Introduction
Chronic lymphocytic leukemia (CLL) is
well-characterized biologically lymphoid malignancy with a
remarkably heterogeneous clinical course, which is reflected in
varied survival times, response to treatment, and the dynamics of
disease progression. To date, much evidence has emerged reporting
several novel alterations that elucidate genomic, epigenetic,
immunogenetic, and tumor microenvironmental mechanisms, which might
drive the evolution of the disease (1–6).
Although these advances have vastly expanded the knowledge of CLL
pathogenesis, the link between the considerable molecular
heterogeneity of this malignancy and the clinical outcome of
patients remains elusive.
For the last years it has been speculated that
immunologic and inflammatory factors, including antigen
stimulation, could be involved in the processes determining the
development and progression of CLL (7). The prevalence of CLL increases
noticeably with age, implying that a persistent exposure to a self
and foreign antigen might be considered as a predisposing factor.
Since CLL patients present progressive immunodeficiency, recurrent
infections are a common clinical feature of this disease (8). Early studies suggested that gut
microbiota plays a key role in defining the B cell receptor (BCR)
repertoire (9), thus stimulation of
alloantigens derived from distinct microbial species might be
involved in the development and proliferation of CLL-specific B
cell clones, and thereby might potentially stand behind the
interindividual variability of clinical outcomes. Furthermore,
certain types of bacteria release factors that might indirectly
contribute to neoplasia by maintaining a proinflammatory
environment (10).
The crosstalk between the microbiome and immune
system takes place at numerous sites, including the skin and
mucosal surfaces. Commensal species within the gastrointestinal
mucosa have been shown to contribute to innate as well as adaptive
immunity at multiple levels (11).
Our previous report documented an accumulation of CD5+CD19+ cells
in tonsillar tissue during chronic antigenic stimulation
accompanying chronic or recurrent tonsillitis in children (12). Recently, low intestinal microbial
diversity and increased abundance of specific bacterial community
members have both been reported to be implicated in the induction
of gene mutations and host immune response (13).
In the last years, gut microbiota has been proven to
modulate the activity of the immune system by creating an imbalance
between cell proliferation and apoptosis (14). To date, significant alterations in
microbiota diversity and composition have been documented in many
cancers, including hematological malignancies, such as acute
lymphoblastic leukemia (ALL) (15,16),
acute myeloid leukemia (AML) (17,18),
and CLL (19). Notably, compelling
evidence shows that host microbiota not only influence cellular
homeostasis or tumor susceptibility, but also is implicated in
disease prognosis and modulation of the efficacy and toxicity of
different anti-tumor therapeutic approaches, including
chemotherapy, radiotherapy, and immunotherapy (20–23).
Therefore, the effects of structural imbalance of host microbiota
might contribute to the interindividual variability in treatment
response as for immunocompromised patients.
Since the activation of cellular proinflammatory
signaling pathways driven by somatic mutations and an increased
release of proinflammatory cytokines is associated with CLL
development, it seems relevant to investigate the role of chronic
inflammation in the pathogenesis of this disease. Notably,
analyzing both oral cavity and gut microbiota community structure
in CLL patients will allow identifying the microbiota profile
related to this proinflammatory environment and define CLL-specific
bacterial strains that might recognize microbial patterns that
distinguish those patients who do not require treatment and could
serve as biomarkers for predicting disease progression and
treatment initiation.
Materials and methods
Study design and participants
The study was approved by the local Ethics Committee
of the Medical University of Lublin (KE-0254/7/2019), and written
informed consent forms were obtained from all participants,
including CLL patients and healthy volunteers (HVs). The study was
performed in accordance with the principles of the Declaration of
Helsinki.
Throat swabs, stool, and peripheral blood samples
from 81 newly diagnosed and untreated CLL patients were collected.
The clinical characteristics of CLL individuals are presented in
Table I. As controls, oral and
fecal samples from 21 HVs [12 females and 9 males at a median age
of 57 years (range 50–84)] were used. The exclusion criteria
included antibiotics therapy within four weeks and a history of
diarrhea and/or vomiting within 72 h. For HVs, exclusion criteria
also included cancer, diabetes, gastrointestinal diseases, and
other conditions that could be affecting the microbiome. There were
no significant differences in body mass index (BMI) value between
CLL patients and HVs (median 27.34, range 20.02–48.46 vs. median
27.30, range 20.07–32.00, P=0.39).
 | Table I.Clinical characteristics of patients
with chronic lymphocytic leukemia. |
Table I.
Clinical characteristics of patients
with chronic lymphocytic leukemia.
| Characteristic | No. of
patients |
|---|
| Age (median,
range) | 65 (33–85) |
| Sex, n (%) |
|
|
Female | 32 (40%) |
|
Male | 49 (60%) |
| Binet stage, n
(%) |
|
| A | 30 (37%) |
| B | 23 (28%) |
| C | 25 (31%) |
| Not
available | 3 (4%) |
| CD38 (cut-off 30%),
n (%) |
|
|
Positive | 35 (43%) |
|
Negative | 43 (53%) |
| Not
available | 3 (4%) |
| ZAP-70 (cut-off
20%), n (%) |
|
|
Positive | 18 (22%) |
|
Negative | 60 (74%) |
| Not
available | 3 (4%) |
| IGHV mutation
status, n (%) |
|
|
Mutated | 38 (47%) |
|
Unmutated | 32 (40%) |
| Not
available | 11 (13%) |
| BCR immunoglobulin
stereotypy, n (%) |
|
|
Stereotyped subsets (major and
minor) | 25 (47%) |
| High
risk stereotyped subsets (#2, #5, #8b) | 5 (7%) |
| Not
available | 11 (14%) |
| TP53
mutation status, n (%) |
|
|
Mutated | 5 (6%) |
|
Wild-type | 74 (92%) |
| Not
available | 2 (2%) |
| Cytogenetics, n
(%) |
|
|
del11q | 19 (24%) |
|
del13q | 48 (60%) |
|
isolated del13q | 30 (37%) |
|
del17p | 6 (7%) |
|
tri12 | 9 (11%) |
|
del6q | 3 (4%) |
| Not
available | 1 (1%) |
Peripheral blood mononuclear cells
isolation
Peripheral blood mononuclear cells (PBMCs) were
isolated from whole blood using density gradient centrifugation on
Biocoll (Biochrom, Germany). They were then cryopreserved in RPMI
1640 medium (Biochrom, Germany) supplemented with 20% fetal bovine
serum (Biochrom, Germany) and 10% dimethyl sulfoxide (Sigma
Aldrich, Germany) and stored at −80°C until further analyses were
performed.
CD38 and ZAP-70 expression
analysis
The expression of CD38 and ZAP-70 on CLL cells was
assessed by flow cytometry after incubation with monoclonal mouse
antihuman antibodies: anti-CD5 PE-Cy5, anti-CD19 FITC, anti-CD38
PE, and anti-ZAP-70 PE (all BD Biosciences, San Jose, CA, USA).
Cells were analyzed by FACSuite (BD Biosciences, San Jose, CA, USA)
on BD FACS Lyric (BD Biosciences, San Jose, CA, USA). Results were
compared to negative control cells without antibodies, and FMO
(fluorescence minus one) control in the absence of anti-CD38
PE/anti-ZAP-70 PE monoclonal antibodies. Cut-off points to define
CD38+ CLL and ZAP-70+ CLL patients' populations were 30 and 20%,
respectively. The gating strategy and representative CD38-positive
and ZAP-70-positive samples have been presented in Fig. S1.
DNA isolation
The QIAamp DNA Bood Mini Kit (Qiagen, Hilden,
Germany) for DNA isolation from PBMCs was used according to the
manufacturer's instructions. DNA quality and quantity were
determined through 260/280 nm absorbance measures using the
BioSpec-Nano spectrophotometer (Shimadzu, Kyoto, Japan).
IGHV and TP53 mutation status
assessment
The TP53 mutation status was determined by
PCR amplification of exons 4 to 10 followed by bidirectional Sanger
sequencing. The obtained sequences were analyzed using GLASS
software (24) according to the
ERIC guidelines (25). For IGHV
somatic hypermutation status determination, the
IGHV–IGHD-IGHJ gene rearrangement was amplified using
framework region (FR1) primers following BIOMED-2 protocol
(26). Then, heteroduplex analysis
and bidirectional Sanger sequencing were performed. IMGT/V-Quest
software (27,28) was used to analyze the obtained
sequence following ERIC guidelines (29). A 98% germline homology cut-off was
used to determine IGHV mutational status. The sequences with a
germline homology of 98% or higher were considered unmutated, and
those with a homology <98% were considered mutated. A subset
analysis was performed using ARRest/AssignSubsets software
(30).
Cytogenetic aberrations
Cytogenetic aberrations (del17p, del11q, del13q,
tri12, del6q) were assessed by fluorescence in situ hybridization
(FISH) in the diagnostic laboratory according to the routine
procedures.
Oral and fecal sample collection,
storage, and preparation for microbiome profiling
Throat swabs were collected and stabilized using
OMNIgene•ORAL kit (DNA Genotek Inc, Canada). The OMNIgene•GUT kit
(DNA Genotek Inc, Canada) was used to self-collect fecal samples by
study participants. Both oral and stool samples were stored until
shipment to the laboratory according to the manufacturer's
recommendations. DNA extraction, amplicon libraries preparation,
and 16S rRNA gene sequencing were performed by Eurofins Genomics
Europe Sequencing GmbH (Ebersberg, Germany). For target-specific
PCR amplification of V3-V5 hypervariable regions of the 16S rRNA
gene, the primers V3F (5′-CCTACGGGNGGCWGCAG-3′) and V5R
(5′-CCGYCAATTYMTTTRAGTTT-3′) were used. Amplicon libraries covering
the specified regions were sequenced on the high-throughput
Illumina MiSeq platform (Illumina).
Microbiome profiling and
statistics
Following the quality check with the use of fastqc
(31), the dataset was normalized
by a subsampling-based strategy using seqtk (32). Reads across all samples were
randomly down-sampled to the lowest read count in the cohort.
Low-quality ends of the reads were trimmed and filtered using a
value of 3 for the maximal error rate parameter. Next, the paired
reads were merged. The taxonomic classification for microbiome
analysis was determined using the SILVA reference database version
138.1 (33). All the above steps
(including reads trimming, filtering, merging, and taxonomic
assignment) were performed using the dada2 R package (34). The microbial phylogenetic tree was
reconstructed from the obtained sequences using IQ-TREE maximum
likelihood phylogeny stochastic algorithm (35).
For data exploration and visualization, including
alpha and beta diversity metrics, the set of R packages: phyloseq
(36), microbiome (37), microbiomeutilities (38), microbial (39), and microViz (40) was used. Faith's Phylogenetic
Diversity (PD) was estimated using picante (41). For comparing microbial communities
divided into different sample groups, the Unifrac algorithm
(42) and the Bray-Curtis
dissimilarity approach (43) were
used, for which also non-metric multidimensional scaling (NMDS)
plots were generated. Differences in beta diversity were assessed
using Permutational multivariate analysis of variance (PERMANOVA)
implemented into the vegan R package (44). Linear discriminant analysis (LDA)
was taken advantage of to compare the relative abundance of the
different taxa between sub-groups (45). The log_FB metric was defined for
each sample as the log of Firmicutes to Bacteroidota
relative abundance ratio. The survival package (46) was used to perform the Cox
Proportional Hazards Regression models for the assessment of the HR
and 95% CI to test the association of selected factors with time to
first treatment (TTFT). Cutpoints for continuous variable metrics
were evaluated by maximally selected rank statistics with the use
of the maxstat R package (47)
implemented by the survminer R tool (48). Testing group differences included a
two-tailed Wilcoxon test. Survival probabilities were estimated
with the use of Kaplan-Meier method and compared using the
long-rank test. P-value <0.05 was considered statistically
significant. Statistical analyses were performed using R software
version 4.1.3 (49).
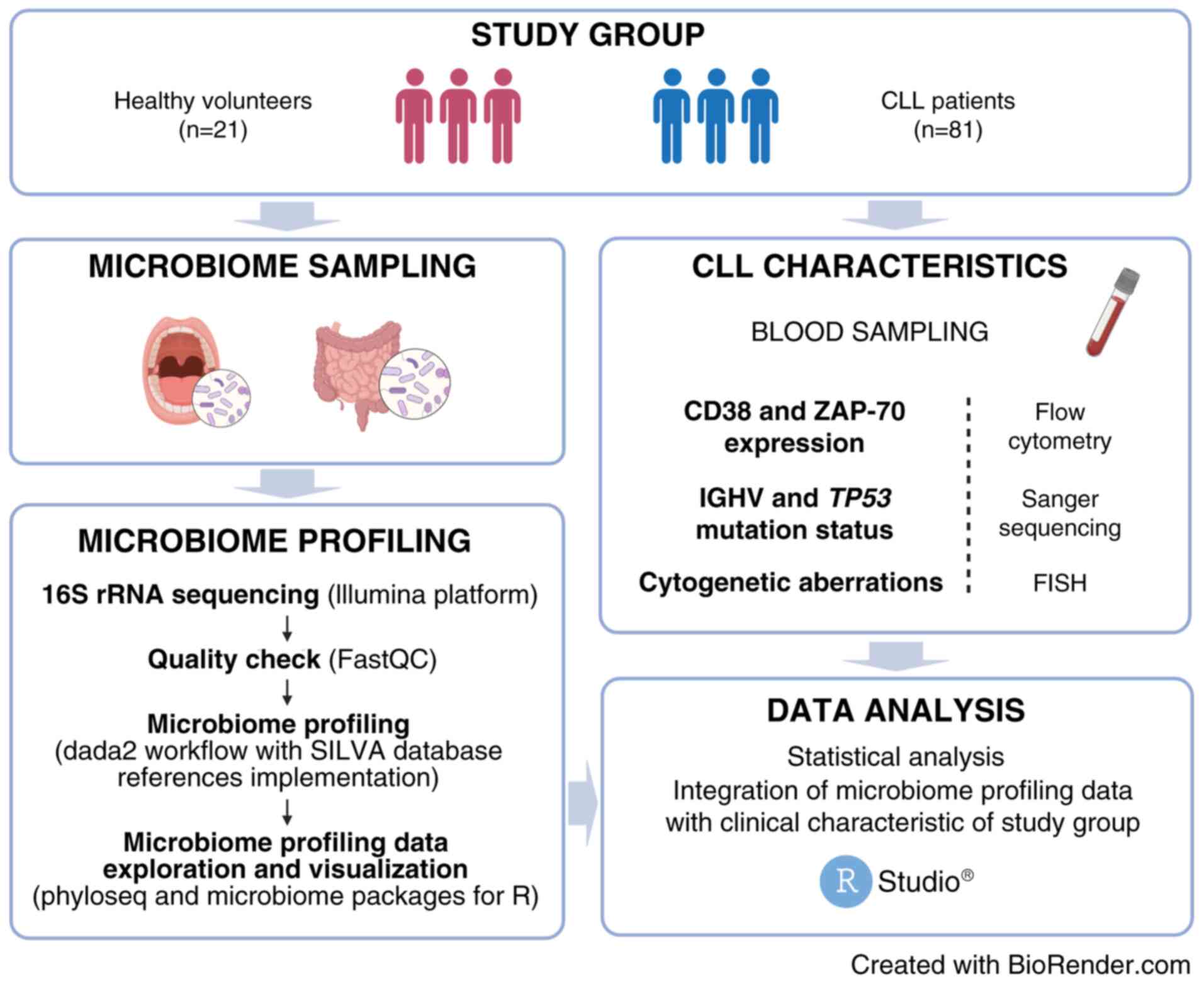
A flowchart illustrating workflow for oral and gut
microbiome analysis in CLL patients and HVs in our study has been
presented in Fig. 1.
Results
Microbiota structure in patients with
CLL and HVs
The microbiota composition of 69 oral and 75 fecal
samples from 81 CLL patients and 17 oral and 21 stool samples from
21 HVs were all analyzed. The optimized sequences were obtained
through data quality control and read preprocessing, and a total of
17.8 k operational taxonomic units (OTUs) were annotated. Among
these identified OTUs, unique annotations were used for further
analysis and classified into 23 phyla, 46 classes, 103 orders, 211
families, 585 genera, and 957 species.
Alpha-diversity and beta-diversity
analysis
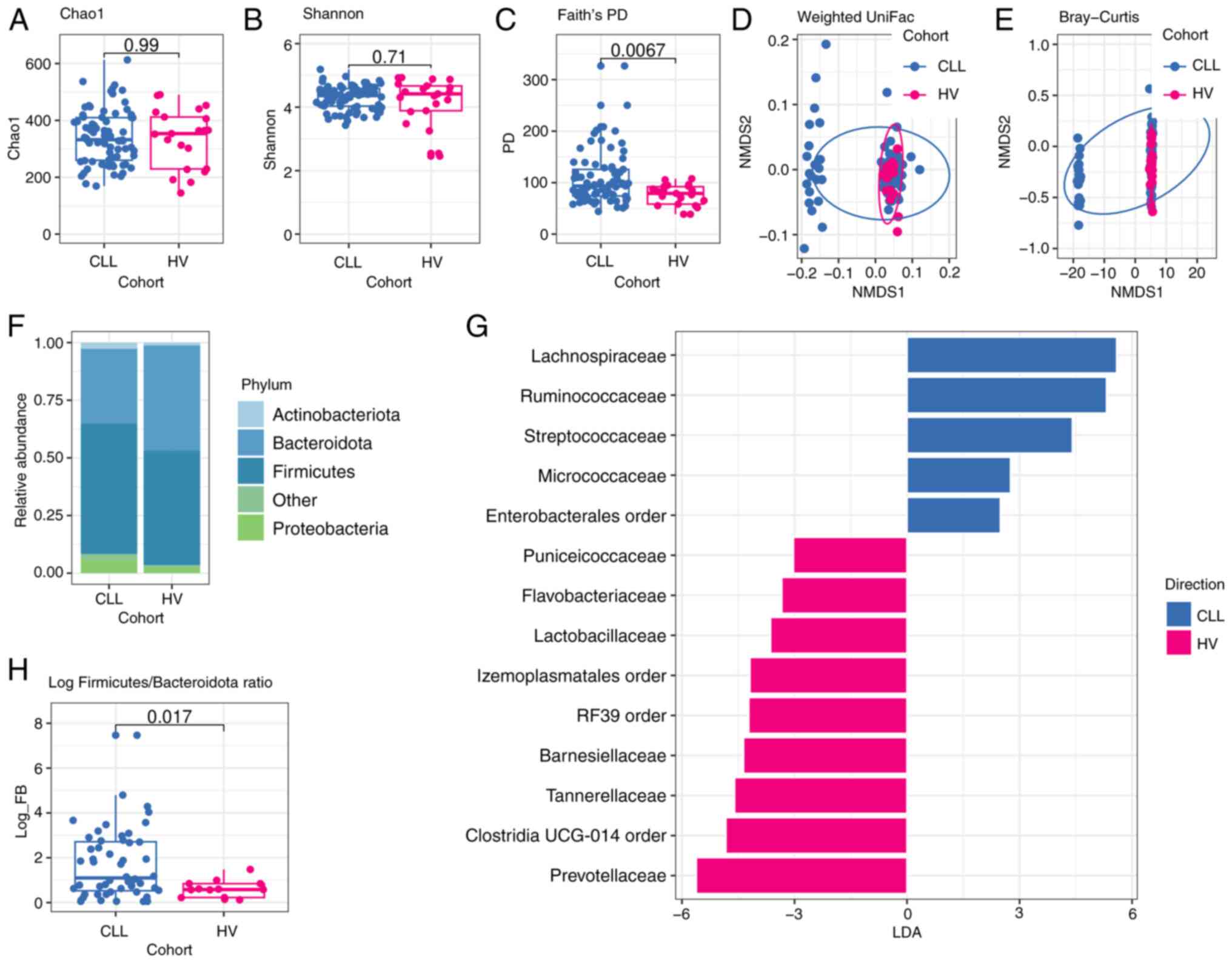
The microbiota diversity within a single sample is
reflected by alpha-diversity, specifically Chao1 and Shannon
indexes. These non-phylogenetic metrics revealed that CLL oral
samples are characterized by a lower richness and evenness than
matched control (Chao1 index median 173.0 vs. median 209.5,
P=0.027; Shannon index median 3.62 vs. median 3.85, P=0.055).
According to Faith's phylogenetic diversity (PD) metric, which is
based on the phylogenetic relationships of microbial taxa, there
were no significant differences in the diversity of oral microbiome
between CLL patients and HVs (median 91.41 vs. median 87.28,
P=0.17), (Fig. 2A-C).
Furthermore, no significant differences in species
richness and evenness between CLL and HVs stool samples (Chao1
index median 332.06 vs. median 353.50 P=0.99, Shannon index median
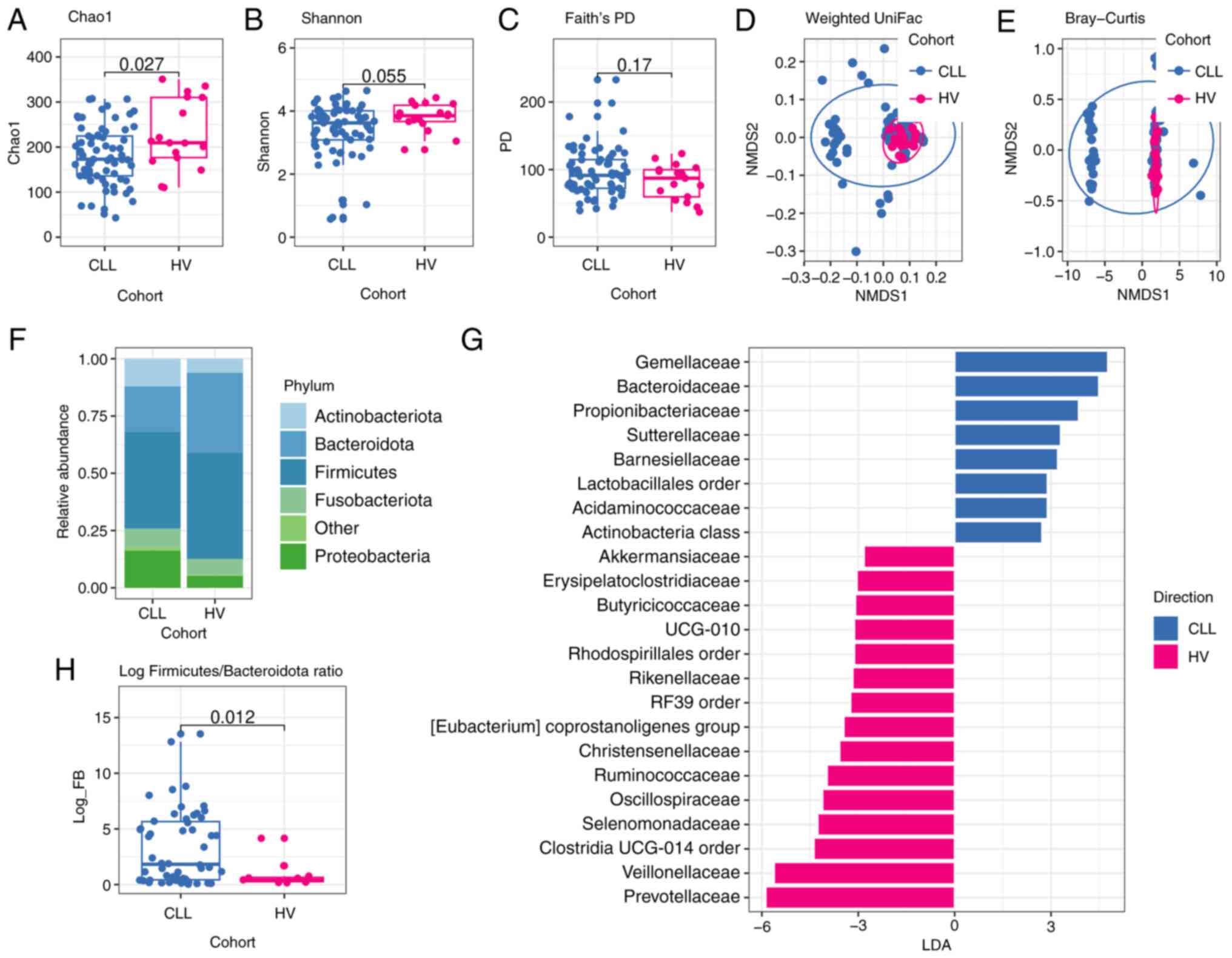
4.36 vs. median 4.42 P=0.71) were found. However, Faith's
phylogenetic diversity (PD) metric revealed that the gut microbial
community of CLL patients is more evolutionarily distinct in
comparison to HVs (median 93.42 vs. median 79.09 P=0.0067)
(Fig. 3A-C).
 | Figure 3.Comparison of gut microbiota of
patients with CLL and HVs. (A) Microbial richness index of Chao1;
(B) Microbial diversity index of Shannon; (C) Microbial diversity
index of Faith's PD. P-values shown in Fig. A-C were calculated by
a two-sided Wilcoxon rank-sum test without adjustment of multiple
comparisons for CLL (n=75) vs. HVs (n=21). NMDS analysis based on
(D) weighted UniFrac distance (R2=0.038; P=0.023) and
(E) Bray-Curtis dissimilarity (R2=0.037; P=0.009).
P-values corresponding to D and E figures were analyzed using the
PERMANOVA test (as implemented by the vegan R package), whereas
dots represent samples. (F) Phylogenetic composition of stool
samples at the phylum level; phyla with a relative abundance
<0.1% in each sample are merged into ‘Other’; (G) LEfSe analysis
indicates enriched bacterial families associated either with CLL
(blue, n=75) or HVs (magenta, n=21). The length of the bar column
represents the LDA score. The logarithmic LDA scores threshold was
2.0, P<0.05 (a two-sided Wilcoxon rank-sum test without
adjustment of multiple comparisons was used for P-value
calculation). (H) Log Firmicutes/Bacteroidota (log FB)
ratio. The P-value was calculated by a two-sided Wilcoxon rank-sum
test without adjustment of multiple comparisons for CLL (n=75) vs.
HVs (n=21). CLL, chronic lymphocytic leukemia; HVs, healthy
volunteers; PD, phylogenetic diversity; NMDS, non-metric
multidimensional scaling; LDA, Linear discriminant analysis; LEfSe,
LDA effect size. |
Next, NMDS was performed for beta-diversity analysis
of oral and gut microbial community structure. The result indicated
that the structure of the oral microbiome in CLL patients was
significantly different from that of HVs group based on Bray-Curtis
dissimilarity (R2=0.081, P=0.002) and on unweighted
uniFrac distance (R2=0.06, P=0.003). Moreover, the
differences were significant based on weighted uniFrac distance
(R2=0.093, P=0.002) (Fig.
2D, E). For beta-diversity of the gut microbiome as determined
by Bray-Curtis dissimilarity and UniFrac distances, significant
differences between CLL and HVs were found, P=0.009 for Bray-Curtis
(R2=0.037), P=0.001 for unweighted uniFrac
(R2=0.061), and P=0.023 for weighted uniFrac
(R2=0.038) (Fig. 3D and
E).
A significant change in the
composition and abundance of oral and gut microbiome in CLL
patients
The representative sequences of OTUs were compared
with the SILVA microbial reference database as to obtain
information on the species classification corresponding to each
OTU. CLL patients differ from HVs in the observed community
structure. The predominant phylum among CLL oral microbiome was
Firmicutes (42.25%), followed by Bacteroidota
(19.89%), Proteobacteria (16.22%), Actinobacteriota
(12.05%), and Fusobacteriota (8.35%) (Figs. 2F, S2A), while almost 90% of the CLL
fecal-derived bacteria were classified into two dominant phyla:
Firmicutes (56.62%) and Bacteroidota (32.43%),
followed by Proteobacteria (6.01%) and Actinobacteria
(2.71%) (Figs. 3F, S2C).
The structure of the oral microbiota
in CLL patients and HVs
Interestingly, remarkable differences in the
relative abundances of specific bacterial phyla in both oral and
intestinal microbiome between CLL patients and HVs were observed.
The Proteobacteria, a common feature of dysbiosis, was
significantly more abundant in CLL oral samples in comparison to
HVs oral samples (P=0.022), whereas the abundance of
Bacteroidota was significantly lower in CLL oral samples
compared to HVs oral samples (P=0.0015) (Table SI). Furthermore, a significant
difference in the value of log Firmicutes and
Bacteroidota (log F/B) ratio was found between CLL patients
and HVs (0.81 vs. 0.28, P=0.012) (Fig.
2H).
Linear discriminant analysis (LDA) effect size
(LEfSe) analysis revealed significant bacterial differences in oral
microbiota between the CLL patients and HVs. In particular, at the
family level, a significantly higher abundance of Gemellaceae,
Bacteroidaceae, Propionibacteriaceae, and
Sutterellaceae, as well as depletion of Prevotellaceae,
Veillonellaceae, Oscillospiraceae, and Ruminococcaceae,
were observed among oral CLL samples in comparison to HVs (Fig. 2G). As Prevotellaceae,
Veillonellaceae, and Ruminococcaceae have been reported
to produce short-chain fatty acids (SCFA) involved in
immunomodulation, the differences in the relative abundance of
genera belonging to these taxa in the oral microbiome between CLL
and HVs were analyzed. CLL oral samples demonstrated a
significantly lower abundance of Prevotella and
Veilonella genera (P=0.0011 and P=0.0016, respectively) in
comparison to HVs. Additionally, a tendency to higher relative
abundance of Rothia (Micrococcaceae family) and
Fusobacteria (Fusobacteriaceae family) in CLL oral samples
in comparison to HVs oral samples (P=0.067 and P=0.097,
respectively) was ascertained.
The structure of the gut microbiota in
CLL patients and HVs
Similarly to oral samples, fecal samples from CLL
patients exhibited an increased abundance of Proteobacteria
and a decreased abundance of Bacteroidota in comparison to
fecal samples collected from HVs (P=0.045 and P=0.026,
respectively) (Table SII).
Consequently, the value of the log F/B ratio was significantly
higher in fecal samples from CLL compared to HVs (0.62 vs. 0.22,
P=0.017) (Fig. 3H).
LEfSe analysis of gut microbiota indicated
significant differences in the abundance of SCFA producers between
CLL patients and HVs. CLL fecal samples exhibited an enrichment of
Lachnospiraceae, Ruminococcaceae, and
Streptococcaceae families, while HVs fecal samples were
enriched in Prevotellaceae, Tannerellaceae and
Barnesiellaceae families (Fig.
3G). At the genus level, CLL fecal samples showed a
significantly higher abundance of Roseburia
(Lachnospiraceae family) in comparison to HVs (P=0.011).
A significant change in the
composition and abundance of oral and gut microbiome in CLL
patients with respect to the selected prognostic and predictive
features
Of note, specific alterations in the oral and
intestinal microbiome of CLL patients with different status of
selected prognostic features, such as Binet stage, mutation status
of TP53 and IGHV, the presence of cytogenetic aberrations,
and expression levels of CD38 and ZAP-70, were found (Tables SIII and SIV).
Microbial diversity in oral microbiota
in CLL patients with respect to the selected prognostic and
predictive features
Oral samples from CLL patients with Binet stage A
showed a lower relative abundance of Actinobacteriota and
Fusobacteriota and a tendency to present a higher relative
abundance of Bacteroidetes compared to CLL patients with
Binet stage B (P=0.041, P=0.047, P=0.06 respectively). At the
family level, oral samples from CLL patients with Binet stage A
were more abundant in Prevotellaceae compared to CLL
patients with Binet stage B (P=0.022) and C (P=0.07), and less
abundant in Lachnospiraceae and Fusobacteriaceae in
comparison to oral samples from CLL patients with Binet stage B
(P=0.022, P=0.035). Moreover, Actinobacteriota showed a
tendency to a higher abundance in oral microbiota in CD38+ CLL
patients in comparison to CD38-CLL patients (P=0.061). CLL patients
with unmutated IGHV showed a tendency to the decreased relative
abundance of Bacteroidetes phylum and Prevotellaceae
(Bacteroidetes phylum), as well as Veillonellaceae
(Firmicutes phylum) families in oral samples compared to CLL
patients with mutated IGHV (P=0.077, P=0.087, and P=0.053,
respectively). Interestingly, CLL patients with stereotyped subsets
exhibited enrichment in Proteobacteria in comparison to
non-stereotyped CLL patients (P=0.016).
However, there were no significant alterations in
oral microbiome composition of CLL patients with distinct
TP53 mutation status or the presence of del13q and del17p.
Nevertheless, an increased relative abundance of
Proteobacteria and a tendency to the higher relative
abundance of Fusobacteriota was found in oral samples from
CLL patients with del11q compared to samples from patients with no
del11q (P=0.019 and P=0.071, respectively). Moreover, CLL patients
with tri12 exhibited an increased abundance of
Actinobacteriota and Firmicutes (P=0.048 and P=0.028,
respectively) phyla and a tendency to lower abundance of
Fusobacteriaceae family (P=0.088) as related to patients
with no tri12.
Microbial diversity in gut microbiota
in CLL patients with respect to the selected prognostic and
predictive features
Stool samples from patients with Binet stage A
exhibited an increased abundance of Bacteroidetes and a
decreased abundance of Firmicutes in comparison to stool
samples from patients with Binet stage B (P=0.0069 and P=0.047,
respectively) and C (P=0.038 and P=0.067, respectively). At the
family level, Bacteroidaceae was more abundant in stool
samples from patients with Binet stage A compared to Binet stage B
(P=0.051) and C (P=0.022), whereas Prevotellaceae was less
abundant in stool samples from patients with Binet stage A compared
to Binet stage B (P=0.02).
Notably, the gut microbiome of CD38+ CLL patients
exhibited a significant increase of Firmicutes phylum
(P=0.045) and a decrease in the Bacteroidaceae family
(P=0.045) in comparison to CD38-CLL. Moreover, a tendency to the
decreased relative abundance of Bacteroidetes phylum and
Bacteroidetes family was found in fecal samples from CLL
patients with unmutated IGHV compared to mutated IGHV (P=0.079,
P=0.089, respectively). There were no significant differences in
gut microbiome composition of CLL patients with distinct
TP53 mutation status or the presence of del17p, del11q, and
tri12.
Log F/B ratio of the gut microbiota as
a potential prognostic feature
Notably, there was a significant increase in log F/B
ratio in CLL patients with Binet stage B and Binet stage C compared
to Binet stage A (P=0.012 and P=0.038, respectively). Furthermore,
a tendency to a higher log F/B ratio was found in CLL patients with
unmutated IGHV in comparison to mutated IGHV (P=0.08) and with
CD38+ compared to CD38- (P=0.062) (Fig. S3).
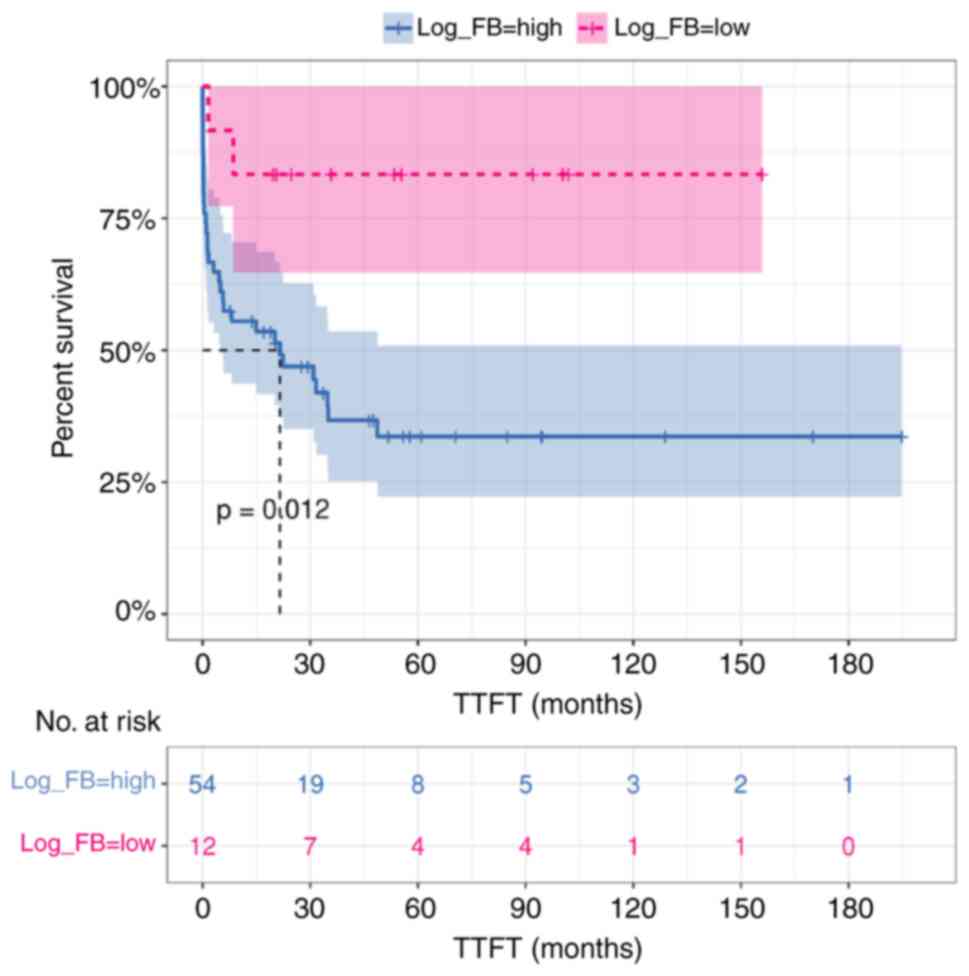
In the univariate model of Cox regression analysis,
intestinal log F/B ratio <-0.39 (as calculated by maximally
selected rank statistics) and corresponding to the 17th percentile,
was a significant predictor of longer TTFT in CLL patients (HR
5.20, 95% CI 1.25–21.72, P=0.024) (Table II). However, the multivariate
analysis, including established risk factors (Binet stage, IGHV
mutation status, CD38 expression, del11q, isolated del13q), showed
that fecal log F/B ratio had no impact on TTFT (HR 1.16, 95% CI
0.12–10.93, P=0.897). Kaplan-Meier estimate confirmed that
intestinal microbiota dysbiosis, with log F/B ratio >-0.39, was
associated with a significantly shorter TTFT (median 21.5 vs. NA,
P=0.012) (Fig. 4).
| Table II.Univariate and multivariate analyses
of time to first treatment in the cohort of 66 patients with
chronic lymphocytic leukemia. |
Table II.
Univariate and multivariate analyses
of time to first treatment in the cohort of 66 patients with
chronic lymphocytic leukemia.
|
| Univariate | Multivariate |
|---|
|
|
|
|
|---|
| Variable | HR (95% CI) | P-value | HR (95% CI) | P-value |
|---|
| Binet stage, A vs.
B | 11.60
(2.63–51.08) | 0.001 | 13.82
(1.44–132.53) | 0.023 |
| Binet stage, A vs.
C | 27.89
(6.40–121.45) | <0.001 | 54.56
(5.22–568.32) | <0.001 |
| CD38 expression,
positive vs. negativea | 3.33
(1.66–6.67) | <0.001 | 0.53
(0.16–1.80) | 0.311 |
| IGHV, unmutated vs.
mutated | 0.14
(0.06–0.034) | <0.001 | 0.52
(0.15–1.84) | 0.307 |
| Del11q, present vs.
absent | 4.49
(2.22–9.10) | <0.001 | 2.51
(0.99–6.38) | 0.052 |
| Isolated del13q,
present vs. absent | 0.39
(0.18–0.85) | 0.019 | 0.48
(0.14–1.65) | 0.243 |
| Intestinal log
FBb | 5.20
(1.25–21.72) | 0.024 | 1.16
(0.12–10.93) | 0.897 |
Discussion
A growing body of research has proved the
significance of microbiome alterations and the potential role of
specific microbial taxa in hematological malignancies (15–18,50,51).
Analysis of oral and intestinal microbiota of newly diagnosed CLL
patients in parallel with HVs allowed for the first time, to
discover significant differences in bacterial composition in CLL
patients. Loss of microbiome complexity in our CLL cohort was
observed as a decreased abundance of Bacteroidota and,
consequently, altered Firmicutes/Bacteroidota (F/B) ratio.
Our findings are in line with previous reports of the reduced
bacterial diversity and intestinal F/B ratio imbalance in other
inflammatory conditions, including obesity, type 2 diabetes,
inflammatory bowel disease, and cancers (52,53).
An enrichment of the Proteobacteria phylum
was revealed, which is a marker of dysbiosis associated with
intestinal inflammatory diseases, such as Crohn's disease and
ulcerative colitis (54). While an
increase in Proteobacteria may be linked to B cell
differentiation, the underlying mechanism is still unclear
(55). Lipopolysaccharide (LPS),
which is a component of the outer membrane of
Proteobacteria, through activation of Toll-like receptor 4
(TLR4), triggers downstream signaling pathways, including NF-κB
activation and dysregulates BCR signaling that represents a stimuli
factor driving CLL cells into proliferation (56). Moreover, LPS-induced activation of
TLR4 promotes inflammation by stimulating the production and
release of pro-inflammatory cytokines (such as TNF-α, IL-1β, and
IL-6), thereby indirectly contributing to neoplasia through
maintaining proinflammatory microenvironment (57,58).
Thus, increased LPS levels from Proteobacteria may
contribute to sustained immune activation and inflammation,
potentially affecting the pathogenesis of CLL. Yuan et al
(59) showed an increased relative
abundance of Proteobacteria and a continuous evolutionary
relationship of gut microbiota, from Proteobacteria phylum
to Escherichia-Shigella genus, in untreated diffuse large
B-cell lymphoma (DLBCL) patients. Moreover, Proteobacteria
was more abundant in the intestinal microbiome of multiple myeloma
(MM) patients as compared to healthy controls (60). Interestingly, this study also showed
an enrichment of nitrogen-recycling bacteria from the
Proteobacteria phylum, such as Klebsiella, in the
microbial community structure of MM patients. These bacteria are
involved in the hydrolysis of urea, which accumulates in excessive
amounts in the blood and intestines in MM patients, and the
synthesis of L-glutamine, which is taken up by MM cells, thereby
may promote tumor progression.
SCFAs, which include acetate, propionate, and
butyrate, are generated through the fermentation of non-digestible
carbohydrates by anaerobic bacteria (phylum Bacteroidota for
propionate and acetate; phylum Firmicutes for butyrate) in
the colon (61). These biologically
active microbial metabolites are key regulators of host physiology,
including immune system balance via promoting both immune response
and tolerance (62). SCFAs suppress
nuclear factor κB and inhibit the production of proinflammatory
cytokines such as IL-6 and TNF-α (63). Moreover, SCFAs upregulate
anti-inflammatory IL-10 via activation of aryl hydrocarbon receptor
(AhR)-dependent gene transcription in B cells and promotion of
regulatory B cells differentiation (64). SCFAs promote the generation of Th1,
Th17, and Treg through the inhibition of histone deacetylase (HDAC)
activity (65). The HDAC mechanism
is also involved in initiating Fas-mediated T cell apoptosis by
butyrate. At lower concentrations, butyrate inhibits T cell
proliferation, while at higher concentrations, it induces apoptosis
of activated T cells. Consequently, the accumulation of T cells
within the inflamed colonic mucosa is inhibited, which eliminates
potential antigenic stimulation and results in inflammation
(66,67). Dysregulation of the innate and
adaptive immune system is a crucial feature in CLL patients.
Immunosuppressive signatures include expansion of anti-inflammatory
cells such as Treg and myeloid-derived suppressor cells, production
of immunosuppressive soluble factors such as IL-10 and TGF-β, and
functional exhaustion of CD8+ effector T cells through expression
of inhibitory receptors (programmed death receptor-1 (PD-1),
cytotoxic T lymphocyte antigen-4 (CTLA-4), lymphocyte activation
gene 3 (LAG-3), CD244 and CD160) (68).
Notably, alterations in microbiota composition in
CLL patients in our study were related mainly to
Lachnospiraceae, Ruminococcaceae, Prevotellaceae, and
Veillonellaceae families, which are among the main producers
of SCFAs. In the recent study by Faitová et al (19), the abundances of SCFAs-producers
belonging to Lachnospiraceae and Ruminococcaceae
families in the gut microbiota of CLL patients were found to be
significantly lower in comparison to healthy control. However, this
observation was limited to a small group of CLL individuals. Since
CLL itself is a heterogeneous disease with varying clinical course
and progression rates, the disease stage may differentially impact
the microbiota composition. In our study, oral and gut microbiota
composition in 81 CLL patients at different stages of the disease
was analyzed: 30 CLL patients (37%) were at Binet stage A, 23 CLL
patients (29%) were at Binet stage B, and 25 CLL patients (31%)
were at Binet stage C. For 3 CLL patients (3%), Binet stage data
were not available. In contrast, the CLL cohort in Faitová's study
(19) included 70% of CLL patients
with Binet stage A, 20% of CLL patients with Binet stage B, and 10%
of CLL patients with Binet stage C. In our CLL cohort, we found
significant differences in oral and gut microbiome structure
depending on the disease stage. In Faitová's study (19), the size of the CLL cohort was
limited, and microbiome alterations between CLL patients at
different disease stages were not analyzed. Additionally, our CLL
cohort consisted of 47% of patients with mutated IGHV and 40% with
unmutated IGHV, while in Faitová's study (19), 70% of patients were unmutated-CLL,
and 30% were mutated-CLL. Therefore, the molecular prognostic
features, which include IGHV mutation status, indicate that our CLL
patients group is the diagnostic cohort, and the CLL patients group
in Faitová's study (19) is the
cohort requiring treatment initiation. As we excluded patients who
had a course of antibiotics within 4 weeks before sampling, it is
noteworthy that non-antibiotic drugs like non-steroidal
anti-inflammatory drugs (69),
calcium-channel blockers (70), and
antipsychotics (71) can also
affect the human microbiome. Moreover, dietary habits and
geographical location may shape the diversity and composition of
the microbiome (72,73). An important issue in microbiome
analysis remains methodological differences such as variations in
DNA extraction methods, library preparation, sequencing platforms,
and pipelines (74,75).
In addition to modulation of tumor growth, the gut
microbiota can affect the response to treatment. MM patients with
minimal/measurable residual disease negative response after
completion of upfront therapy showed a higher relative abundance of
SCFAs-producers Eubacterium hallii and Faecalibacterium
prausnitzi (76). Additionally,
a decreased risk of MM relapse/progression after allogeneic
hematopoietic cell transplantation was associated with the
enrichment of Eubacterium limosum according to the study by
Peled et al (77). Yoon
et al (78) showed a
positive correlation of Escherichia, Klebsiella,
Lactobacillus, and Weissella genera abundance in the gut
microbiome of DLBCL patients with indicators predicted to be
associated with disease burden, such as Ann Arbor stage and
international prognostic index, as well as a higher susceptibility
to side effects of chemotherapy.
Nevertheless, this study suggests the multi-faceted
role of microbiome in the pathology of CLL. For instance,
SCFA-producing bacteria, whose abundance was significantly changed
in our CLL cohort, have been previously reported to be implicated
in the maintenance of the intestinal barrier integrity (18,79,80),
regulation of macrophage balance in the intestine (81), and modulation of NK cell
cytotoxicity activity (82).
Moreover, by analyzing both oral and gut microbiota in CLL patients
and observing a loss of complexity and remarkable differences in
the relative abundances of specific bacterial phyla, a CLL-specific
microbiome profile characterized by enrichment of
Proteobacteria, depletion of Bacteroidota, and
impaired F/B ratio was identified. We observed a higher value of
log F/B ratio in CLL patients with more advanced disease stage and
negative prognostic features (high CD38 expression and unmutated
IGHV) and the association of an increased value of this parameter
with a significantly shorter TTFT, which is in line with Faitova
et al study (83) showing
that lower diversity of the gut microbiome in CLL patients is
associated with more aggressive and/or more progressive disease
development. Although we aimed for our CLL cohort to be as
homogenic as possible, not all alterations observed in the oral and
intestinal microbiome were the same. These differences may be
related to specific factors associated with each anatomical site.
The oral cavity plays a key role in host defense against invading
antigens and is directly exposed to external factors such as diet
and oral hygiene practices (84,85).
In contrast, the gut microbiome is more complex and diverse than
the oral microbiome (86).
Furthermore, CLL is characterized by immune dysregulation, which
can lead to alterations in the local immune environment of both the
oral cavity and the gut. However, this immune dysregulation may
affect microbial communities differently in each site.
In conclusion, the structure of oral and gut
microbiota of CLL patients exhibits specific alterations in
comparison to healthy individuals and is associated with distinct
prognostic features. Furthermore, our findings suggest that altered
microbiota might be implicated in CLL pathogenesis through
SCFA-related metabolic pathways, thus intestinal microflora
modulation might provide a novel approach to improve the efficacy
of CLL treatment.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank Dr Piotr Flieger
(Department of Foreign Languages, Medical University of Lublin,
Lublin, Poland) for editing the manuscript for English
language.
Funding
This study was funded by a grant from the Polish National
Science Centre (grant no. 2018/29/B/NZ5/02706) and a grant from the
Medical University of Lublin (grant no. DS 462).
Availability of data and materials
The raw 16S rRNA sequencing data have been
deposited in the European Nucleotide Archive at EMBL-EBI under
accession number PRJEB67303 (www.ebi.ac.uk/ena/browser/view/PRJEB67303). All
other data generated in the present study may be requested from the
corresponding author.
Authors' contributions
KG conceived and directed the project and analyses.
MP and MS wrote the manuscript with input and comments from MK, JZ,
LB and KG. PBMC from recruited patients with CLL were isolated by
MP, MS, and JZ. MP coordinated oral and fecal sample collection and
their shipment to Eurofins Genomics Europe Sequencing GmbH for
microbiome sequencing. KG and MP confirm the authenticity of all
the raw data. MP, MS, JZ and MMa performed molecular
characteristics of CLL cells. EW, AK, and MSR conducted cytogenetic
analyses. MK performed bioinformatical analysis including data
processing, taxonomy assignment, statistics and visualization. MS,
MP, MK, JZ, LB and KG interpreted the results. MMo, EK, PJ, TW,
AW-W, JZB and TR organized patient recruitment and sampling. All
authors revised the article and read and approved the final version
of the manuscript.
Ethics approval and consent to
participate
Our study was approved by the local Ethics
Committee of the Medical University of Lublin (approval no.
KE-0254/7/2019) and written informed consent forms were obtained
from all participants, including patients with CLL and HVs.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Beekman R, Chapaprieta V, Russiñol N,
Vilarrasa-Blasi R, Verdaguer-Dot N, Martens JHA, Duran-Ferrer M,
Kulis M, Serra F, Javierre BM, et al: The reference epigenome and
regulatory chromatin landscape of chronic lymphocytic leukemia. Nat
Med. 24:868–880. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Oakes CC, Seifert M, Assenov Y, Gu L,
Przekopowitz M, Ruppert AS, Wang Q, Imbusch CD, Serva A, Koser SD,
et al: DNA methylation dynamics during B cell maturation underlie a
continuum of disease phenotypes in chronic lymphocytic leukemia.
Nat Genet. 48:253–264. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ten Hacken E and Burger JA:
Microenvironment interactions and B-cell receptor signaling in
Chronic Lymphocytic Leukemia: Implications for disease pathogenesis
and treatment. Biochim Biophys Acta. 1863:401–413. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nadeu F, Clot G, Delgado J, Martín-García
D, Baumann T, Salaverria I, Beà S, Pinyol M, Jares P, Navarro A, et
al: Clinical impact of the subclonal architecture and mutational
complexity in chronic lymphocytic leukemia. Leukemia. 32:645–653.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nadeu F, Diaz-Navarro A, Delgado J, Puente
XS and Campo E: Genomic and epigenomic alterations in chronic
lymphocytic leukemia. Annu Rev Pathol Mech Dis. 15:149–177. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Baliakas P, Moysiadis T, Hadzidimitriou A,
Xochelli A, Jeromin S, Agathangelidis A, Mattsson M, Sutton LA,
Minga E, Scarfò L, et al: Tailored approaches grounded on
immunogenetic features for refined prognostication in chronic
lymphocytic leukemia. Haematologica. 104:360–369. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stevenson FK, Forconi F and Kipps TJ:
Exploring the pathways to chronic lymphocytic leukemia. Blood.
138:827–835. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hilal T, Gea-Banacloche JC and Leis JF:
Chronic lymphocytic leukemia and infection risk in the era of
targeted therapies: Linking mechanisms with infections. Blood Rev.
32:387–399. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Benckert J, Schmolka N, Kreschel C, Zoller
MJ, Sturm A, Wiedenmann B and Wardemann H: The majority of
intestinal IgA+ and IgG+ plasmablasts in the human gut are
antigen-specific. J Clin Invest. 121:1946–1955. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jain T, Sharma P, Are AC, Vickers SM and
Dudeja V: New insights into the cancer-microbiome-immune axis:
Decrypting a decade of discoveries. Front Immunol. 12:6220642021.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zheng D, Liwinski T and Elinav E:
Interaction between microbiota and immunity in health and disease.
Cell Res. 30:492–506. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wlasiuk P, Niedzielski A, Skorka K,
Karczmarczyk A, Zaleska J, Zajac M, Putowski M, Pac-Kozuchowska E
and Giannopoulos K: Accumulation of CD5+CD19+
B lymphocytes expressing PD-1 and PD-1L in hypertrophied pharyngeal
tonsils. Clin Exp Med. 16:503–509. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yuan D, Tao Y, Wang H, Wang J, Cao Y, Cao
W, Pan S and Yu Z: A comprehensive analysis of the microbiota
composition and host driver gene mutations in colorectal cancer.
Invest New Drugs. 40:884–894. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lazar V, Ditu LM, Pircalabioru GG,
Gheorghe I, Curutiu C, Holban AM, Picu A, Petcu L and Chifiriuc MC:
Aspects of gut microbiota and immune system interactions in
infectious diseases, immunopathology, and cancer. Front Immunol.
9:18302018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bai L, Zhou P, Li D and Ju X: Changes in
the gastrointestinal microbiota of children with acute
lymphoblastic leukaemia and its association with antibiotics in the
short term. J Med Microbiol. 66:1297–1307. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nearing JT, Connors J, Whitehouse S, Van
Limbergen J, Macdonald T, Kulkarni K and Langille MGI: Infectious
complications are associated with alterations in the gut microbiome
in pediatric patients with acute lymphoblastic leukemia. Front Cell
Infect Microbiol. 9:282019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Galloway-Peña JR, Smith DP, Sahasrabhojane
P, Ajami NJ, Wadsworth WD, Daver NG, Chemaly RF, Marsh L, Ghantoji
SS, Pemmaraju N, et al: The role of the gastrointestinal microbiome
in infectious complications during induction chemotherapy for acute
myeloid leukemia. Cancer. 122:2186–2196. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang R, Yang X, Liu J, Zhong F, Zhang C,
Chen Y, Sun T, Ji C and Ma D: Gut microbiota regulates acute
myeloid leukaemia via alteration of intestinal barrier function
mediated by butyrate. Nat Commun. 13:25222022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Faitová T, Svanberg R, Da Cunha-Bang C,
Ilett EE, Jørgensen M, Noguera-Julian M, Paredes R, MacPherson CR
and Niemann CU: The gut microbiome in patients with chronic
lymphocytic leukemia. Haematologica. 107:2238–2243. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gopalakrishnan V, Spencer CN, Nezi L,
Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman
K, Wei SC, et al: Gut microbiome modulates response to anti-PD-1
immunotherapy in melanoma patients. Science. 359:97–103. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Viaud S, Saccheri F, Mignot G, Yamazaki T,
Daillère R, Hannani D, Enot DP, Pfirschke C, Engblom C, Pittet MJ,
et al: The intestinal microbiota modulates the anticancer immune
effects of cyclophosphamide. Science. 342:971–976. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Uribe-Herranz M, Rafail S, Beghi S,
Gil-de-Gómez L, Verginadis I, Bittinger K, Pustylnikov S, Pierini
S, Perales-Linares R, Blair IA, et al: Gut microbiota modulate
dendritic cell antigen presentation and radiotherapy-induced
antitumor immune response. J Clin Invest. 130:466–479. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Routy B, Le Chatelier E, Derosa L, Duong
CPM, Alou MT, Daillère R, Fluckiger A, Messaoudene M, Rauber C,
Roberti MP, et al: Gut microbiome influences efficacy of PD-1-based
immunotherapy against epithelial tumors. Science. 359:91–97. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pal K, Bystry V, Reigl T, Demko M, Krejci
A, Touloumenidou T, Stalika E, Tichy B, Ghia P, Stamatopoulos K, et
al: GLASS: Assisted and standardized assessment of gene variations
from Sanger sequence trace data. Bioinformatics. 33:3802–3804.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Malcikova J, Tausch E, Rossi D, Sutton LA,
Soussi T, Zenz T, Kater AP, Niemann CU, Gonzalez D, Davi F, et al:
ERIC recommendations for TP53 mutation analysis in chronic
lymphocytic leukemia-update on methodological approaches and
results interpretation. Leukemia. 32:1070–1080. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
van Dongen JJM, Langerak AW, Brüggemann M,
Evans PA, Hummel M, Lavender FL, Delabesse E, Davi F, Schuuring E,
García-Sanz R, et al: Design and standardization of PCR primers and
protocols for detection of clonal immunoglobulin and T-cell
receptor gene recombinations in suspect lymphoproliferations:
Report of the BIOMED-2 concerted action BMH4-CT98-3936. Leukemia.
17:2257–2317. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Giudicelli V, Brochet X and Lefranc MP:
IMGT/V-QUEST: IMGT standardized analysis of the immunoglobulin (IG)
and T cell receptor (TR) nucleotide sequences. Cold Spring Harb
Protoc. 2011:695–715. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Brochet X, Lefranc M-P and Giudicelli V:
IMGT/V-QUEST: The highly customized and integrated system for IG
and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids
Res. 36:W503–W508. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rosenquist R, Ghia P, Hadzidimitriou A,
Sutton LA, Agathangelidis A, Baliakas P, Darzentas N, Giudicelli V,
Lefranc MP, Langerak AW, et al: Immunoglobulin gene sequence
analysis in chronic lymphocytic leukemia: updated ERIC
recommendations. Leukemia. 31:1477–1481. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bystry V, Agathangelidis A, Bikos V,
Sutton LA, Baliakas P, Hadzidimitriou A, Stamatopoulos K and
Darzentas N; European Research Initiative on CLL, :
ARResT/AssignSubsets: A novel application for robust
subclassification of chronic lymphocytic leukemia based on B cell
receptor IG stereotypy. Bioinformatics. 31:3844–3846. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Andrews S: FastQC: A quality control tool
for high throughput sequence data. www.bioinformatics.babraham.ac.uk/projects/fastqc21–Feb.
2023
|
|
32
|
Li H: Seqtk: Toolkit for processing
sequences in FASTA/Q formats. github.com/lh3/seqtk21–Feb. 2023
|
|
33
|
Quast C, Pruesse E, Yilmaz P, Gerken J,
Schweer T, Yarza P, Peplies J and Glöckner FO: The SILVA ribosomal
RNA gene database project: Improved data processing and web-based
tools. Nucleic Acids Res. 41:D590–D596. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Callahan BJ, McMurdie PJ, Rosen MJ, Han
AW, Johnson AJA and Holmes SP: DADA2: High-resolution sample
inference from Illumina amplicon data. Nat Methods. 13:581–583.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nguyen LT, Schmidt HA, von Haeseler A and
Minh BQ: IQ-TREE: A fast and effective stochastic algorithm for
estimating maximum-likelihood phylogenies. Mol Biol Evol.
32:268–274. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
McMurdie PJ and Holmes S: Phyloseq: An R
package for reproducible interactive analysis and graphics of
microbiome census data. PLoS One. 8:e612172013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lahti L and Shetty S: Tools for microbiome
analysis in R. Microbiome package version 1.23.1.
github.com/microbiome. 21–Feb. 2023
|
|
38
|
Shetty S and Lahti L: Microbiomeutilities:
Utilities for microbiome analytics.
github.com/microsud/microbiomeutilities. 21–Feb. 2023
|
|
39
|
Guo K and Gao P: Microbal-an R package for
microbial community analysis with dada2 and phyloseq.
github.com/guokai8/microbial. 21–Feb. 2023
|
|
40
|
Barnett D, Arts I and Penders J: microViz:
An R package for microbiome data visualization and statistics. J
Open Source Softw. 6:32012021. View Article : Google Scholar
|
|
41
|
Kembel SW, Cowan PD, Helmus MR, Cornwell
WK, Morlon H, Ackerly DD, Blomberg SP and Webb CO: Picante: R tools
for integrating phylogenies and ecology. Bioinformatics.
26:1463–1464. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lozupone CA and Knight R: The unifrac
significance test is sensitive to tree topology. BMC
Bioinformatics. 16:2112015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bray JR and Curtis JT: An ordination of
the upland forest communities of southern Wisconsin. Ecol Monogr.
27:325–349. 1957. View Article : Google Scholar
|
|
44
|
Oksanen J, Simpson G, Blanchet F, Kindt R,
Legendre P, Minchin PR, O'Hara RB, Solymos P, Stevens HH, Szoecs E,
et al: Vegan: Community ecology package. R package version 2.6–7.
github.com/vegandevs/vegan. 21–Feb. 2023
|
|
45
|
Segata N, Izard J, Waldron L, Gevers D,
Miropolsky L, Garrett WS and Huttenhower C: Metagenomic biomarker
discovery and explanation. Genome Biol. 12:R602011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Allignol A and Latouche A: CRAN task view:
Survival analysis. github.com/cran-task-views/Survival. 21–Feb.
2023
|
|
47
|
Hothorn T: maxstat: Maximally selected
rank statistics. rdrr.io/cran/maxstat. 21–Feb. 2023
|
|
48
|
Kassambara A: survminer R package:
Survival analysis and visualization.
github.com/kassambara/survminer. 21–Feb. 2023
|
|
49
|
RStudio Team, . RStudio: Integrated
development environment for R. www.rstudio.com
|
|
50
|
Kawari M, Akhtar M, Sager M, Basbous Z,
Baydoun I, Kabanja J, Darweesh M, Mokhtar N, Kanfar S, Mutahar E,
et al: Alterations of gut microbiome in untreated chronic
lymphocytic leukemia (CLL); future therapeutic potentials. Blood.
134:5455. 2019. View Article : Google Scholar
|
|
51
|
Shi Z and Zhang M: Emerging roles for the
gut microbiome in lymphoid neoplasms. Clin Med Insights Oncol.
15:1179554921102412021. View Article : Google Scholar
|
|
52
|
Madhogaria B, Bhowmik P and Kundu A:
Correlation between human gut microbiome and diseases. Infect Med.
1:180–191. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
An J, Kwon H and Kim YJ: The
Firmicutes/bacteroidetes ratio as a risk factor of breast cancer. J
Clin Med. 12:22162023. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Khorsand B, Asadzadeh Aghdaei H,
Nazemalhosseini-Mojarad E, Nadalian B, Nadalian B and Houri H:
Overrepresentation of Enterobacteriaceae and Escherichia
coli is the major gut microbiome signature in Crohn's disease
and ulcerative colitis; a comprehensive metagenomic analysis of
IBDMDB datasets. Front Cell Infect Microbiol. 12:10158902022.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Mirpuri J, Raetz M, Sturge CR, Wilhelm CL,
Benson A, Savani RC, Hooper LV and Yarovinsky F:
Proteobacteria-specific IgA regulates maturation of the intestinal
microbiota. Gut Microbes. 5:28–39. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hajishengallis G and Lambris JD: Microbial
manipulation of receptor crosstalk in innate immunity. Nat Rev
Immunol. 11:187–200. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kawai T and Akira S: The role of
pattern-recognition receptors in innate immunity: Update on
Toll-like receptors. Nat Immunol. 11:373–384. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ferrer G and Montserrat E: Critical
molecular pathways in CLL therapy. Mol Med. 24:92018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yuan L, Wang W, Zhang W, Zhang Y, Wei C,
Li J and Zhou D: Gut microbiota in untreated diffuse large B cell
lymphoma patients. Front Microbiol. 12:6463612021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Jian X, Zhu Y, Ouyang J, Wang Y, Lei Q,
Xia J, Guan Y, Zhang J, Guo J, He Y, et al: Alterations of gut
microbiome accelerate multiple myeloma progression by increasing
the relative abundances of nitrogen-recycling bacteria. Microbiome.
8:742020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Mirzaei R, Afaghi A, Babakhani S, Sohrabi
MR, Hosseini-Fard SR, Babolhavaeji K, Khani Ali Akbari S,
Yousefimashouf R and Karampoor S: Role of microbiota-derived
short-chain fatty acids in cancer development and prevention.
Biomed Pharmacother. 139:1116192021. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Kim CH: Control of lymphocyte functions by
gut microbiota-derived short-chain fatty acids. Cell Mol Immunol.
18:1161–1171. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Singh N, Thangaraju M, Prasad PD, Martin
PM, Lambert NA, Boettger T, Offermanns S and Ganapathy V: Blockade
of dendritic cell development by bacterial fermentation products
butyrate and propionate through a transporter (Slc5a8)-dependent
inhibition of histone deacetylases. J Biol Chem. 285:27601–27608.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Rosser EC, Piper CJM, Matei DE, Blair PA,
Rendeiro AF, Orford M, Alber DG, Krausgruber T, Catalan D, Klein N,
et al: Microbiota-derived metabolites suppress arthritis by
amplifying aryl-hydrocarbon receptor activation in regulatory B
cells. Cell Metab. 31:837–851.e10. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Park J, Kim M, Kang SG, Jannasch AH,
Cooper B, Patterson J and Kim CH: Short-chain fatty acids induce
both effector and regulatory T cells by suppression of histone
deacetylases and regulation of the mTOR-S6K pathway. Mucosal
Immunol. 8:80–93. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zimmerman MA, Singh N, Martin PM,
Thangaraju M, Ganapathy V, Waller JL, Shi H, Robertson KD, Munn DH
and Liu K: Butyrate suppresses colonic inflammation through
HDAC1-dependent Fas upregulation and Fas-mediated apoptosis of T
cells. Am J Physiol Liver Physiol. 302:G1405–G1415. 2012.PubMed/NCBI
|
|
67
|
Bailón E, Cueto-Sola M, Utrilla P,
Rodríguez-Cabezas ME, Garrido-Mesa N, Zarzuelo A, Xaus J, Gálvez J
and Comalada M: Butyrate in vitro immune-modulatory effects might
be mediated through a proliferation-related induction of apoptosis.
Immunobiology. 215:863–873. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Griggio V, Perutelli F, Salvetti C,
Boccellato E, Boccadoro M, Vitale C and Coscia M: Immune
dysfunctions and immune-based therapeutic interventions in chronic
lymphocytic leukemia. Front Immunol. 11:5945562020. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Rogers MAM and Aronoff DM: The influence
of non-steroidal anti-inflammatory drugs on the gut microbiome.
Clin Microbiol Infect. 22:178.e1–178.e9. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zheng T and Marques FZ: Gut microbiota:
Friends or foes for blood pressure-lowering drugs. Hypertension.
79:1602–1604. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Minichino A, Preston T, Fanshawe JB,
Fusar-Poli P, McGuire P, Burnet PWJ and Lennox BR:
Psycho-pharmacomicrobiomics: A systematic review and meta-analysis.
Biol Psychiatry. 95:611–628. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Statovci D, Aguilera M, MacSharry J and
Melgar S: The impact of Western diet and nutrients on the
microbiota and immune response at mucosal interfaces. Front
Immunol. 8:8382017. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Gupta VK, Paul S and Dutta C: Geography,
ethnicity or subsistence-specific variations in human microbiome
composition and diversity. Front Microbiol. 8:11622017. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Fernández-Pato A, Sinha T, Gacesa R,
Andreu-Sánchez S, Gois MFB, Gelderloos-Arends J, Jansen DBH, Kruk
M, Jaeger M, Joosten LAB, et al: Choice of DNA extraction method
affects stool microbiome recovery and subsequent phenotypic
association analyses. Sci Rep. 14:39112024. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
González A, Fullaondo A and Odriozola A:
Techniques, procedures, and applications in microbiome analysis.
pp81–115. 2024.
|
|
76
|
Pianko MJ, Devlin SM, Littmann ER,
Chansakul A, Mastey D, Salcedo M, Fontana E, Ling L, Tavitian E,
Slingerland JB, et al: Minimal residual disease negativity in
multiple myeloma is associated with intestinal microbiota
composition. Blood Adv. 3:2040–2044. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Peled JU, Devlin SM, Staffas A, Lumish M,
Khanin R, Littmann ER, Ling L, Kosuri S, Maloy M, Slingerland JB,
et al: Intestinal microbiota and relapse after hematopoietic-cell
transplantation. J Clin Oncol. 35:1650–1659. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Yoon SE, Kang W, Chalita M, Lim J, Kim WS
and Kim SJ: Comprehensive understanding of gut microbiota in
treatment naïve diffuse large B cell lymphoma patients. Blood.
138:2409. 2021. View Article : Google Scholar
|
|
79
|
Ruth MR and Field CJ: The immune modifying
effects of amino acids on gut-associated lymphoid tissue. J Anim
Sci Biotechnol. 4:272013. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Ma L, Ni Y, Wang Z, Tu W, Ni L, Zhuge F,
Zheng A, Hu L, Zhao Y, Zheng L and Fu Z: Spermidine improves gut
barrier integrity and gut microbiota function in diet-induced obese
mice. Gut Microbes. 12:18328572020. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Nakamura A, Kurihara S, Takahashi D,
Ohashi W, Nakamura Y, Kimura S, Onuki M, Kume A, Sasazawa Y,
Furusawa Y, et al: Symbiotic polyamine metabolism regulates
epithelial proliferation and macrophage differentiation in the
colon. Nat Commun. 12:21052021. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Lai HC, Chang CJ, Yang CH, Hsu YJ, Chen
CC, Lin CS, Tsai YH, Huang TT, Ojcius DM, Tsai YH and Lu CC:
Activation of NK cell cytotoxicity by the natural compound
2,3-butanediol. J Leukoc Biol. 92:807–814. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Faitova T, Coelho M, Da Cunha-Bang C,
Ozturk S, Kartal E, Bork P, Seiffert M and Niemann CU: The
diversity of the microbiome impacts chronic lymphocytic leukemia
development in mice and humans. Haematologica. May 9–2024.doi:
10.3324/haematol.2023.284693 (Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Akimbekov NS, Digel I, Yerezhepov AY,
Shardarbek RS, Wu X and Zha J: Nutritional factors influencing
microbiota-mediated colonization resistance of the oral cavity: A
literature review. Front Nutr. 9:10293242022. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Kilian M, Chapple ILC, Hannig M, Marsh PD,
Meuric V, Pedersen AM, Tonetti MS, Wade WG and Zaura E: The oral
microbiome-an update for oral healthcare professionals. Br Dent J.
221:657–666. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Tan X, Wang Y and Gong T: The interplay
between oral microbiota, gut microbiota and systematic diseases. J
Oral Microbiol. 15:22131122023. View Article : Google Scholar : PubMed/NCBI
|