Introduction
High-risk early-stage triple-negative breast cancer
(TNBC) is often associated with early recurrence and high mortality
rates (1). In addition to
potentially increasing the likelihood of tumor resectability and
breast conservation rate, patients who achieve pathologic complete
response (pCR) after preoperative chemotherapy have longer
event-free and overall survival (2). The recent KEYNOTE-522 trial (3) demonstrated a significantly higher pCR
rate in patients who received pembrolizumab plus preoperative
chemotherapy (64.8%) compared to that in those who received placebo
plus preoperative chemotherapy (51.2%). However, the follow-up
period for clinical trials of preoperative chemotherapy, including
immune checkpoint inhibitor (ICI), is still short, and little is
known about the overall picture of immune-related adverse events
(irAEs).
In a randomized phase III trial of pembrolizumab or
placebo as preoperative systemic chemotherapy (PST) in patients
with stage II or III TNBC, grade 3 or higher irAEs occurred in
12.9% of patients in the pembrolizumab chemotherapy group (4). The most frequent irAEs were
hypothyroidism (15.7%), severe skin reactions (5.7%),
hyperthyroidism (5.2%), and adrenal insufficiency (2.6%), with no
hemophagocytic lymphohistiocytosis (HLH) reported. Fatal irAEs
occur in a small number of patients, ranging from 0.36% in patients
receiving anti-PD-1 monotherapy to 1.23% in those receiving
combination therapy (5).
Hematological irAEs (hemirAEs) are difficult to
treat and have a high mortality rate. In a retrospective study, the
incidence of hemirAEs was reported to be 0.6%, with higher
frequencies of anemia, neutropenia, and thrombocytopenia (6). Among the hemophagocytic irAEs,
hemophagocytic syndrome is relatively rare, with only a few case
reports of hemophagocytic syndrome with ICIs; however, it can be a
life-threatening complication. Here, we report a case of
pembrolizumab-induced HLH in a patient with locally advanced
TNBC.
Case report
A 38-year-old woman presented to National Center for
Global Health and Medicine (Tokyo, Japan) in November 2022 with
locally advanced left breast cancer, cT1N3M0, cStage IIIC, which
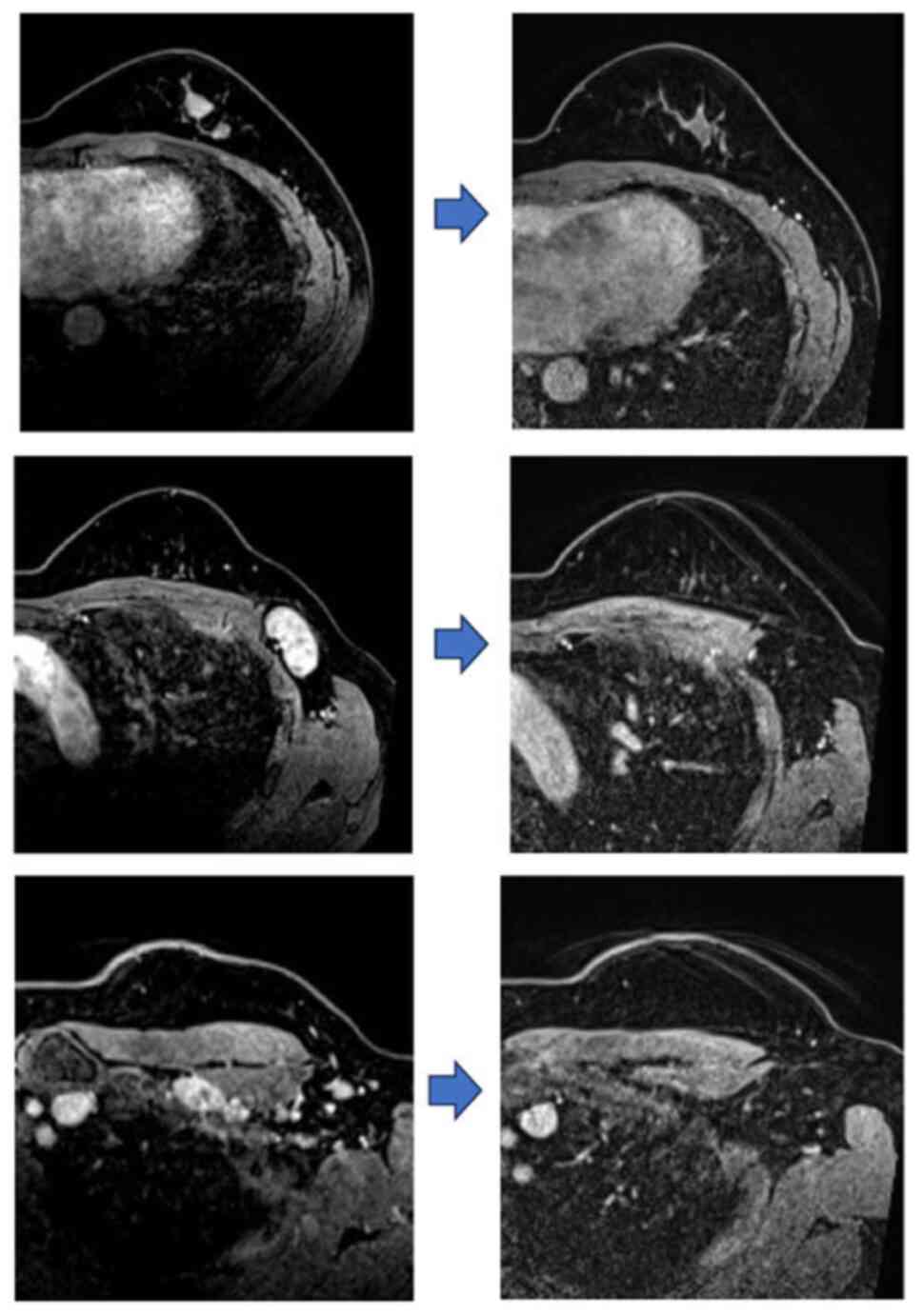
was HR-negative- and HER2-negative (Fig. 1). The patient had no relevant
medical history. The patient exhibited a pathogenic mutation in the
germline BRCA1 (breast cancer susceptibility gene) as identified by
the BRACAnalysis CDx® (Germline Companion Diagnostic
Test). PST with pembrolizumab was considered appropriate for this
young patient, who had a poor prognosis based on the clinical stage
and biomarkers.
The patient was initially treated with carboplatin,
paclitaxel, and pembrolizumab without severe adverse events until
the third course, at which point the mass became non-palpable
(cT0). On day 8 of the third course (day 62 from the onset of
chemotherapy), she experienced persistent grade 1 diarrhea without
infection symptoms for two weeks, raising concerns for
immune-related colitis, prompting a colonoscopy. Owing to mild
mucosal inflammation and localized sigmoid edema observed on CT, a
5-ASA preparation was administered, improving her colitis to grade
0 within two weeks. However, diarrhea recurred on day 96, 19 days
post-colonoscopy, escalating to grade 2. Consequently, steroids
(PSL; prednisone) were initiated at a dosage of 0.5 mg/kg/day (30
mg/day), following the guidelines of the Japanese Society of
Clinical Oncology. The introduction of PSL led to an improvement in
diarrhea to grade 0, and the steroid dosage was subsequently
tapered by 5 mg per week.
Owing to immune-related adverse events (irAEs)
manifesting as colitis, pembrolizumab was discontinued on day 70,
and the patient continued chemotherapy with carboplatin and
paclitaxel. On day 114, liver function deteriorated to grade 2,
despite treatment with 20 mg/day of steroids. IrAE hepatitis was
suspected, leading to hospitalization. A high fever was also
observed during this period. The steroid dosage was increased to 30
mg/day (0.5 mg/kg) on day 118, five days following the onset of
fever and liver dysfunction, and on the fifth-day
post-hospitalization. Liver function temporarily improved but
deteriorated again to grade 3, five days after the dosage increase
(day 122; post-hospitalization day 9). Consequently, the steroid
dosage was elevated to 60 mg/day (1 mg/kg). Although liver function
showed slight improvement, it worsened to grade 4 on day 127
(post-hospitalization day 14). The patient then commenced treatment
with mycophenol mofetil (MMF) at 2,000 mg/day, as per the Japanese
guidelines for irAEs.
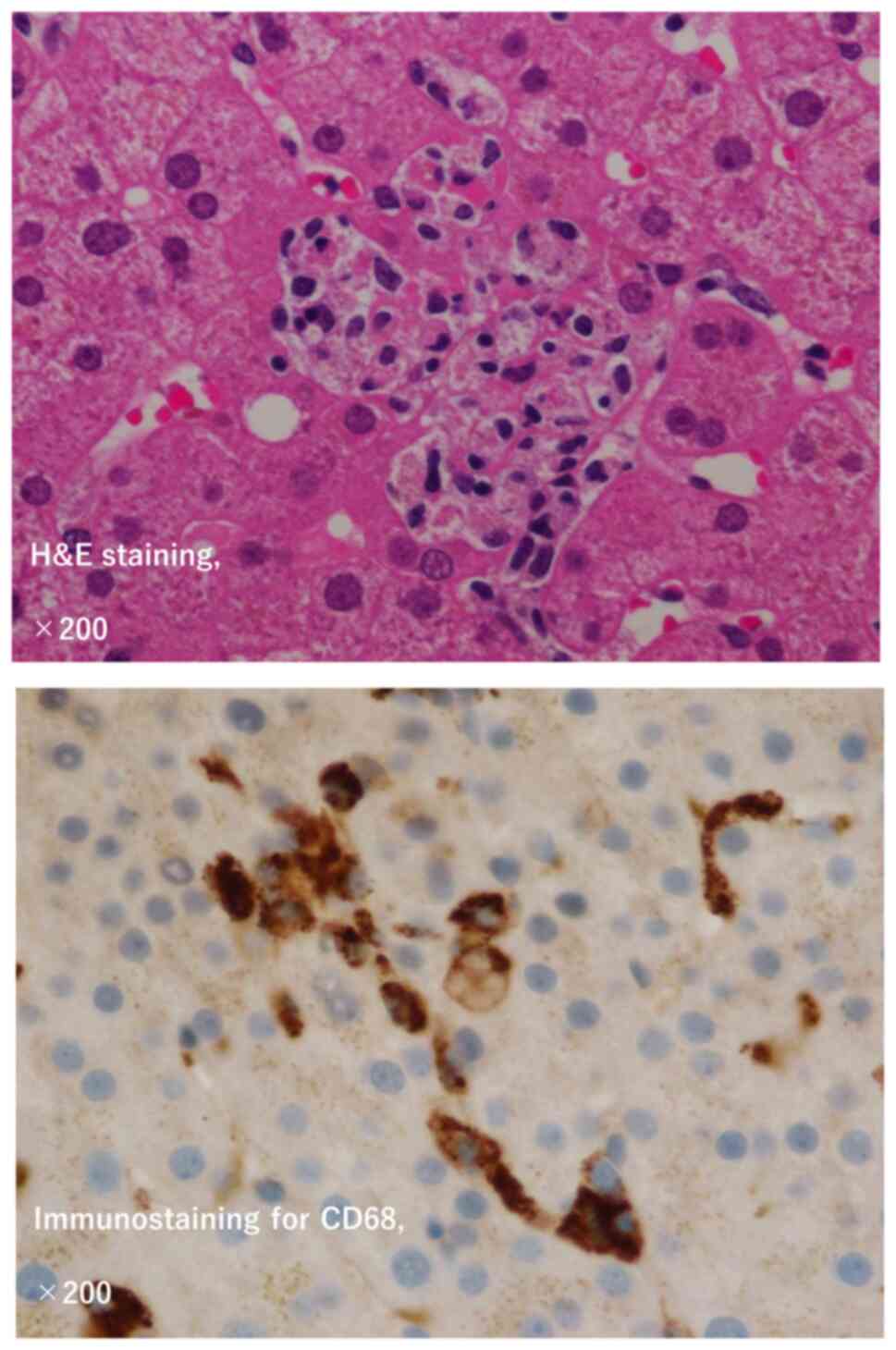
A hepatologist performed a liver biopsy on day 126
for further assessment. The subsequent day, continuous bleeding
from the biopsy site was noted, with unmeasurably high
fibrinogen/fibrin degradation products (FDP) and D-dimer levels. No
hematopenia was observed. Given the persistent fever, coagulation
abnormalities, liver damage, elevated lactate dehydrogenase (LDH)
levels, hyperferritinemia, and the hemophagocytic profile observed
in the liver biopsy pathology (Fig.
2), a comprehensive diagnosis of secondary HLH was made. The
diagnostic score for reactive hemophagocytic syndrome in adult HLH
[HScore (7)] is shown in Table I. Levels of interleukin-6 (IL-6) and
soluble IL-2 receptor (sIL2R) were elevated to 325.0 pg/ml and 2180
U/ml, respectively. She was diagnosed with disseminated
intravascular coagulation (DIC) with a predominantly fibrinolytic
system, and treatment with nafamostat and fresh frozen plasma (FFP)
was initiated. The day after MMF management began, liver function
and coagulation abnormalities deteriorated. Methylprednisolone (1
g/day) was administered as a steroid pulse for three days (day 128,
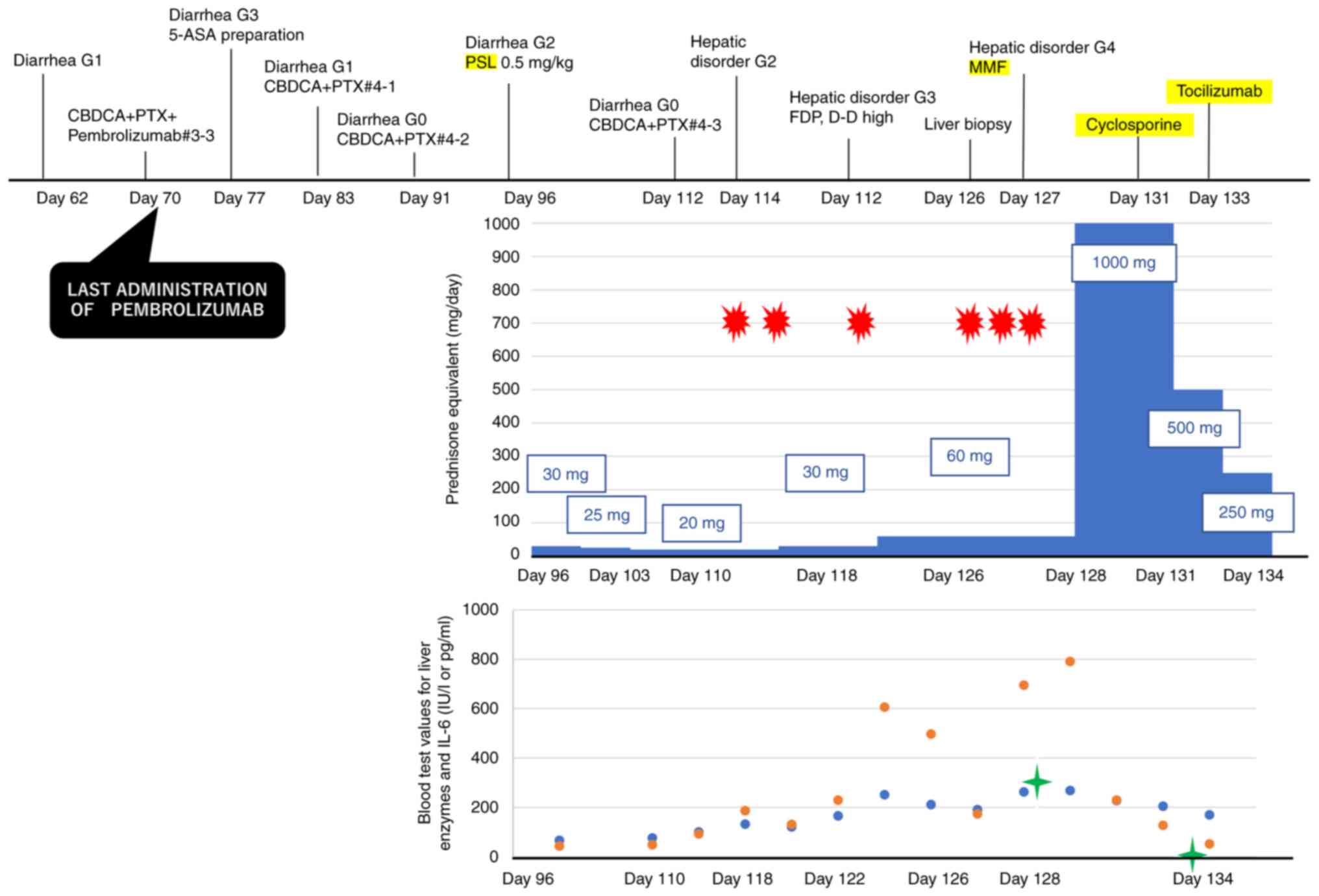
post-hospitalization day 15). The course of the patient's symptoms,
liver function, and medications administered are detailed in
Fig. 3.
 | Table I.HScore. |
Table I.
HScore.
| Parameter | No. of points
(criteria for scoring) |
|---|
| Known underlying
immunosuppressiona | 0 (no) or 18
(yes) |
| Temperature, °C | 0 (<38.4),
33 (38.4–39.4), or 49 (>39.4) |
| Organomegaly | 0 (no), 23
(hepatomegaly or splenomegaly) or 38 (hepatomegaly and
splenomegaly) |
| No. of
cytopeniasb | 0 (1 lineage),
24 (2 lineages), or 34 (3 lineages) |
| Ferritin, ng/ml | 0 (<2,000), 35
(2,000–6,000), or 50 (>6,000) |
| Triglyceride,
mmol/l | 0 (<1.5),
44 (1.5–4), or 64 (>4) |
| Fibrinogen, g/l | 0 (<2.5) or
30 (>2.5) |
| Serum glutamic
oxaloacetic transaminase, IU/l | 0 (<30) or
19 (>30) |
| Hemophagocytosis
features on bone marrow aspirate | 0 (no) or 35
(yes) |
Liver function and coagulation abnormalities
improved after the completion of steroid pulse therapy, and gradual
tapering was planned. Given the onset of HLH, whereas she was on 60
mg PSL, we considered the need for further correction of
hypercytokinemia before tapering the steroids with tocilizumab, an
anti-interleukin (IL)-6 antibody, 8 mg/kg once on the third day
after the end of the pulse (day 133, post-hospitalization day 20).
The levels of IL-6 decreased to 1.8 pg/ml after the steroid pulse.
We also administered cyclosporine after the pulse steroid therapy.
The doses of MMF and cyclosporine were tapered, as with the
steroids.
The differential diagnoses included coronavirus
disease 2019 (COVID-19), tuberculosis, bacterial pneumonia, viral
hepatitis, adrenal insufficiency, and irAEs such as CRS and HLH.
The patient tested negative for COVID-19. Extensive tests for
infectious diseases, including interferon (IFN)-gamma release
assay, human immunodeficiency virus, Aspergillus antigen, and
urine/sputum/blood cultures on multiple occasions, were unrevealed.
The respiratory viral panel was negative (for influenza A/B,
parainfluenza 1/2/3/4, adenovirus, coronavirus [not COVID-19], and
rhinovirus/enterovirus) before hospitalization. Before the
initiation of PST, Epstein-Barr virus (EBV) demonstrated a
pre-existing infection pattern (EBV-EBNA 40, EBV–VCA IgG 160,
VCA-IgM <10), and an EBV DNA level of 4.0×104
copies/ml on day 127 (the day before the steroid pulse) indicated
either reactivation of EBV owing to ICI use or potential EBV
hepatitis. Upon detailed analysis, the EBV DNA level decreased
during steroid tapering, and liver function consistently improved.
Therefore, whereas EBV may influence pathogenesis, its precise role
remains unclear. The patients were discharged on
post-hospitalization day 69 with a PSL dosage of 25 mg/day and
attended weekly outpatient follow-ups. The PSL dose was reduced to
2 mg/day (with MMF completed and cyclosporine at 100 mg/day) by the
day of breast cancer surgery (day 209). In the surgical specimen, a
pCR was confirmed (pathological stage ypT0ypN0). PSL was
discontinued 130 days post-initiation of steroid therapy
(post-hospitalization day 108). The patient was monitored without
additional cancer-directed treatments and remained
recurrence-free.
Discussion
HLH is a rare and severe disease characterized by
the hyperactivation of immune cells (such as T lymphocytes,
monocytes, and macrophages). They can be of primary or secondary
origin, mainly owing to infection, cancer, or autoimmune diseases
(Table II). Primary HLH is most
frequently observed in infants and children. The diagnosis of
secondary hemophagocytic HLH is based on exclusion rather than
direct evidence of specific triggers and is associated with
aberrant immunomodulatory mechanisms such as hypercytokinemia.
Consequently, once HLH pathogenesis is established, it is essential
to confirm the presence of potential underlying factors, including
infections, malignancies, and autoimmune diseases. In this case, a
genetic predisposition seemed unlikely, given the absence of family
history or immunodeficiency. Moreover, considering the observed
reduction in breast cancer size following chemotherapy, the
likelihood of secondary HLH resulting from the tumor was also
deemed unlikely. Importantly, no evidence of rheumatic disease,
such as arthralgia or skin rash, was observed, and antinuclear
antibodies were negative prior to the initiation of PST. Thus,
considering the exclusion of infectious diseases, a diagnosis of
secondary HLH resulting from ICI was considered. Although not
previously conceptualized, knowledge of ICI-related HLH is
essential for oncologists treating patients with ICI. HLH is
generally life-threatening, with a mortality rate of up to 66%
(3). In clinical practice, the
rapid progression of HLH often leads to multiple organ failure and
death, which are difficult to treat. Therefore, accurate diagnosis
and early intervention are critical to improve the prognosis of
HLH.
| Table II.Classification of HLH. |
Table II.
Classification of HLH.
| Primary (inherited)
HLH | Secondary (reactive)
HLH |
|---|
| •FHL | •Infection-associated
hemophagocytic syndrome |
|
| Virus-associated
hemophagocytic syndrome |
|
| Bacteria-associated
hemophagocytic syndrome |
|
| Fungus-associated
hemophagocytic syndrome |
|
| Parasite-associated
hemophagocytic syndrome |
| •Immune deficiency
syndrome |
•Malignancy-associated hemophagocytic
syndrome |
| Griscelli
syndrome | •Lymphoma-associated
hemophagocytic syndrome |
| Chedial-Higashi
syndrome, etc. |
|
|
|
•Autoimmune-associated hemophagocytic
syndrome |
|
| •Immune checkpoint
inhibitors-associated hemophagocytic syndrome |
|
| •Other
(transplanting-associated hemophagocytic syndrome, etc.) |
Patients with HLH typically present with recurrent
high fever and pancytopenia. Blood tests reveal hyperferritinemia,
hypertriglyceridemia, and hypofibrinogenemia. Features of
hemophagocytosis may be observed in a bone marrow smear
examination. Diagnostic criteria for children are well established
(HLH-2004) (8), and a probability
score (HScore) for adults has recently been proposed (Table I) (7). The onset of HLH can range from 5 days
to 1 year following the initial dose of ICI (9), with a median time of 6 weeks (10). Although rare, HPS should be
suspected when persistent high fever and blood cell loss in two or
more systems are observed, with or without associated
hypertriglyceridemia, hyperferritinemia, hypofibrinogenemia, and
elevated sIL2R levels. In this instance, the HScore was 199
(90–250), and establishing a diagnosis was challenging owing to the
absence of clear thrombocytopenia evidence.
Case reports of HLH during ICI treatment for solid
tumors are summarized in Table
III. The mean time to symptom onset was after eight doses. Bone
biopsies were conducted in approximately half of the patients and
were not universally performed despite numerous reports where HLH
was clinically diagnosed. We performed a liver biopsy, although it
was not obligatory. However, the phagocytic images provided
sufficient evidence to suggest that HLH developed in these
patients. Steroids were administered in all cases, and nonsteroidal
immunosuppressive therapies (NSIT) were used in four cases, as in
the current case. Of the 14 patients, 11 showed improvement, three
(one of whom was receiving nonsteroidal immunosuppressive therapy)
died, and the outcome of one patient was not documented.
| Table III.List of prior cases of HLH secondary
to ICI therapy. |
Table III.
List of prior cases of HLH secondary
to ICI therapy.
| First author,
year | Type of ICI | Timing/cycles of
therapy | Primary
malignancy | Method of
diagnosis | BM
biopsy/pathology | Treatment | Clinical outcome | (Refs.) |
|---|
| Present case | Pembrolizumab | 3 doses | Breast cancer | Liver biopsy, fever,
coagulation abnormality, elevation of ferritin, LDH, iver enzyme
levels l and IL-6 | Liver biopsy for
HLH | Steroids,
mycophenolate mofetil, tocilizumab, cyclosporine | Improvement | - |
| Kurozumi, 2021 (case
1) | Pembrolizumab | 1 dose | NSCLC | BM biopsy, fever,
cytopenia, coagulation abnormality, elevation of ferritin and liver
enzyme levels | N/A | Steroids | Improvement | (15) |
| Kurozumi, 2021 (case
2) | Pembrolizumab | 16 cycles of
durvalumab, 2 of doses pemetrexed + pembrolizumab | NSCLC | Cytopenia,
coagulation abnormalities, and elevation of ferritin, liver enzyme
levels | N/A | Steroids | Improvement | (15) |
| Sackstein, 2021 | Pembrolizumab | 3 doses | NSCLC | Fever, cytopenia,
elevation of ferritin, soluble IL-2R, LDH and liver enzyme
levels | N/A | Steroids,
tocilizumab | Improvement | (16) |
| Okawa, 2019 | Pembrolizumab | 1 dose | NSCLC | BM biopsy, liver
biopsy, soluble IL-2R, ferritin elevation, cytopenias | BM biopsy for
HLH | Steroids | Improvement | (17) |
| Laderian, 2019 | Pembrolizumab | 12 months | Thymic cancer | BM biopsy, liver
biopsy, soluble IL-2R, ferritin elevation, cytopenias | BM biopsy for
HLH | Steroids,
IVIG, | Death | (18) |
|
|
|
|
|
|
|
|
anakinraa |
|
| Honjo, 2019 | Nivolumab | 4 doses | NSCLC | Ferritin, soluble
IL-2R, triglyceride elevation | N/A | Steroids,
mycophenolate mofetil | Improvement | (19) |
| Hantel, 2018 | Ipilimumab and
nivolumab | 4 doses of
ipilimumab, 1 dose of ipilimumab and nivolumab | Melanoma | BM biopsy, soluble
IL-2R elevation | BM biopsy for
HLH | Steroids | Improvement | (20) |
| Satzger, 2018 | Ipilimumab and
nivolumab | 4 doses | Melanoma | Liver biopsy,
ferritin, triglyceride, soluble IL-2 elevation, cytopenias | N/A | Steroids,
mycophenolate mofetil | Improvement | (21) |
| Sadaat, 2018 | Pembrolizumab | 6 doses | Melanoma | NK cell functional
assay, soluble CD163 elevation | N/A | Steroids | Improvement | (22) |
| Takeshita,
2017 | Nivolumab | 2 doses | NSCLC | BM biopsy | BM biopsy for
HLH | Steroids | Improvement | (23) |
| Malissen, 2017
(case 1) | Nivolumab | 17 months | Melanoma | BM biopsy | BM biopsy for
HLH | Steroids | Death | (24) |
| Shah, 2017 | Pembrolizumab | 9 months | Bladder cancer | BM biopsy, NK cell
functional assay, soluble IL-2R elevation | BM biopsy for
HLH | Steroids and
etoposide (HLH 2004) | Unknown | (25) |
| Malissen, 2017
(case 2) | Ipilimumab | 1 dose of
ipilimumab; prior history of 9 months of nivolumab | Melanoma | Ferritin,
triglyceride elevation, cytopenias | BM biopsy negative
for HLH | Steroids | Improvement | (24) |
| Malissen, 2017
(case 3) | Avelumab | 1 dose | Merkel cell
carcinoma | Ferritin,
triglyceride elevation, cytopenias | N/A | Steroids | Death | (24) |
HLH is an umbrella term for hyperinflammatory
conditions that involve supramaximal activation of the immune
system. The overproduction of inflammatory cytokines by abnormally
activated T lymphocytes and macrophages is one of the main factors
in disease pathogenesis (11).
There are many reports of increased blood levels of inflammatory
cytokines such as tumor necrosis factor α, IFN-γ, IL-1, IL-6,
IL-12, IL-18, sIL-2R, and FasL in patients with HLH. It is also
widely known that such hypercytokinemia is accompanied by abnormal
activation of the blood coagulation system/fibrinolytic system
(fibrinolytic system), a condition known as DIC. Therefore, the
prognosis of HLH can be improved by suppressing inflammatory
cytokine expression.
Pembrolizumab is a monoclonal antibody that inhibits
the programmed cell death-1 (PD-1) receptor. PD-1 is expressed by
immune cells and plays a role in regulating self-tolerance by
downregulating immune responses. The interaction of PD-1 with PD-L1
in the tumor microenvironment compromises normal T cell function
and promotes the conversion of cytotoxic T cells into regulatory T
cells (12). Inhibition of PD-1 and
PD-L1 signaling through checkpoint inhibitors enhances T-cell
cytotoxicity and induces tumor regression. However, given the
comprehensive nature of these interactions, there are significant
implications for both cancer cells and the host's normal tissues.
It has been hypothesized that the hyperinflammatory state caused by
immunotherapy-induced T-cell activation may lead to HLH. In the
absence of definitive physical evidence, it is crucial to exclude
other causes of secondary HLH.
In this case, we suspected HLH based on the clinical
findings and measured IL-6 levels before administering the steroid
pulse, which confirmed that it decreased with treatment. Although
IL-6 measurement was not necessary for the diagnosis of HLH, it was
helpful in understanding the pathophysiology while awaiting the
results of the liver biopsy, allowing us to initiate treatment.
Hyperactivation of immune cells by EBV reactivation is also
considered to have had an effect.
Although the recent irAE guidelines from the Society
of Immunotherapy for Cancer discuss HLH as an irAE with potentially
high lethality, no specific treatment recommendations have been
made (13). Hence, real-world data
and case reports of rare irAEs are needed to understand their
frequency and severity and improve clinical management. A review of
the management of blood-related irAEs recommends the prompt
administration of high-dose corticosteroids (2–5 mg/kg/day, IV) and
anti-IL-6 inhibitors such as tocilizumab for cytokine storms
associated with hemophagocytic syndrome. Should corticosteroids
fail to elicit an adequate response, the use of cyclosporine or a
single dose of etoposide (150 mg/kg, IV) should be considered in
collaboration with a hematologist (14). Earlier case reports of
ICI-associated HLH have utilized anti-inflammatory cytokines,
including tocilizumab, alongside steroids (Table III). Importantly, recent evidence
underscores the significance of early intervention with
nonsteroidal immunosuppressive therapies over steroids for treating
irAEs (15). In this case, the
onset of HLH during lymphocyte suppressive therapy involving
high-dose steroids (1 mg/kg) and MMF, indicates a cytokine storm.
Steroid pulses were administered to inhibit the cytokine cascade,
and the anti-IL-6 antibody, tocilizumab, was used during steroid
tapering. Additionally, cyclosporine was employed to suppress T
cells through a mechanism distinct from that of steroids,
contributing to successful treatment.
In conclusion, we encountered cases of HLH and DIC
associated with ICI administration during preoperative systemic
chemotherapy for breast cancer. HLH is a rare but severe
life-threatening complication of checkpoint inhibitor therapy that
underscores the need for vigilance and preparedness. Our
understanding of the full spectrum of side effects associated with
ICIs that have a relatively short history of use is incomplete.
Anticipation of the underlying pathophysiology and identification
of appropriate treatment strategies are of paramount importance.
Prospective data are crucial for assessing the efficacy of
nonsteroidal immunosuppressive therapies in conjunction with
steroids.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YK wrote the manuscript. YK, AS, TT and HS analyzed
and interpreted the patient's clinical data for the manuscript. YK,
AS, TT, HH, KH, YH, DK, RN, HS, AH and CS contributed to collecting
the relevant literature and to data analysis, and reviewed and
critically interpreted the information. AS and CS confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
The patient provided written informed consent for
the publication of this case report.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hudis CA and Gianni L: Triple-negative
breast cancer: An unmet medical need. Oncologist. 16 (Suppl
1):S1–S11. 2011. View Article : Google Scholar
|
|
2
|
Cortazar P, Zhang L, Untch M, Mehta K,
Costantino JP, Wolmark N, Bonnefoi H, Cameron D, Gianni L,
Valagussa P, et al: Pathological complete response and long-term
clinical benefit in breast cancer: The CTNeoBC pooled analysis.
Lancet. 384:164–172. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schmid P, Cortes J, Pusztai L, McArthur H,
Kümmel S, Bergh J, Denkert C, Park YH, Hui R, Harbeck N, et al:
Pembrolizumab for early triple-negative breast cancer. N Engl J
Med. 382:810–821. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schmid P, Cortes J, Dent R, Pusztai L,
McArthur H, Kümmel S, Bergh J, Denkert C, Park YH, Hui R, et al:
Event-free survival with pembrolizumab in early triple-negative
breast cancer. N Engl J Med. 386:556–567. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang DY, Salem JE, Cohen JV, Chandra S,
Menzer C, Ye F, Zhao S, Das S, Beckermann KE, Ha L, et al: Fatal
toxic effects associated with immune checkpoint inhibitors: A
systematic review and meta-analysis. JAMA Oncol. 4:1721–178. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kramer R, Zaremba A, Moreira A, Ugurel S,
Johnson DB, Hassel JC, Salzmann M, Gesierich A, Weppler A, Spain L,
et al: Hematological immune related adverse events after treatment
with immune checkpoint inhibitors. Eur J Cancer. 147:170–181. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fardet L, Galicier L, Lambotte O, Marzac
C, Aumont C, Chahwan D, Coppo P and Hejblum G: Development and
validation of the HScore, a score for the diagnosis of reactive
hemophagocytic syndrome. Arthritis Rheumatol. 66:2613–2620. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Henter JI, Horne A, Aricó M, Egeler RM,
Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski
J and Janka G: HLH-2004: Diagnostic and therapeutic guidelines for
hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer.
48:124–131. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dupré A, Michot JM, Schoeffler A,
Frumholtz L, Baroudjian B, Delyon J, Lebbe C and Lambotte O:
Haemophagocytic lymphohistiocytosis associated with immune
checkpoint inhibitors: A descriptive case study and literature
review. Br J Haematol. 189:985–992. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Noseda R, Bertoli R, Müller L and Ceschi
A: Haemophagocytic lymphohistiocytosis in patients treated with
immune checkpoint inhibitors: Analysis of WHO global database of
individual case safety reports. J Immunother Cancer. 7:1172019.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Planas R, Felber M, Vavassori S and
Pachlopnik Schmid J: The hyperinflammatory spectrum: From defects
in cytotoxicity to cytokine control. Front Immunol. 14:11633162023.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brahmer JR, Abu-Sbeih H, Ascierto PA,
Brufsky J, Cappelli LC, Cortazar FB, Gerber DE, Hamad L, Hansen E,
Johnson DB, et al: Society for Immunotherapy of Cancer (SITC)
clinical practice guideline on immune checkpoint inhibitor-related
adverse events. J Immunother Cancer. 9:e0024352021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Michot JM, Lazarovici J, Tieu A, Champiat
S, Voisin AL, Ebbo M, Godeau B, Michel M, Ribrag V and Lambotte O:
Haematological immune-related adverse events with immune checkpoint
inhibitors, how to manage? Eur J Cancer. 122:72–90. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Faleck DM, Dougan M, Tello M, Grossman JE,
Moss AC and Postow MA: Accelerating the evolution of immune-related
enterocolitis management. J Clin Oncol. 41:3110–3115. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kurozumi A, Takahashi H, Watanabe T and
Iwasaki Y: Two cases of lung cancer with hemophagocytic
lymphohistiocytosis caused by immune checkpoint inhibitors. Thorac
Cancer. 12:1625–168. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sackstein P, Zaemes J and Kim C:
Pembrolizumab-induced cytokine release syndrome in a patient with
metastatic lung adenocarcinoma: A case report. J Immunother Cancer.
9:e0028552021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Okawa S, Kayatani H, Fujiwara K, Ozeki T,
Takada K, Iwamoto Y, Minami D, Sato K and Shibayama T:
Pembrolizumab-induced autoimmune hemolytic anemia and
hemophagocytic lymphohistiocytosis in non-small cell lung cancer.
Intern Med. 58:699–702. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Laderian B, Koehn K, Holman C, Lyckholm L
and Furqan M: Association of hemophagocytic lymphohistiocytosis and
programmed death 1 checkpoint inhibitors. J Thorac Oncol.
14:e77–e78. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Honjo O, Kubo T, Sugaya F, Nishizaka T,
Kato K, Hirohashi Y, Takahashi H and Torigoe T: Severe cytokine
release syndrome resulting in purpura fulminans despite successful
response to nivolumab therapy in a patient with pleomorphic
carcinoma of the lung: A case report. J Immunother Cancer.
7:972019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hantel A, Gabster B, Cheng JX, Golomb H
and Gajewski TF: Severe hemophagocytic lymphohistiocytosis in a
melanoma patient treated with ipilimumab + nivolumab. J Immunother
Cancer. 6:732018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Satzger I, Ivanyi P, Länger F, Kreipe HH,
Schaper-Gerhardt K, Beutel G, Cornberg M and Gutzmer R:
Treatment-related hemophagocytic lymphohistiocytosis secondary to
checkpoint inhibition with nivolumab plus ipilimumab. Eur J Cancer.
93:150–153. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sadaat M and Jang S: Hemophagocytic
lymphohistiocytosis with immunotherapy: Brief review and case
report. J Immunother Cancer. 6:492018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Takeshita M, Anai S, Mishima S and Inoue
K: Coincidence of immunotherapy-associated hemophagocytic syndrome
and rapid tumor regression. Ann Oncol. 28:186–189. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Malissen N, Lacotte J, Du-Thanh A,
Gaudy-Marqueste C, Guillot B and Grob JJ: Macrophage activation
syndrome: A new complication of checkpoint inhibitors. Eur J
Cancer. 77:88–89. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shah D, Shrestha R, Ramlal R, Hatton J and
Saeed H: Pembrolizumab associated hemophagocytic
lymphohistiocytosis. Ann Oncol. 28:14032017. View Article : Google Scholar : PubMed/NCBI
|