021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fu XW and Song CQ: Identification and
validation of pyroptosis-related gene signature to predict
prognosis and reveal immune infiltration in hepatocellular
carcinoma. Front Cell Dev Biol. 9:7480392021. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Guo DZ, Huang A, Wang YP, Cao Y, Fan J,
Yang XR and Zhou J: Development of an eight-gene prognostic model
for overall survival prediction in patients with hepatocellular
carcinoma. J Clin Transl Hepatol. 9:898–908. 2021.PubMed/NCBI
|
|
51
|
Lei D, Chen Y, Zhou Y, Hu G and Luo F: A
starvation-Based 9-mRNA signature correlates with prognosis in
patients with hepatocellular carcinoma. Front Oncol. 11:7167572021.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tian D, Yu Y, Zhang L, Sun J and Jiang W:
A five-gene-based prognostic signature for hepatocellular
carcinoma. Front Med (Lausanne). 8:6813882021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zheng S, Xie X, Guo X, Wu Y, Chen G, Chen
X, Wang M, Xue T and Zhang B: Identification of a
pyroptosis-related gene signature for predicting overall survival
and response to immunotherapy in hepatocellular carcinoma. Front
Genet. 12:7892962021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Qi LN, Xiang BD, Wu FX, Ye JZ, Zhong JH,
Wang YY, Chen YY, Chen ZS, Ma L, Chen J, et al: Circulating tumor
cells undergoing EMT provide a metric for diagnosis and prognosis
of patients with hepatocellular carcinoma. Cancer Res.
78:4731–4744. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Schilling D, Todenhöfer T, Hennenlotter J,
Schwentner C, Fehm T and Stenzl A: Isolated, disseminated and
circulating tumour cells in prostate cancer. Nat Rev Urol.
9:448–463. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhou H, Zhu L, Song J, Wang G, Li P, Li W,
Luo P, Sun X, Wu J, Liu Y, et al: Liquid biopsy at the frontier of
detection, prognosis and progression monitoring in colorectal
cancer. Mol Cancer. 21:862022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Diamantopoulou Z, Castro-Giner F, Schwab
FD, Foerster C, Saini M, Budinjas S, Strittmatter K, Krol I,
Seifert B, Heinzelmann-Schwarz V, et al: The metastatic spread of
breast cancer accelerates during sleep. Nature. 607:156–162. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Magri V, Marino L, Nicolazzo C, Gradilone
A, De Renzi G, De Meo M, Gandini O, Sabatini A, Santini D, Cortesi
E and Gazzaniga P: Prognostic role of circulating tumor cell
trajectories in metastatic colorectal cancer. Cells. 12:11722023.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Xie J, Wang B, Luo W, Li C and Jia X:
Upregulation of KIF18B facilitates malignant phenotype of
esophageal squamous cell carcinoma by activating CDCA8/mTORC1
pathway. J Clin Lab Anal. 36:e246332022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Xiang C, Sun WH, Ke Y, Yu X and Wang Y:
CDCA8 contributes to the development and progression of thyroid
cancer through regulating CDK1. J Cancer. 13:2322–2335. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ci C, Tang B, Lyu D, Liu W, Qiang D, Ji X,
Qiu X, Chen L and Ding W: Overexpression of CDCA8 promotes the
malignant progression of cutaneous melanoma and leads to poor
prognosis. Int J Mol Med. 43:404–412. 2019.PubMed/NCBI
|
|
62
|
Jeon T, Ko MJ, Seo YR, Jung SJ, Seo D,
Park SY, Park KU, Kim KS, Kim M, Seo JH, et al: Silencing CDCA8
suppresses hepatocellular carcinoma growth and stemness via
restoration of ATF3 tumor suppressor and inactivation of
AKT/β-catenin signaling. Cancers (Basel). 13:10552021. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wang Y, Martin TA and Jiang WG: HAVcR-1
expression in human colorectal cancer and its effects on colorectal
cancer cells in vitro. Anticancer Res. 33:207–214. 2013.PubMed/NCBI
|
|
64
|
Zheng X, Xu K, Chen L, Zhou Y and Jiang J:
Prognostic value of TIM-1 expression in human non-small-cell lung
cancer. J Transl Med. 17:1782019. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Cuadros T, Trilla E, Sarró E, Vilà MR,
Vilardell J, de Torres I, Salcedo M, López-Hellin J, Sánchez A,
Ramón y Cajal S, et al: HAVCR/KIM-1 activates the IL-6/STAT-3
pathway in clear cell renal cell carcinoma and determines tumor
progression and patient outcome. Cancer Res. 74:1416–1428. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ye L, Zhang Q, Cheng Y, Chen X, Wang G,
Shi M, Zhang T, Cao Y, Pan H, Zhang L, et al: Tumor-derived
exosomal HMGB1 fosters hepatocellular carcinoma immune evasion by
promoting TIM-1+ regulatory B cell expansion. J
Immunother Cancer. 6:1452018. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Gu MM, Gao D, Yao PA, Yu L, Yang XD, Xing
CG, Zhou J, Shang ZF and Li M: p53-inducible gene 3 promotes cell
migration and invasion by activating the FAK/Src pathway in lung
adenocarcinoma. Cancer Sci. 109:3783–3793. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Li M, Li S, Liu B, Gu MM, Zou S, Xiao BB,
Yu L, Ding WQ, Zhou PK, Zhou J and Shang ZF: PIG3 promotes NSCLC
cell mitotic progression and is associated with poor prognosis of
NSCLC patients. J Exp Clin Cancer Res. 36:392017. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Qin XY, Suzuki H, Honda M, Okada H, Kaneko
S, Inoue I, Ebisui E, Hashimoto K, Carninci P, Kanki K, et al:
Prevention of hepatocellular carcinoma by targeting MYCN-positive
liver cancer stem cells with acyclic retinoid. Proc Natl Acad Sci
USA. 115:4969–4974. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Qin XY, Su T, Yu W and Kojima S: Lipid
desaturation-associated endoplasmic reticulum stress regulates MYCN
gene expression in hepatocellular carcinoma cells. Cell Death Dis.
11:662020. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Lee D, Xu IMJ, Chiu DKC, Leibold J, Tse
APW, Bao MHR, Yuen VWH, Chan CYK, Lai RKH, Chin DWC, et al:
Induction of oxidative stress through inhibition of thioredoxin
reductase 1 is an effective therapeutic approach for hepatocellular
carcinoma. Hepatology. 69:1768–1786. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Muñoz-Gámez JA, López Viota J, Barrientos
A, Carazo Á, Sanjuán-Nuñez L, Quiles-Perez R, Muñoz-de-Rueda P,
Delgado Á, Ruiz-Extremera Á and Salmerón J: Synergistic
cytotoxicity of the poly (ADP-ribose) polymerase inhibitor ABT-888
and temozolomide in dual-drug targeted magnetic nanoparticles.
Liver Int. 35:1430–1441. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wang S, Liu J, Wu H, Jiang A, Zhao K, Yan
K, Wu W, Han H, Zhang Y and Yang W: All-trans retinoic acid (ATRA)
inhibits insufficient radiofrequency ablation (IRFA)-induced
enrichment of tumor-initiating cells in hepatocellular carcinoma.
Chin J Cancer Res. 33:694–707. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Sun J, Liu C, Shi J, Wang N, Jiang D, Mao
F, Gu J, Zhou L, Shen L, Lau WY and Cheng S: A novel chemotherapy
strategy for advanced hepatocellular carcinoma: A multicenter
retrospective study. Chin Med J (Engl). 135:2338–2343. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Augello G, Emma MR, Cusimano A, Azzolina
A, Mongiovì S, Puleio R, Cassata G, Gulino A, Belmonte B,
Gramignoli R, et al: Targeting HSP90 with the small molecule
inhibitor AUY922 (luminespib) as a treatment strategy against
hepatocellular carcinoma. Int J Cancer. 144:2613–2624. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X,
Li Z, Traugh N, Bu X, Li B, et al: Signatures of T cell dysfunction
and exclusion predict cancer immunotherapy response. Nat Med.
24:1550–1558. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Lee JO, Lee KW, Oh DY, Kim JH, Im SA, Kim
TY and Bang YJ: Combination chemotherapy with capecitabine and
cisplatin for patients with metastatic hepatocellular carcinoma.
Ann Oncol. 20:1402–1407. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Li L, Zhang Y, Zhou Y, Hu H, Hu Y,
Georgiades C, Mao HQ and Selaru FM: Quaternary nanoparticles enable
sustained release of bortezomib for hepatocellular carcinoma.
Hepatology. 76:1660–1672. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zhu AX, Blaszkowsky LS, Ryan DP, Clark JW,
Muzikansky A, Horgan K, Sheehan S, Hale KE, Enzinger PC, Bhargava P
and Stuart K: Phase II study of gemcitabine and oxaliplatin in
combination with bevacizumab in patients with advanced
hepatocellular carcinoma. J Clin Oncol. 24:1898–1903. 2006.
View Article : Google Scholar : PubMed/NCBI
|