Introduction
Approximately 2.2 million new lung cancer cases and
1.8 million deaths of patients with lung cancer were reported
worldwide in 2020 (1). Although
considerable progress has been achieved in lung cancer diagnosis
and treatment, the five-year survival rate for this disease remains
low (2). The survival rate for lung
cancer is closely associated with the disease stage, and the
five-year survival rate of patients with stage I lung cancer can
reach 85.5–90.2% (3). Therefore,
the identification of novel and reliable biomarkers for the early
diagnosis of lung cancer is critical.
Cancer stem cells (CSCs) are cells that exhibit stem
cell properties, have the ability for self-renewal, and promote the
invasion and growth of tumor cells in cancer (4). The presence of CSCs leads to cellular
heterogeneity in malignancies as well as innate antibiotic
resistance and enhanced invasive capacity, which contribute to
cancer progression and metastasis (5). Several conventional CSC markers,
including CD44, CD90 and SRY-box transcription factor 2, have been
identified in lung cancer; however, these markers are not used
universally in clinical practice (6). The mRNA expression-based stemness
index (mRNAsi) is a quantitative measure that indicates the degree
of similarity between tumors and stem cells (7,8). This
index is based on mRNA expression profiles and reflects the
stemness characteristics of the transcriptome (9). Previous studies have revealed that
lung cancer tissues exhibit higher mRNAsi values than normal
tissues and suggest that high mRNAsi values are associated with
poor survival in patients with lung adenocarcinoma (LUAD) (10,11).
Although circulating tumor DNA (ctDNA) exhibits
potential for the detection of lung cancer, its sensitivity and
specificity in the diagnosis of early lung cancer is not optimal
(12). Circulating cell-free RNA
(cfRNA) is actively secreted by cells or released by apoptotic and
necrotic cells into the blood (13). Plasma cfRNAs have been shown to
reflect the systemic response to a growing tumor and to indicate
the tissue origin of the tumor (14). The release of cfRNA into the blood
circulation by early tumors allows upregulated, tumor-specific and
tumor-derived RNAs to be identified in the blood at an early stage,
which may facilitate the early diagnosis of tumors (15). Studies have indicated that the
combined analysis of cfRNA and ctDNA is complementary, improving
the early diagnosis of lung cancer (16,17).
As most studies on mRNAsi are typically conducted
using tumor tissues, whether cfRNA-based mRNAsi can be used for the
early diagnosis of lung cancer remains unclear. In the present
study, primary lung cancer tissues, adjacent non-cancerous tissues
and lymph nodes were collected for RNA sequencing and analysis to
identify gene expression signatures stratified by mRNAsi.
Furthermore, paired blood samples were collected for cfRNA
sequencing and the determination of a stemness gene expression
signature of cfRNAs based on tissue stemness genes. Subsequently,
the genes and pathways associated with tissue and blood stemness
were analyzed. Finally, correlations of tissue mRNAsi and the cfRNA
stemness index with immune infiltration were examined.
Materials and methods
Tissue and blood samples
Tumor tissue, paracancerous tissue (>5 cm away
from the tumor edge), peripheral blood and lymph node samples were
obtained from 22 patients with stage I–III non-small cell lung
cancer (NSCLC) between October 2021 and March 2022 at the Shanghai
Chest Hospital, Shanghai Jiao Tong University School of Medicine
(Shanghai, China). None of the patients had received any
chemotherapy, targeted therapy or immunotherapy before surgery. The
clinicopathological data of the patients, including their sex, age,
pathological and molecular pathological characteristics (based on
TNM staging) (18), and recurrence,
were collected.
The freshly collected tissues were snap-frozen and
stored in liquid nitrogen. Blood samples were temporarily stored at
4°C. Plasmapheresis was completed within 4 h of the blood being
drawn from the patient, and the plasma was stored immediately in a
refrigerator at −80°C.
RNA sequencing of samples
Tissue total RNA was extracted from frozen tissues
using a MagMAX FFPE DNA/RNA Ultra Kit (Thermo Fisher Scientific,
Inc.) following the standard protocol provided by the manufacturer
but skipping the deparaffinization process. The concentration of
the RNA was measured by NanoDrop™ 2000 spectrophotometer (Thermo
Fisher Scientific, Inc.) and the quality of the RNA was evaluated
using TapeStation 4200 system (Agilent Technologies, Inc.), and RNA
with DV200 ≥40% was considered qualified. Qualified RNA was
reverse-transcribed with random hexamers for first strand
synthesize and underwent second strand synthesizing, end
modification and polyadenylation, sequencing adapter ligation and
library amplification using a Hieff NGS® Ultima
Dual-mode RNA Library Prep Kit [Yeasen Biotechnology (Shanghai)
Co., Ltd.]. Purified libraries were pooled and sequenced using a
NovaSeq 6000 system (Illumina, Inc.) with PE150 mode. Low-quality
sequences were filtered out prior to downstream analysis.
Cell free RNA was extracted from plasma samples
using SpCap-cfRNA extraction and library preparation technology,
which is a modified from the method by Jacobsen et al
(19) specifically for low
abundance plasma cell free RNA extraction and library preparation.
In brief, the SpCap-cfRNA technology utilizes poly-T oligo-coated
magnatic beads to capture cell free RNA from the plasma, and after
extensive wash, captured RNA is reverse-transcribed on beads to
make RNA libraries for sequencing. The cfRNA libraries were pooled
and sequenced using NovaSeq 6000 system (Illumina, Inc.) with PE150
mode. The sequencing data were subjected to bioinformatics
analysis.
Calculation of mRNAsi
The R package TCGAbiolinks (2.18.0) was used to
calculate the mRNAsi of different sample types (20). Stemness signatures were analyzed
using Progenitor Cell Biology Consortium stemSig (version 2.18.0)
(21). The mRNAsi, a continuous
variable, was used to categorise the patients into high- and
low-mRNAsi groups for different sample types, based on the
median.
Weighted gene co-expression network
analysis (WGCNA) and identification of differentially expressed
genes (DEGs)
The WGCNA package (version 1.71) (22) was used construct coexpression
networks. In this analysis, the Pearson's correlation coefficients
of genes were calculated to obtain correlation matrices, and
appropriate thresholds calculated by the function pickSoftThreshold
in WGCNA were selected to measure the connectivity between genes.
Adjacency matrices were converted to topological overlap matrices
(TOM), and the corresponding differences (1-TOM) were calculated.
Genes with similar expression patterns were then grouped into
co-expression modules. First, WGCNA of the tumor tissues was
performed. In addition, the DEGs of tumor and paracancerous tissues
were screened using the EdgeR package (version 3.34.0) (23) with the criteria of fold change >2
and adjusted P-value <0.05. Correlation analyses were then
performed to determine the WGCNA modules most relevant to the
mRNAsi of tumor tissues and blood. The tissue and peripheral blood
samples were categorized into high and low mRNAsi score groups
using the median value as a cutoff. DEGs between the high and low
mRNAsi groups were screened using the aforementioned EdgeR package.
Pathway enrichment analysis of the identified genes was performed
using the Kyoto Encyclopedia of Genes and Genomes (KEGG)
orthology-based annotation system (version 3.0) (https://www.genome.jp/kegg/) with default
parameters.
Identification of fusion
transcripts
The STAR-Fusion tool (version 1.8.1) was used to
detect fusion transcripts based on the plasma cfRNA sequencing data
(24). In the gene expression
analysis, STAR was used to map the sequencing data to the reference
genome (hg38) and define the transcript coordinates based on the
gene annotation format file from GENCODE (version 27; GRCh38)
(https://www.gencodegenes.org/human/release_27.html).
Gene expression abundance was quantified as reads per kilobase per
million mapped reads by using the Cufflinks package (version 2.2.1)
(25). The EdgeR package was used
to identify DEGs between the fusion-positive and normal
samples.
Correlation between stemness index and
immune cell infiltration
The gene expression profiles were uploaded to the
CIBERSORT website (https://cibersortx.stanford.edu/) to analyze the
immune cells infiltrating the tumor tissue. Using LM22 as a
reference, 1,000 random permutation operations were performed, and
the results were screened based on a significance threshold of
P<0.05. The ggplot2 package (version 3.4.4) (https://github.com/tidyverse/ggplot2)
was used to analyze differences in immune cell populations between
normal paracancerous tissues and tumors. The packages limma
(version 3.54.2) (https://bioconductor.org/packages/limma), reshape2
(version 1.4.4) (https://github.com/hadley/reshape) and ggExtra
(version 0.10.1) (https://github.com/daattali/ggExtra) were used to
evaluate the relationships between the mRNAsi-related genes and
immune infiltrating cells. Spearman's rank correlation tests were
used for analysis, and the ggplot2 package (version 3.4.4)
(https://github.com/tidyverse/ggplot2)
was used to generate correlation heat maps.
Statistical analysis
R software (version 4.1.2) (https://www.r-project.org/) was used to statistically
analyze all data. Differences in mRNAsi values between different
types of matched samples were assessed using the Friedman test
followed by Wilcoxon signed-rank tests with Bonferroni correction.
An unpaired t-test was used to analyze the statistical significance
of differences in normally distributed variables between two
groups, and the Wilcoxon rank-sum test was used to compare
non-normally distributed variables between two groups. Kaplan-Meier
(K-M) analysis and log-rank tests were used to compare survival
between two groups. P<0.05 was considered to indicate a
statistically significant result.
Results
Clinical data
A total of 22 patients were included in the present
study (Table I). The age of the
patients ranged from 46 to 74 years, with an average of 63.8 years.
Among all patients, 63.6% were women, 90.9% had adenocarcinoma,
86.3% had stage I or II cancer, and 40.9% had poorly differentiated
tumors.
 | Table I.Clinical characteristics of the
enrolled patients. |
Table I.
Clinical characteristics of the
enrolled patients.
|
Characteristics | Values |
|---|
| Mean age,
years | 63.8 |
| Sex, n (%) |
|
|
Female | 14 (63.6) |
|
Male | 8 (36.4) |
| Histology, n
(%) |
|
|
Adenocarcinoma | 20 (90.9) |
|
Squamous cell carcinoma | 2 (9.1) |
| Differentiation
degree, n (%) |
|
|
Poor | 9 (40.9) |
|
Moderate | 13 (59.1) |
| T stage, n (%) |
|
| T1 | 11 (50.0) |
| T2 | 7 (31.8) |
| T3 | 3 (13.6) |
| T4 | 1 (4.5) |
| N stage, n (%) |
|
| N0 | 20 (90.9) |
| N1 | 0 (0.0) |
| N2 | 2 (9.1) |
| N3 | 0 (0.0) |
| M stage |
|
| M0 | 22 (100.0) |
| M1 | 0 (0.0) |
| pTMN stage |
|
| I | 16 (72.7) |
| II | 3 (13.6) |
|
III | 3 (13.6) |
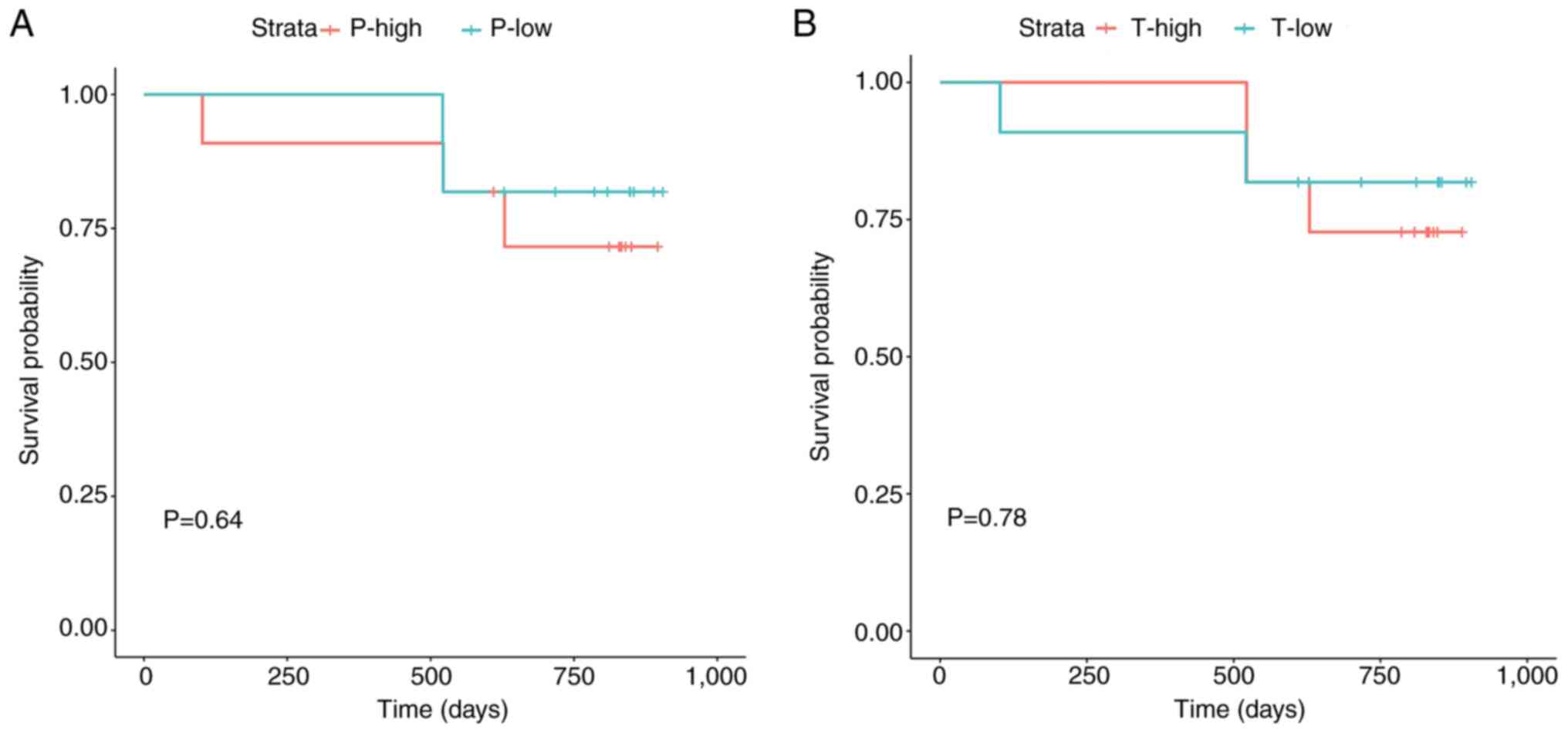
mRNAsi correlates with the prognosis
of patients with lung cancer
By evaluating the median value of mRNAsi in the
peripheral blood and cancer tissue samples of patients with lung
cancer, the patients were categorized into high and low mRNAsi
groups for each sample type. K-M survival analysis revealed no
significant difference in overall survival between the high and low
mRNAsi groups for peripheral blood or cancer tissue samples
(Fig. 1). This may be attributed to
the short duration of follow-up, as the median follow-up time was
only 820 days. To investigate the relationship between mRNAsi and
disease recurrence, the recurrence rates of patients in the groups
over two years were calculated. In both the peripheral blood and
cancer tissue samples, the two-year recurrence rate of patients in
the high mRNAsi group was 27.3%, whereas that of patients in the
low mRNAsi group was 18.2%. Notably, when the peripheral blood and
cancer tissue samples both had high mRNAsi values, the two-year
recurrence rate of the patients was 18.2%, which was twice that of
patients with low mRNAsi values.
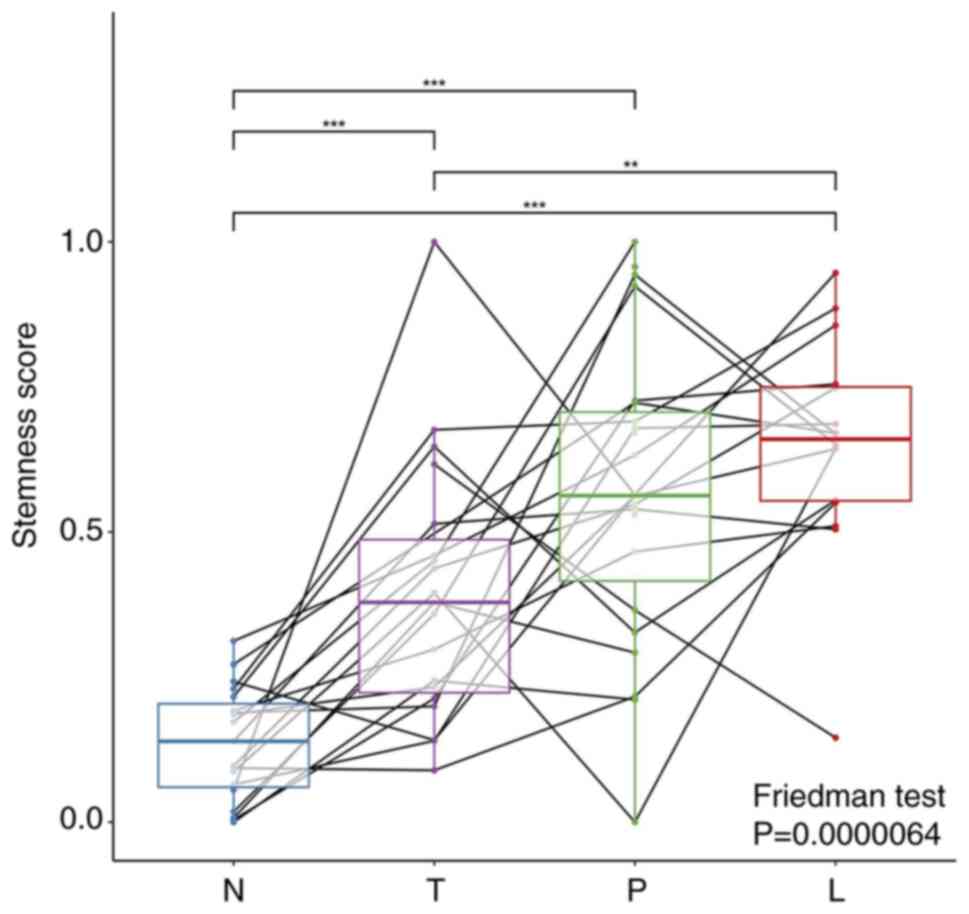
Comparison of mRNAsi values in
peripheral blood and tissues
To investigate the association between tumor
stemness and metastasis, the mRNAsi values of paracancerous
tissues, cancer tissues, lymph nodes and peripheral blood were
compared. The results revealed that the mRNAsi value of tumor
tissues was significantly higher than that of paracancerous normal
tissues, and the mRNAsi value of lymph nodes was significantly
higher than that of tumor tissues. Also, the mRNAsi value of cfRNA
in peripheral blood was higher than that in cancer tissues, albeit
not significantly, and was significantly higher than that in
paracancerous tissues (Fig. 2).
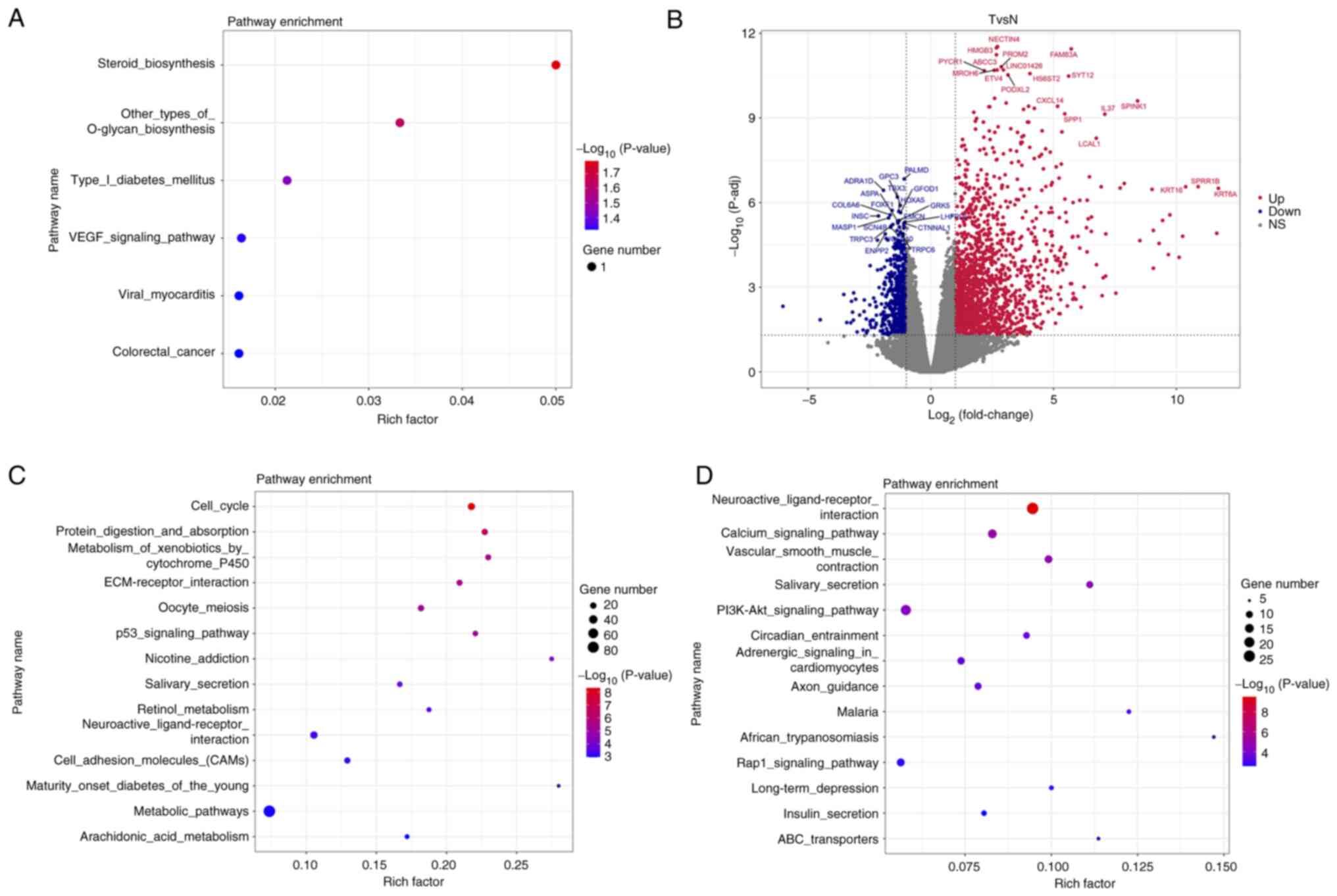
Genes and pathways associated with
lung cancer development
WGCNA of the tumor tissues revealed that several
genes, including ALKBH3, MED19, protein tyrosine phosphatase
receptor type C associated protein (PTPRN), ERAL1 and PPME1 are
associated with lung cancer (Table
SI). KEGG analysis indicated that these genes are enriched in
the pathways ‘steroid biosynthesis’ and ‘other types of O-glycan
biosynthesis’ (Fig. 3A). The DEGs
associated with tumorigenesis, identified by the comparison of gene
expression in tumor tissue with that in paracancerous tissue, are
presented as a volcano plot (Fig.
3B; Table SII). Upregulated
genes are enriched in the ‘cell cycle’, ‘protein digestion and
absorption’ and ‘metabolism of xenobiotics by cytochrome P450’
pathways (Fig. 3C). By contrast,
downregulated genes are enriched in the ‘neuroactive
ligand-receptor interaction’, ‘calcium signaling pathway’ and
‘vascular smooth muscle contraction’ pathways (Fig. 3D).
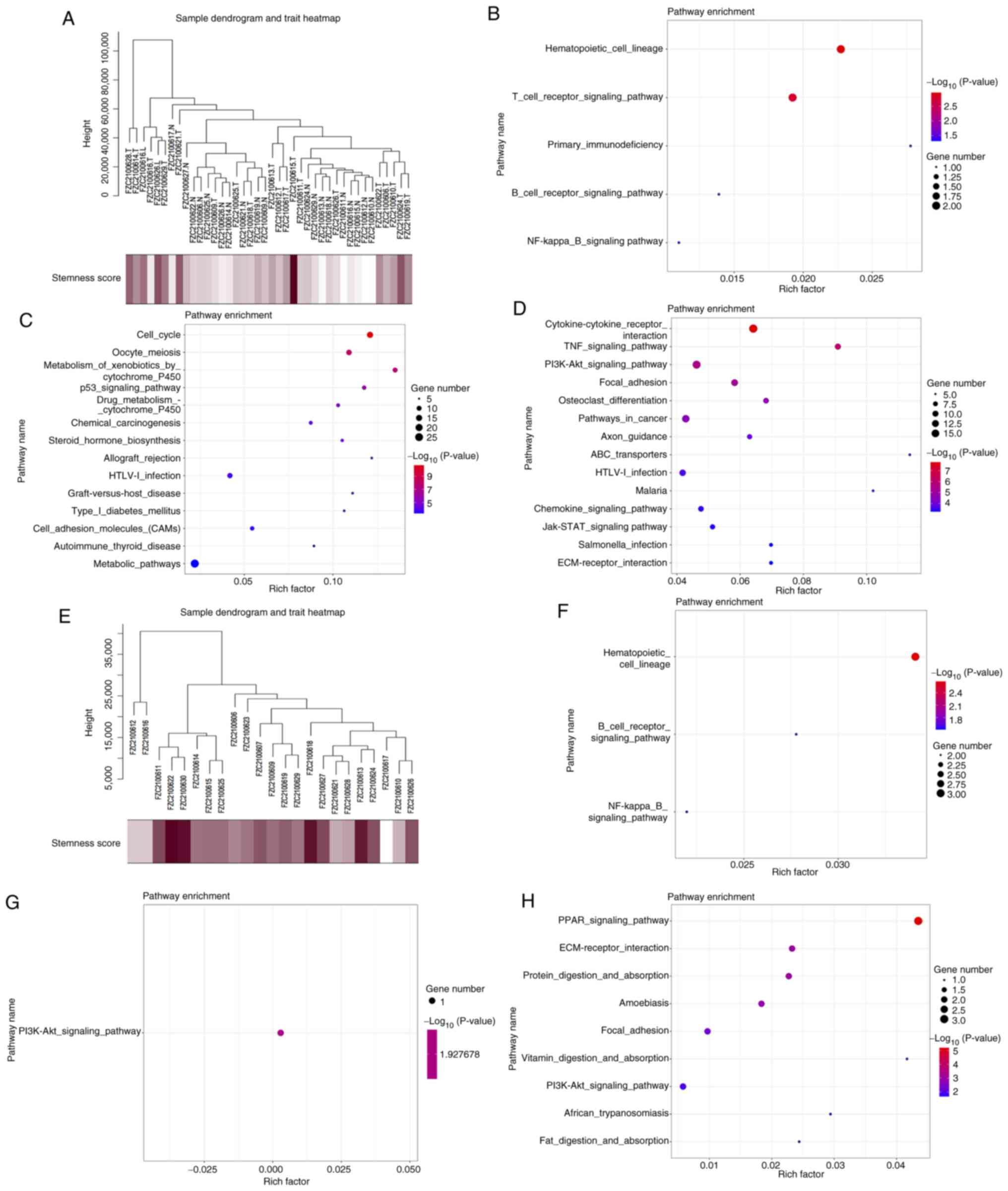
Genes and pathways associated with the
mRNAsi of tumor tissues and peripheral blood
WGCNA and DEG analyses were conducted to identify
genes associated with the mRNAsi of tumor tissues and peripheral
blood. First, WGCNA was used to perform the cluster analysis of
genes associated with the mRNAsi of tumor tissues (Fig. 4A). The results indicated that CYBC1,
DEF6, LIME1, CD3D and CD7 are among the genes associated with the
mRNAsi of tumor tissues (Table
SIII). The pathways found to be associated with this mRNAsi
include ‘hematopoietic cell lineage’ and ‘T-cell receptor signaling
pathway’ (Fig. 4B). The stemness
index of the tissues was used to categorise the tumors into high
and low mRNAsi groups, and the DEGs between the two groups were
determined (Table SIV). The
pathways enriched in the upregulated genes are ‘cell cycle’,
‘oocyte meiosis’ and ‘metabolism of xenobiotics by cytochrome P450’
(Fig. 4C). The pathways enriched
with the downregulated genes are ‘cytokine-cytokine receptor
interaction’, ‘TNF signaling pathway’ and ‘PI3K-Akt signaling
pathway’ (Fig. 4D).
The WGCNA results revealed that AEN, BCL7A, BLNK,
CCT3 and CD22 are among the genes associated with the mRNAsi of
peripheral blood (Fig. 4E, Table SV). These genes are enriched in
‘hematopoietic cell lineage’, ‘B-cell receptor signaling pathway’
and ‘NF-kappa B signaling pathway’ (Fig. 4F). PTPRCAP was the only gene found
to be associated with the stemness index in both tissues and
peripheral blood (Fig. S1). The
DEGs associated with the stemness index in the peripheral blood are
presented in Table SVI. The
upregulated genes are enriched in ‘PI3K-Akt signaling pathway’
(Fig. 4G), whereas the
downregulated genes are enriched in ‘PPAR signaling pathway’, ‘ECM
receptor interaction’ and ‘protein digestion and absorption’
(Fig. 4H).
Identification of rare fusion variants
through cfRNA sequencing
Seven rare fusion variants, specifically
IGSF9-SCAMP3, SEC62-ALCAM, CHD1L-ARHGAP26, EML4-ALK, ZFAND3-FKBP5,
RASSF3-RHPN2 and PPOX-KHDC4, were identified by cfRNA sequencing
(Table II). These results
demonstrate that rare fusion variants can be identified using cfRNA
sequencing.
| Table II.Rare fusion variants in tissues
identified by RNA sequencing. |
Table II.
Rare fusion variants in tissues
identified by RNA sequencing.
| Fusion variant | Junction Read
Count | Spanning Frag
Count |
|---|
| IGSF9-SCAMP3 | 37 | 8 |
| SEC62-ALCAM | 27 | 13 |
| CHD1L-ARHGAP26 | 17 | 11 |
| EML4-ALK | 11 | 13 |
| ZFAND3-FKBP5 | 39 | 19 |
| RASSF3-RHPN2 | 68 | 21 |
| PPOX-KHDC4 | 9 | 17 |
Correlation between mRNAsi and immune
cell infiltration
Correlations between immune cell levels and the
mRNAsi in tumor tissues or peripheral blood were analyzed. The
results indicate that M2 macrophages and CD4+ memory T
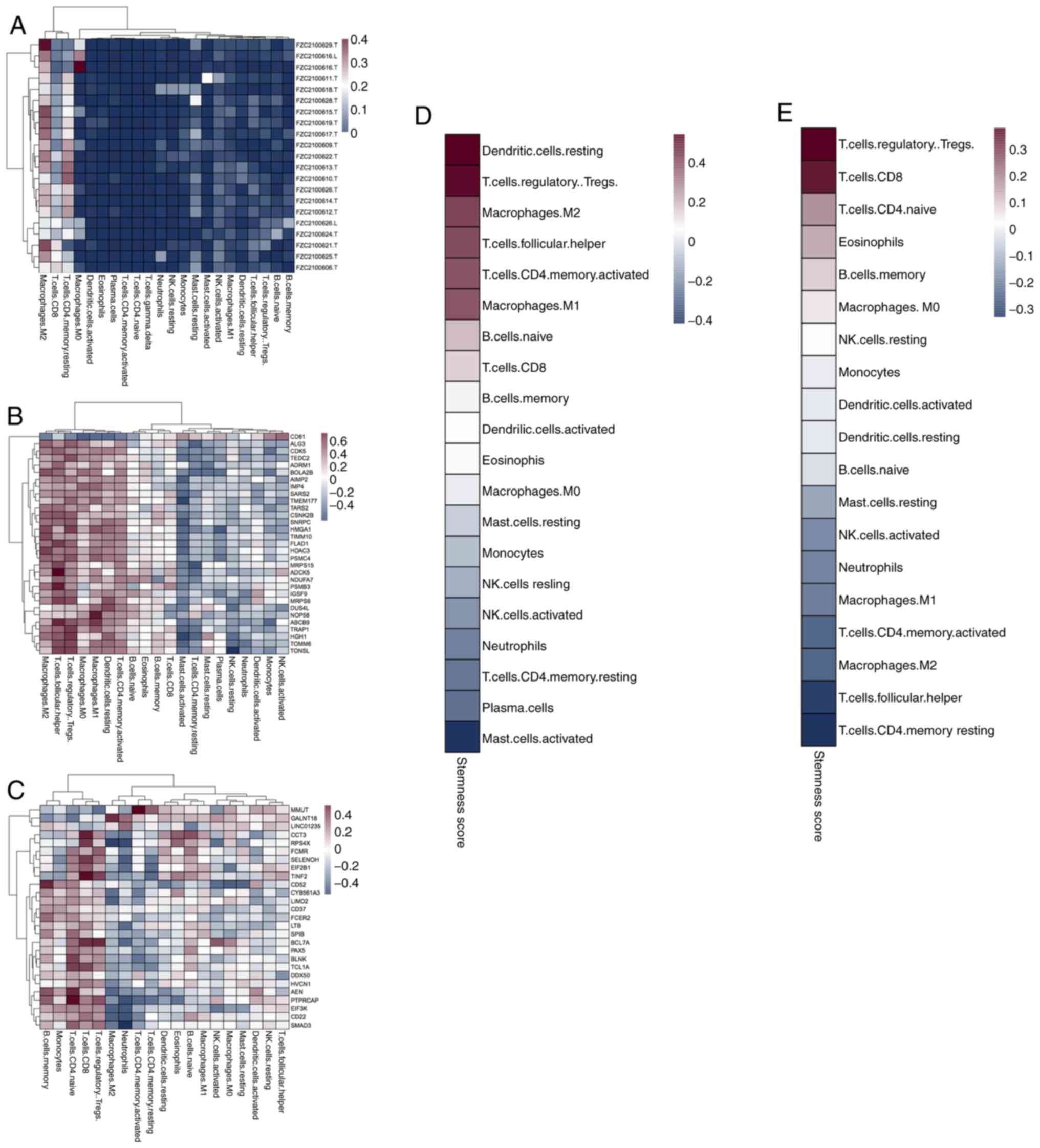
cells are activated in the cancer tissue microenvironment (Fig. 5A). In addition, TMEM177, MPRS15,
PSMB3, NOP58 and TONSL genes correlate with mRNAsi in the immune
microenvironment of the cancer tissues (Fig. 5B). By contrast, the genes that
correlate with cfRNA stemness in immune cells include RPS4X, CD52,
BCL7A, HVCN1, AEN and SMAD3 (Fig.
5C). The mRNAsi values of dendritic cells, regulatory T cells
(Tregs), and M2 macrophages in the tumor tissues are higher than
those in other types of immune cells (Fig. 5D). The cfRNA stemness index values
of Tregs and CD8+ T cells are higher than those of other
types of immune cells (Fig. 5E). In
addition, the cfRNA stemness of dendritic cells and M2 macrophages
is lower than the stemness of the cancer tissue. Finally, the
associations between the degree of stemness and various immune
cells were analyzed. The results revealed that Tregs are closely
associated with tissue and blood stemness (Figs. S2 and S3). Notably, the mRNAsi-associated immune
cells and genes differ between the tumor tissues and peripheral
blood.
Discussion
The treatment and prognosis of lung cancer vary
according to the extent of the disease at diagnosis (26). Early detection and diagnosis
methods, particularly non-invasive approaches, are crucial for
improving the overall survival rate and prognosis of patients with
lung cancer (27). Numerous studies
have focused on the identification of biomarkers for the early
detection of lung cancer based on cfDNA analysis (28–30).
In the present study, cfRNA sequencing was used to explore the
genes, pathways and immune cells associated with lung cancer cell
stemness.
The mRNAsi value of metastatic lymph nodes was found
to be significantly higher than that of tumor tissues. In addition,
a previous study has reported that the mRNAsi value of metastatic
tumors is considerably higher than that of primary tumors,
particularly in prostate and pancreatic cancers (7). Furthermore, a comparison of two-year
recurrence rates in the current study revealed that high tumor and
blood mRNAsi values are associated with a poorer prognosis, which
is consistent with the results of previous studies (31,32).
Studies of cfRNAs in lung cancer have typically focused on miRNAs
or a few known cancer-associated mRNAs (33). Plasma-based cfRNA transcriptome
analysis has been demonstrated to be useful for the early
detection, diagnosis and monitoring of NSCLC (17). In the present study, the stemness
index of cfRNA was higher than that of tumor tissues. Additional
studies are necessary to elucidate the relationship between cfRNAs
and tissue stemness in lung cancer.
WGCNA revealed that the ‘hematopoietic cell lineage’
pathway is enriched in mRNAsi-related genes in lung cancer tissue
and blood. Hematopoietic cell lineages have previously been
indicated to be involved in the proliferation, differentiation and
adhesion of lung cancer cells (34). The present study also revealed that
PTPRCAP is the only gene associated with the stemness index in
peripheral blood as well as in tumor tissues. PTPRCAP is an
immunological marker of triple-negative breast cancer, the
expression of which positively correlates with disease-free
survival (35). Additionally,
PTPRCAP is a natural killer cell-associated gene that is associated
with the response of patients with chronic myeloid leukemia to
tyrosine kinase inhibitor (TKI) treatment (36). However, the role of PTPRCAP in lung
cancer, particularly its effect on lung cancer cell stemness, is
unknown. Therefore, additional studies are necessary to investigate
the mechanism of action of PTPRCAP in lung cancer progression.
Seven rare fusion variants, namely IGSF9-SCAMP3,
SEC62-ALCAM, CHD1L-ARHGAP26, EML4-ALK, ZFAND3-FKBP5, RASSF3-RHPN2
and PPOX-KHDC4, were identified in the present study using cfRNA
sequencing. Anaplastic lymphoma kinase (ALK) fusion has been found
in 3–7% of patients with NSCLC (37). Considerable progress has been
achieved in the treatment of ALK+ NSCLC using TKIs, as
demonstrated by clinical trials (38,39).
EML4-ALK is the most frequently observed fusion variant resulting
from ALK rearrangement in NSCLC (40). For patients with
EML4-ALK+ NSCLC, the National Comprehensive Cancer
Network guidelines recommend crizotinib, ceritinib or alectinib as
first-line therapies (41).
However, to the best of our knowledge, the other fusion variants
have not been reported in lung cancer and require validation.
Immune cells are a crucial part of the tumor
microenvironment (TME) and play a crucial role in tumor
immunotherapy (42). The present
study indicated that M2 macrophages and CD4+ memory T
cells are activated in the microenvironment of lung cancer tissues.
Tumor-associated macrophages (TAMs) are derived from peripheral
blood monocytes that infiltrate solid tumor tissues (43). TAMs acquire an M2-type macrophage
phenotype in response to signals from the TME and contribute to
tumor metastasis (44). In
addition, the CSC-mediated accumulation of M2 macrophages is
reported to contribute to the generation of an immunosuppressive,
pro-tumorigenic TME (42). A high
invasive abundance of CD4+ memory-activated T cells has
been demonstrated to be associated with poor survival in patients
with LUAD (45). Furthermore, in
the present study, high mRNAsi values, indicating the upregulated
expression of stemness-associated genes, were observed in the
dendritic and Treg cells in tissues and in Treg and CD8+
T cells in the blood, which may be attributed to the environments
in which the cells are located. CSCs are able to alter the
composition and functional characteristics of tumor-specific
effector T cells and promote the expansion of immunosuppressive
tumor-promoting Tregs (46). In
addition to regulating the immune environment, CSCs exhibit various
properties that allow them to directly evade the effector
mechanisms of immune cells (47).
Since a high cfRNA stemness index was associated with increased
Treg enrichment, an elevated cfRNA stemness index is indicative of
an immunosuppressive state.
In summary, the present study reveals the unique
stemness gene expression characteristics of blood cfRNAs, offering
novel insights for the development of non-invasive early lung
cancer detection methods. However, the study has some limitations.
First, only 22 lung cancer samples were analyzed. The results of
such a small sample could be biased; therefore, future studies with
more samples or patients from multiple centers are necessary to
support the findings. Second, as the study is based on
bioinformatics analysis, it has certain shortcomings in terms of
experimental validation. Therefore, well-designed basic experiments
and clinical trials are critical for confirmation of the
results.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was funded by the National Natural Science Foundation
of China (grant no. 81773273) and The Special Project of Clinical
Research in Health Field of Shanghai Municipal Health Commission
(grant no. 202240019).
Availability of data and materials
The data generated in the present study may be found
in the CNGB Sequence Archive of China National GeneBank DataBase
(https://db.cngb.org/) under accession number
CNP0005912.
Authors' contributions
RL, BY and YC conceived and designed the study. BY
drafted the manuscript. RL and ZW reviewed and revised the
manuscript. YC and XP performed the experiments. ZW, JL, RW and BL
conducted the data analysis and prepared the figures. RL, BY and BL
confirm the authenticity of all the raw data. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
Shanghai Chest Hospital (Shanghai, China; approval no. KS1740), and
complied with the principles of the Declaration of Helsinki. All
patients who participated in the study signed informed consent
forms.
Patient consent for publication
Not applicable.
Competing interests
ZW, JL, and RW are employees of Berry Oncology
Corporation, and some of the materials used in the study were
provided by Berry Oncology Corporation. BL is an employee of
Liaoning Kanghui Biotechnology Corporation, and assays and analyses
in this study were provided free of charge by Liaoning Kanghui
Biotechnology Corporation. The other authors declare that they have
no competing interests.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
Globocan estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Samarth N, Gulhane P and Singh S:
Immunoregulatory framework and the role of miRNA in the
pathogenesis of NSCLC-A systematic review. Front Oncol.
12:10893202022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shi JF, Wang L, Wu N, Li JL, Hui ZG, Liu
SM, Yang BY, Gao SG, Ren JS, Huang HY, et al: Clinical
characteristics and medical service utilization of lung cancer in
China, 2005–2014: Overall design and results from a multicenter
retrospective epidemiologic survey. Lung Cancer. 128:91–100. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shi X, Liu Y, Cheng S, Hu H, Zhang J, Wei
M, Zhao L and Xin S: Cancer stemness associated with prognosis and
the efficacy of immunotherapy in adrenocortical carcinoma. Front
Oncol. 11:6516222021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bhuvaneswari MS, Priyadharsini S,
Balaganesh N, Theenathayalan R and Hailu TA: Investigating the lung
adenocarcinoma stem cell biomarker expressions using machine
learning approaches. Biomed Res Int. 2022:35181902022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wan R, Liao H, Liu J, Zhou L, Yin Y, Mu T
and Wei J: Development of a 5-gene signature to evaluate lung
adenocarcinoma prognosis based on the features of cancer stem
cells. Biomed Res Int. 2022:44044062022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Su C, Zheng J, Chen S, Tuo J, Su J, Ou X,
Chen S and Wang C: Identification of key genes associated with
cancer stem cell characteristics in Wilms' tumor based on
bioinformatics analysis. Ann Transl Med. 10:12042022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Malta TM, Sokolov A, Gentles AJ,
Burzykowski T, Poisson L, Weinstein JN, Kamińska B, Huelsken J,
Omberg L, Gevaert O, et al: Machine learning identifies stemness
features associated with oncogenic dedifferentiation. Cell.
173:338–354.e15. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liao Y, Xiao H, Cheng M and Fan X:
Bioinformatics analysis reveals biomarkers with cancer stem cell
characteristics in lung squamous cell carcinoma. Front Genet.
11:4272020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hou S, Xu H, Liu S, Yang B, Li L, Zhao H
and Jiang C: Integrated bioinformatics analysis identifies a new
stemness index-related survival model for prognostic prediction in
lung adenocarcinoma. Front Genet. 13:8602682022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li N, Li Y, Zheng P and Zhan X: Cancer
stemness-based prognostic immune-related gene signatures in lung
adenocarcinoma and lung squamous cell carcinoma. Front Endocrinol
(Lausanne). 12:7558052021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li RY and Liang ZY: Circulating tumor DNA
in lung cancer: Real-time monitoring of disease evolution and
treatment response. Chin Med J (Engl). 133:2476–2485. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Heitzer E, Haque IS, Roberts CES and
Speicher MR: Current and future perspectives of liquid biopsies in
genomics-driven oncology. Nat Rev Genet. 20:71–88. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Roskams-Hieter B, Kim HJ, Anur P, Wagner
JT, Callahan R, Spiliotopoulos E, Kirschbaum CW, Civitci F,
Spellman PT, Thompson RF, et al: Plasma cell-free RNA profiling
distinguishes cancers from pre-malignant conditions in solid and
hematologic malignancies. NPJ Precis Oncol. 6:282022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Larson MH, Pan W, Kim HJ, Mauntz RE,
Stuart SM, Pimentel M, Zhou Y, Knudsgaard P, Demas V, Aravanis AM
and Jamshidi A: A comprehensive characterization of the cell-free
transcriptome reveals tissue- and subtype-specific biomarkers for
cancer detection. Nat Commun. 12:23572021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sorber L, Zwaenepoel K, Jacobs J, De Winne
K, Goethals S, Reclusa P, Van Casteren K, Augustus E, Lardon F,
Roeyen G, et al: Circulating cell-free DNA and RNA analysis as
liquid biopsy: Optimal centrifugation protocol. Cancers (Basel).
11:4582019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Seneviratne C, Shetty AC, Geng X,
McCracken C, Cornell J, Mullins K, Jiang F and Stass S: A pilot
analysis of circulating cfRNA transcripts for the detection of lung
cancer. Diagnostics (Basel). 12:28972022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Goldstraw P, Chansky K, Crowley J,
Rami-Porta R, Asamura H, Eberhardt WE, Nicholson AG, Groome P,
Mitchell A, Bolejack V, et al: The IASLC lung cancer staging
project: Proposals for revision of the TNM stage groupings in the
forthcoming (Eighth) edition of the TNM classification for lung
cancer. J Thorac Oncol. 11:39–51. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jacobsen N, Nielsen PS, Jeffares DC,
Eriksen J, Ohlsson H, Arctander P and Kauppinen S: Direct isolation
of poly(A)+ RNA from 4 M guanidine thiocyanate-lysed cell extracts
using locked nucleic acid-oligo(T) capture. Nucleic Acids Res.
32:e642004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Colaprico A, Silva TC, Olsen C, Garofano
L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM,
Castiglioni I, et al: TCGAbiolinks: An R/Bioconductor package for
integrative analysis of TCGA data. Nucleic Acids Res. 44:e712016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen D, Liu J, Zang L, Xiao T, Zhang X, Li
Z, Zhu H, Gao W and Yu X: Integrated machine learning and
bioinformatic analyses constructed a novel stemness-related
classifier to predict prognosis and immunotherapy responses for
hepatocellular carcinoma patients. Int J Biol Sci. 18:360–373.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: A Bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang X, Wang F, Yan F, Huang D, Wang H,
Gao B, Gao Y, Hou Z, Lou J, Li W and Yan J: Identification of a
novel HOOK3-FGFR1 fusion gene involved in activation of the
NF-kappaB pathway. Cancer Cell Int. 22:402022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Trapnell C, Roberts A, Goff L, Pertea G,
Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL and Pachter L:
Differential gene and transcript expression analysis of RNA-seq
experiments with TopHat and Cufflinks. Nat Protoc. 7:562–578. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao Y, Jia S, Zhang K and Zhang L: Serum
cytokine levels and other associated factors as possible
immunotherapeutic targets and prognostic indicators for lung
cancer. Front Oncol. 13:10646162023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shai S, Patolsky F, Drori H, Scheinman EJ,
Davidovits E, Davidovits G, Tirman S, Arber N, Katz A and Adir Y: A
novel, accurate, and non-invasive liquid biopsy test to measure
cellular immune responses as a tool to diagnose early-stage lung
cancer: A clinical trials study. Respir Res. 24:522023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mathios D, Johansen JS, Cristiano S,
Medina JE, Phallen J, Larsen KR, Bruhm DC, Niknafs N, Ferreira L,
Adleff V, et al: Detection and characterization of lung cancer
using cell-free DNA fragmentomes. Nat Commun. 12:50602021.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sugimoto A, Matsumoto S, Udagawa H,
Itotani R, Usui Y, Umemura S, Nishino K, Nakachi I, Kuyama S, Daga
H, et al: A large-scale prospective concordance study of plasma-
and tissue-based next-generation targeted sequencing for advanced
non-small cell lung cancer (LC-SCRUM-Liquid). Clin Cancer Res.
29:1506–1514. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang S, Meng F, Li M, Bao H, Chen X, Zhu
M, Liu R, Xu X, Yang S, Wu X, et al: Multidimensional cell-free DNA
fragmentomic assay for detection of early-stage lung cancer. Am J
Respir Crit Care Med. 207:1203–1213. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen M, Wang X, Wang W, Gui X and Li Z:
Immune- and stemness-related genes revealed by comprehensive
analysis and validation for cancer immunity and prognosis and its
nomogram in lung adenocarcinoma. Front Immunol. 13:8290572022.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang H, Wang Y, Luo W, Zhang X, Cao R,
Yang Z, Duan J and Wang K: Integrative stemness characteristics
associated with prognosis and the immune microenvironment in lung
adenocarcinoma. BMC Pulm Med. 22:4632022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Müller S, Janke F, Dietz S and Sültmann H:
Circulating MicroRNAs as potential biomarkers for lung cancer.
Recent Results Cancer Res. 215:299–318. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Narayanan S, Wu ZX, Wang JQ, Ma H,
Acharekar N, Koya J, Yoganathan S, Fang S, Chen ZS and Pan Y: The
spleen tyrosine kinase inhibitor, entospletinib (GS-9973) restores
chemosensitivity in lung cancer cells by modulating ABCG2-mediated
multidrug resistance. Int J Biol Sci. 17:2652–2665. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Marchetti P, Antonov A, Anemona L,
Vangapandou C, Montanaro M, Botticelli A, Mauriello A, Melino G and
Catani MV: New immunological potential markers for triple negative
breast cancer: IL18R1, CD53, TRIM, Jaw1, LTB, PTPRCAP. Discov
Oncol. 12:62021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Park H, Kang S, Kim I, Kim S, Kim HJ, Shin
DY, Kim DY, Lee KH, Ahn JS, Sohn SK, et al: The association of
genetic alterations with response rate in newly diagnosed chronic
myeloid leukemia patients. Leuk Res. 114:1067912022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wu R, Liu S, Lv G, Deng C, Wang R, Zhang
S, Zhu D, Wang L, Lei Y and Luo Z: First-line crizotinib therapy is
effective for a novel SEC31A-anaplastic lymphoma kinase fusion in a
patient with stage IV lung adenocarcinoma: A case report and
literature reviews. Anticancer Drugs. 34:294–301. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zou Z, Gu Y, Liang L, Hao X, Fan C, Xin T,
Zhao S, Liu Z, Guo Y, Ma K, et al: Alectinib as first-line
treatment for advanced ALK-positive non-small cell lung cancer in
the real-world setting: Preliminary analysis in a Chinese cohort.
Transl Lung Cancer Res. 11:2495–2506. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shaw AT, Solomon BJ, Chiari R, Riely GJ,
Besse B, Soo RA, Kao S, Lin CC, Bauer TM, Clancy JS, et al:
Lorlatinib in advanced ROS1-positive non-small-cell lung cancer: A
multicentre, open-label, single-arm, phase 1–2 trial. Lancet Oncol.
20:1691–1701. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Guo W, Liang J, Zhang D, Huang X and Lv Y:
Lung adenocarcinoma harboring complex EML4-ALK fusion and BRAF
V600E co-mutation responded to alectinib. Medicine (Baltimore).
101:e309132022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ettinger DS, Wood DE, Aisner DL, Akerley
W, Bauman JR, Bharat A, Bruno DS, Chang JY, Chirieac LR, D'Amico
TA, et al: NCCN guidelines insights: Non-small cell lung cancer,
version 2.2021. J Natl Compr Canc Netw. 19:254–266. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Steven A, Fisher SA and Robinson BW:
Immunotherapy for lung cancer. Respirology. 21:821–833. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu P, Liu Y, Chen L, Fan Z, Luo Y and Cui
Y: Anemoside A3 inhibits macrophage M2-like polarization to prevent
triple-negative breast cancer metastasis. Molecules. 28:16112023.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Najafi M, Goradel NH, Farhood B, Salehi E,
Nashtaei MS, Khanlarkhani N, Khezri Z, Majidpoor J, Abouzaripour M,
Habibi M, et al: Macrophage polarity in cancer: A review. J Cell
Biochem. 120:2756–2765. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen H, Lin R, Lin W, Chen Q, Ye D, Li J,
Feng J, Cheng W, Zhang M and Qi Y: An immune gene signature to
predict prognosis and immunotherapeutic response in lung
adenocarcinoma. Sci Rep. 12:82302022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Fridman WH, Zitvogel L, Sautès-Fridman C
and Kroemer G: The immune contexture in cancer prognosis and
treatment. Nat Rev Clin Oncol. 14:717–734. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Müller L, Tunger A, Plesca I, Wehner R,
Temme A, Westphal D, Meier F, Bachmann M and Schmitz M:
Bidirectional crosstalk between cancer stem cells and immune cell
subsets. Front Immunol. 11:1402020. View Article : Google Scholar : PubMed/NCBI
|