Introduction
Among malignant tumors, esophageal carcinoma (EC)
ranks seventh in terms of the global incidence and sixth in terms
of mortality (1). This type of
cancer includes two main pathological types: Esophageal squamous
cell carcinoma (ESCC) and esophageal adenocarcinoma (2). China accounts for approximately half
of all ESCC cases worldwide (3). In
China, esophageal cancer ranks sixth in terms of incidence rate
among malignant tumors and fourth in terms of the number of deaths
(4), and ESCC accounts for >90%
of all EC cases (5).
As part of the tumor microenvironment,
microorganisms may participate in tumor development by inducing
chronic or persistent inflammation (6). The human microbiota includes trillions
of bacteria, archaea, fungi and viruses that interact with the
human body (2), and are distributed
in the skin, respiratory tract, oral cavity and gastrointestinal
tract (3), with >70% of the
human microbiota located in the gastrointestinal tract (7). However, the microecological
composition of each part is not uniform, and different parts of the
gastrointestinal tract may have specific microecological
communities (8). Sex, obesity, age,
food, host genetic background, environment, antibacterial drugs and
other factors affect microbial structures (9–13).
Furthermore, different methods of material extraction may affect
research results on the digestive tract flora (14). Given the close relationship between
gut microbiota and human health, studying gut microbiota is helpful
for the diagnosis, assessment and prognosis evaluation of diseases
(15). The microflora in the
digestive tract is related to the occurrence and development of
ESCC (16). The changes in the
esophageal flora should be studied or specific bacterial changes
should be detected, and these studies may be beneficial for the
early diagnosis, evaluation and favorable prognosis of ESCC
(17–19). The sampling methods for research on
the flora that causes esophageal diseases include saliva
collection, oropharyngeal swabs, esophageal mucosal swabs,
endoscopic biopsies, endoscopic mucosal resection specimens,
surgical biopsies after esophageal surgeries, esophageal string
tests and Cytosponge devices (18,20–23).
The microbial composition may vary depending on the
sampling method and tissue source, and the microbial community
composition of the different segments of the digestive tract may
exhibit variations (24).
Therefore, the selection of samples for microbial analysis is
crucial for research, and the sampling method may affect the
results of gastrointestinal microbiota research. Studies on the
esophageal flora of patients with ESCC remain in their infancy and,
to the best of our knowledge, the most suitable type of samples for
this disease is unknown (25–27).
The advantages and disadvantages of different
sampling methods, and their effects on exploring the relationship
between esophageal microbiota and different esophageal diseases
still require further research. The aim of the present study was to
provide a theoretical basis for the selection of standard sampling
methods in the study of esophageal microbiota in patients with ESCC
by comparing differences in the bacterial flora between surgical
and endoscopic esophageal mucosa tissues.
Materials and methods
Sample source
A total of 72 patients with ESCC who were diagnosed
via digestive endoscopy and thoracic surgery at Taihe Hospital
(Shiyan, China) between July 2018 and July 2019 were selected to
participate in the present study. The patients were divided into
the postoperative tissue group (Group A) and the esophageal mucosa
group (Group B) based on the different sample sources of esophageal
cancer tissue. Group A comprised 27 esophageal cancer postoperative
tissue samples, and Group B comprised 45 esophageal mucosa samples.
Patients in group A ranged in age from 36 to 77 years (median, 62.5
years), while patients in group B ranged in age from 37 to 85 years
(median, 65.4 years) (Table I).
 | Table I.Basic information of included
patients with esophageal squamous cell carcinoma. |
Table I.
Basic information of included
patients with esophageal squamous cell carcinoma.
|
|
| Sex | Age, years |
|---|
|
|
|
|
|
|---|
| Group | No. | Male, n | Female, n |
P-valuea | Minimum | Maximum | Mean |
P-valueb |
|---|
| Group A | 27 | 23 | 4 | 0.607 | 36 | 77 | 62.5 | 0.251 |
| Group B | 45 | 38 | 7 |
| 37 | 85 | 65.4 |
|
For patients with ESCC, the following inclusion
criteria were applied: Age ≥18 years; pathological diagnosis of
ESCC; without metabolic diseases (such as diabetes), hyperlipidemia
or other infectious diseases; good general condition; no intake of
antibiotics, acid suppressants or probiotics within the past 2
months; balanced diet and no special dietary habits; and no serious
liver, kidney and immunodeficiency diseases. The exclusion criteria
were as follows: Use of drugs affecting the microecology of the
esophagus in the past 2 months; complications of metabolic or
infectious diseases; presence of tumors other than ESCC; incomplete
data; and not considered suitable for inclusion by the researchers
(such as individuals with severe picky eating, long-term alcohol
abuse and recent oral disease).
The study protocol was reviewed and approved by the
Taihe Hospital Ethics Committee (approval no. 2018KS020; Shiyan,
China), and written informed consent was obtained from all patients
before they were allowed to participate in the present study.
Furthermore, the present study was conducted in accordance with the
provisions of The Declaration of Helsinki.
Sample collection
Esophageal mucosal tissue samples were obtained
during endoscopic examination. Gastroscopy was performed 6–8 h
after fasting and warm water was used for gargling before
examination. After the esophageal tumor lesions were found, four to
eight specimens were collected with sterile biopsy forceps for
examination. Two specimens were marked, placed in sterile
cryopreservation tubes and frozen in −196°C liquid nitrogen for
temporary storage, and then transferred to a −80°C refrigerator for
long-term storage. The remaining tissues were fixed in 10% neutral
buffered formalin at room temperature for 24–48 h, and sent to the
pathology. Fixed tissue samples were dehydrated using a series of
graded alcohol solutions (70, 95 and 100% ethanol) to remove
moisture from the tissue. Alcohol was removed from dehydrated
tissues with xylene to make the tissue transparent, and then the
tissue was embedded and placed in paraffin. The treatment of
surgical specimens was the same as for endoscopic mucosal tissue,
and appropriate samples were chosen for follow-up studies in
accordance with the inclusion criteria. The selected samples were
quickly transferred to a −196°C liquid nitrogen tank for temporary
storage, and then transferred to a −80°C refrigerator for long-term
storage.
DNA extraction
The DNA of the sample was extracted with an
UltraClean® Microbial DNA Isolation Kit (15,800; Mo Bio
Laboratories, Inc.) using the sodium dodecyl sulfate lysate
freeze-thaw method. The purity and quantity of the DNA were
determined using a NanoDrop spectrophotometer (Thermo Fisher
Scientific, Inc.). The sample was frozen at −20°C for later
use.
16S ribosomal DNA sequencing
The V4 region of the 16S ribosomal RNA
gene was amplified by PCR. The primers included 515F
(5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′).
The PCR system (50 µl) comprised the following: 25 µl Phusion
High-Fidelity PCR Master Mix (M0531; New England BioLabs, Inc.), 3
µl each of forward/reverse primers (10 µM), 10 µl DNA template and
9 µl double-distilled water. The thermocycling conditions were as
follows: Pre-denaturation at 98°C for 30 sec, followed by 25 cycles
of denaturation at 98°C for 15 sec, annealing at 58°C for 15 sec
and extension at 72°C for 15 sec, and a final extension at 72°C for
1 min. The amplification products of each sample were detected by
electrophoresis on a 1% agarose gel at 100 V for 40 min. The UVI
gel imaging system (Thermo Fisher Scientific, Inc.) was used for
image capture and recording, and DNA electrophoresis did not show
mixed bands and tails, indicating that the purity of DNA fragments
was good and there was no obvious degradation. The gel recovery kit
(DP219-03; Tiangen Biotech Co., Ltd.) was used to recover and
purify the DNA of the target strip. The Qubit® dsDNA HS
Assay kit (Q32854; Invitrogen; Thermo Fisher Scientific, Inc.) was
used to accurately quantify the recovered DNA, and parallel
sequencing was performed following mixing of the samples (the same
amount of library was taken from each sample). The library
amplification products were analyzed for fragment length using an
Agilent 2,100 Bioanalyzer (Agilent Technologies, Inc.) and High
Sensitivity DNA Kit (5,067–4,626; Agilent Technologies, Inc.), and
a Qubit 3.0 Fluorometer (Invitrogen; Thermo Fisher Scientific,
Inc.) was used to measure the library concentration. The final
concentration of the library on the machine was 1.8 pM. Paired-end
150-bp mode sequencing was performed on the library using an
Illumina HiSeq 4,000 platform (Illumina, Inc.) and a HiSeg
3,000/4,000 SBS Kit (300 cycles; FC-410-1003; Illumina, Inc.).
Sequencing was completed at Shanghai Biotecan Pharmaceuticals Co.,
Ltd.
Operational taxonomic units (OTUs)
clustering and species annotation
OTUs were analyzed with V search version 2.4.4
(28) and clustered with a
similarity of 97%. Representative sequences were annotated on the
basis of the SILVA128 database (29). The abundance and classification of
the OTUs were recorded.
Bioinformatics analysis and
statistical analysis
Quantitative insights into microbial ecology
(version 1.8.0; http://qiime.org/) and R (www.r-project.org; version 3.2.0) were used to analyze
the data. α diversity indices, including Chao1, Shannon, Simpson
and abundance-based coverage estimator, were calculated. The
abundance and uniformity of OTUs were compared, and the UniFrac
distance was calculated (30).
Principal coordinates analysis and nonmetric multidimensional
scaling (NMDS) plots were generated for the β analysis of the
sample flora structure. The Vegan package (version 2.5-3;
http://github.com/vegandevs/vegan/releases) in R
(v3.2.0) software, MEGAN 4 (31,32)
and Graphical Phylogenetic Analysis (version 1.1.3) were used to
visualize the groups and abundances (33). Venn diagrams were generated using
the Venn Diagram module of the R software (v3.2.0) to visualize
common and unique OTUs between groups.
The Wilcoxon rank-sum test in the R 3.2.0 software
package was used to compare the differences in microbial
communities at various taxonomic levels between two groups. Species
bearing significant differences between groups were selected using
linear discriminant analysis (LDA) effect size (LEfSe) analysis
(34) and an LDA value ≥2 was
considered statistically significant with P<0.05. Random forest
analysis was performed using the default settings of the random
forest module in R 3.2.0 to compare the differences between groups,
and the p ROC package was used for receiver operating
characteristic (ROC) curve analysis (35,36).
The BugBase tool can be used for the prediction of
the microbial phenotype, using OTU tables as input files to
standardize the OTU tables. Subsequently, pre-processed databases
and BugBase tools were used to automatically select thresholds to
predict microbial phenotypes, and the abundance of each phenotype
in each group was calculated and compared (37). The BugBase database was employed to
predict the phenotypes of esophageal bacteria (38).
For intergroup comparison involving phenotypic
content prediction, the Wilcoxon rank-sum test was used to compare
the abundance information among group samples, and the P-value was
obtained.
Statistical analysis was performed using SPSS 21.0
(IBM Corp.). Normally distributed continuous variables are
presented as the mean ± standard deviation, nonnormally distributed
continuous data are presented as the median (lower quartile, upper
quartile) and microbial abundance was conveyed as a percentage.
Fisher's exact test, unpaired Student's t-test and nonparametric
Wilcoxon rank-sum tests were conducted for comparison. P<0.05
was considered to indicate a statistically significant
difference.
Results
Sample sequencing data
After clustering was performed with 97% similarity,
3,656 OTUs, including 2,926 in the esophageal cancer postoperative
tissue group (Group A) and 2,772 in the esophageal mucosa group
(Group B), and 2,042 in both groups, were obtained. A total of 884
OTUs were unique to Group A and 730 OTUs were unique to Group B
(Fig. S1).
α diversity analysis
The Shannon and Chao indices of the postoperative
tissue samples (Group A) were significantly higher than those of
the esophageal mucosa tissue samples (Group B) (P<0.05). The
Simpson index of Group A was higher than that of Group B, but the
difference was not significant (P>0.05). These findings
indicated that the diversity of the microbial flora in
postoperative tissues was higher than that in the esophageal mucosa
group (Fig. 1A).
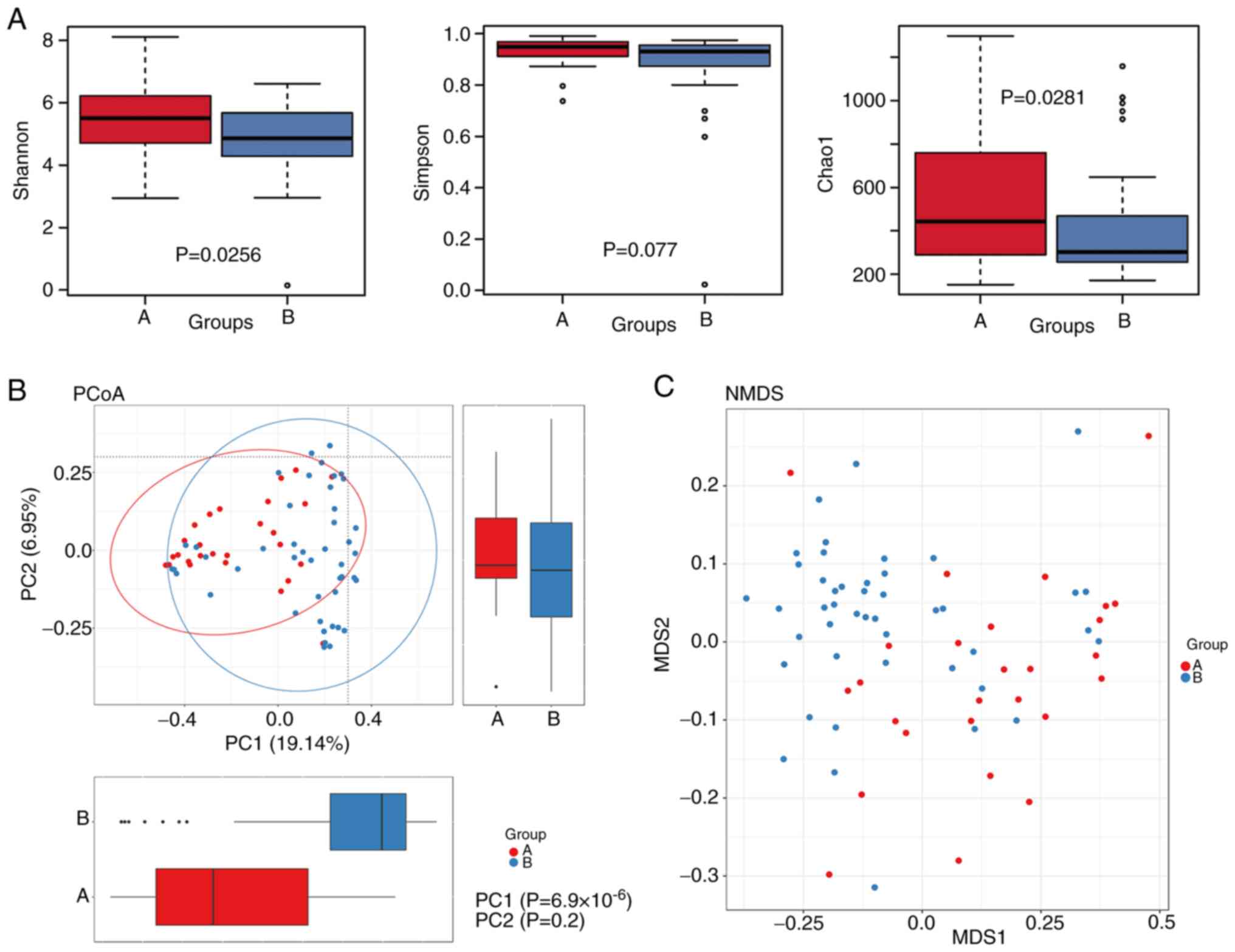 | Figure 1.Comparison of α and β diversity of
the esophageal flora after esophageal surgery (group A) and in the
esophageal mucosa group (group B). (A) (A-1) Shannon, (A-2) Simpson
and (A-3) Chao1 indices. The P-value is indicated at the top of
each image. The abscissa indicates the name of the group, and the
ordinate shows the α diversity index of the different groups. The
box chart shows five statistics (minimum value, first quartile,
median, third quartile and maximum value) as five lines from the
bottom to the top. Outliers are indicated as ‘o’. The P-value was
calculated using the Wilcoxon rank sum test. (B) PCoA plots of the
unweighted UniFrac distances of the variation in microbiota
composition detected in the postoperative tissue group (Group A)
and the esophageal mucosa group (Group B). The values of the two
vectors are marked in the lower right corner. Each point in the
figure represents a sample, red represents Group A, blue represents
Group B, and the distance reflects the similarity of the samples.
(C) Scatter plot of two groups of NMDS analysis results. Each point
represents a sample, and points of the same color are from the same
group. The distance reflects the similarity of the samples. In the
plot, blue represents the esophageal mucosa group, and red
corresponds to the esophageal cancer postoperative tissue group.
NMDS, nonmetric multidimensional scaling; PC, principal component;
PCoA, principal coordinate analysis. |
β diversity analysis
Principal component (PC)1 and PC2 represented the
potential factors influencing the deviation of the microbial
composition of the two groups. For the two groups, PC1=19.14%,
suggesting that the bacterial composition in the two groups was not
significantly different (Fig. 1B).
NMDS analysis showed that the overall flora of the two groups could
not be clearly distinguished. This result demonstrated that the
overall composition of the flora of the two groups was not markedly
different (Fig. 1C).
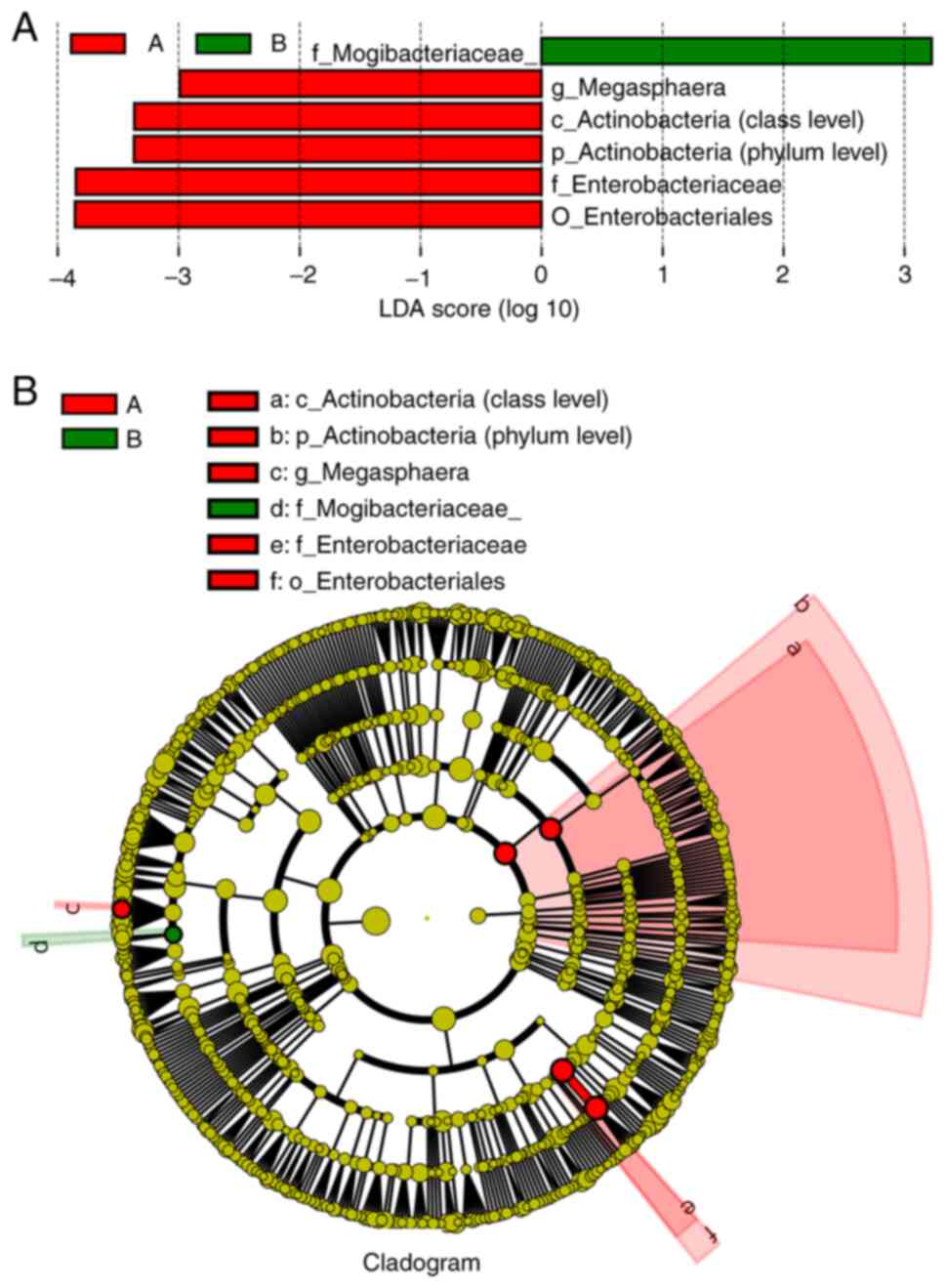
Differential LEfSe analysis
The abundance of Megasphaera, Actinobacteria
(class level), Actinobacteria (phylum level),
Enterobacteriaceae and Enterobacteriales in the
esophageal postoperative tissue samples (Group A) was higher than
that in the esophageal mucosal tissue samples (Group B), but the
abundance of Mogibacteriaceae in the esophageal mucosa
tissue samples (Group B) was higher than that in the postoperative
samples (Group A). The difference in microbial abundance between
the two different tissues was statistically significant (P<0.05;
Fig. 2A and B).
Characteristics of the esophageal
flora of the two groups
There were differences in microbial composition
between the two groups at the phylum and genus levels, as well as
differences in classes, orders and families (Table SI, Table SII, Table SIII).
Analysis of the microbial flora
composition at the phylum level
The two groups of samples were considerably
different at the phylum level, and the five phyla with the most
significant differences were identified. Actinobacteria and
Verrucomicrobiae were more abundant in the postoperative
tissue group than in the esophageal mucosa group. The abundance of
Fusobacteria, SR1 and Spirochaetes was significantly
lower in the postoperative tissue group than in the esophageal
mucosa group (P<0.05; Table
II).
| Table II.Significant differences in phylum
levels between the two groups. |
Table II.
Significant differences in phylum
levels between the two groups.
|
|
|
| IQR |
|
|---|
|
|
|
|
|
|
|---|
|
| Median (%) | P 25 (%) | P75 (%) |
|
|---|
|
|
|
|
|
|
|---|
| Name | Group A | Group B | Group A | Group B | Group A | Group B |
P-valuea |
|---|
| Actinobacteria | 2.330 | 1.319 | 1.318 | 0.504 | 3.727 | 3.074 | 0.030 |
| Fusobacteria | 0.683 | 4.017 | 0.246 | 1.358 | 1.345 | 7.050 | <0.001 |
| SR1 | 0.000 | 0.021 | 0.000 | 0.000 | 0.004 | 0.131 | 0.003 |
| Spirochaetes | 0.007 | 0.189 | 0.000 | 0.022 | 0.161 | 1.464 | 0.007 |
|
Verrucomicrobia | 0.276 | 0.000 | 0.001 | 0.000 | 0.591 | 0.097 | 0.005 |
Microbial flora composition analysis
at the genus level
At the genus level, Bifidobacterium, Collinsella,
Bacteroides, Parabacteroides, Butyricimonas, Paraprevotella,
Gemella, Enterococcus, Blautia, Coprococcus, Lachnospira,
Roseburia, Faecalibacterium, Oscillospira, Ruminococcus, Megamonas,
Megasphaera, Ruminococcus, Phascolarctobacterium, Sutterella
and Akkermansia were more abundant in the postoperative
tissue group than in the esophageal mucosa group, whereas the
abundance of Porphyromonas, Prevotella, [Prevotella], Catonella,
Oribacterium, Peptostreptococcus, Selenomonas, Parvimonas,
Fusobacterium, Leptotrichia, Ralstonia, Campylobacter,
Actinobacillus and Treponema in the former was
significantly lower than that in the latter (P<0.05; Table III).
| Table III.Significant differences in genus
levels between the two groups. |
Table III.
Significant differences in genus
levels between the two groups.
|
|
|
| IQR |
|
|---|
|
|
|
|
|
|
|---|
|
| Median (%) | P 25 (%) | P75 (%) |
|
|---|
|
|
|
|
|
|
|---|
| Name | Group A | Group B | Group A | Group B | Group A | Group B |
P-valuea |
|---|
|
Bifidobacterium | 0.849 | 0.063 | 0.437 | 0.003 | 1.496 | 0.620 | 0.001 |
|
Collinsella | 0.039 | 0.000 | 0.001 | 0.000 | 0.187 | 0.011 | 0.001 |
|
Bacteroides | 12.194 | 0.907 | 7.543 | 0.176 | 27.594 | 4.786 | <0.001 |
|
Parabacteroides | 0.697 | 0.021 | 0.145 | 0.001 | 1.673 | 0.362 | <0.001 |
|
Porphyromonas | 0.043 | 0.518 | 0.015 | 0.095 | 0.218 | 3.505 | <0.001 |
|
Prevotella | 5.757 | 12.227 | 2.510 | 4.770 | 8.104 | 22.868 | 0.001 |
|
Butyricimonas | 0.053 | 0.001 | 0.012 | 0.000 | 0.229 | 0.011 | <0.001 |
|
Paraprevotella | 0.156 | 0.000 | 0.015 | 0.000 | 0.235 | 0.025 | <0.001 |
|
[Prevotella] | 0.231 | 2.475 | 0.063 | 0.599 | 0.490 | 5.784 | <0.001 |
| Gemella | 0.002 | 0.000 | 0.000 | 0.000 | 0.012 | 0.002 | 0.011 |
|
Enterococcus | 0.075 | 0.010 | 0.002 | 0.000 | 0.177 | 0.069 | 0.020 |
| Blautia | 0.218 | 0.001 | 0.011 | 0.000 | 0.531 | 0.024 | <0.001 |
|
Catonella | 0.000 | 0.159 | 0.000 | 0.009 | 0.001 | 0.319 | <0.001 |
|
Coprococcus | 0.112 | 0.001 | 0.016 | 0.000 | 0.257 | 0.028 | <0.001 |
|
Lachnospira | 0.398 | 0.032 | 0.080 | 0.001 | 1.214 | 0.382 | 0.007 |
|
Oribacterium | 0.001 | 0.052 | 0.000 | 0.000 | 0.031 | 0.215 | 0.013 |
|
Roseburia | 0.314 | 0.002 | 0.038 | 0.000 | 0.893 | 0.272 | 0.002 |
|
Ruminococcus | 0.276 | 0.000 | 0.001 | 0.000 | 0.434 | 0.069 | 0.003 |
|
Peptostreptococcus | 0.007 | 0.220 | 0.002 | 0.044 | 0.109 | 0.945 | <0.001 |
|
Faecalibacterium | 0.436 | 0.007 | 0.089 | 0.000 | 1.320 | 0.117 | 0.001 |
|
Oscillospira | 0.223 | 0.002 | 0.009 | 0.000 | 0.779 | 0.051 | <0.001 |
|
Ruminococcus | 0.604 | 0.030 | 0.124 | 0.000 | 0.969 | 0.390 | <0.001 |
|
Megamonas | 0.305 | 0.003 | 0.104 | 0.000 | 1.095 | 0.234 | <0.001 |
|
Megasphaera | 0.071 | 0.005 | 0.005 | 0.000 | 0.199 | 0.062 | 0.013 |
|
Phascolarctobacterium | 1.037 | 0.001 | 0.278 | 0.000 | 2.109 | 0.329 | <0.001 |
|
Selenomonas | 0.013 | 0.679 | 0.000 | 0.069 | 0.209 | 4.561 | <0.001 |
|
Parvimonas | 0.001 | 0.032 | 0.000 | 0.004 | 0.013 | 0.239 | 0.001 |
|
Fusobacterium | 0.408 | 2.577 | 0.101 | 0.996 | 1.251 | 5.611 | <0.001 |
|
Leptotrichia | 0.009 | 0.220 | 0.003 | 0.021 | 0.097 | 0.887 | <0.001 |
|
Sutterella | 0.773 | 0.015 | 0.224 | 0.000 | 1.290 | 0.434 | <0.001 |
|
Ralstonia | 0.000 | 0.001 | 0.000 | 0.000 | 0.002 | 0.020 | 0.014 |
|
Campylobacter | 0.034 | 0.228 | 0.009 | 0.066 | 0.381 | 1.088 | 0.007 |
|
Actinobacillus | 0.001 | 0.087 | 0.000 | 0.008 | 0.072 | 0.694 | 0.003 |
|
Treponema | 0.007 | 0.189 | 0.000 | 0.022 | 0.161 | 1.458 | 0.006 |
|
Akkermansia | 0.190 | 0.000 | 0.000 | 0.000 | 0.591 | 0.074 | 0.005 |
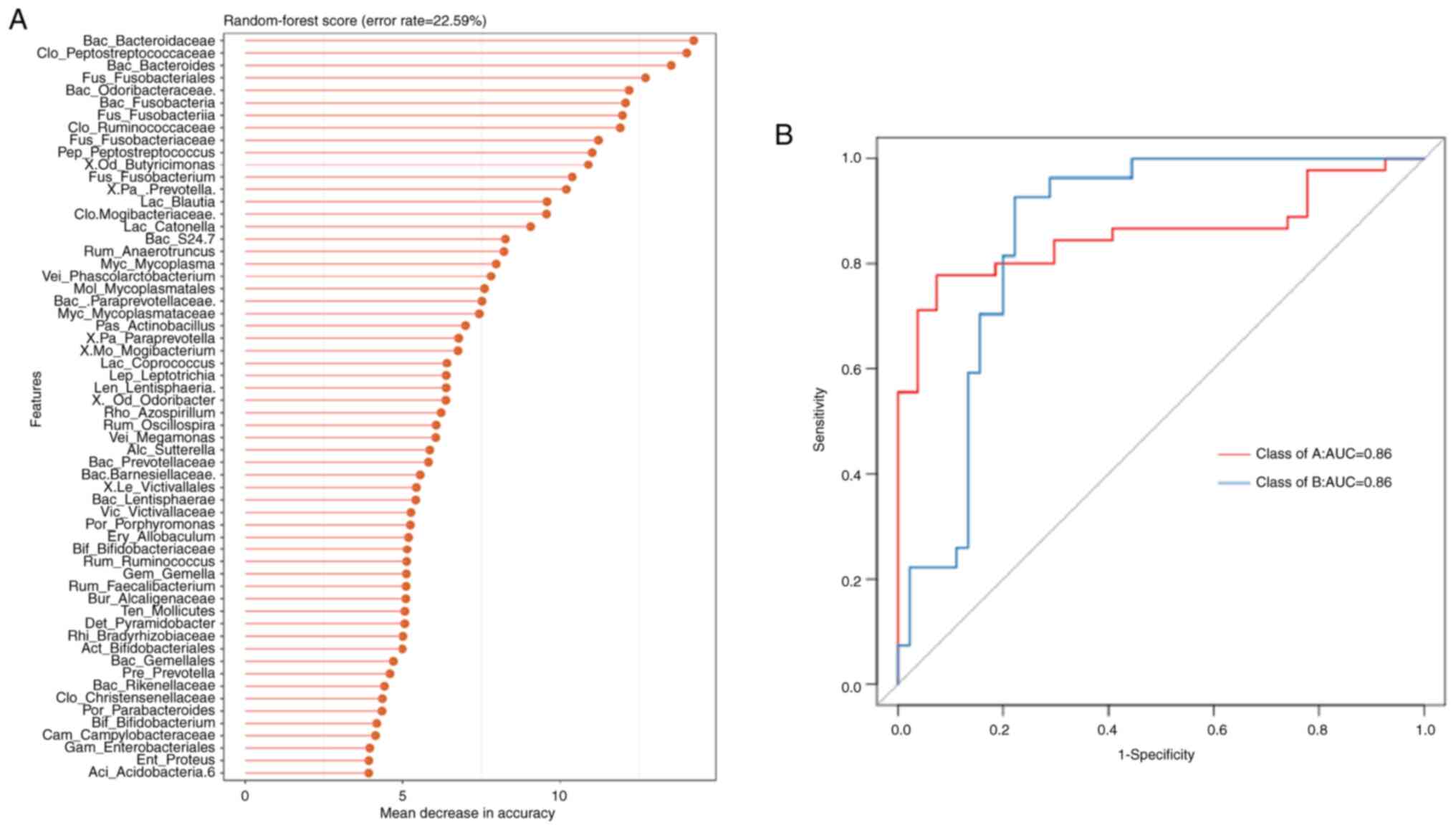
Predictive performance of the
esophageal microbiome in two groups of patients (genus level)
The random forest method is a machine learning
method that can effectively classify and predict grouped samples.
The bacterial genera that serve a major role in the classification
performance in the classifier were arranged in descending order of
their effects (Fig. 3A). The top 60
species were selected for the random forest method to establish a
model. The error rate refers to the error rate of using the
characteristics of the microbial community for random forest method
prediction classification. The higher the error rate, the lower the
accuracy of classification based on bacterial genus features, which
may result in unclear bacterial genus features between groups. The
error rate was 22.59% (Fig. 3A).
The ROC curve confirmed that the forecasting model constructed by
the random forest method was reliable and could effectively
distinguish between the two groups of samples (area under the
curve, 0.86; Fig. 3B).
Comparison of phenotype classification
based on BugBase
The phenotype prediction using BugBase showed that
the relative abundance of Gram-positive bacteria was higher in the
postoperative tissue group than in the mucosal tissue group. By
contrast, the relative abundance of Gram-negative bacteria in the
postoperative tissue group was significantly lower than that in the
mucosal tissue group (Fig. S2;
P<0.05, Table IV). The two
groups were similar under the following conditions: Aerobic,
anaerobic, presence of mobile elements, facultatively anaerobic,
forms biofilms, potentially pathogenic and stress-tolerant
conditions, and the differences were not significant (P>0.05;
Table IV).
| Table IV.Comparison of phenotype
classification based on BugBase. |
Table IV.
Comparison of phenotype
classification based on BugBase.
|
|
|
| IQR |
|
|---|
|
|
|
|
|
|
|---|
|
| Relative abundance
with trait (median) | Relative abundance
with trait (P25) | Relative abundance
with trait (P75) |
|
|---|
| Phenotype of
prokaryotic microorganisms |
|
|
|
|
|---|
| Group A | Group B | Group A | Group B | Group A | Group B |
P-valuea |
|---|
| Aerobic | 0.080 | 0.103 | 0.038 | 0.041 | 0.153 | 0.213 | 0.603 |
| Anaerobic | 0.551 | 0.579 | 0.342 | 0.465 | 0.768 | 0.763 | 0.418 |
| Contains mobile
elements | 0.362 | 0.348 | 0.244 | 0.247 | 0.409 | 0.496 | 0.945 |
| Facultatively
anaerobic | 0.175 | 0.172 | 0.132 | 0.089 | 0.278 | 0.263 | 0.314 |
| Forms biofilms | 0.265 | 0.236 | 0.192 | 0.150 | 0.376 | 0.447 | 0.890 |
| Gram-negative | 0.589 | 0.667 | 0.482 | 0.513 | 0.687 | 0.785 | 0.039 |
| Gram-positive | 0.411 | 0.333 | 0.313 | 0.215 | 0.518 | 0.487 | 0.039 |
| Potentially
pathogenic | 0.174 | 0.129 | 0.108 | 0.059 | 0.225 | 0.295 | 0.555 |
| Stress
tolerant | 0.169 | 0.174 | 0.108 | 0.063 | 0.231 | 0.317 | 0.936 |
Discussion
The normal human microbiota serves a role in human
nutrition, drug metabolism, maintenance of the integrity of the
intestinal mucosal barrier, immunomodulation and protection against
pathogens (39). Changes in
microbial community composition are related to numerous diseases,
including tumors (40,41). Bacteria were first found in tumors
over a century ago (42). Different
tumor types have a unique flora; however, the characterization of
tumor microbiomes is often challenging because of their low biomass
(43). The microbiota, as a part of
the tumor microenvironment, serves an important role in
tumorigenesis and metastasis (44).
However, the composition of microbial communities in different
parts of the human body is not consistent
The amount of bacteria in the digestive tract is 10
times the total amount of human cells (45). Most bacteria have a specific spatial
distribution and are not cultivable (46,47).
The microbial communities in the mouth, esophagus and rectum vary
in type and quantity (48). The
composition of microbial communities may vary between different
organs of the same individual and different parts of the same organ
(41,48–51).
Therefore, in microbial community research, the influence of organs
and tissues on microbial communities needs to be considered. At
present, the gut microbiota is the most extensively explored
component of the digestive tract microbiota (52,53).
Different sampling methods may affect the results of research
examining microbial communities. In order to identify more
reasonable sampling methods, scholars have conducted extensive
research (54–58).
The esophagus contains numerous types of bacteria,
and abundant florae can be found between the oropharynx and the
stomach. Some esophageal florae in the stomach are similar to those
in the oral cavity, and the three different parts of the esophagus
have no specific bacteria (20,59).
The abundance of archaea and phages in a normal esophagus is low,
and a normal esophagus also contains Streptococcus, Prevotella,
Veillonella, Clostridium, Haemophilus, Neisseria, Porphyromonas
and other bacteria (17,60). Shao et al (61) found that the microbial environment
of ESCC is composed of Firmicutes, Bacteroidetes and
Proteobacteria. The abundance of Fusobacterium in tumors is
increased (3.2 vs. 1.3 %), whereas the abundance of
Streptococcus is decreased (12 vs. 30.2%) compared with that
in nontumor tissues (61).
Studies have been performed to improve the sampling
methods of esophageal flora. Liu et al (15) reported that swabs and biopsies of
patients with ESCC had similar microbial profiles. However, Gall
et al (20) suggested that
the amount of DNA recovered from a mucosal chip brush was greater
than that from mucosal samples in esophageal adenocarcinoma.
Okereke et al (62) studied
Barrett's esophagus and confirmed that swabs obtained from the
oropharynx or an endoscope could not replace biopsies of esophageal
mucosa. Further research also demonstrated that mucosal biopsy
should be used for the analysis of the esophageal flora (21).
α diversity can reflect the diversity of a microbial
community (63). The Chao1 index
describes the richness of a community and reflects the number of
microbial members, such as OTUs, in a community. The Shannon and
Simpson indices reflect the uniformity of a community and the
abundance of its members (63). The
present study revealed that the Chao1 and Shannon indices of the
postoperative tissue group were increased compared with those of
the mucosal tissue group. Although the Simpson index of the
postoperative tissue group was higher than that of the mucosal
tissue group, the difference between the two groups was not
statistically significant, suggesting that the postoperative tissue
flora was richer than the mucosal tissue flora, and the uniformity
was good, indicating that the distribution of bacteria in the
postoperative tissue group was uniform. The β diversity of the
microbiome refers to the differences between samples in colony
structures, which can be investigated at two sample sites,
ecological communities or populations (64). The two groups of bacteria had a
P-value >0.05, indicating that the diversity of the two groups
was not significantly different.
LEfSe analysis revealed that the flora of the two
groups included different species. Megasphaera, Actinobacteria,
Enterobacteriaceae and Enterobacteriales were more
abundant in the postoperative esophagus tissues than in the mucosal
tissues. Mogibacteriaceae was more abundant in the mucosal
tissue group than in the postoperative tissue group. The bacterial
species of the two groups were compared at the phylum and genus
levels. The predominant phyla in the postoperative tissue group
were Actinobacteria and Verrucomicrobiae. The
dominant phyla in the mucosal tissue group were Fusobacteria,
SR1 and Spirochaetes.
Analysis at the genus level revealed different
dominant bacteria in the two groups of flora. The different
distributions of flora in the esophageal tissues can be explained
as follows: The flora may participate in the occurrence and
development of ESCC, and the abundance of bacteria changes with the
tumor progression and invasion of ESCC (65,66).
The differences between the two groups might be caused by
variations in pH gastric acid, bile reflux, and other undetermined
factors (61,67).
The random forest method was adopted in the present
study, and the top 60 species were selected to establish a model.
The reliability of the model was verified using ROC curve analysis,
and the model could effectively distinguish between the two groups
of samples. BugBase is a microecological component analysis tool
that can identify high-level phenotypes present in microecological
samples and make phenotype predictions. Phenotypic types include
Gram-positive, Gram-negative, biofilm formation, pathogenicity,
mobile elements, oxygen demand (including anaerobic bacteria,
aerobic bacteria and facultative bacteria), and oxidative stress
tolerance (39). The comparison of
the BugBase phenotypes of the two groups showed differences in
Gram-negative and Gram-positive bacteria, and this finding might be
related to the aforementioned variation in the distribution of
bacterial groups. In the human body, by understanding the microbial
phenotype, more targeted treatments can be selected (68). The present study may provide a
reference for the study of the microbiota of esophageal cancer.
Although flora activity is not the only factor in
the pathogenesis of ESCC, dysbacteriosis may serve an important
role in the occurrence and development of ESCC (69). The present study demonstrated that
there were differences in the microbial composition between
postoperative esophageal cancer tissues and esophageal mucosal
tissues. The source of the sample should be considered in studies
on the esophageal flora. Considering the increased richness and
improved uniformity of postoperative tissue microbiota compared
with the mucosal group, it was predicted that postoperative tissue
may be more conducive to the study of esophageal cancer
microbiota.
The present study had some limitations that can
affect the interpretation of the results. First, the florae of
different parts of the esophagus and postoperative tissues were not
compared. Second, other sampling methods, such as endoscopic smear,
were not applied. Third, as aforementioned, two types of sources of
esophageal cancer tissue were included in the present study.
However, the postoperative tissues and endoscopic biopsy tissues
included in the study were not from the same patients. After the
esophageal mucosal tissue was sampled, it was divided into two
parts. One part was sent to the pathology department for further
pathological examination, and the other part was frozen for further
investigation. Only tissues confirmed by pathologists as esophageal
cancer were included in the present study. Similarly, the patients
included in the postoperative tissue group were all diagnosed with
ESCC by pathologists. In the present study, the esophageal mucosal
and postoperative esophageal cancer tissues were not obtained from
the same individuals for two main reasons. First, some patients are
diagnosed with esophageal cancer after they have completed
gastroscopy and pathological examination, but they may no longer be
suitable for direct surgery and instead choose radiotherapy,
chemotherapy or immunotherapy. For these patients, only endoscopic
tissue can be obtained and postoperative tissue cannot be obtained.
Second, some patients diagnosed with esophageal cancer may receive
further surgical treatment at a hospital near where they reside
instead, so it may not be possible to obtain postoperative samples.
Similarly, some patients who have been diagnosed with esophageal
cancer in other hospitals choose to undergo surgery at Shiyan Taihe
Hospital (Shiyan, China). As these patients did not undergo
gastroscopy examination at Shiyan Taihe Hospital, endoscopic
esophageal mucosal tissues from these patients could not be
obtained. Finally, the sample size of the present study was small
and the study included only two types of tissue. Future studies
should use a larger sample size and more types of esophageal tissue
to determine the best collection method for evaluating esophageal
samples. The present study included an analysis of the composition
of esophageal microbiota in postoperative tissues and mucosal
tissues of ESCC, and found that there were differences in microbial
composition between the two types of tissues. The optimal potential
biomarkers for distinguishing between the two tissues were
screened. This may provide a reference for sample selection in
future studies on the esophageal microbiome of patients with
ESCC.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank Dr Zi-Wei Fan and Dr
Jiang-Man Zhao from Shanghai Biotecan Pharmaceuticals Co., Ltd.
(Shanghai, China) for their assistance in the interpretation of
sequencing reports.
Funding
The present study was supported by the Health Commission of
Hubei Province scientific research project (grant nos. WJ2021M046
and WJ2023Q022), the Shiyan City Science and Technology Bureau
Guiding Research Project (grant no. 21Y19), and the Key Research
and Development Program of Shaanxi (grant no. 2021ZDLSF02-06).
Availability of data and materials
The data generated in the present study may be found
in the Sequence Read Archive database under accession number
Bioproject PRJNA779607 or at the following URL: https://www.ncbi.nlm.nih.gov/bioproject/?term=779607.
Authors' contributions
XBL, ZYG, QT and SXH contributed to the
conceptualization of the study, and reviewed and edited the
manuscript. XBL, JCM and ZYG wrote the manuscript. ZYG and JCM
performed statistical analyses. JRZ, WX and HW collected clinical
data and samples. QT and SXH contributed to funding acquisition and
editing. XBL and QT confirm the authenticity of all the raw data.
All authors revised the manuscript, and read and approved the final
manuscript.
Ethics approval and consent to
participate
The study protocol was reviewed and approved by the
Taihe Hospital Ethics Committee (approval no. 2018KS020; Shiyan,
China), and all patients received information concerning their
participation in the study and provided written informed
consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ESCC
|
esophageal squamous cell carcinoma
|
|
EC
|
esophageal carcinoma
|
|
OTUs
|
operational taxonomic units
|
|
NMDS
|
nonmetric multidimensional scaling
|
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Baba Y, Iwatsuki M, Yoshida N, Watanabe M
and Baba H: Review of the gut microbiome and esophageal cancer:
Pathogenesis and potential clinical implications. Ann Gastroenterol
Surg. 1:99–104. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Abnet CC, Arnold M and Wei WQ:
Epidemiology of esophageal squamous cell carcinoma.
Gastroenterology. 154:360–373. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen R, Zheng RS, Zhang SW, Zeng HM, Wang
SM, Sun KX, Gu XY, Wei WW and He J: Analysis of incidence and
mortality of esophageal cancer in China, 2015. Zhonghua Yu Fang Yi
Xue Za Zhi. 53:1094–1097. 2019.(In Chinese). PubMed/NCBI
|
|
5
|
Wen X, Wen D, Yang Y, Chen Y, Wang G and
Shan B: Urban-rural disparity in helicobacter pylori
infection-related upper gastrointestinal cancer in China and the
decreasing trend in parallel with socioeconomic development and
urbanization in an endemic area. Ann Glob Health. 83:444–462. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang Q, Rao Y, Guo X, Liu N, Liu S, Wen P,
Li S and Li Y: Oral microbiome in patients with oesophageal
squamous cell carcinoma. Sci Rep. 9:190552019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kwon YJ, Kwak HJ, Lee HK, Lim HC and Jung
DH: Comparison of bacterial community profiles from large intestine
specimens, rectal swabs, and stool samples. Appl Microbiol
Biotechnol. 105:9273–9284. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zoetendal EG, Raes J, van den Bogert B,
Arumugam M, Booijink CC, Troost FJ, Bork P, Wels M, de Vos WM and
Kleerebezem M: The human small intestinal microbiota is driven by
rapid uptake and conversion of simple carbohydrates. ISME J.
6:1415–1426. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cuevas-Sierra A, Riezu-Boj JI, Guruceaga
E, Milagro FI and Martinez JA: Sex-Specific associations between
gut prevotellaceae and host genetics on adiposity. Microorganisms.
8:9382020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
La-Ongkham O, Nakphaichit M, Nakayama J,
Keawsompong S and Nitisinprasert S: Age-related changes in the gut
microbiota and the core gut microbiome of healthy Thai humans. 3
Biotech. 10:2762020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Eck A, Rutten N, Singendonk M, Rijkers GT,
Savelkoul P, Meijssen CB, Crijns CE, Oudshoorn JH, Budding AE and
Vlieger AM: Neonatal microbiota development and the effect of early
life antibiotics are determined by two distinct settler types. PLoS
One. 15:e2281332020. View Article : Google Scholar
|
|
12
|
Neckovic A, van Oorschot R, Szkuta B and
Durdle A: Investigation of direct and indirect transfer of
microbiomes between individuals. Forensic Sci Int Genet.
45:1022122020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pan HW, Du LT, Li W, Yang YM, Zhang Y and
Wang CX: Biodiversity and richness shifts of mucosa-associated gut
microbiota with progression of colorectal cancer. Res Microbiol.
171:107–114. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Matsumoto H, Kuroki Y, Higashi S, Goda K,
Fukushima S, Katsumoto R, Oosawa M, Murao T, Ishii M, Oka K, et al:
Analysis of the colonic mucosa associated microbiota (MAM) using
brushing samples during colonic endoscopic procedures. J Clin
Biochem Nutr. 65:132–137. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu AQ, Vogtmann E, Shao DT, Abnet CC, Dou
HY, Qin Y, Su Z, Wei WQ and Chen W: A comparison of biopsy and
mucosal swab specimens for examining the microbiota of upper
gastrointestinal carcinoma. Cancer Epidemiol Biomarkers Prev.
28:2030–2037. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Peters BA, Wu J, Pei Z, Yang L, Purdue MP,
Freedman ND, Jacobs EJ, Gapstur SM, Hayes RB and Ahn J: Oral
microbiome composition reflects prospective risk for esophageal
cancers. Cancer Res. 77:6777–6787. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Deshpande NP, Riordan SM,
Castaño-Rodríguez N, Wilkins MR and Kaakoush NO: Signatures within
the esophageal microbiome are associated with host genetics, age,
and disease. Microbiome. 6:2272018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Elliott D, Walker AW, O'Donovan M,
Parkhill J and Fitzgerald RC: A non-endoscopic device to sample the
oesophageal microbiota: A case-control study. Lancet Gastroenterol
Hepatol. 2:32–42. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yu G, Gail MH, Shi J, Klepac-Ceraj V,
Paster BJ, Dye BA, Wang GQ, Wei WQ, Fan JH, Qiao YL, et al:
Association between upper digestive tract microbiota and
cancer-predisposing states in the esophagus and stomach. Cancer
Epidemiol Biomarkers Prev. 23:735–741. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gall A, Fero J, Mccoy C, Claywell BC,
Sanchez CA, Blount PL, Li X, Vaughan TL, Matsen FA, Reid BJ and
Salama NR: Bacterial composition of the human upper
gastrointestinal tract microbiome is dynamic and associated with
genomic instability in a Barrett's esophagus cohort. PLoS One.
10:e1290552015. View Article : Google Scholar
|
|
21
|
Okereke IC, Miller AL, Hamilton CF, Booth
AL, Reep GL, Andersen CL, Reynolds ST and Pyles RB: Microbiota of
the oropharynx and endoscope compared to the esophagus. Sci Rep.
9:102012019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fillon SA, Harris JK, Wagner BD, Kelly CJ,
Stevens MJ, Moore W, Fang R, Schroeder S, Masterson JC, Robertson
CE, et al: Novel device to sample the esophageal microbiome-the
esophageal string test. PLoS One. 7:e429382012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kageyama S, Takeshita T, Takeuchi K,
Asakawa M, Matsumi R, Furuta M, Shibata Y, Nagai K, Ikebe M, Morita
M, et al: Characteristics of the salivary microbiota in patients
with various digestive tract cancers. Front Microbiol. 10:17802019.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lederer AK, Rasel H, Kohnert E, Kreutz C,
Huber R, Badr MT, Dellweg P, Bartsch F and Lang H: Gut microbiota
in diagnosis, therapy and prognosis of cholangiocarcinoma and
gallbladder carcinoma-a scoping review. Microorganisms.
11:23632023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zeng R, Gou H, Lau HC and Yu J: Stomach
microbiota in gastric cancer development and clinical implications.
Gut. 17:gutjnl-2024-332815. 2024.
|
|
26
|
Li Z, Shi C, Zheng J, Guo Y, Fan T, Zhao
H, Jian D, Cheng X, Tang H and Ma J: Fusobacterium nucleatum
predicts a high risk of metastasis for esophageal squamous cell
carcinoma. BMC Microbiol. 21:3012021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li M, Shao D, Zhou J, Gu J, Qin J, Chen W
and Wei W: Signatures within esophageal microbiota with progression
of esophageal squamous cell carcinoma. Chin J Cancer Res.
32:755–767. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rognes T, Flouri T, Nichols B, Quince C
and Mahé F: VSEARCH: A versatile open source tool for metagenomics.
PeerJ. 4:e25842016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Quast C, Pruesse E, Yilmaz P, Gerken J,
Schweer T, Yarza P, Peplies J and Glöckner FO: The SILVA ribosomal
RNA gene database project: Improved data processing and web-based
tools. Nucleic Acids Res. 41:D590–D596. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Caporaso JG, Kuczynski J, Stombaugh J,
Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich
JK, Gordon JI, et al: QIIME allows analysis of high-throughput
community sequencing data. Nat Methods. 7:335–336. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huson DH and Weber N: Microbial community
analysis using MEGAN. Methods Enzymol. 531:465–485. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mitra S, Stärk M and Huson DH: Analysis of
16S rRNA environmental sequences using MEGAN. BMC Genomics. 12
(Suppl 3):S172011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Asnicar F, Weingart G, Tickle TL,
Huttenhower C and Segata N: Compact graphical representation of
phylogenetic data and metadata with GraPhlAn. PeerJ. 3:e10292015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Segata N, Izard J, Waldron L, Gevers D,
Miropolsky L, Garrett WS and Huttenhower C: Metagenomic biomarker
discovery and explanation. Genome Biol. 12:R602011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Breiman L: Random forests. Machine
Learning. 45:5–32. 2001. View Article : Google Scholar
|
|
36
|
Liaw A and Wiener M: Classification and
regression by randomforest. R News. 2:18–22. 2002.
|
|
37
|
Ward T, Larson J, Meulemans J, Hillmann B,
Lynch J, Sidiropoulos D, Spear J, Caporaso G, Blekhman R, Knight R,
et al: BugBase predicts organism level microbiome phenotypes:
bioRxiv. 2:doi.org/10.1101/133462. 2017.
|
|
38
|
Thomas AM, Jesus EC, Lopes A, Aguiar S Jr,
Begnami MD, Rocha RM, Carpinetti PA, Camargo AA, Hoffmann C,
Freitas HC, et al: Tissue-associated bacterial alterations in
rectal carcinoma patients revealed by 16S rRNA community profiling.
Front Cell Infect Microbiol. 6:1792016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Human Microbiome Project Consortium, .
Structure, function and diversity of the healthy human microbiome.
Nature. 486:207–214. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lloyd-Price J, Abu-Ali G and Huttenhower
C: The healthy human microbiome. Genome Med. 8:512016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Vuik F, Dicksved J, Lam SY, Fuhler GM, van
der Laan L, van de Winkel A, Konstantinov SR, Spaander M,
Peppelenbosch MP, Engstrand L and Kuipers EJ: Composition of the
mucosa-associated microbiota along the entire gastrointestinal
tract of human individuals. United European Gastroenterol J.
7:897–907. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rius-Rocabert S, Llinares PF, Pozuelo MJ,
Garcia A and Nistal-Villan E: Oncolytic bacteria: Past, present and
future. Fems Microbiol Lett. 366:fnz1362019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nejman D, Livyatan I, Fuks G, Gavert N,
Zwang Y, Geller LT, Rotter-Maskowitz A, Weiser R, Mallel G, Gigi E,
et al: The human tumor microbiome is composed of tumor
type-specific intracellular bacteria. Science. 368:973–980. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang H, Fu L, Leiliang X, Qu C, Wu W, Wen
R, Huang N, He Q, Cheng Q, Liu G and Cheng Y: Beyond the gut: The
intratumoral microbiome's influence on tumorigenesis and treatment
response. Cancer Commun (Lond). 44:1130–1167. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Langille MG, Zaneveld J, Caporaso JG,
Mcdonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega
Thurber RL, Knight R, et al: Predictive functional profiling of
microbial communities using 16S rRNA marker gene sequences. Nat
Biotechnol. 31:814–821. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Garcia MD, Sanabria J, Wist J and Holmes
E: Effect of operational parameters on the cultivation of the gut
microbiome in continuous bioreactors inoculated with feces: A
systematic review. J Agric Food Chem. 71:6213–6225. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang S, Cao X and Huang H: Sampling
strategies for three-dimensional spatial community structures in
IBD microbiota research. Front Cell Infect Microbiol. 7:512017.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Jandhyala SM, Talukdar R, Subramanyam C,
Vuyyuru H, Sasikala M and Reddy DN: Role of the normal gut
microbiota. World J Gastroenterol. 21:8787–8803. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kashiwagi S, Naito Y, Inoue R, Takagi T,
Nakano T, Inada Y, Fukui A, Katada K, Mizushima K, Kamada K, et al:
Mucosa-associated microbiota in the gastrointestinal tract of
healthy Japanese subjects. Digestion. 101:107–120. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Saffarian A, Mulet C, Regnault B, Amiot A,
Tran-Van-Nhieu J, Ravel J, Sobhani I, Sansonetti PJ and Pedron T:
Crypt- and mucosa-associated core microbiotas in humans and their
alteration in colon cancer patients. mBio. 10:e01315–e01319. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Vasapolli R, Schütte K, Schulz C, Vital M,
Schomburg D, Pieper DH, Vilchez-Vargas R and Malfertheiner P:
Analysis of transcriptionally active bacteria throughout the
gastrointestinal tract of healthy individuals. Gastroenterology.
157:1081–1092. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Richard ML and Sokol H: The gut mycobiota:
Insights into analysis, environmental interactions and role in
gastrointestinal diseases. Nat Rev Gastroenterol Hepatol.
16:331–345. 2019.PubMed/NCBI
|
|
53
|
Yuan X, Chang C, Chen X and Li K: Emerging
trends and focus of human gastrointestinal microbiome research from
2010–2021: A visualized study. J Transl Med. 19:3272021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Budding AE, Grasman ME, Eck A, Bogaards
JA, Vandenbroucke-Grauls CM, van Bodegraven AA and Savelkoul PH:
Rectal swabs for analysis of the intestinal microbiota. PLoS One.
9:e1013442014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Araujo-Perez F, Mccoy AN, Okechukwu C,
Carroll IM, Smith KM, Jeremiah K, Sandler RS, Asher GN and Keku TO:
Differences in microbial signatures between rectal mucosal biopsies
and rectal swabs. Gut Microbes. 3:530–535. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kotar T, Pirš M, Steyer A, Cerar T, Soba
B, Skvarc M, Poljsak PM and Lejko ZT: Evaluation of rectal swab use
for the determination of enteric pathogens: A prospective study of
diarrhoea in adults. Clin Microbiol Infect. 25:733–738. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Fair K, Dunlap DG, Fitch A, Bogdanovich T,
Methé B, Morris A, Mcverry BJ and Kitsios GD: Rectal swabs from
critically Ill patients provide discordant representations of the
gut microbiome compared to stool samples. mSphere. 4:e00358–e00319.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hernandez-Arriaga A, Baumann A, Witte OW,
Frahm C, Bergheim I and Camarinha-Silva A: Changes in oral
microbial ecology of C57BL/6 mice at different ages associated with
sampling methodology. Microorganisms. 7:2832019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Dong L, Yin J, Zhao J, Ma SR, Wang HR,
Wang M, Chen W and Wei WQ: Microbial similarity and preference for
specific sites in healthy oral cavity and esophagus. Front
Microbiol. 9:16032018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
El-Zimaity H, Di Pilato V, Novella RM,
Brcic I, Rajendra S, Langer R, Dislich B, Tripathi M, Guindi M and
Riddell R: Risk factors for esophageal cancer: Emphasis on
infectious agents. Ann N Y Acad Sci. 1434:319–332. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Shao D, Vogtmann E, Liu A, Qin J, Chen W,
Abnet CC and Wei W: Microbial characterization of esophageal
squamous cell carcinoma and gastric cardia adenocarcinoma from a
high-risk region of China. Cancer. 125:3993–4002. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Okereke I, Hamilton C, Reep G, Krill T,
Booth A, Ghouri Y, Jala V, Andersen C and Pyles R: Microflora
composition in the gastrointestinal tract in patients with
Barrett's esophagus. J Thorac Dis. 11:S1581–S1587. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Li W, Huang Y, Tong S, Wan C and Wang Z:
The characteristics of the gut microbiota in patients with
pulmonary tuberculosis: A systematic review. Diagn Microbiol Infect
Dis. 109:1162912024. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Maki KA, Kazmi N, Barb JJ and Ames N: The
oral and gut bacterial microbiomes: Similarities, differences, and
connections. Biol Res Nurs. 23:7–20. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Gao S, Li S, Ma Z, Liang S, Shan T, Zhang
M, Zhu X, Zhang P, Liu G, Zhou F, et al: Presence of porphyromonas
gingivalis in esophagus and its association with the
clinicopathological characteristics and survival in patients with
esophageal cancer. Infect Agent Cancer. 11:32016. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Liu Y, Lin Z, Lin Y, Chen Y, Peng XE, He
F, Liu S, Yan S, Huang L, Lu W, et al: Streptococcus and prevotella
are associated with the prognosis of oesophageal squamous cell
carcinoma. J Med Microbiol. 67:1058–1068. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Soroush A, Etemadi A and Abrams JA:
Non-acid fluid exposure and esophageal squamous cell carcinoma. Dig
Dis Sci. 67:2754–2762. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Song X, Greiner-Tollersrud OK and Zhou H:
Oral microbiota variation: A risk factor for development and poor
prognosis of esophageal cancer. Dig Dis Sci. 67:3543–3556. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Gao S, Zhang Z, Sun K, Li MX and Qi YJ:
Upper gastrointestinal tract microbiota with oral origin in
relation to oesophageal squamous cell carcinoma. Ann Med.
55:22954012023. View Article : Google Scholar : PubMed/NCBI
|