Introduction
Pleuropulmonary blastoma (PPB) predominantly
manifests in infants, particularly those <6 years old (1). According to the data of the
International PPB registry, the incidence of PPB is 0.35–0.65/100
000 (2). According to the
pathological findings (3), PPB can
be divided into three types: Type I is cystic without solid
component; Type II is a cystic-solid mass; and type III is a
complete solid mass with liquefaction and necrosis. The prognosis
of PPB is significantly correlated with the pathological type and
stage of the tumor. Type I PPB can progress to Type II and III, and
the prognosis of type I PPB is significantly more favorable than
that of Type II and III PPB, since type II and III PPB often have
recurrence and distant metastasis. According to a study (4), the 5-year survival rate of patients
with Type I PPB is 82%, and that of Type II and III PPB is 71 and
53%, respectively. Therefore, early diagnosis of PPB is necessary,
which is of great significance to improve the survival rate of
patients.
The primary clinical manifestations are respiratory
symptoms, including cough, fever, dyspnea and respiratory distress.
Imaging examinations can enable in the diagnostic process; however,
pathological examination remains the ‘gold standard’ for the
diagnosis of PPB. The pathological classification of PPB is highly
linked to both the treatment approach and the prognosis of the
patient. Therefore, accurate pathological classification is
crucial. Type I and Type Ir PPB (also known as degenerative or
static PPB, it is so named since only well-differentiated fibers
and cartilage can be seen in the tumor capsule wall and the lack of
primitive childish embryonic components) are primarily managed with
surgical intervention, supplemented by radiotherapy if required,
whereas Types II and III necessitate chemotherapy following
surgical resection. The exact pathogenesis of PPB remains unknown
(5).
The present report describes the case of PPB and
analyses its clinical manifestations, imaging features,
pathological features and molecular genetic changes to improve the
understanding of this disease.
Case report
A 3-year-old female patient was admitted to the
Affiliated Hospital of Zunyi Medical University (Zunyi, China) in
May 2023 for further evaluation after presenting with a 2-month
history of cough without obvious inducement and without symptoms
including sputum, fever, chills, shortness of breath, dyspnea,
abdominal pain, abdominal distension, nausea, vomiting, urinary
frequency, and urgency. The symptoms had not markedly improved
after oral anti-inflammatory treatment. A chest X-ray revealed
bronchitis and a mass in the right thoracic cavity (data not
shown), prompting a recommendation for a contrast-enhanced chest
computed tomography (CT) scan. Physical examination showed a good
mental state, no evident three concave signs, a stable and regular
breathing rhythm, symmetrical thorax bilaterally, no pleural
friction rub and no subcutaneous crepitus. The right lung exhibited
hyperresonance with distinct fine moist rales. A chest CT plain
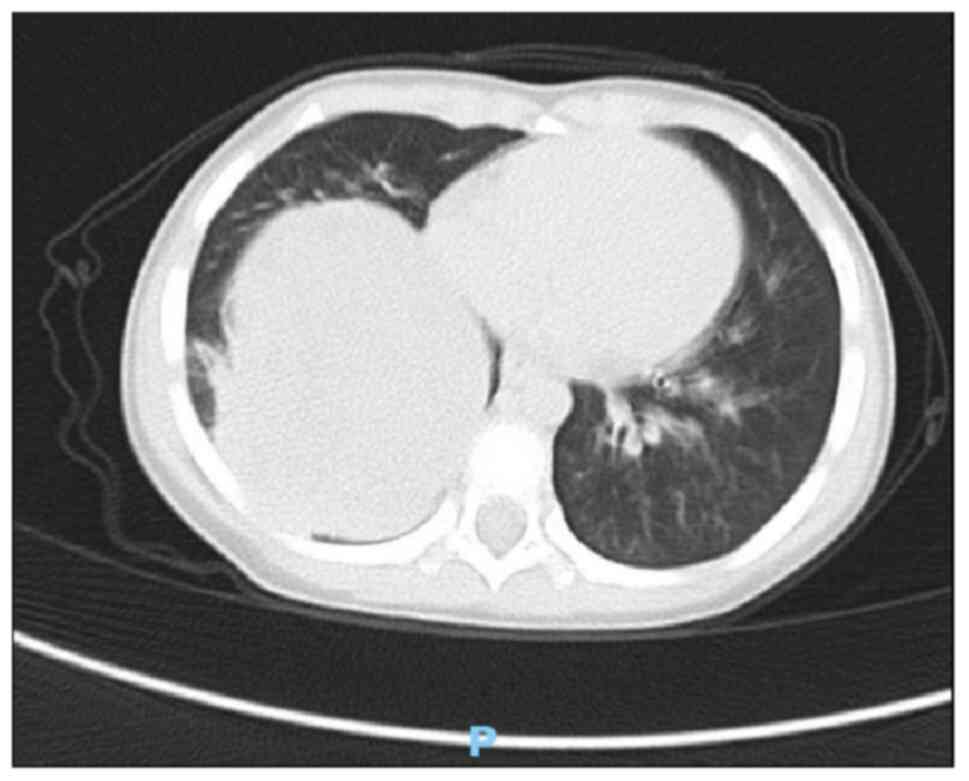
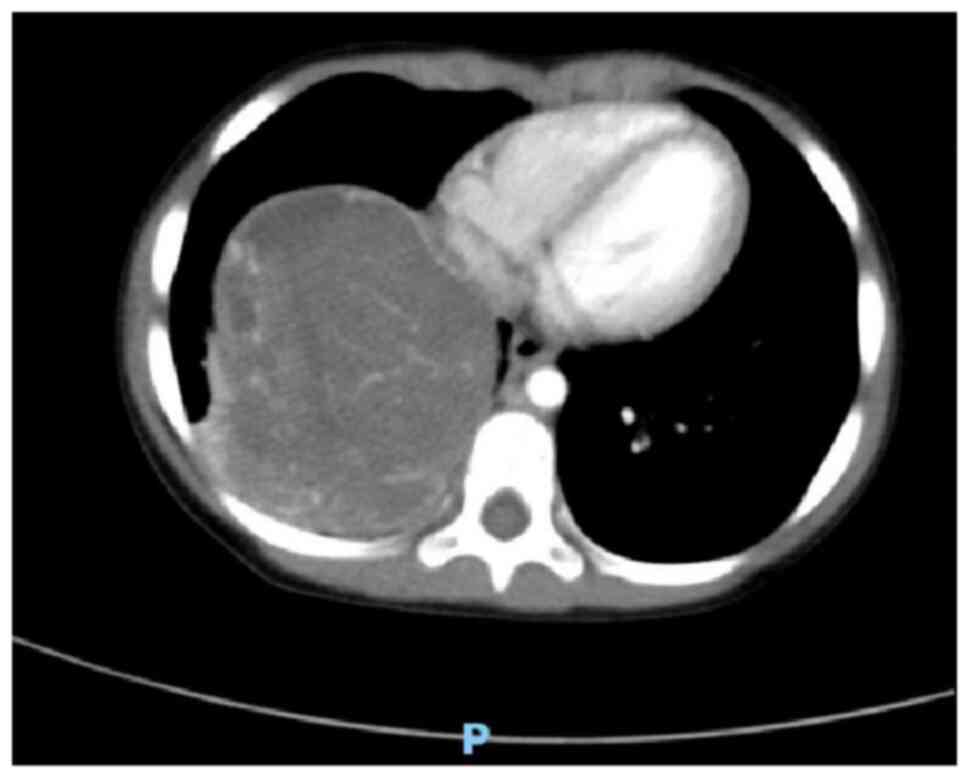
scan and enhanced scan revealed a mass measuring ~85×74×68 mm in
the right lower thoracic cavity with uneven density, clear
boundaries, and significant heterogeneous enhancement on the
contrast-enhanced scan (Figs. 1 and
2). Multiple tortuous vascular
shadows were noted within the mass. The right lower lobe was
compressed, and flaky increased density shadows were observed in
the right middle and lower lobes. The mediastinum remained
centered, with no enlarged lymph nodes in the mediastinum or
bilateral pulmonary hila. The size and morphology of the heart
appeared normal. These findings indicated a space-occupying lesion
in the right thoracic cavity, which was considered a neoplastic
lesion likely representing PPB.
Following the completion of relevant examinations
(including routine blood work, liver function test, coagulation
function test, HIV+, syphilis+ hepatitis C
and other infection indicators, stool and urine test, heart color
Doppler ultrasound and electrocardiogram) and the exclusion of
surgical contraindications, a thoracoscopic right middle and lower
lobectomy with thoracic lesion resection was performed.
Intraoperative findings revealed firmer lung tissue in the right
middle and lower lobes, with the tumor occupying nearly the entire
middle and lower lobes, measuring 80×70×60 mm, with indistinct
boundaries and slightly thickened interlobar fissures.
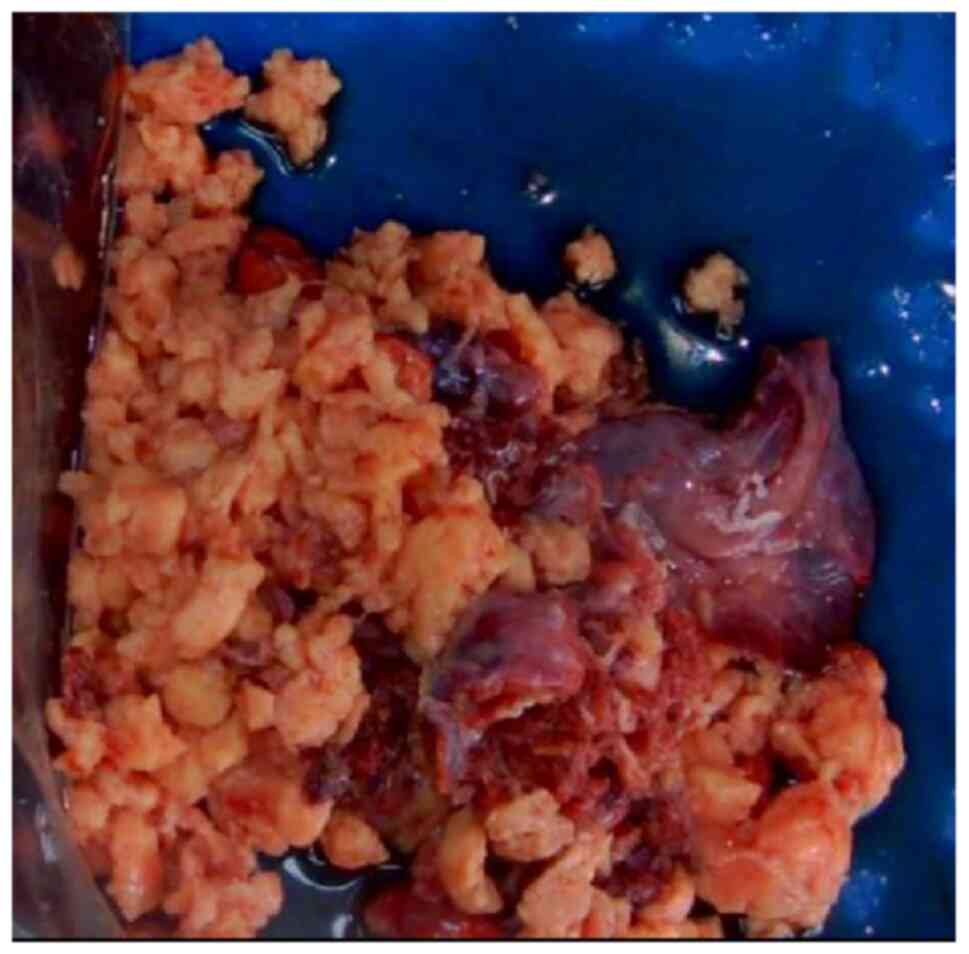
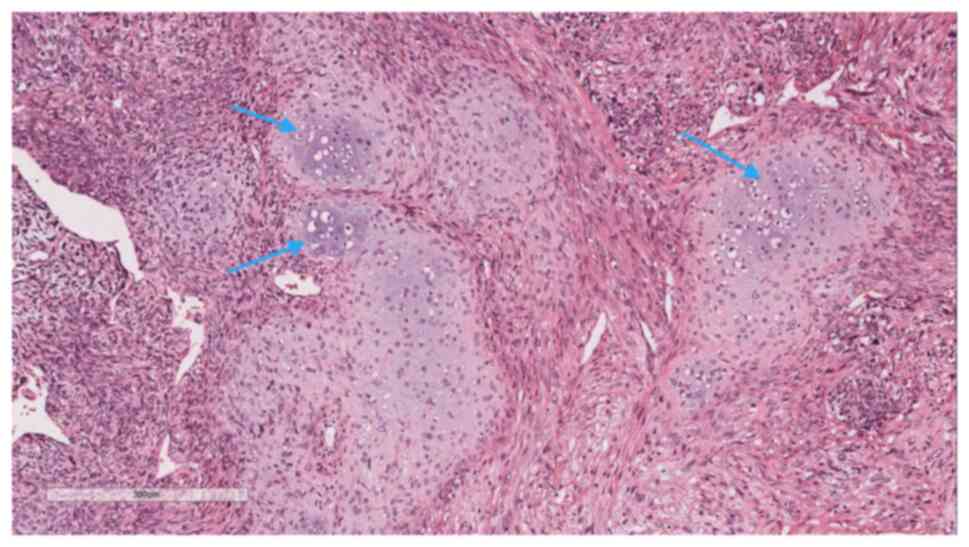
A pathological biopsy examination was performed on
the lung tissue specimen, measuring 92×85×76 mm. Gross examination
revealed a mass of fragmented gray-white to gray-brown tissue,
~81×72×58 mm in size (Fig. 3). The
cut surface appeared gray-white and solid, with a delicate cut
surface. The boundary between the mass and the surrounding lung
tissue was relatively distinct, with certain areas exhibiting tight
adhesion to the visceral pleura. The specimens were fixed in 4%
neutral formalin at room temperature for 12 h, followed by routine
dehydration, paraffin embedding and sectioning at a thickness of 5
µm. Hematoxylin and eosin staining was then performed at room
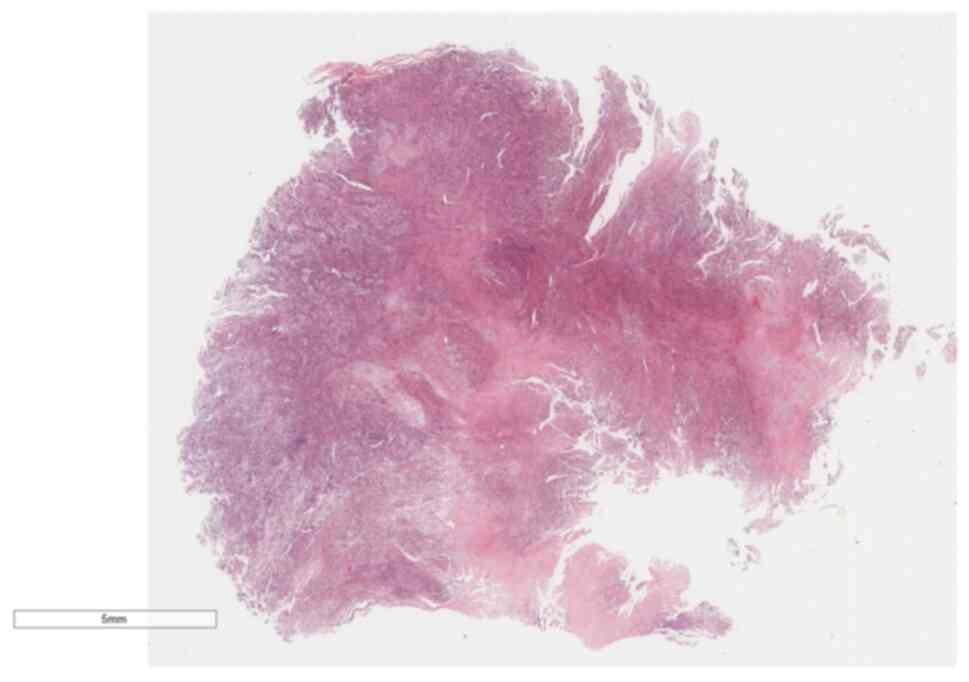
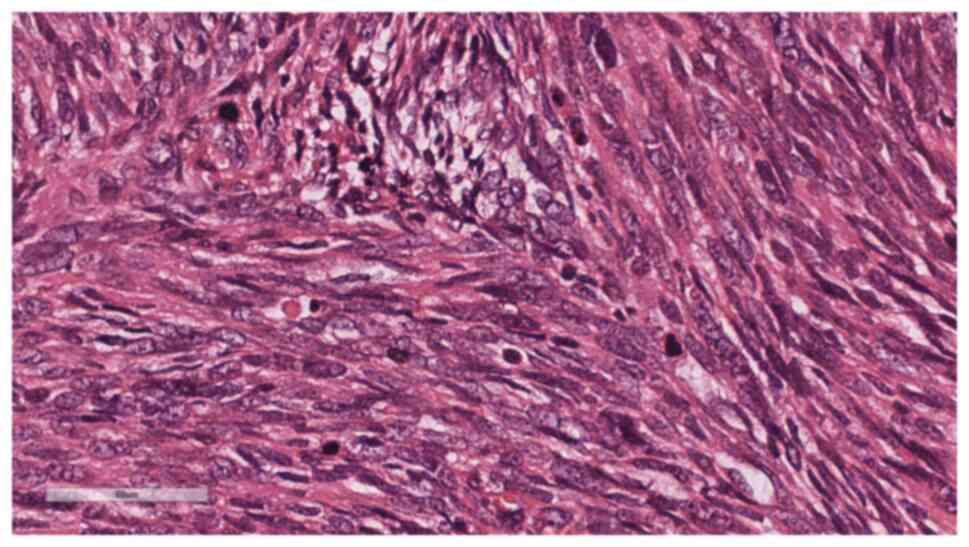
temperature for 5 min each. Microscopic examination (Leica
Biosystems) at low magnification revealed tumor cells diffusely
distributed in sheets with high cellular density, and a few
glandular-like structures were visible between the spindle-shaped
solid sheets of tissue (Fig. 4). At
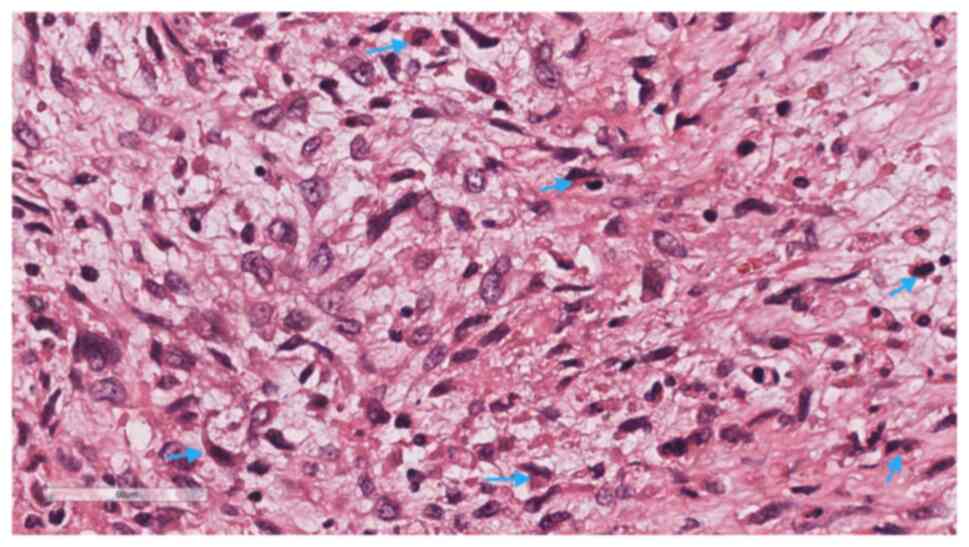
high magnification, the solid part was composed of immature
primitive embryonic components (Fig.
5), lacking malignant epithelial elements. In certain areas,
embryonal rhabdomyosarcoma (ERMS)-like differentiation (Fig. 6) and immature chondrosarcoma-like
differentiation (Fig. 7) were
observed. The tumor cells exhibited anaplastic changes, with
hyperchromatic enlarged nuclei, an increased nucleo-cytoplasmic
ratio, and the presence of tumor giant cells and pathological
mitotic figures.
For immunohistochemistry, the specimens were fixed
in 10% neutral formalin (12 h) at 28°C., followed by routine
dehydration, paraffin embedding and sectioning at a thickness of 3
µm. Immunohistochemistry using the Envision twostep method was
employed to assess the expression of relevant proteins in the tumor
tissue. The staining procedures were performed strictly according
to the manufacturer's instructions (all primary antibodies used
were rabbit or mouse anti-human monoclonal antibodies, purchased
from Fuzhou Maixin Biotechnology Development Co. Ltd., and were
used at a working concentration of 1:100). The primary antibodies
were added to the sections and incubated overnight (12 h) at 4°C.
The sections were then prewarmed at 37°C for 30 min, washed 3 times
(5 min/time) with PBS (cat. no. TW-0821), incubated in an oven at
37°C for 15 min with secondary antibody (cat. no. DNS-0811) and
washed 3 times (5 min/time) with PBS. DAB color development (cat.
no. TT-0801; 1:20 preparation) was performed. Microscopic
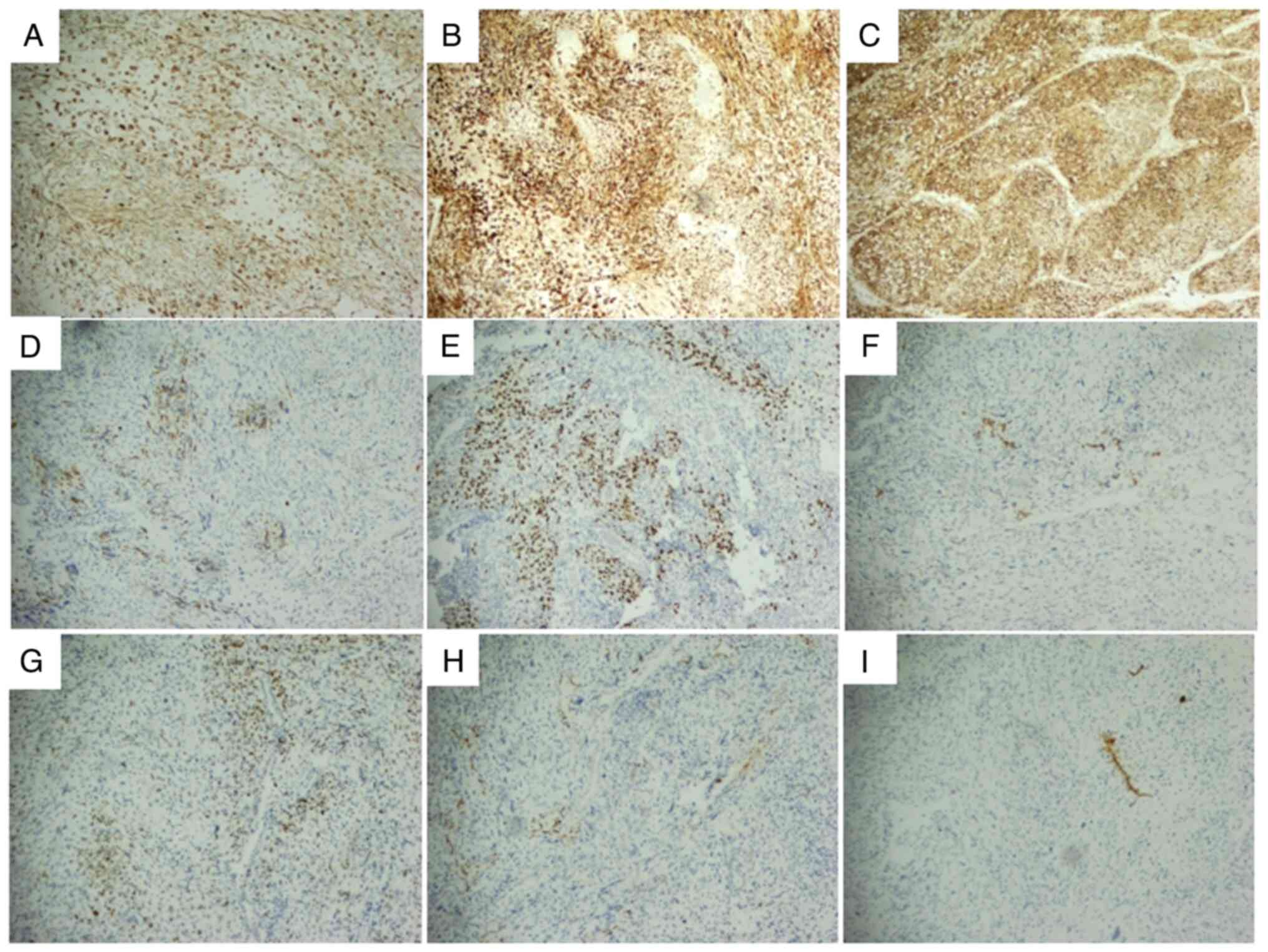
examination (Leica Biosystems) results demonstrated the following:
The tumor cells exhibited positivity for Vimentin (cat. no.
RMA0547), CD10 (cat. no. MAB0668) and CD56 (cat. no. MAB0743)
(Fig. 8A-C). Desmin, myogenic
differentiation 1 (MyoD1) (cat. no. MAB0822) and CD99 (cat. no.
MAB0059) were focally or sporadically positive (Fig. 8D-F). Spalt like transcription factor
4 (cat. no. MAB0691) demonstrated focal positivity (Fig. 8G), whilst smooth muscle actin (SMA;
cat. no. Kit0006) showed local focal positivity (Fig. 8H). Myogenin (cat. no. MAB0866)
exhibited local scattered focal positivity (Fig. 8I). Human Melanoma Black 45 (cat. no.
MAB0098) was sporadically positive in certain regions and CD34
(cat. no. Kit0004) displayed local positivity (Fig. S1). CK-Pan (AE1/AE3; cat. no.
Kit0009), epithelial membrane antigen (cat. no. Kit0011), thyroid
transcription factor-1 (TTF-1; cat. no. MAB0677) and Catenin (cat.
no. MAB0754) were positive in the mature epithelium entrapped
within the tumor, and signal transducer and activator of
transcription 6 (cat. no. MRA0845), anaplastic lymphoma kinase
(ALK; cat. no. MAB0848), CD21 (cat. no. MAB0708), Chromogranin A
(cat. no. MAB0707), OCT3/4 (cat. no. MAB0874), S-100 (cat. no.
Kit0007) and Synaptophysin (cat. no. MAB0742) were all negative
(Fig. S2).
Based on the clinical history, pathological
morphological characteristics and immunohistochemical results, the
diagnosis was as follows: PPB (Type III) of the right middle and
lower lobes, accompanied by rhabdomyosarcoma, chondrosarcoma and
other differentiation. The tumor involved the visceral pleura,
whilst the bronchial and pulmonary stumps were not affected by the
tumor.
After surgery, the patient was treated with IVADo
(ifosfamide + vincristine + actinomycin D + adriamycin) regimen for
12 courses in Chongqing Children's Hospital (Chongqing, China).
IVADo chemotherapy consisted of ifosfamide 3 g/m2/dose
IV on days 1 and 2 (6 g/m2/cycle) with MESNA,
vincristine 1.5 mg/m2 IV on day 1 (maximum 2 mg),
actinomycin-D 1.5 mg/m2 IV on day 1 (maximum 2 mg) and
doxorubicin 30 mg/m2/dose IV on days 1 and 2 (60
mg/m2/cycle) for four 21-day cycles followed by IVA
(ifosfamide, vincristine and actinomycin-D) on day 1 for eight
21-day cycles. The patient was reexamined by chest and abdominal
imaging every 3 months. At present, the patient has recovered well
and has had no recurrence.
Discussion
Characteristics of the disease
Malignant lung tumors in infants are rare,
constituting only 0.5–1.0% of primary lung neoplasms at the global
level (6). PPB, first characterized
in 1988, is identified as a malignant tumor arising within the
pulmonary interstitium of infants and is the most prevalent primary
lung tumor in this demographic, typically originating from the lung
and/or pleura (7). It predominantly
comprises malignant primitive embryonic mesenchymal components
along with benign pulmonary epithelial elements. The clinical
manifestations of PPB are nonspecific, primarily including
respiratory symptoms such as cough, sputum and dyspnea.
Consequently, PPB is often missed or misdiagnosed (5). Furthermore, the majority of cases
documented in the literature pertain to infants <6 years old
(4,8), though instances have been noted in
adolescents (9) and even adults
(10–12). Moreover, the incidence rate shows no
significant difference between sexes. Common metastatic sites
encompass the brain, bone, spleen, lymph nodes, kidneys, pancreas
and adrenal glands (12–14). In the present case, PPB manifested
in the lung of a 3-year-old patient, with cough as the initial
symptom.
Imaging characteristics
Imaging examinations, particularly chest CT scans,
are valuable in differentiating the characteristics of lung
lesions. These scans can not only identify the heterogeneity of PPB
but also provide diagnostic insights into the presence of pleural
effusion and chest wall invasion. For larger masses, a biopsy can
be performed under CT guidance to confirm the diagnosis (15). In the present case, CT revealed a
right lung mass identified as a neoplastic lesion, possibly
indicative of PPB. Differentiation between neurogenic tumors and
germ cell-derived tumors is necessary and should be corroborated
with clinical and pathological findings. Typically, PPB presents as
a solitary, well-defined mass that can exceed 10 cm (13). Based on histological variations, CT
scans display different imaging characteristics. Type I PPB lesions
often appear as single or multiple subpleural or intrapulmonary
cystic masses, requiring differential diagnosis from other cystic
lesions such as bronchogenic cysts, pulmonary cysts, pulmonary
bullae and interstitial emphysema. Type II PPB frequently presents
as intrapulmonary cystic and solid masses, whilst type III PPB
predominantly manifests as solid lesions with uniform density and
clear boundaries. The differential diagnosis of type II and III
PPB, especially when locally invasive, often necessitates
differentiation from other malignant tumors, such as neuroblastoma,
Ewing's sarcoma and rhabdomyosarcoma. Although CT provides valuable
reference data for clinicians and pathologists in diagnosing PPB,
it is not definitive, and the final diagnosis relies on
pathological examination (3,4).
Pathological characteristics
According to the Dehner classification (14), PPB is categorized into type I
(completely cystic, with a better prognosis), type II (cystic and
solid) and type III (completely solid, with a poor prognosis).
Certain studies further classify PPB into four types (Ir, I, II and
III) based on gross pathological appearance and morphological
characteristics. Type I cystic PPB may progress to invasive types
II and III but may also regress to type Ir, where ‘r’ denotes
regression, characterized by the absence of malignant tumor cells.
Types II and III PPB are invasive (14,16,17).
Types I and Ir exhibit a lower degree of malignancy, whereas types
II and III are highly malignant and invasive. Type I PPB is
typically unilateral, solitary, peripheral and >5 cm (4). Microscopically, cystic structures with
fibrous septa containing immature mesenchymal cells and benign
respiratory epithelium are observed. In type Ir, the cyst wall
fibrous septa lack immature cells compared with type I. Type II PPB
is characterized by both cystic and solid areas, with nodular solid
regions containing undifferentiated ovoid and stellate cells
growing in sheets, whilst the cystic areas resemble those in type I
PPB. Type III is purely solid and microscopically appears as a
mixture of blast cells with sarcomatoid areas (chondrosarcomatous,
fibrosarcomatous, rhabdomyosarcomatous and anaplastic components)
present, often with frequent mitotic figures. Types II and III PPB
are histologically similar in the solid areas, necessitating
adequate sampling for evaluation (14,18).
The immunophenotype of PPB is non-specific. Tumor cells express
Vimentin and, in most cases, muscle-specific actin. Depending on
differentiation, rhabdomyoblastic areas express desmin, MyoD1 and
Myogenin, whilst cartilaginous areas express S-100. CK and TTF-1
mark cystic benign epithelial cells and benign epithelial cells
entrapped within the tumor substance (19).
Differential diagnosis
Type III PPB must be distinguished from the
following tumors: i) Classic pulmonary blastoma, which is a
biphasic highly malignant tumor containing both malignant
epithelial and mesenchymal components, whereas PPB comprises benign
epithelial and malignant mesenchymal components. The primary
distinguishing factor between the two is the presence or absence of
malignant epithelial cells observed microscopically; ii) ERMS, as
both ERMS and PPB exhibit concurrent mutations in Dcr-1 homolog and
ribonuclease type III (DICER1), making immunohistochemistry crucial
for differentiation. ERMS is a highly malignant soft tissue sarcoma
originating from mesenchymal stem cells during the embryonic
period, characterized by differentiation towards rhabdomyosarcoma.
It presents primitive undifferentiated stellate cells, small round
cells and well-differentiated cells that stain strongly with eosin,
displaying tadpole-shaped, spindle-shaped, ribbon-like, tennis
racket-like and large round tumor cells. ERMS shows diffuse strong
expression of MyoD1 and Myogenin, whereas PPB expresses these
markers focally when rhabdomyoblastic differentiation is present
(19). Furthermore, a previous
study reported that insulin-like growth factor is overexpressed in
ERMS but under-expressed in PPB (20). Immunohistochemistry is essential for
differentiating between these two entities; iii) mesenchymal
chondrosarcoma (MC), a highly malignant biphasic tumor comprising
undifferentiated primitive round cells and well-differentiated
hyaline cartilage. The tumor cells are small to medium-sized round
cells that express CD99, unlike PPB. Cartilage differentiation in
MC can vary from loosely arranged small lesions to
well-differentiated large areas of mature cartilage, often
containing large granular calcifications. MC is characterized by
the HEY1-NCOA2 fusion gene, whereas PPB primarily exhibits
heterozygous germline mutations in DICER1 (21); iv) inflammatory myofibroblastic
tumor (IMT), which the World Health Organization, in 2020, defined
as a unique, rarely metastatic tumor composed of spindle-shaped
myofibroblasts and fibroblasts accompanied by plasma cells,
lymphocytes, eosinophils and other inflammatory cells (22). The immunophenotype of IMT expresses
Vimentin and shows varying degrees of expression of SMA, calponin
and desmin. A total of 50–60% of cases are ALK-positive, with about
2/3 of IMTs exhibiting ALK gene rearrangements. By contrast, PPB
does not express ALK or have ALK gene rearrangements; and v)
primitive neuroectodermal tumor (PNET), which is primarily composed
of small round and short spindle-shaped primitive cells with
diffuse distribution. Homer-Wright pseudorosettes may be observed.
PNET expresses neuron-specific enolase and synaptophysin, which PPB
does not express (23).
The patient in the present case had type III PPB
with rhabdomyosarcoma, chondrosarcoma and other differentiations,
accompanied by pleural invasion. During diagnosis, it is crucial to
differentiate PPB from classic pulmonary blastoma, ERMS and MC.
Moreover, differentiation should be based on clinical history,
characteristic pathological morphology and immunohistochemical
results.
Genetic modification
PPB and other tumors exhibit familial
characteristics. Whole-exome sequencing has identified heterozygous
germline mutations in DICER1 in infants with PPB. The DICER1
protein belongs to the ribonuclease III enzyme family (24) and the DICER1 gene is a highly
conserved gene located on chromosome 14q32.13, encoding 1,992 amino
acids and comprising 27 exons. This gene controls the production of
the ribonuclease enzyme, Dicer1, which serves a critical role in
regulating protein translation (25). Data from the International PPB
Registry indicate that ~70% of patients with PPB possess DICER1
gene mutations (8). Common
pathogenic mutations in the DICER1 gene include loss-of-function
(LOF) mutations and hotspot missense mutations in the RNase IIIb
domain, with LOF mutations being the predominant mode of familial
inheritance. There is a marked association between DICER1 and
Sertoli-Leydig cell tumors (and ovarian germ cell tumors),
suggesting that patients with these tumors should be tested for
germline or somatic DICER1 mutations. Thyroid nodular hyperplasia,
with or without papillary thyroid carcinoma, may be the most common
associated pathology in DICER1 germline mutation carriers. Other
DICER1-related tumors include cervical (non-vaginal) ERMS, ciliated
müllerian mixed tumor, nasal chondromesenchymal hamartoma, small
intestinal hamartomatous polyps and pituitary blastoma. Any of
these tumors, individually or in combination, may indicate
DICER1-related syndrome. Therefore, it is particularly important
for clinicians and pathologists to raise this concern and recommend
genetic counseling and testing (14). From the clinical history, typical
morphology and immunohistochemical results in the present case, the
diagnosis was clear; however, genetic testing was not available,
which was a limitation.
Treatment and prognosis
The treatment of PPB includes surgery, chemotherapy,
radiotherapy and neoadjuvant therapy. Complete surgical resection
is the main goal of treatment for children with PPB. It has also
been reported that neural cell adhesion molecule 1/fibroblast
growth factor module can be used as a therapeutic target for
pleuropulmonary blastoma (26).
Type I and Ir PPB have not been reported to have metastases and are
treated with complete resection with wide negative margins.
Systemic chemotherapy and surgical resection are the key to the
treatment of type II and type III PPB. Neoadjuvant chemotherapy
after biopsy can be used for large tumors that are difficult to
resect and with unpredictable negative margins. Furthermore, the
prognosis of PPB is related to the pathological classification. The
5-year overall survival rate of type I and Ir PPB has been reported
to be 98.0%, and that of type II and III PPB is 71 and 53%,
respectively (4).
The present report describes a rare case of PPB
(type III) with rhabdomyosarcoma and chondrosarcoma differentiation
occurring in the middle and lower lobe of the right lung. Regular
chemotherapy was performed after surgery, and the patient was
followed up for 12 months. Therefore, the present report
demonstrates that a comprehensive description of PPB, including
clinical manifestations, imaging features, pathological features
and molecular genetic changes provide a good reference value for
clinical diagnosis and treatment.
PPB is the most common pulmonary malignant tumor in
children, its incidence is low, it is difficult to distinguish from
other lung space occupying lesions, it is difficult to diagnose
early, and it often progresses into a pathological type with a poor
prognosis. The disease is closely related to a DICER1 gene
variation, and children with DICER1 syndrome and their relatives
are at risk of developing multisystem tumors. The clinical
manifestations of children with PPB should be paid attention to,
lung imaging and pathological examinations in the early stage
should be performed, as well as genetic testing if necessary and
long-term monitoring of the family of a patient with DICER1
syndrome.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SL and JW analyzed the data, conducted the
histopathological evaluation and assisted in writing the
manuscript.. XT acquired the CT scan images. TX performed the
immunohistochemical staining. XT analyzed the patient data. JJW and
SL confirm the authenticity of all the raw data. All authors have
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The present case report was approved by the Ethics
Committee of the Affiliated Hospital of Zunyi Medical University
(Zunyi, China; approval no. KLLY-2024-354).
Patient consent for publication
Written informed consent was obtained from the
parents of the patient for the publication of the present case
report and any accompanying images.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
PPB
|
pleuropulmonary blastoma
|
|
DICER1
|
Dcr-1 homolog and ribonuclease type
III
|
|
CT
|
computed tomography
|
References
|
1
|
Masarweh K, Mordechai O, Gur M, Bar-Yoseph
R, Bentur L and Ilivitzki A: Challenges in DICER1-associated lung
disease. J Clin Med. 12:19182023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nasr A, Himidan S, Pastor AC, Taylor G and
Kim PC: Is congenital cystic adenomatoid malformation a
premalignant lesion for pleuropulmonary blastoma? J Pediatr Surg.
45:1086–1089. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dehner LP: Pleuropulmonary blastoma is THE
pulmonary blastoma of childhood. Semin Diagn Pathol. 11:144–151.
1994.PubMed/NCBI
|
|
4
|
Messinger YH, Stewart DR, Priest JR,
Williams GM, Harris AK, Schultz KA, Yang J, Doros L, Rosenberg PS,
Hill DA and Dehner LP: Pleuropulmonary blastoma: A report on 350
central pathology-confirmed pleuropulmonary blastoma cases by the
International Pleuropulmonary Blastoma Registry. Cancer.
121:276–285. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hemead H, Aly RG, Kotb M and Abdelaziz A:
Pediatric pleuropulmonary blastoma: Analysis of four cases. BMC
Cancer. 24:12682024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hussain M, Baig FA and Hussain S:
Pulmonary blastoma. J Coll Physicians Surg Pak. 17:438–440.
2007.PubMed/NCBI
|
|
7
|
Bownes LV, Hutchins SC, Cardenas AM, Kelly
DR and Beierle EA: Pleuropulmonary blastoma in an adolescent. J
Pediatr Surg Case Rep. 59:1014822020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schultz KAP, Harris AK, Nelson AT, Watson
D, Lucas JT Jr, Miniati D, Stewart DR, Hagedorn KN, Mize W,
Kamihara J, et al: Outcomes for children with type II and type III
pleuropulmonary blastoma following chemotherapy: A report from the
international PPB/DICER1 registry. J Clin Oncol. 41:778–789.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gbande P, Abukeshek T, Bensari F and
El-Kamel S: Pleuropulmonary blastoma, a rare entity in childhood.
Case Reports. 7:202002062021.PubMed/NCBI
|
|
10
|
Berean K, Truong LD, Dudley AW Jr and
Cagle PT: Immunohistochemical characterization of pulmonary
blastoma. Am J Clin Pathol. 89:773–777. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bhalerao S, Adhav A, Gandhe S and Nagarkar
R: Metachronous pleuropulmonary blastoma in an adult patient with
endometrial cancer: A case report. Oxf Med Case Reports.
2019:omz0562019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu AH, Zheng WY and Wu L: Pleuropulmonary
blastoma in an adult women with pleurorrhea as the major clinical
manifestation: Report of a case. Nan Fang Yi Ke Da Xue Xue Bao.
28:2241–2243. 2008.(In Chinese). PubMed/NCBI
|
|
13
|
Koss MN, Hochholzer L and O'Leary T:
Pulmonary blastomas. Cancer. 67:2368–2381. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dehner LP, Messinger YH, Schultz KA,
Williams GM, Wikenheiser-Brokamp K and Hill DA: Pleuropulmonary
blastoma: Evolution of an entity as an entry into a familial tumor
predisposition syndrome. Pediatr Dev Pathol. 18:504–511. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Engwall-Gill AJ, Chan SS, Boyd KP, Saito
JM, Fallat ME, St Peter SD, Bolger-Theut S, Crotty EJ, Green JR,
Hulett Bowling RL, et al: Accuracy of chest computed tomography in
distinguishing cystic pleuropulmonary blastoma from benign
congenital lung malformations in children. JAMA Netw Open.
5:e22198142022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bisogno G, Sarnacki S, Stachowicz-Stencel
T, Colin VM, Ferrari A, Godzinski J, Villars MG, Bien E, Hameury F,
Helfre S, et al: Pleuropulmonary blastoma in children and
adolescents: The EXPeRT/PARTNER diagnostic and therapeutic
recommendations. Pediatr Blood Cancer. 68 (Suppl 1):e290452021.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Landry-Truchon K, Houde N, Lhuillier M,
Charron L, Hadchouel A, Delacourt C, Foulkes WD, Galmiche-Rolland L
and Jeannotte L: Deletion of Yy1 in mouse lung epithelium unveils
molecular mechanisms governing pleuropulmonary blastoma
pathogenesis. Dis Model Mech. 13:dmm0459892020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Knight S, Knight T, Khan A and Murphy J:
Current management of pleuropulmonary blastoma: A surgical
perspective. Children (Basel). 6:862019.PubMed/NCBI
|
|
19
|
Han LM, Weiel JJ, Longacre TA and Folkins
AK: DICER1-associated tumors in the female genital tract: Molecular
basis, clinicopathologic features, and differential diagnosis. Adv
Anat Pathol. 29:297–308. 2022.PubMed/NCBI
|
|
20
|
Venkatramani R, Triche TJ, Wang L, Shimada
H and Mascarenhas L: Insulin-like growth factor 2 gene expression
molecularly differentiates pleuropulmonary blastoma and embryonal
rhabdomyosarcoma. J Pediatr Hematol Oncol. 37:e356–360. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Folpe A, Graham RP, Martinez A,
Schembri-Wismayer D, Boland J and Fritchie KJ: Mesenchymal
chondrosarcomas showing immunohistochemical evidence of
rhabdomyoblastic differentiation: A potential diagnostic pitfall.
Hum Pathol. 77:28–34. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gros L, Dei Tos AP, Jones RL and Digklia
A: Inflammatory myofibroblastic tumour: State of the Art. Cancers
(Basel). 14:36622022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Onoda T, Kanno M, Sato H, Takahashi N,
Izumino H, Ohta H, Emura T, Katoh H, Ohizumi H, Ohtake H, et al:
Identification of novel ALK rearrangement A2M-ALK in a neonate with
fetal lung interstitial tumor. Genes Chromosomes Cancer.
53:865–874. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dehner LP, Schultz KA and Hill DA:
Pleuropulmonary blastoma: More than a lung neoplasm of childhood.
Mo Med. 116:206–210. 2019.PubMed/NCBI
|
|
25
|
González IA, Stewart DR, Schultz KAP,
Field AP, Hill DA and Dehner LP: DICER1 tumor predisposition
syndrome: An evolving story initiated with the pleuropulmonary
blastoma. Mod Pathol. 35:4–22. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shukrun R, Golan H, Caspi R, Pode-Shakked
N, Pleniceanu O, Vax E, Bar-Lev DD, Pri-Chen S, Jacob-Hirsch J,
Schiby G, et al: NCAM1/FGF module serves as a putative
pleuropulmonary blastoma therapeutic target. Oncogenesis. 8:482019.
View Article : Google Scholar : PubMed/NCBI
|