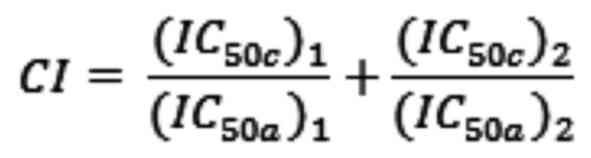Introduction
Cholangiocarcinoma (CCA) is one of the most
challenging carcinomas to manage, given its inherent heterogeneity
and the limited efficacy of available therapeutic options.
Epidemiological data reveals that CCA constitutes ~15% of primary
liver cancers and ~3% of gastrointestinal malignancies,
contributing to nearly 2% of all annual cancer-associated
fatalities globally (1,2). Most patients present with advanced
CCA, as the early stages are typically asymptomatic. Modern
radiographic diagnostic techniques, including abdominal magnetic
resonance imaging, abdominal computerized tomography and magnetic
resonance cholangiopancreatography, are able to detect CCA.
However, these techniques generally require verification via
histological or cytological analysis.
Depending on the characteristics of the CCA,
neoadjuvant chemotherapy may be used to reduce the size of large
tumors prior to surgery (3). If the
tumor is considered to be resectable, surgical resection is
typically the initial approach, followed by adjuvant chemotherapy
to reduce the risk of recurrence (2). In cases where surgical removal is not
feasible, palliative chemotherapy can be administered to improve
the quality of life of the patient. Currently, patients with CCA
are treated with conventional DNA-damaging chemotherapies. These
include platinum-based drugs such as cisplatin, gemcitabine,
oxaliplatin and capecitabine, in addition to a combination of
5-fluorouracil and folinic acid (4,5).
However, it is necessary to improve the efficacy of these
treatments, as CCA can develop resistance and become unresponsive
to chemotherapy.
In the age of personalized medicine, the molecular
profiling of CCA facilitates the use of targeted treatments
(6,7). This approach leverages the concept of
synthetic lethality, in which the concurrent inhibition of two
associated genes results in cell death (8). Notably, the DNA damage and repair
pathways have been identified as promising targets for the
inhibition of cancer growth (9).
Poly(ADP-ribose) polymerase (PARP), an enzyme involved in DNA
repair, exhibits synthetic lethality with breast and ovarian cancer
susceptibility protein 1/2 (BRCA1/2), which is key to homologous
recombination (HR) repair (10).
Targeting PARP in tumors with a defective BRCA1/2 gene
causes the accumulation of DNA damage and subsequently induces cell
death. The US Food and Drug Administration (FDA) has approved
numerous PARP inhibitors, including olaparib (11), rucaparib (12) and niraparib (13) and talazoparib (14), whereas fluzoparib (15) and pamiparib (16) were approved in China. Ataxia
telangiectasia and Rad3-related protein (ATR) serves as a DNA
damage sensor and plays a crucial role in the response to
replicative stress (17). A
functional loss of ATR can sensitize cancer cells to DNA-damaging
chemotherapeutics, while ATR activation contributes to the
development of resistance to PARP inhibitors via the mitigation of
replicative stress (10). Notably,
ATR has been reported to exhibit synthetic lethality with PARP
(17,18), and currently, at least eight ATR
inhibitors are undergoing clinical trials (19). Efforts to combine ATR inhibitors
with PARP inhibitors are ongoing, with the aim of overcoming
resistance, enhancing therapeutic effectiveness, and potentially
reducing the dosage of each drug, thereby decreasing toxicity.
A number of clinical trials have explored the
combined use of ATR and PARP inhibitors in the treatment of ovarian
cancer (20), breast cancer
(21) and prostate cancer (22). In the context of CCA, one such trial
is currently suspended (23), while
another is evaluating the combination of the ATR inhibitor AZD6738
with the PARP inhibitor olaparib (24). Several in vitro, in vivo, and
ex vivo studies have investigated the effects of PARP or ATR
inhibitors, and their combinations with DNA-damaging agents on CCA
(25–27). For example, Serra-Camprubí et
al (25) assessed the effects
of the PARP inhibitors olaparib, pamiparib and niraparib on
patient-derived xenograft (PDX) cell lines from patients with CCA
and diverse genetic profiles. The study concluded that patients
with advanced CCA and pathogenic BRCA2 mutations could
potentially benefit from PARP inhibitor treatment. Similarly,
Bezrookove et al (26)
evaluated the impact of olaparib and niraparib on PDX and
established CCA cell lines, all with various DNA damage repair gene
mutation profiles. The study showed that niraparib was more potent
than olaparib, and the combination of niraparib with gemcitabine
synergistically inhibited tumor growth. Additionally, Moolmuang and
Ruchirawat (27) investigated the
cytotoxic effects of the ATR inhibitor VE-821, both alone and in
combination with the ATM serine/threonine kinase inhibitor
KU-55933, on various CCA cell lines. The combination of the two
inhibitors had a greater effect on growth inhibition than either
inhibitor alone in all the cell lines tested. However, research on
the effects of combining ATR with different PARP inhibitors on CCA
cell lines is limited (28).
In the present study, the aim was to investigate the
effects of various PARP inhibitors, namely, olaparib, veliparib and
talazoparib, and the ATR inhibitor AZD6738, both individually and
in combination, on established cell lines with diverse genetic
backgrounds. Additionally, the mechanism of the DNA damage response
(DDR) elicited by these treatments was examined, and the
synergistic activity between AZD6738 and the various PARP
inhibitors was assessed to determine their efficacy via the
combination index (CI).
Materials and methods
Drugs
Olaparib (cat. no. HY-10162), veliparib (cat. no.
HY-10129), talazoparib (cat. no. HY-16106) and AZD6738 (cat. no.
HY-19323) were purchased from MedChemExpress. Olaparib and
veliparib were dissolved in 100% dimethyl sulfoxide (DMSO; cat. no.
A3672,0250; PanReac AppliChem; ITW Reagents Division) to create a
100 mM stock solution, while talazoparib and AZD6738 were dissolved
in DMSO to a concentration of 50 mM. The drug stocks were kept at
−80°C until used.
Cell lines and culture
The MMNK-1 (cat. no. JCRB1554; immortalized human
cholangiocyte) (29) and HuH-28
(cat. no. JCRB0426; cholangiocarcinoma) (30) cell lines were obtained from the
Japanese Collection of Research Bioresources Cell Bank (JCRB),
while the TFK-1 (cell no. RCB2537; cholangiocarcinoma) cell line
(31) was received from the RIKEN
BioResource Center. SiSP-K01 and SiSP-K05 primary cell lines were a
gift from Professor Seiji Okada of Kumamoto University (Kumamoto,
Japan) (32). SiSP-K01 was derived
from a 64-year-old female with intrahepatic, moderately
differentiated CCA and was used at passage 51. SiSP-K05 was derived
from another female patient, age 53 years, with intrahepatic,
moderately differentiated CCA and was also used at passage 51. The
origin of each cell line is summarized in Table SI. The experimental protocol was
approved by the Human Research Ethics Committee of the Faculty of
Medicine, Ramathibodi Hospital, Mahidol University (Bangkok,
Thailand; approval no. MURA2023/155).
The MMNK-1 and TFK-1 cell lines were maintained in
DMEM/F12 (cat. no. 12400024; Gibco; Thermo Fisher Scientific, Inc.)
and RPMI-1640 medium (cat. no. 11875093; Gibco; Thermo Fisher
Scientific, Inc.), respectively. The HuH-28, SiSP-K01 and SiSP-K05
cell lines were cultured in DMEM (cat. no. 12800017; Gibco; Thermo
Fisher Scientific, Inc.). All cell lines were supplemented with 1%
penicillin/streptomycin (cat. no. 15140122; Invitrogen; Thermo
Fisher Scientific, Inc.) and 10% fetal bovine serum (FBS; cat. no.
ES-009-B; Merck KGaA), with the exception of MMNK-1, which was
supplemented with 15% FBS. The cells were incubated at 37°C with 5%
CO2.
Mutation analysis of CCA cell
lines
The genetic profiles of the TFK-1 and HuH-28 cell
lines were obtained by next-generation sequencing, as described in
Jamnongsong et al (33). The
genetic profiles of SiSP-K01 (https://www.ncbi.nlm.nih.gov/sra/SRR31111387), and
SiSP-K05 (https://www.ncbi.nlm.nih.gov/sra/SRR31111386) were
also obtained by next-generation sequencing and are available under
BioProject ID PRJNA1176211 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1176211/).
The set of 27 DDR genes was based on those in the study by
Bezrookove et al (26);
AT-rich interaction domain 1A/B (ARID1A and ARID1B),
ataxia telangiectasia mutated (ATM), ATR, ATRX
chromatin remodeler (ATRX), BRCA1 associated deubiquitinase
1 (BAP1), BRCA1 associated RING domain 1 (BARD1), BLM
RecQ like helicase (BLM), BRCA1, BRCA2, BRCA1
interacting DNA helicase 1 (BRIP1), checkpoint kinase 2
(CHEK2), FA complementation group A/C/D2/E/F/G/L (FANCA,
FANCC, FANCD2, FANCE, FANCF, FANCG and FANCL), MRE11
homolog, double strand break repair nuclease (MRE11), nibrin
(NBN), partner and localizer of BRCA2 (PALB2), RAD50
double strand break repair (RAD50), RAD51 recombinase
(RAD51), RAD51 paralog B/C (RAD51B and RAD51C)
and WRN RecQ like helicase (WRN). To assess the
pathogenicity of the identified exonic mutations that led to
changes in amino acids, the Franklin tool by Genoox (http://franklin.genoox.com; accessed on September 26,
2023) was employed. This analysis was conducted in accordance with
the Association for Molecular Pathology (AMP) classification
guidelines. These guidelines stratify variants into four tiers
based on their clinical relevance to bile duct cancer, as discussed
by Li et al (34). Tier 1
includes variants with strong clinical significance, tier 2
comprises variants with potential significance, tier 3 encompasses
variants of uncertain significance, and tier 4 contains variants
that are benign or likely to be benign.
Sensitivity assay and CI
calculation
Cells were seeded into 96-well plates at a final
concentration of 3,000 cells/well and incubated for 24 h before the
medium was replaced with that containing the drugs of interest.
Treatments were with either a PARP inhibitor (olaparib, veliparib,
and talazoparib) or the ATR inhibitor AZD6738 alone, or a
combination of PARP inhibitor and AZD6738, for a duration of 120 h
at 37°C. A mock treatment was also conducted, in which the
concentration of DMSO was >0.016%. Following treatment, cell
viability was assessed using the CellTiter-Glo®
Luminescent Cell Viability Assay according to the manufacturer's
protocol (cat. no. G7572; Promega Corporation). Survival
percentages were determined by normalizing the luminescent signal
to that of untreated cells. These percentages were then plotted and
half maximal inhibitory concentration (IC50) values were
calculated using GraphPad Prism software, version 9.5.1
(Dotmatics). The CI values were subsequently calculated according
to the Chou-Talalay method (35) as
shown below:
In this formula, (IC50c)1 is
the IC50 of AZD6738 used in combination;
(IC50a)1 is the IC50 of AZD6738
used alone; (IC50c)2 is the IC50
of the PARP inhibitor used in combination and
(IC50a)2 is the IC50 of the PARP
inhibitor used alone.
The CI values were categorized as follows (36): 0.1–0.3, strong synergism; 0.3–0.7,
synergism; 0.7–0.85, moderate synergism; 0.85–0.9, slight
synergism; 0.9–1.1, nearly additive; 1.1–1.2, slight antagonism;
1.2–1.45, moderate antagonism. The experiments were conducted in
three biological replicates.
Clonogenic survival assays
Cells were seeded in 6-well plates at a density of
600 cells/well for MMNK-1 or 1,000 cells/well for SiSP-K01 and
SiSP-K05 and incubated at 37°C with 5% CO2 for 24 h.
They were then exposed to AZD6738, various concentrations of PARP
inhibitors, or a combination of both, for 120 h at 37°C. The media
was subsequently replaced, and the cells were incubated for 7–10
days until colonies formed. For visualization, a 0.5% w/v solution
of crystal violet (cat. no. C077; Sigma-Aldrich; Merck KGaA) in 40%
v/v methanol in water was added and incubated for 10 min at room
temperature. The plates were then washed and air-dried, and images
were acquired using a ChemiDoc™ MP Imaging System
(Bio-Rad Laboratories, Inc.). All the images were exported as tif
files, and the intensity of each well was measured using ImageJ
1.53n software (37) as previously
described (38). The experiments
were performed on three biological replicates.
Micronuclei and γ-H2A histone family
member X (γ-H2AX) foci formation assays
Cells were seeded in a slide chamber
(Lab-Tek™, cat. no. 154526; Thermo Fisher Scientific,
Inc.) and allowed to grow until they reached 80% confluence.
Subsequently, the medium was replaced with fresh media containing
AZD6738, PARP inhibitor or a combination of AZD6738 and PARP
inhibitor. The cells were then incubated for an additional 24 h at
37°C in a 5% CO2 atmosphere. Fluorescence staining was
performed using a method modified from that in previous studies
(39,40). Briefly, cells were washed with PBS
and fixed with 4% paraformaldehyde for 10 min at room temperature.
After another wash with PBS, cells were permeabilized with 0.5%
Triton X-100 in PBS for 15 min. Non-specific binding was blocked
using Intercept® (PBS) blocking buffer (cat. no.
927-70001; LI-COR Biosciences) for 1 h at room temperature. For the
detection of γ-H2AX foci, cells were incubated with a mouse
monoclonal antibody against γ-H2AX (Ser139; 1:1,000; cat. no.
80312; Cell Signaling Technology, Inc.) at 4°C overnight. Alexa
Fluor® 488 donkey anti-mouse IgG (1:500; cat. no.
A21202; Thermo Fisher Scientific, Inc.) was used as the secondary
antibody and was incubated with the cells for 1 h at room
temperature. Micronuclei and nuclei were visualized with Hoechst
33342 solution (1:2,000; cat. no. H3570; Thermo Fisher Scientific,
Inc.) for 5 min at room temperature. Images were acquired with a
fluorescence microscope (ECLIPSE Ci; Nikon Corporation). All
samples were visualized using the same intensity and exposure time,
and images were analyzed using ImageJ 1.53n software (37). At least 225 nuclei were analyzed for
both micronuclei and γ-H2AX foci formation. The experiments were
performed with at least two biological replicates.
Statistical analysis
The IC50 and CI results were reported as
the mean ± standard deviation. The IC50 values and
relative intensities of colonies across each CCA cell line were
statistically compared with those of the MMNK-1 cholangiocyte cell
line, utilizing multiple t-tests with the Holm-Sidak method in
GraphPad Prism software, version 9.5.1 (Dotmatics). P<0.05 was
considered to indicate a statistically significant difference. The
same statistical analysis was applied to assess significant
differences in the average number of micronuclei per cell between
treatments with AZD6738 or PARP inhibitors alone or in combination.
The results of the γ-H2AX foci formation assay are presented as
medians and were analyzed using the Mann-Whitney test.
Results
Genetic profiling of DDR in CCA cell
lines and AMP classification
As shown in Table I,
the numbers of mutated genes differed among the CCA cell lines.
Among the four CCA cell lines, TFK-1 had the fewest DDR mutated
genes (n=9), whereas HuH-28 had the same number of DDR mutated
genes as SiSP-K01 (n=11). SiSP-K05 contained the highest number of
mutated genes (n=15). Fig. 1
demonstrates the distribution of DDR mutated genes in each CCA cell
line. In all CCA cell lines analyzed, mutations in ATR,
BRCA2 and WRN were detected. In addition, a subset of
three CCA cell lines exhibited mutations in eight different genes:
ARID1A, ARID1B, ATM, ATRX, BARD1, BRIP1, FANCA and
PALB2. Three other mutated genes, BLM, BRCA1 and
RAD51B, were found in two CCA cell lines; HuH-28 and
SiSP-K05 for BLM and BRCA1, and RAD51B for
SiSP-K01 and SiSP-K05. There were four mutated genes, namely
BAP1, CHEK2, FANCF and RAD50, that were only observed
in a single CCA cell line. However, there were nine genes
associated with DDR, specifically FANCC, FANCD2, FANCE, FANCG,
FANCL, MRE11, NBN, RAD51 and RAD51C, that were not
detected in any of the CCA cell lines. Details of the genetic
variants in DDR genes that were identified are presented in
Table SII. Next, the
classification of genetic variants in DDR genes was determined
according to the guidelines of the AMP, as indicated in Table II. SiSP-K01 exhibited a range of
variants spanning the clinically relevant tiers 2–4, whereas
SiSP-K05 had a range of variants spanning tiers 2 and 3. Notably,
the TFK-1 and HuH-28 cell lines only demonstrated variants
exclusively from tiers 3 and 4. Additionally, it was observed that
variants in BRCA1 and PALB2 genes were confined to
tiers 2 and 3, whereas those in the BRCA2 and BRIP1
genes spanned tiers 2–4. Variants in other genes fell into tier 3
and/or tier 4. Collectively, these findings suggest that the CCA
cell lines are promising candidates for further testing with PARP
inhibitors and AZD6738.
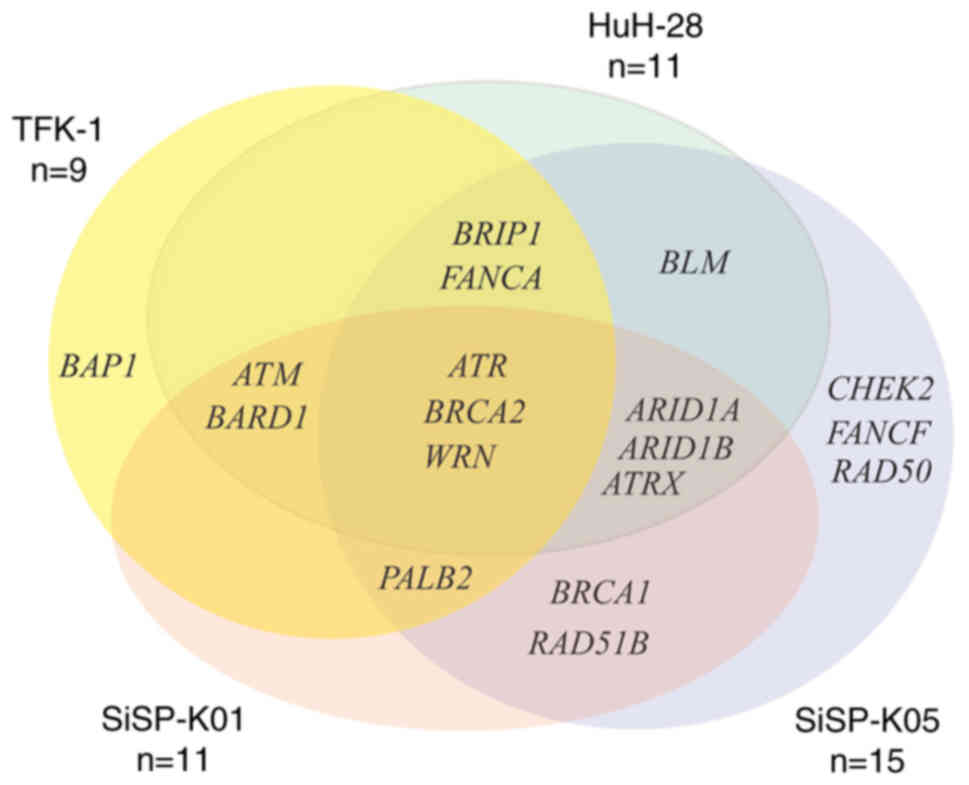 | Figure 1.Venn diagram of mutated DNA damage
response genes in cholangiocarcinoma cell lines. ARID1A/B, AT-rich
interaction domain 1A/B; ATM, ataxia telangiectasia mutated; ATR,
ataxia telangiectasia and Rad3-related protein; ATRX, ATRX
chromatin remodeler; BAP1, BRCA1 associated deubiquitinase 1;
BARD1, BRCA1 associated RING domain 1; BLM, BLM RecQ like helicase;
BRIP1, BRCA1 interacting DNA helicase 1; CHEK2, checkpoint kinase
2; FANCA/F, FA complementation group A/F; PALB2, partner and
localizer of BRCA2; RAD51B, RAD51 recombinase paralog B; WRN, WRN
RecQ like helicase. |
 | Table I.List of DDR mutated genes in CCA cell
lines. |
Table I.
List of DDR mutated genes in CCA cell
lines.
| CCA cell lines | DDR mutated
genes |
|---|
| TFK-1 | ATM, ATR, BAP1,
BARD1, BRCA2, BRIP1, FANCA, PALB2, WRN |
| HuH-28 | ARID1A, ARID1B,
ATM, ATR, ATRX, BARD1, BLM, BRCA2, BRIP1, FANCA, WRN |
| SiSP-K01 | ARID1A, ARID1B,
ATM, ATR, ATRX, BARD1, BRCA1, BRCA2, PALB2, RAD51B, WRN |
| SiSP-K05 | ARID1A, ARID1B,
ATR, ATRX, BLM, BRCA1, BRCA2, BRIP1, CHEK2, FANCA, FANCF, PALB2,
RAD50, RAD51B, WRN |
| Table II.Classification of genetic variants in
cholangiocarcinoma cell lines according to AMP guidelines. |
Table II.
Classification of genetic variants in
cholangiocarcinoma cell lines according to AMP guidelines.
|
| AMP classification,
tier |
|---|
|
|
|
|---|
| Genes | TFK-1 | HuH-28 | SiSP-K01 | SiSP-K05 |
|---|
| ARID1A | - | 3 | 3 | 3 |
| ARID1B | - | 3 | NA | NA |
| ATM | 4 | 4 | NA | - |
| ATR | 4 | 4 | 3 | 3 |
| ATRX | - | 4 | 3 | 3 |
| BAP1 | 3 | - | - | - |
| BARD1 | 4 | 4 | NA | - |
| BLM | - | 3 and 4 | - | 3 |
| BRCA1 | - | - | 2 and 3 | NA |
| BRCA2 | 4 | 4 | 2 and 3 | 2 and 3 |
| BRIP1 | 4 | 4 | - | 2 and 3 |
| CHEK2 | - | - | - | NA |
| FANCA | 4 | 4 | - | 3 |
| FANCF | - | - | - | 3 |
| PALB2 | 3 | - | 2 and 3 | 2 |
| RAD50 | - | - | - | NA |
| RAD51B | - | - | 3 | 3 |
| WRN | 4 | 4 | 4 | 3 |
Evaluating the impacts of AZD6738 and
PARP inhibitors on cell viability and clonogenic survival
To assess cell viability in the four CCA cell lines
with distinct DDR mutated backgrounds, TFK-1, HuH-28, SiSP-K01 and
SiSP-K05 cells were treated with AZD6738, PARP inhibitors alone, or
their combinations, and their responses to treatment were compared
with those of the immortalized cholangiocyte cell line MMNK-1.
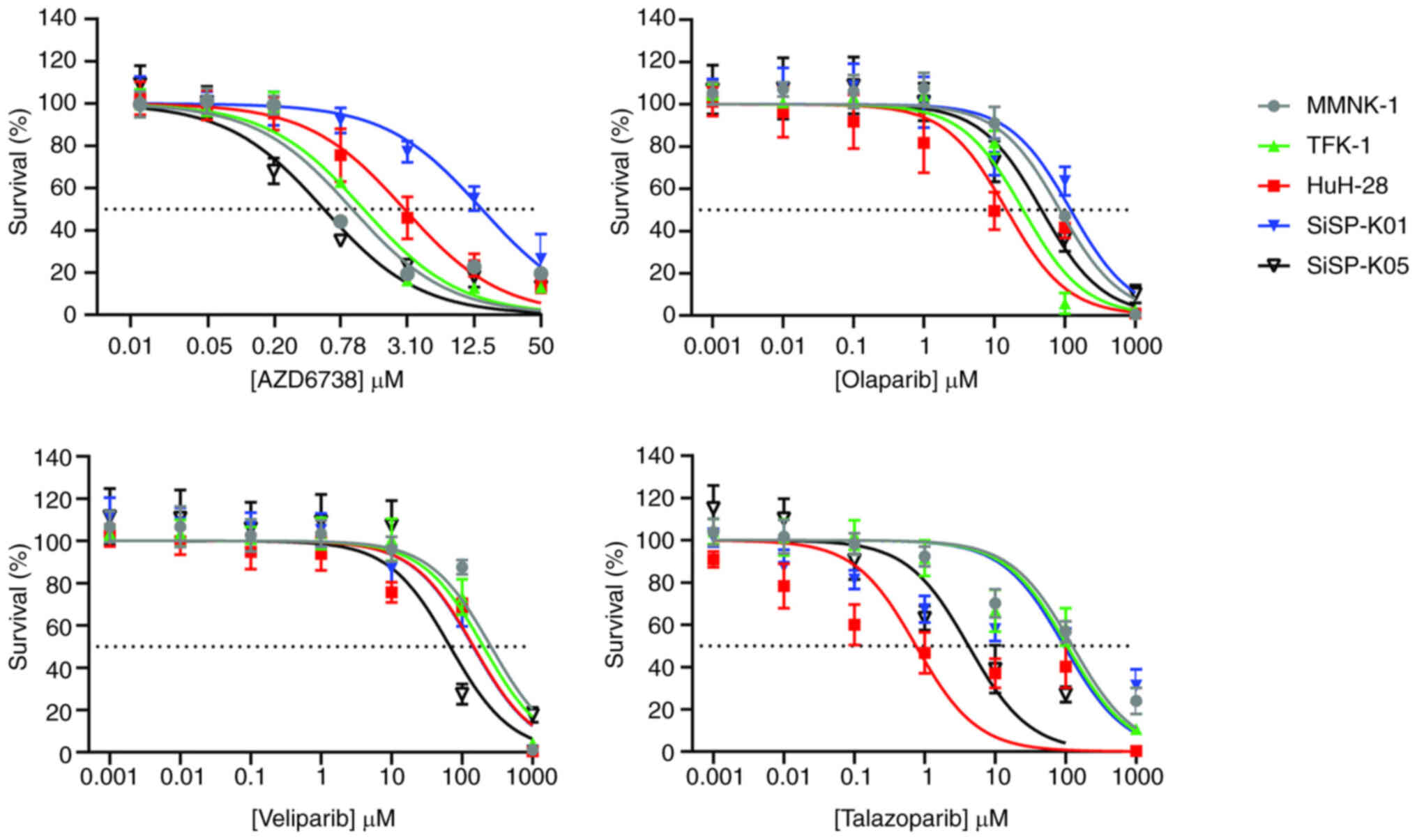
Fig. 2 and Table III display the dose-response
curves from the sensitivity assay and the IC50a profiles
of all the cell lines in response to AZD6738 and the various PARP
inhibitors, respectively.
| Table III.IC50a profiles from the
sensitivity assay of cholangiocarcinoma and cholangiocyte cell
lines in response to AZD6738 or various poly(ADP-ribose) polymerase
inhibitors. |
Table III.
IC50a profiles from the
sensitivity assay of cholangiocarcinoma and cholangiocyte cell
lines in response to AZD6738 or various poly(ADP-ribose) polymerase
inhibitors.
| Drug | Cell line | IC50a,
µM | P-value |
|---|
| AZD6738 | MMNK-1 | 0.997±0.141 | - |
|
| TFK-1 | 1.263±0.073 | 0.126 |
|
| HuH-28 | 3.204±1.587 | 0.074 |
|
| SiSP-K01 | 15.633±5.324 | 0.018 |
|
| SiSP-K05 | 0.554±0.020 | 0.017 |
| Olaparib | MMNK-1 | 85.033±5.664 | - |
|
| TFK-1 | 25.667±3.661 | <0.001 |
|
| HuH-28 | 19.740±16.283 | 0.010 |
|
| SiSP-K01 | 121.067±5.140 | 0.005 |
|
| SiSP-K05 | 46.790±16.939 | 0.021 |
| Veliparib | MMNK-1 | 256.800±14.127 | - |
|
| TFK-1 | 202.067±47.561 | 0.241 |
|
| HuH-28 | 145.950±10.112 | 0.010 |
|
| SiSP-K01 | 143.833±20.857 | 0.005 |
|
| SiSP-K05 | 67.607±3.466 | <0.001 |
| Talazoparib | MMNK-1 | 127.767±39.302 | - |
|
| TFK-1 | 105.880±60.491 | 0.627 |
|
| HuH-28 | 1.095±0.920 | 0.010 |
|
| SiSP-K01 | 100.003±35.553 | 0.416 |
|
| SiSP-K05 | 4.378±1.977 | 0.017 |
Comparison of AZD6738 and the PARP inhibitors
indicated that AZD6738 was the most toxic to the cell lines. This
is evidenced by AZD6738 having the lowest IC50a values,
both minimal and maximal, in comparison with the PARP inhibitors.
The IC50a values for AZD6738 ranged from 0.554±0.020 µM
(in SiSP-K05) to 15.633±5.324 µM (in SiSP-K01). By contrast, the
ranges of the IC50a values for the PARP inhibitors were
as follows: Olaparib, from 19.740±16.283 µM (in HuH-28) to
121.067±5.140 µM (in SiSP-K01); veliparib, from 67.607±3.466 µM (in
SiSP-K05) to 256.800±14.127 µM (in MMNK-1); and talazoparib, from
1.095±0.920 µM (in HuH-28) to 127.767±39.302 µM (in MMNK-1).
Next, the IC50a profiles of CCA cell
lines when treated with AZD6738 or various PARP inhibitors alone
were compared with those of MMNK-1 cholangiocytes (Table III). For AZD6738, only SiSP-K05
(IC50a, 0.554±0.020 µM) was significantly more sensitive
(P=0.017) than the MMNK-1 cholangiocytes (IC50a,
0.997±0.141 µM). By contrast, SiSP-K01 cells (IC50a,
15.633±5.324 µM; P=0.018) exhibited significantly lower sensitivity
than MMNK-1 cholangiocytes to AZD6738, while TFK-1 and HuH-28 cells
both displayed sensitivity levels similar to those of the
cholangiocytes. For the PARP inhibitors, three CCA cell lines,
namely TFK-1 (IC50a, 25.667±3.661 µM; P<0.001),
HuH-28 (IC50a, 19.740±16.283 µM; P=0.010) and SiSP-K05
(IC50a, 46.790±16.939 µM; P=0.021), demonstrated
significantly greater sensitivity than MMNK-1 cells
(IC50a, 85.033±5.664 µM) to olaparib, while HuH-28
(IC50a, 145.950±10.112 µM; P=0.010), SiSP-K01
(IC50a, 143.833±20.857 µM; P=0.005) and SiSP-K05 cells
(IC50a, 67.607±3.466 µM; P<0.001) were significantly
more sensitive than MMNK-1 cells (IC50a, 256.800±14.127
µM) to veliparib. Additionally, the sensitivity of HuH-28 and
SiSP-K05 cells to talazoparib was heightened compared with that of
MMNK-1 cells. SiSP-K01 cells (IC50a, 121.067±5.140 µM;
P=0.005) were significantly less sensitive than MMNK-1 cells to
olaparib, whereas TFK-1 cells displayed veliparib sensitivity
comparable to that of MMNK-1 cells. The sensitivity to talazoparib
of TFK-1 and SiSP-K01 cells was similar to that of MNNK-1 cells.
Among the cell lines with significantly greater sensitivity than
MNNK-1 cells to PARP inhibitors, talazoparib was the most potent,
as evidenced by its low IC50a. The IC50a
values for the PARP inhibitors were as follows: Olaparib, MMNK-1
(85.033±5.664 µM) compared with TFK-1 (25.667±3.661 µM), HuH-28
(19.740±16.283 µM) and SiSP-K05 (46.790±16.939 µM); veliparib,
MMNK-1 (256.800±14.127 µM) compared with HuH-28 (145.950±10.112
µM), SiSP-K01 (143.833±20.857 µM) and SiSP-K05 (67.607±3.466 µM);
and talazoparib, MMNK-1 (127.767±39.302 µM) compared with HuH-28
(1.095±0.920 µM) and SiSP-K05 (4.378±1.977 µM).
The clonogenicity of the two primary CCA cell lines
and MMNK-1 cholangiocytes when treated individually with AZD6738
and of PARP inhibitors at the respective IC50a was
evaluated (Fig. S1). The results
demonstrate that all inhibitors inhibited clonogenicity. The
inhibitory effect on clonogenic survival was >50% when compared
with the clonogenicity of the untreated group. These results
confirm that at the IC50a, these drugs are capable of
inhibiting the clonogenicity of primary cell lines.
Collectively, these observations suggest that,
compared with AZD6738, PARP inhibitors exhibit a broader range of
effectiveness in CCA cell lines with diverse genetic backgrounds
and less toxicity. Among the tested PARP inhibitors, talazoparib is
the most potent.
Drug combinations accelerate DNA
damage as indicated by micronuclei and γ-H2AX formation
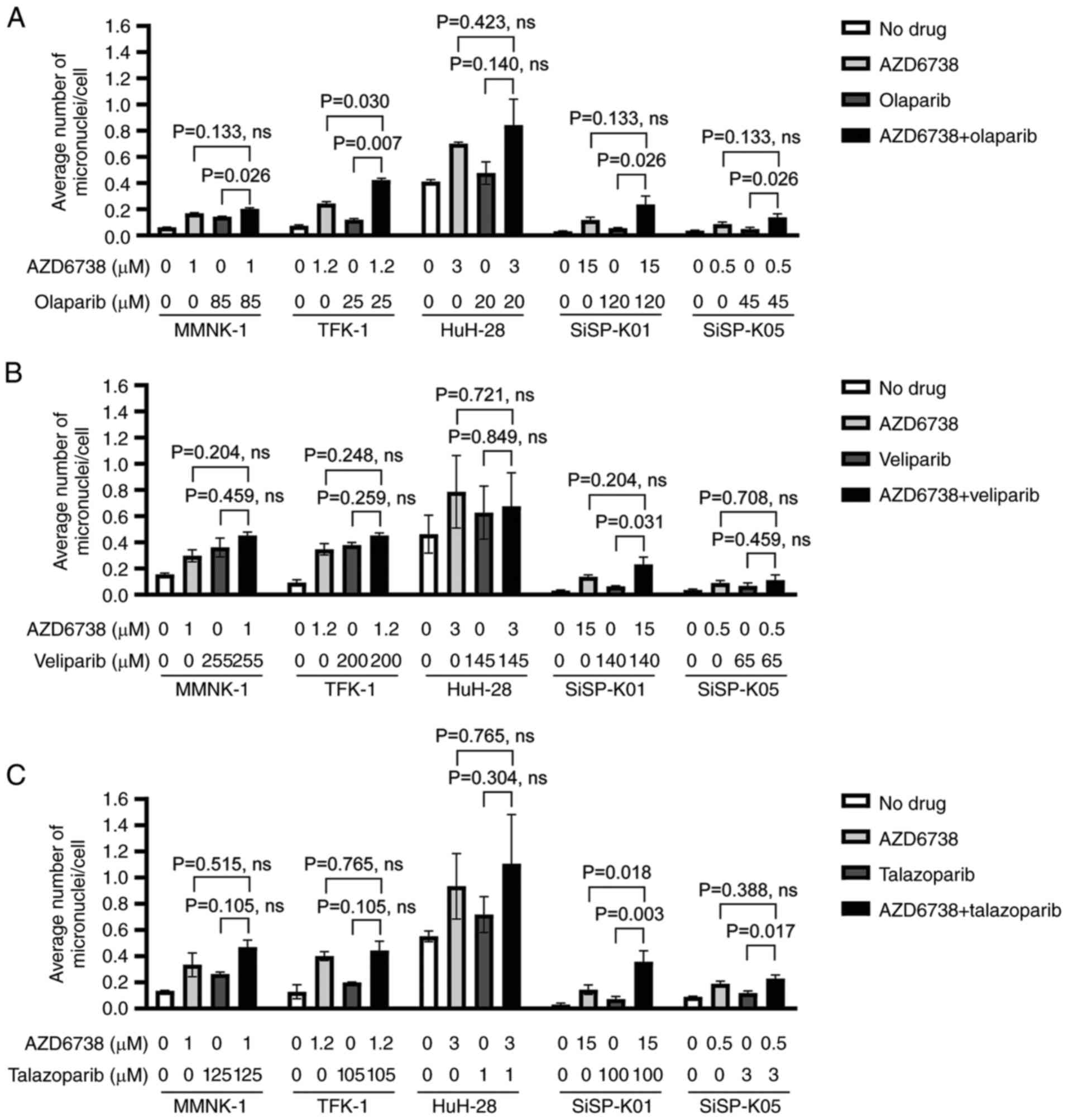
To determine whether combining AZD6738 with PARP
inhibitors increases DNA damage compared with the individual effect
of each drug at its IC50a, micronuclei formation was
evaluated in the CCA cell lines subjected to these treatments. The
results showed that the drug combinations induced more DNA damage
than each drug did on its own, particularly when cells were treated
with the olaparib (Fig. 3A) and
talazoparib (Fig. 3C) combinations.
Specifically, in the case of olaparib (Fig. 3A), there were significant
differences in the extent of damage when MMNK-1, TFK-1, SiSP-K01
and SiSP-K05 cell lines were treated with the combination of
olaparib and AZD6738 compared with olaparib alone. For the TFK-1
cell line, a significant difference was also observed when the
effect of combination treatment was compared with that of AZD6738
alone. With talazoparib (Fig. 3C),
the SiSP-K01 cell line exhibited a significantly higher average
number of micronuclei per cell when treated with the combination
than with individual AZD6738 or talazoparib treatments. By
contrast, SiSP-K05 presented a significant increase only when the
combination was compared with talazoparib alone; no such increase
was observed when compared with AZD6738 alone. However, in the
context of veliparib (Fig. 3B),
combining AZD6738 with veliparib did not result in a significant
increase in micronuclei across all cell lines tested, with the
exception of the SiSP-K01 cell line, in which a significant
difference was found between the combination treatment and
veliparib alone. Representative images of micronuclei for each
condition are presented in Fig.
S2, Fig. S3, Fig. S4.
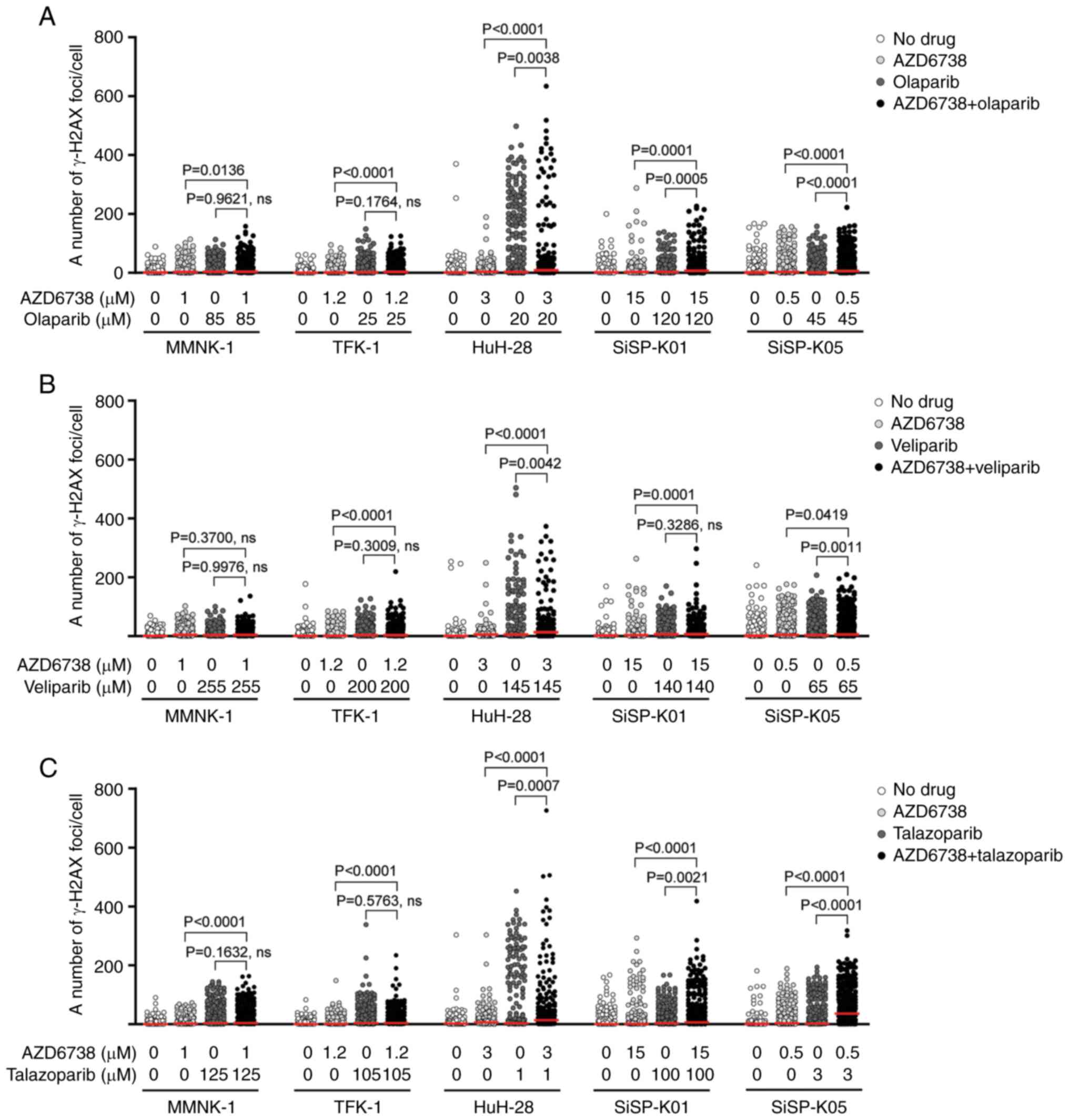
The investigation was expanded to determine if the
increased DNA damage seen following treatment with drug
combinations could be attributed to a rise in DNA double-strand
breaks, as compared with the individual effects of the drugs at
their IC50a (Fig. 4).
The number of cell lines with significant increases in γ-H2AX foci,
a marker of DNA double-strand breaks, following combination
treatments when compared with AZD6738 alone was greater than that
when compared with PARP inhibitor alone. Specifically, with
olaparib and talazoparib (Fig. 4A and
C), a significant increase in γ-H2AX foci was observed in all
CCA cell lines and in normal cholangiocytes when these were
compared with the effects of AZD6738 alone. However, with
veliparib, the significant increase was noted only in CCA cell
lines, not in cholangiocytes. Furthermore, the CCA cell lines
HuH-28, SiSP-K01 and SiSP-K05 exhibited a significant increase in
γ-H2AX when treated with combinations of olaparib or talazoparib,
compared with these PARP inhibitors alone. With regard to
veliparib, two CCA cell lines, HuH-28 and SiSP-K05, demonstrated a
significant increase in γ-H2AX for the combination compared with
veliparib alone. Representative images of γ-H2AX foci formation,
indicative of DNA damage, are presented in Fig. S5, Fig.
S6, Fig. S7.
These observations highlight the enhanced efficacy
of combination treatments in inducing DNA damage, as compared with
the effects of individual drug treatments. The pronounced increase
in DNA double-strand breaks observed across various cell lines
suggests the potential for synergistic interactions between AZD6738
and the PARP inhibitors.
Evaluating the impacts of AZD6738 and
PARP inhibitor combinations on cell viability and CI
The effects of combinations of AZD6738 and different
PARP inhibitors on cell viability were investigated, to determine
which combinations yielded synergistic effects and dose reductions.
The CI, a quantitative tool, was used to assess whether the drug
interactions were synergistic, antagonistic or additive (36). First, the IC50c profile
of AZD6738 when combined with the PARP inhibitors olaparib,
veliparib and talazoparib was determined as shown in Table IV, and the dose-response curves are
presented in Fig. S8. Following
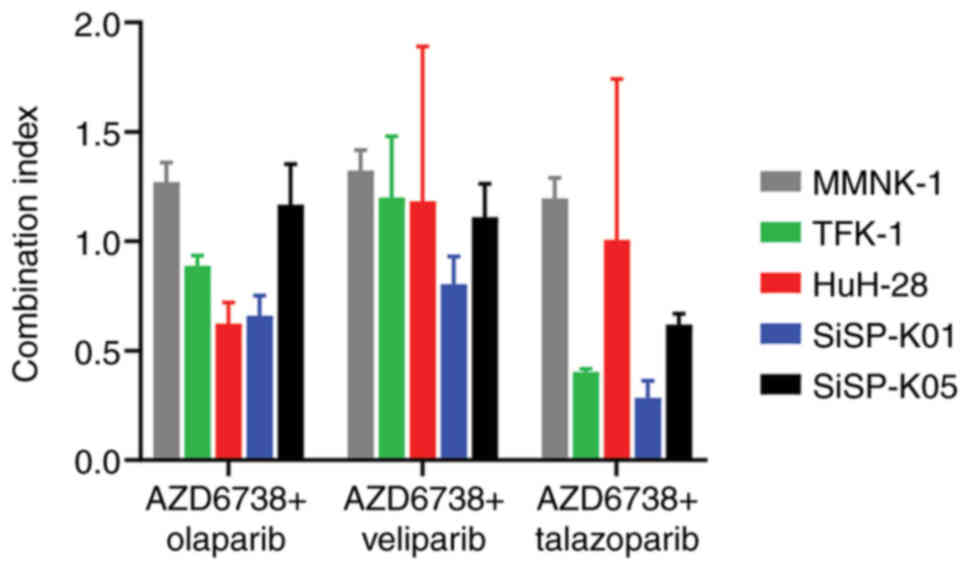
this, the CIs were calculated, which are presented in Table V and visualized in Fig. 5.
| Table IV.IC50c profiles of
cholangiocarcinoma and cholangiocyte cell lines in response to
various combinations of AZD6738 and PARP inhibitors. |
Table IV.
IC50c profiles of
cholangiocarcinoma and cholangiocyte cell lines in response to
various combinations of AZD6738 and PARP inhibitors.
|
|
| AZD6738 | PARP inhibitor |
|---|
|
|
|
|
|
|---|
| Drug
combination | Cell line | IC50c,
µM |
P-valuea | IC50c,
µM |
P-valuea |
|---|
| AZD6738 +
olaparib | MMNK-1 | 1.238±0.083 | - | 1.655±0.161 | - |
|
| TFK-1 | 1.043±0.070 | 0.070 | 1.540±0.133 | 0.394 |
|
| HuH-28 | 1.417±0.590 | 0.630 | 2.699±1.611 | 0.327 |
|
| SiSP-K01 | 5.804±1.415 | 0.005 | 33.953±17.481 | 0.033 |
|
| SiSP-K05 | 0.641±0.116 | 0.003 | 0.538±0.106 | 0.002 |
| AZD6738 +
veliparib | MMNK-1 | 1.307±0.118 | - | 1.438±0.179 | - |
|
| TFK-1 | 1.51±0.419 | 0.462 | 2.625±1.033 | 0.228 |
|
| HuH-28 | 2.377±0.122 | 0.004 | 6.104±0.599 | 0.003 |
|
| SiSP-K01 | 6.802±0.172 | <0.001 | 46.230±2.546 | <0.001 |
|
| SiSP-K05 | 0.612±0.103 | 0.003 | 0.495±0.132 | 0.002 |
| AZD6738 +
talazoparib | MMNK-1 | 1.178±0.084 | - | 0.633±0.090 | - |
|
| TFK-1 | 0.501±0.031 | <0.001 | 0.425±0.030 | 0.056 |
|
| HuH-28 | 0.451±0.098 | 0.002 | 0.424±0.167 | 0.241 |
|
| SiSP-K01 | 2.703±0.373 | 0.005 | 9.195±1.692 | 0.002 |
|
| SiSP-K05 | 0.319±0.007 | <0.001 | 0.158±0.009 | 0.002 |
| Table V.CI values of various combinations of
AZD6738 and poly(ADP-ribose) polymerase inhibitors. |
Table V.
CI values of various combinations of
AZD6738 and poly(ADP-ribose) polymerase inhibitors.
| Drug
combination | Cell line | CI |
|---|
| AZD6738 +
olaparib | MMNK-1 | 1.269±0.091 |
|
| TFK-1 | 0.887±0.047 |
|
| HuH-28 | 0.623±0.097 |
|
| SiSP-K01 | 0.659±0.092 |
|
| SiSP-K05 | 1.167±0.185 |
| AZD6738 +
veliparib | MMNK-1 | 1.323±0.094 |
|
| TFK-1 | 1.200±0.280 |
|
| HuH-28 | 1.182±0.709 |
|
| SiSP-K01 | 0.804±0.126 |
|
| SiSP-K05 | 1.109±0.153 |
| AZD6738 +
talazoparib | MMNK-1 | 1.195±0.094 |
|
| TFK-1 | 0.403±0.015 |
|
| HuH-28 | 1.007±0.736 |
|
| SiSP-K01 | 0.283±0.079 |
|
| SiSP-K05 | 0.619±0.051 |
The data reveal that all drug combinations were most
effective against the SiSP-K05 cell line, with the IC50c
for each drug combination being lower for SiSP-K05 cells than for
MMNK1 cells, as detailed in Table
IV. Specifically, the combinations of AZD6738 with olaparib,
veliparib and talazoparib consistently showed lower
IC50c values in SiSP-K05 cells compared with MMNK-1
cells. AZD6738 and olaparib had the following IC50c
values: MMNK-1, 1.238±0.083 and 1.655±0.161 µM; SiSP-K05,
0.641±0.116 µM (P=0.003) and 0.538±0.106 µM (P=0.002),
respectively. For AZD6738 and veliparib, the IC50c
values were: MMNK-1, 1.307±0.118 and 1.438±0.179 µM; SiSP-K05,
0.612±0.103 µM (P=0.003) and 0.495±0.132 µM (P=0.002),
respectively. AZD6738 and talazoparib had the following
IC50c values; MMNK-1, 1.178±0.084 and 0.633±0.090 µM;
SiSP-K05, 0.319±0.007 µM (P<0.001) and 0.158±0.009 µM (P=0.002),
respectively. By contrast, the SiSP-K01 cell line exhibited lower
sensitivity than MMNK-1 cells to all these drug combinations, with
IC50c values as follows: AZD6738 and olaparib,
5.804±1.415 µM (P=0.005) and 33.953±17.481 µM (P=0.033); AZD6738
and veliparib; 6.802±0.172 µM (P<0.001) and 46.230±2.546 µM
(P<0.001); AZD6738 and talazoparib; 2.703±0.373 µM (P=0.005) and
9.195±1.692 µM (P=0.002), respectively. Furthermore, HuH-28 cells
were less sensitive than MMNK-1 cells to the combination of AZD6738
and veliparib, but both cell lines displayed a similar response to
the combination of AZD6738 and olaparib. For the AZD6738 and
talazoparib combination, the IC50c of AZD6738 for both
TFK-1 and HuH-28 cells differed significantly from that for MMNK-1
cells. However, no such difference was observed for talazoparib
between these two cell lines. Notably, the IC50c values
for all PARP inhibitors were considerably lower when used in
combination than when administered alone.
The CI analysis (Table
V), revealed that synergistic effects were more prevalent in
CCA cell lines when AZD6738 was combined with either olaparib or
talazoparib, in comparison to its combination with veliparib. These
findings are visually represented in Fig. 5. Specifically, the combination of
AZD6738 and olaparib displayed synergistic effects in TFK-1, HuH-28
and SiSP-K01 cell lines. These effects varied, with values ranging
from 0.623±0.097 in HuH-28 cells to 0.887±0.047 in TFK-1 cells. The
combination of AZD6738 and talazoparib produced even stronger
synergistic effects, with values spanning from 0.283±0.079 in
SiSP-K01 cells to 0.619±0.051 in SiSP-K05 cells. Notably, the
combinations comprising AZD6738 with either olaparib or talazoparib
both showed synergistic effects in TFK-1 and SiSP-K01 cells.
However, synergism in the SiSP-K05 cell line was only observed for
the AZD6738 and talazoparib combination. Notably, all tested
combinations exhibited synergistic effects in the SiSP-K01 cell
line, with particularly strong synergism observed for the AZD6738
and talazoparib combination.
Overall, while synergistic effects were observed in
SiSP-K01 cells for all drug combinations, the IC50c
profile indicates potential toxicity from these combinations. By
contrast, the combination of AZD6738 and talazoparib appears to
offer better efficacy for SiSP-K05 cells. This is evidenced by the
IC50c values of AZD6738 and talazoparib in SiSP-K05
cells being lower than those in MMNK-1 cells, and the observed
synergistic effect of this combination.
Discussion
Since the first PARP inhibitor was approved by the
FDA for the treatment of germline BRCA mutated advanced
ovarian cancer, its indications have expanded to other types of
cancers (11,41,42).
Additionally, numerous ATR inhibitors have entered clinical trials,
aiming to target replication stress and combat PARP inhibitor
resistance (43). Currently,
clinical trials are evaluating the treatment of various cancers
with ATR inhibitors, particularly in combination with PARP
inhibitors (44,45). However, only one ongoing trial is
specifically addressing advanced CCA, focusing on the combination
of AZD6738 and olaparib (24). The
decreasing cost of sequencing has enabled more extensive genetic
profiling to be performed in numerous types of cancer. This has
increased research into how DDR-mutated profiles influence
sensitivity to DNA damage and the response to repair-targeted
drugs. In this context, the present study examined the response of
cell lines with different DDR-mutated profiles to the ATR inhibitor
AZD6738, various PARP inhibitors and their combinations. The
results suggest that cell lines with a higher number of DDR
mutations are more sensitive to AZD6738, PARP inhibitors and their
combinations. Notably, among the PARP inhibitors, talazoparib
exhibited the highest potency, both as a standalone treatment and
in combination with AZD6738, for the treatment of CCA cell lines.
Furthermore, combining PARP inhibitors with AZD6738 may reduce the
toxicity associated with higher concentrations of ATR and PARP
inhibitors.
It has been reported that alterations in DDR genes
can be identified in up to 20% of patients with CCA, with
extrahepatic CCA exhibiting a higher incidence than other CCA types
(7). Several studies have
investigated these alterations in CCA genes to expand the
therapeutic use of ATR and PARP inhibitors. For example, a study by
Bezrookove et al (26)
analyzed the mutational profiles of DDR genes in 195 CCA samples
using cBioPortal. They discovered that mutations in ARID1A,
BAP1 and ATM genes were particularly prevalent, being
found in 20.51, 13.3 and 7.7% of cases, respectively. Based on
these findings, the authors used four cell lines with different DDR
mutation profiles to evaluate the response to the PARP inhibitors
niraparib and olaparib. Niraparib inhibited cell growth more
effectively than olaparib, but the response varied according to the
mutational profile. In addition, a study by Serra-Camprubí et
al (25) tested tumoroids
derived from PDXs with confirmed pathogenic mutations in ARID1A,
BAP1 or BRCA2 for sensitivity to olaparib, pamiparib and
niraparib. The study revealed that tumoroids with a BRCA2
defect exhibited sensitivity to olaparib and pamiparib, while other
tumoroids did not respond to these PARP inhibitors. By comparison,
the results of the present study indicate that mutations in ATR,
BRCA2 and WRN were present in all four CCA cell lines,
while ARID1A, ARID1B and ATRX mutations were found in
HuH-28, SiSP-K01 and SiSP-K05, and ATM mutations in TFK-1,
HuH-28 and SiSP-K01. Among the tier 2 genes identified, BRCA1,
BRCA2 and PALB2 have been reviewed by an expert panel in
ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/, accessed on
September 15, 2023). However, only certain variants in BRCA1
and BRCA2 were acknowledged by the panel, while PALB2
variants were not. Notably, the SiSP-K05 cell line, in which 15
mutated genes were detected, exhibited high sensitivity to both ATR
and PARP inhibitors. While SiSP-K01 and HuH-28 were found to share
the same number of mutated genes (n=11), they displayed contrasting
sensitivity profiles. SiSP-K01 exhibited low sensitivity, whereas
the sensitivity pattern of HuH-28 was found to be similar to that
of SiSP-K05. Notably, the only mutated gene exclusively shared
between HuH-28 and SiSP-K05 is BLM, which could potentially
contribute to the increased sensitivity of these cell lines to PARP
and ATR inhibitors. This observation warrants further
investigation. These findings suggest that an accumulation of
mutations in DDR genes could potentially be a more accurate
predictor of CCA sensitivity to PARP inhibitors than a single
gene.
Targeting ATR has become an attractive therapeutic
strategy since it was observed that cancer cells are vulnerable to
replication stress (46). This
finding has been supported by studies focusing on CCA cell lines.
Nam et al (47) investigated
the response to AZD6738 of nine CCA cell lines with varying
expression levels of ATR, ATM and p53. Their findings revealed that
cell lines with low levels of both ATM and p53 were sensitive to
AZD6738, while those with low ATM but high p53 levels exhibited
resistance. In another study, Moolmuang and Ruchirawat (27) reported the sensitivity of three CCA
cell lines to the ATR inhibitor VE-821. The study highlighted the
effectiveness of VE-821 in inhibiting the colony formation of CCA
cells, but it did not provide information on DDR-related molecular
profiles that might explain the varying drug responses. By
comparison, in the present study, only the SiSP-K05 cell line
showed a high sensitivity to AZD6738. SiSP-K01 was less sensitive,
with an IC50a value >10 mM, which was higher than
that in MMNK-1 cells. Several studies have suggested that the level
of replicative stress contributes to the sensitivity towards ATR
inhibitors. For instance, Dorado Garcia et al (48) demonstrated that increased
replicative stress in paired box 3-forkhead box O1-expressing
alveolar rhabdomyosarcoma cells heightened their sensitivity to ATR
inhibitors. In addition, a study by King et al (49) showed that high-risk neuroblastomas
with MYCN proto-oncogene-induced replication stress are highly
susceptible to ATR inhibitors VE-821 and AZD6738. Consequently, the
lower sensitivity of SiSP-K01 to AZD6738 may be associated with
reduced levels of replicative stress. However, it is hypothesized
that CCA cells can develop resistance to ATR inhibitors if the
replicative stress is mitigated by other pathways, potentially
decreasing treatment efficacy. Further investigation is warranted
to confirm this notion.
To enhance the efficacy of PARP inhibitors and
prevent resistance to these drugs, combining PARP inhibitors with
ATR inhibitors has been proposed as a promising treatment strategy
for several types of cancer, such as ovarian and prostate cancer
(50). Furthermore, numerous
clinical trials are actively recruiting patients to assess the
effectiveness of ATR inhibitors in combination with various PARP
inhibitors (51). It has been shown
that this type of combination results in increased DNA damage,
leading to mitotic catastrophe and p53-independent cell death
(48). This DNA damage can be
assessed using micronuclei and comet assays as indicators of
genomic instability, with the detection of γ-H2AX formation to
indicate DNA double-strand break formation (52). Nam et al (28) demonstrated that the combination of
AZD6738 and olaparib induced greater DNA damage in CCA cell lines
than either drug alone, as evidenced by a comet assay. Similarly,
King et al (49) observed
increased DNA damage in high-risk neuroblastomas treated with a
combination of VE-821 and olaparib. In addition, a study performed
by Sule et al (53) revealed
that the combination of AZD6738 and olaparib induced DNA damage in
IDH1-mutant cell lines, as shown by the increased formation of
γ-H2AX. Lloyd et al (54)
demonstrated that a similar combination promoted genomic
instability in ATM-defective cell lines, identified through
micronuclei analysis. In the present study, the results regarding
micronuclei and γ-H2AX formation align with these findings,
demonstrating a similar pattern of increased micronuclei formation
and γ-H2AX levels, particularly in cases where AZD6738 is combined
with either olaparib or talazoparib. Notably, these combinations
resulted in more extensive damage compared with the effects of the
drugs used individually. However, it is crucial to consider that
several mechanisms, beyond ATR activation, can contribute to PARP
inhibitor resistance in CCA. These include the restoration of HR,
mutations in PARP that diminish PARP inhibitor binding, and
increased PARP inhibitor efflux (55). Further studies are required to
explore these possibilities in CCA, in order to optimize treatment
strategies and minimize the development of resistance.
In clinical practice, toxicity must be considered,
as a combinational approach could potentially produce severe
effects, particularly with drugs targeting DDR pathways (42). Conducting preclinical drug
combination studies to understand drug interactions through the CI
can be rational for studies in humans (35). The results of the present
demonstrate that combination regimens have the potential to reduce
the required dose of PARP inhibitors from that used as a
monotherapy. The combination of AZD6738 and talazoparib resulted in
a stronger synergistic effect compared with other combinations,
particularly in TFK-1, SiSP-K01 and SiSP-K05 cell lines. Olaparib
and talazoparib, both FDA-approved for several types of cancer,
differ in their mechanisms; talazoparib is more potent than
olaparib in PARP-trapping, which refers to the process of retaining
the PARP-DNA complex, thus enhancing the cytotoxicity of
talazoparib (56,57). Previous studies have demonstrated
that the proteins responding to PARP-trapping differ from those
responding to non-trapping PARP inhibitors, and these responses
vary depending on the type of cancer (58,59).
This may explain why synergistic effects were observed to vary in
CCA cell lines with different molecular backgrounds.
The strength of the present study is the
elucidation of the relationship between DDR-mutated profiles and
the response to AZD6738, various PARP inhibitors, and their
combinations. The study also discovered that the number of mutated
genes contributes to the response to individual drugs and their
combinations. However, it should be noted that while some
combinations exhibited an improved CI, the concentrations used were
still toxic to normal cells. Therefore, the development of a
targeted drug delivery system for this type of CCA may enhance
treatment efficacy. Alternatively, pre-selecting cancers that are
more vulnerable to these drug combinations, for example, by the
measurement of replicative stress, could reduce drug toxicity.
A limitation of the present study is the limited
number of cell lines used, considering that CCA is heterogeneous,
and its mutational profiles vary depending on etiological agents
and tumor locations. Therefore, more diverse cell lines are
required to confirm if the accumulation of certain DDR genes
benefits from ATR and PARP inhibitors, either alone or in
combination. Additionally, while genetic profiling was used,
functional biomarkers such as DNA fiber assays or RAD51 foci
formation were not included. These could provide more insight and
serve as additional criteria for determining the sensitivity to DDR
inhibitors. Moreover, the frequency of genetic variations in the
CCA cell lines was not well established, leaving to uncertainty
about which mutations are predominant in these cell lines. The
study also lacks evaluation of the long-term treatment of CCA using
dose-escalating protocols for a defined duration, for example,
starting with a low dose for 3 months, escalating to a higher dose
for another 6 months and then using a maintenance dose for 9
months. Such evaluation would provide an increased understanding of
the dynamics of genetic alterations associated with the development
of drug resistance. Finally, future research should include in
vivo validation experiments in animal models, such as PDX and
pharmacokinetic studies. This would enable evaluation of the
complex tumor microenvironment and its impact on drug response,
including drug toxicity.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank Professor Seiji
Okada (Division of Hematopoiesis, Joint Research Center for Human
Retrovirus Infection & Graduate School of Medical Sciences,
Kumamoto University, Kumamoto, Japan) for experimental support.
Funding
This research is supported by Mahidol University (Basic Research
Fund: fiscal year 2022) and the National Research Council of
Thailand (grant no. N41A640160). SJ is supported by the National
Research Council of Thailand (grant no. N41A640162) and the
Foundation for Cancer Care Siriraj Hospital.
Availability of data and materials
The next-generation sequencing results generated
for SiSP-K01 and SiSP-K05 in the present study may be found in the
Sequence Read Archive at the following URLs (https://www.ncbi.nlm.nih.gov/sra/SRR31111387 and
http://www.ncbi.nlm.nih.gov/sra/SRR31111386). The
other data generated in the present study may be requested from the
corresponding authors.
Authors' contributions
TL contributed to data interpretation, formal
analysis, experimentation, methodology and visualization, and wrote
the original draft of the manuscript. SP and RW contributed to
methodology, experimentation and manuscript review and editing. SC,
WS and PO contributed to the experimentation, and participated in
the review and editing of the manuscript. SJ was responsible for
conceptualization, resource and funding acquisition, and
contributed to manuscript review and editing. DD supervised the
study, and contributed to conceptualization, funding acquisition
and manuscript review and editing. DD and SJ confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
The protocol was approved by the Human Research
Ethics Committee, Faculty of Medicine Ramathibodi Hospital, Mahidol
University (Bangkok, Thailand; approval no. MURA2023/155). The
study utilized established primary cell lines obtained from
Professor Seiji Okada, Kumamoto University (Kumamoto, Japan) and
handled in accordance with the guidelines and regulations of the
Human Research Ethics Committee of Ramathibodi Hospital.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
During the preparation of this work, ChatGPT 4 was
utilized for spelling and grammar checks. Subsequently, the authors
reviewed and edited the content as necessary, taking full
responsibility for the ultimate content of the present
manuscript.
References
|
1
|
Brindley PJ, Bachini M, Ilyas SI, Khan SA,
Loukas A, Sirica AE, The BT, Wongkham S and Gores GJ:
Cholangiocarcinoma. Nat Rev Dis Primers. 7:652021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Banales JM, Marin JJG, Lamarca A,
Rodrigues PM, Khan SA, Roberts LR, Cardinale V, Carpino G, Andersen
JB, Braconi C, et al: Cholangiocarcinoma 2020: The next horizon in
mechanisms and management. Nat Rev Gastroenterol Hepatol.
17:557–588. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ohaegbulam KC, Koethe Y, Fung A, Mayo SC,
Grossberg AJ, Chen EY, Sharzehi K, Kardosh A, Farsad K, Rocha FG,
et al: The multidisciplinary management of cholangiocarcinoma.
Cancer. 129:184–214. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Squadroni M, Tondulli L, Gatta G, Mosconi
S, Beretta G and Labianca R: Cholangiocarcinoma. Crit Rev Oncol
Hematol. 116:11–31. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gönül Geyik Ö, Anichini G, Ulukaya E,
Marra F and Raggi C: DNA damage response inhibitors in
cholangiocarcinoma: Current progress and perspectives. Cells.
11:14632022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chae H, Kim D, Yoo C, Kim KP, Jeong JH,
Chang HM, Lee SS, Park DH, Song TJ, Hwang S, et al: Therapeutic
relevance of targeted sequencing in management of patients with
advanced biliary tract cancer: DNA damage repair gene mutations as
a predictive biomarker. Eur J Cancer. 120:31–39. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ahn DH and Bekaii-Saab T: Biliary tract
cancer and genomic alterations in homologous recombinant
deficiency: Exploiting synthetic lethality with PARP inhibitors.
Chin Clin Oncol. 9:62020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Beijersbergen RL, Wessels LFA and Bernards
R: Synthetic lethality in cancer therapeutics. Annu Rev Cancer
Biol. 1:141–161. 2017. View Article : Google Scholar
|
|
9
|
Pilié PG, Tang C, Mills GB and Yap TA:
State-of-the-art strategies for targeting the DNA damage response
in cancer. Nat Rev Clin Oncol. 16:81–104. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
D'Andrea AD: Mechanisms of PARP inhibitor
sensitivity and resistance. DNA Repair (Amst). 71:172–176. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fong PC, Boss DS, Yap TA, Tutt A, Wu P,
Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, et
al: Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA
mutation carriers. N Engl J Med. 361:123–134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Balasubramaniam S, Beaver JA, Horton S,
Fernandes LL, Tang S, Horne HN, Liu J, Liu C, Schrieber SJ, Yu J,
et al: FDA approval summary: rucaparib for the treatment of
patients with deleterious BRCA mutation-associated advanced ovarian
cancer. Clin Cancer Res. 23:7165–7170. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mirza MR, Monk BJ, Herrstedt J, Oza AM,
Mahner S, Redondo A, Fabbro M, Ledermann JA, Lorusso D, Vergote I,
et al: Niraparib maintenance therapy in platinum-sensitive,
recurrent ovarian cancer. N Engl J Med. 375:2154–2164. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hoy SM: Talazoparib: First global
approval. Drugs. 78:1939–1946. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li N, Bu H, Liu J, Zhu J, Zhou Q, Wang L,
Yin R, Wu X, Yao S, Gu K, et al: An open-label, multicenter,
single-arm, phase II study of fluzoparib in patients with germline
BRCA1/2 mutation and platinum-sensitive recurrent ovarian cancer.
Clin Cancer Res. 27:2452–2458. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xu B, Yin Y, Dong M, Song Y, Li W, Huang
X, Wang T, He J, Mu X, Li L, et al: Pamiparib dose escalation in
Chinese patients with non-mucinous high-grade ovarian cancer or
advanced triple-negative breast cancer. Cancer Med. 10:109–118.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
da Costa AABA, Chowdhury D, Shapiro GI,
D'Andrea AD and Konstantinopoulos PA: Targeting replication stress
in cancer therapy. Nat Rev Drug Discov. 22:38–58. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Smith G, Alholm Z, Coleman RL and Monk BJ:
DNA damage repair inhibitors-combination therapies. Cancer J.
27:501–505. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mullard A: DNA damage response drugs for
cancer yield continued synthetic lethality learnings. Nat Rev Drug
Discov. 21:403–405. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shah PD, Wethington SL, Pagan C, Latif N,
Tanyi J, Martin LP, Morgan M, Burger RA, Haggerty A, Zarrin H, et
al: Combination ATR and PARP inhibitor (CAPRI): A phase 2 study of
ceralasertib plus olaparib in patients with recurrent,
platinum-resistant epithelial ovarian cancer. Gynecol Oncol.
163:246–253. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Clinical Trials: NCT03330847: To assess
safety and efficacy of agents targeting DNA damage repair with
olaparib versus olaparib monotherapy. https://clinicaltrials.gov/show/NCT03330847June
4–2023
|
|
22
|
Clinical Trials: NCT03787680: Targeting
resistant prostate cancer with ATR and PARP inhibition (TRAP
Trial). https://clinicaltrials.gov/show/NCT03787680June
4–2023
|
|
23
|
Clinical Trials: NCT03878095: Testing
olaparib and AZD6738 in IDH1 and IDH2 mutant tumors. https://clinicaltrials.gov/show/NCT03878095June
4–2023
|
|
24
|
Clinical Trials: NCT04298021: DDR-umbrella
study of DDR targeting agents in advanced biliary tract cancer.
https://clinicaltrials.gov/study/NCT04298021November
13–2023
|
|
25
|
Serra-Camprubí Q, Verdaguer H, Oliveros W,
Lupión-Garcia N, Llop-Guevara A, Molina C, Vila-Casadesús M, Turpin
A, Neuzillet C, Frigola J, et al: Human metastatic
cholangiocarcinoma patient-derived xenografts and tumoroids for
preclinical drug evaluation. Clin Cancer Res. 29:432–445. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bezrookove V, Patino JM, Nosrati M,
Desprez PY, McAllister S, Soroceanu L, Baron A, Osorio R,
Kashani-Sabet M and Dar AA: Niraparib suppresses cholangiocarcinoma
tumor growth by inducing oxidative and replication stress. Cancers
(Basel). 13:44052021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Moolmuang B and Ruchirawat M: The
antiproliferative effects of ataxia-telangiectasia mutated and ATM-
and Rad3-related inhibitions and their enhancements with the
cytotoxicity of DNA damaging agents in cholangiocarcinoma cells. J
Pharm Pharmacol. 73:40–51. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nam AR, Yoon J, Jin MH, Bang JH, Oh KS,
Seo HR, Kim JM, Kim TY and Oh DY: ATR inhibition amplifies
antitumor effects of olaparib in biliary tract cancer. Cancer Lett.
516:38–47. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Maruyama M, Kobayashi N, Westerman KA,
Sakaguchi M, Allain JE, Totsugawa T, Okitsu T, Fukazawa T, Weber A,
Stolz DB, et al: Establishment of a highly differentiated
immortalized human cholangiocyte cell line with SV40T and hTERT.
Transplantation. 77:446–451. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kusaka Y, Tokiwa T and Sato J:
Establishment and characterization of a cell line from a human
cholangiocellular carcinoma. Res Exp Med (Berl). 188:367–375. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Saijyo S, Kudo T, Suzuki M, Katayose Y,
Shinoda M, Muto T, Fukuhara K, Suzuki T and Matsuno S:
Establishment of a new extrahepatic bile duct carcinoma cell line,
TFK-1. Tohoku J Exp Med. 177:61–71. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Suntiparpluacha M, Chanwat R,
Limsrichamrern S, More-krong P, Srifa S, Kongsri K, Aroonpruksakul
S, Sathirareuangchai S, Sampatavanich S, Okada S and Jirawatnotai
S: Establishment of cholangiocarcinoma organoids from long-term
frozen tissues. J Basic Appl Pharmacol. 2:O11–O25. 2022.
|
|
33
|
Jamnongsong S, Kueanjinda P, Buraphat P,
Sakornsakolpat P, Vaeteewoottacharn K, Okada S, Jirawatnotai S and
Sampattavanich S: Comprehensive drug response profiling and
pan-omic analysis identified therapeutic candidates and prognostic
biomarkers for Asian cholangiocarcinoma. iScience. 25:1051822022.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li MM, Datto M, Duncavage EJ, Kulkarni S,
Lindeman NI, Roy S, Tsimberidou AM, Vnencak-Jones CL, Wolff DJ,
Younes A and Nikiforova MN: Standards and guidelines for the
interpretation and reporting of sequence variants in cancer: A
joint consensus recommendation of the association for molecular
pathology, American society of clinical oncology, and college of
american pathologists. J Mol Diagn. 19:4–23. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chou TC: Theoretical basis, experimental
design, and computerized simulation of synergism and antagonism in
drug combination studies. Pharmacol Rev. 58:621–681. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schindelin J, Arganda-Carreras I, Frise E,
Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S,
Schmid B, et al: Fiji: An open-source platform for biological-image
analysis. Nat Methods. 9:676–682. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jang SM, Redon CE, Fu H, Indig FE and
Aladjem MI: RepID-deficient cancer cells are sensitized to a drug
targeting p97/VCP segregase. Mol Cell Toxicol. 17:141–149. 2021.
View Article : Google Scholar
|
|
39
|
Wikiniyadhanee R, Lerksuthirat T,
Stitchantrakul W, Chitphuk S, Sura T and Dejsuphong D: TRIM29 is
required for efficient recruitment of 53BP1 in response to DNA
double-strand breaks in vertebrate cells. FEBS Open Bio.
10:2055–2071. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lerksuthirat T, Wikiniyadhanee R, Chitphuk
S, Stitchantrakul W, Sampattavanich S, Jirawatnotai S, Jumpathong J
and Dejsuphong D: DNA Repair Biosensor-Identified DNA Damage
Activities of Endophyte Extracts from Garcinia cowa.
Biomolecules. 10:16802020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Audeh MW, Carmichael J, Penson RT,
Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN,
Oaknin A, Loman N, et al: Oral poly(ADP-ribose) polymerase
inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and
recurrent ovarian cancer: A proof-of-concept trial. Lancet.
376:245–251. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Martorana F, Da Silva LA, Sessa C and
Colombo I: Everything comes with a price: The toxicity profile of
DNA-damage response targeting agents. Cancers (Basel). 14:9532022.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bradbury A, Hall S, Curtin N and Drew Y:
Targeting ATR as cancer therapy: A new era for synthetic lethality
and synergistic combinations? Pharmacol Ther. 207:1074502020.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Clinical Trials: NCT03462342: Combination
ATR and PARP inhibitor (CAPRI) trial with AZD6738 and olaparib in
recurrent ovarian cancer (CAPRI). https://clinicaltrials.gov/study/NCT03462342November
11–2023
|
|
45
|
Clinical Trials: NCT05269316: Study to
evaluate IMP9064 as a monotherapy or in combination in patients
with advanced solid tumors. https://www.clinicaltrials.gov/study/NCT05269316November
11–2023
|
|
46
|
Lecona E and Fernandez-Capetillo O:
Targeting ATR in cancer. Nat Rev Cancer. 18:586–595. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Nam AR, Jin MH, Park JE, Bang JH, Oh DY
and Bang YJ: Therapeutic targeting of the DNA damage response using
an ATR inhibitor in biliary tract cancer. Cancer Res Treat.
51:1167–1179. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Dorado Garcia H, Pusch F, Bei Y, von
Stebut J, Ibáñez G, Guillan K, Imami K, Gürgen D, Rolff J,
Helmsauer K, et al: Therapeutic targeting of ATR in alveolar
rhabdomyosarcoma. Nat Commun. 13:42972022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
King D, Southgate HED, Roetschke S,
Gravells P, Fields L, Watson JB, Chen L, Chapman D, Harrison D,
Yeomanson D, et al: Increased replication stress determines ATR
inhibitor sensitivity in neuroblastoma cells. Cancers (Basel).
13:62152021. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Bhamidipati D, Haro-Silerio JI, Yap TA and
Ngoi N: PARP inhibitors: Enhancing efficacy through rational
combinations. Br J Cancer. 129:904–916. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yano K and Shiotani B: Emerging strategies
for cancer therapy by ATR inhibitors. Cancer Sci. 114:2709–2721.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sadeghi F, Asgari M, Matloubi M, Ranjbar
M, Karkhaneh Yousefi N, Azari T and Zaki-Dizaji M: Molecular
contribution of BRCA1 and BRCA2 to genome instability in breast
cancer patients: Review of radiosensitivity assays. Biol Proced
Online. 22:232020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sule A, Van Doorn J, Sundaram RK, Ganesa
S, Vasquez Juan C and Bindra Ranjit S: Targeting IDH1/2 mutant
cancers with combinations of ATR and PARP inhibitors. NAR Cancer.
3:zcab0182021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lloyd RL, Wijnhoven PWG, Ramos-Montoya A,
Wilson Z, Illuzzi G, Falenta K, Jones GN, James N, Chabbert CD,
Stott J, et al: Combined PARP and ATR inhibition potentiates genome
instability and cell death in ATM-deficient cancer cells. Oncogene.
39:4869–4883. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lee EK and Matulonis UA: PARP inhibitor
resistance mechanisms and implications for post-progression
combination therapies. Cancers (Basel). 12:20542020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Onji H and Murai J: Reconsidering the
mechanisms of action of PARP inhibitors based on clinical outcomes.
Cancer Sci. 113:2943–2951. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Murai J, Huang SYN, Renaud A, Zhang Y, Ji
J, Takeda S, Morris J, Teicher B, Doroshow JH and Pommier Y:
Stereospecific PARP trapping by BMN 673 and comparison with
olaparib and rucaparib. Mol Cancer Ther. 13:433–443. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Mosler T, Baymaz HI, Gräf JF, Mikicic I,
Blattner G, Bartlett E, Ostermaier M, Piccinno R, Yang J, Voigt A,
et al: PARP1 proximity proteomics reveals interaction partners at
stressed replication forks. Nucleic Acids Res. 50:11600–11618.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zamalloa LG, Pruitt MM, Hermance NM, Gali
H, Flynn RL and Manning AL: RB loss sensitizes cells to
replication-associated DNA damage after PARP inhibition by
trapping. Life Sci Alliance. 6:e2023020672023. View Article : Google Scholar : PubMed/NCBI
|