Introduction
Head and neck cancers, particularly oral squamous
cell carcinoma (OSCC), pose a significant global health challenge
owing to the complex etiology and limited success of treatment for
improving patient survival (1).
Intricate interplay among genetic alterations, environmental
exposure and viral infections has been implicated in the
pathogenesis of OSCC. However, the precise molecular mechanisms
underlying their development have not been fully elucidated
(2). This underscores the need for
innovative investigations to identify novel molecular targets that
aid the development of effective therapeutics.
Based on the molecular landscape, Fanconi anemia
complementation group E (FANCE) has been identified as a key
component of DNA damage response mechanisms, particularly in the
Fanconi anemia (FA) pathway, where it plays a vital role in
repairing DNA interstrand crosslinks and maintaining genomic
stability (3,4). FANCE primarily functions in the repair
of DNA interstrand crosslink-complex forms of DNA damage that, if
not properly repaired, can lead to genomic instability and elevate
the risk of cancer development (5).
By effectively repairing these interstrand crosslinks, FANCE not
only safeguards the integrity of the genetic material of the cell
but also contributes to the maintenance of a stable genomic
environment, which is crucial for cellular health and the
prevention of malignancies (3).
Emerging evidence suggests that FANCE may also influence the tumor
microenvironment (TME) through mechanisms beyond its canonical DNA
repair functions, potentially affecting immune responses within the
tumor (6). This dual role of FANCE,
not only in maintaining genomic stability but also in possibly
modulating immune activity, raises intriguing questions about its
broader impact on tumor progression and therapeutic resistance in
OSCC (7). These considerations
underscore the importance of further research to explore how FANCE
may contribute to both the oncogenic processes and the modulation
of the tumor immune microenvironment in OSCC, which could reveal
new avenues for targeted therapy and prognostic assessment.
The expression of FANCE is aberrant in various types
of cancer (6,8,9),
including head and neck squamous cell carcinoma (HNSCC). Altered
expression of FANCE has been linked to a five-year survival rate in
patients with HNSCC, suggesting its potential use as a prognostic
biomarker (6). In addition,
epigenetic modifications, such as changes in methylation patterns,
have been associated with aberrant FANCE expression in these
malignancies (10). Furthermore,
FANCE expression is associated with immune cell infiltration,
particularly by CD4+ T cells and macrophages, indicating
its potential involvement in modulating the tumor immune
microenvironment (6,7).
The present study aimed to determine the function of
FANCE in the development of OSCC by assessing its expression
profiles and association with immune markers, as well as its
functional implications for tumor cell biology. Furthermore, the
association between FANCE and immune markers was explored to
understand its potential influence on the tumor immune
microenvironment. This comprehensive approach allowed investigation
into how FANCE may modulate both oncogenic processes and immune
responses in OSCC. The findings of the present study could provide
valuable insights into the molecular mechanisms underlying OSCC
progression and potentially inform the development of novel
targeted therapeutic strategies and prognostic assessments.
Material and methods
Data acquisition and
preprocessing
Gene expression data and clinical information for
OSCC were retrieved from The Cancer Genome Atlas (TCGA,
cancer.gov/tcga/) via the Genomic Data Commons Data Portal
(https://portal.gdc.cancer.gov/). HNSCC
data from TCGA, including RNA-Seq and corresponding clinical
metadata, were pre-processed to ensure quality and consistency,
(portal.gdc.cancer.gov/projects/TCGA-HNSC). Such processes
comprised normalizing expression, removing batch effects and
verifying sample annotations using RStudio software v. 3.5.3
(RStudio, Inc.). P<0.05 was considered statistically
significant.
Differential expression and
survival
Differential expression was analyzed to compare
FANCE gene expression between normal and OSCC tissue samples from
TCGA-HNSC and identify significant changes. The association between
FANCE expression and patient survival was assessed using
Kaplan-Meier estimates, with log-rank tests for statistical
comparison. The analysis was performed with RStudio v. 3.5.3
(RStudio, Inc.); P<0.05 was considered statistical
significance.
Unsupervised clustering and
validation
The optimal number of clusters for patient
stratification was identified using the Elbow method (11), which evaluates within-cluster sums
of squares to ascertain the point of diminishing returns in cluster
variance. Thereafter, patient data, which came from TCGA-HNSC
expression profile, were categorized using the k-means clustering
algorithm, which is a machine learning technique that partitions
data based on inherent gene expression profiles. The resulting
clusters were validated using principal component analysis (PCA) to
ensure robust separation of the data into distinct genetic profiles
(12). Analysis was performed using
RStudio v. 3.5.3 (RStudio, Inc.), with P<0.05 regarded as
statistical significance.
Differential expression in clusters
and immune infiltration assessment
To explore the expression profile of FANCE across
clusters identified by the Elbow method and categorized using the
k-means clustering algorithm during unsupervised learning, unpaired
t-tests were used to compare FANCE expression. This comparison
aimed to assess the variability in FANCE expression across distinct
molecular subtypes, thereby laying the groundwork for further
biological interpretation. Having established significant
differences in FANCE expression, the underlying biological
mechanisms were investigated. The ESTIMATE algorithm was used to
quantify and contrast the immune and stromal components of the TME
across clusters. This analysis was instrumental in elucidating the
potential associations between the differential expression of FANCE
and the characteristics of the TME within varying subtypes.
Analysis was performed with RStudio v. 3.5.3 (RStudio, Inc.), where
P<0.05 was taken as the criterion for statistical
significance.
Cross-validation of macrophage
abundance
The abundance of macrophages within the TME was
cross-validated using an array of computational methodologies.
Marker gene-based single-sample Gene Set Enrichment Analysis
(ssGSEA) enrichment and analyses using MCP-counter (version 1.2.0)
and xCell (version 1.1.0), together with the deconvolution
algorithms, CIBERSORT (version v1.03) and TIMER (version 2.17), by
RStudio v. 3.5.3 (RStudio, Inc.), facilitated a comprehensive
assessment of immune cell infiltration within clusters obtained by
the Elbow method and k-means clustering. These diverse
bioinformatics tools enabled a more nuanced quantitation of
macrophage presence and their potential impact on the OSCC
microenvironment. P<0.05 as the benchmark for statistical
significance.
Cell culture and transfection
Human oral keratinocytes (HOKs) were obtained from
Shanghai Meixuan Biological Science and Technology Ltd (cat. no.
MXC1005). The HSC3 and HN6 cells were obtained from the BeNa
Culture Collection (Beijing Beina Chunglian Institute of
Biotechnology; cat. no. BNCC341400) and central laboratory of
Peking University School of Stomatology, respectively. SCC9 (cat.
no. CRL-1629) and SCC25 (cat. no. CRL-1628) cells were purchased
from the American Type Culture Collection. The cell lines were
incubated at 37°C under a humidified 5% CO2 atmosphere
in DMEM (Gibco; Thermo Fisher Scientific, Inc.), supplemented with
10% fetal bovine serum and 1% penicillin/streptomycin (all from
Gibco; Thermo Fisher Scientific, Inc.). The cell lines were
authenticated by short tandem repeat profiling analysis and were
free of mycoplasma contamination. FANCE was knocked down in HSC3
and HN6 cells using siRNA (100 nM) transfection with
Lipofectamine® 3000 (Thermo Fisher Scientific, Inc.).
Following culture at 37°C for 48 h, the knockdown efficiency of the
cells was detected and subsequent experiments were conducted. The
siRNA sequences used for FANCE knockdown and the control siRNA were
as follows: Negative control-sense: 5′-UUCUCCGAACGUGUCACGUTT-3′;
antisense: 5′-ACGUGACACGUUCGGAGAATT-3′; FANCE siRNA-sense:
5′-GCUUCUUCACGAAUGUAGUCC-3′; and antisense:
5′-ACUACAUUCGUGAAGAAGCUG-3′. Control siRNA and siRNA targeting
FANCE were purchased from Shanghai GenePharma Co., Ltd.
Reverse transcription-quantitative PCR
(RT-qPCR)
The mRNA expression of FANCE in HSC3 and HN6 cell
lines was assessed by RT-qPCR. Total RNA was extracted from cells
using TRIzol® (Takara Bio Inc.), and its integrity was
verified by measuring absorbance at 260/280 nm. Complementary DNA
was synthesized from 1 µg RNA using the Evo M-MLV RT Kit Mix
for qPCR [Accurate Biotechnology (Hunan) Co., Ltd.], according to
the manufacturer's instructions. RT-qPCR was performed in
triplicate using SYBR Green [Accurate Biotechnology (Hunan) Co.,
Ltd.] under the following cycling conditions: Denaturation at 95°C
for 10 min, then 40 cycles of 95°C for 5 s and 58°C for 30 s.
Melting curves were analyzed to confirm the product specificity and
FANCE mRNA was quantified using the 2−ΔΔCq method
(13) with the following forward
and reverse (5′3′) primers:
FANCE forward, AGGAGAGACCCGAACATAAGTC and reverse:
CTCGCCAGTCTTAACTGCCA;
β-actin forward: AAACTGGAACGGTGAAGGTG and reverse:
AGTGGGGTGGCTTTTAGGAT, and normalized to β-actin.
Cell proliferation
Cell proliferation was quantified using a cell
counting kit-8 (CCK-8; Shenzhen Aipno Biomedical Technology Co.,
Ltd.). Briefly, cells were seeded at a density of 3×103
cells/well in 96-well plates and left to adhere overnight. The
CCK-8 solution was added to the wells at 0, 24, 48 and 72 h after
seeding and incubated at 37°C for 1 h. Absorbance was measured at
450 nm using a microplate reader. Blank readings were subtracted
from all measurements.
Wound healing
HSC3 and HN6 cells with FANCE knockdown at 100%
confluence were scratched, and adherent cells were rinsed and
cultured in serum-free medium. Images of cell migration were
captured at various intervals thereafter using inverted
fluorescence microscopy (Leica Camera, Germany; DMi8), and wound
area was used to measure with image J (1.48v), the ratio of wound
closure was calculated. Wound healing rate=(initial area-healing
area)/initial area.
Transwell migration
HSC3 (4×104 cells/well) and HN6
(6×104 cells/well) with FANCE knockdown suspended in
serum-free DMEM were seeded into the upper chamber of Transwell
chambers (cat. no. 3422; Corning, Inc.), and DMEM containing 10%
FBS was added to the lower chamber, incubated at 37°C for 24 h,
then fixed with 4% paraformaldehyde at room temperature for 30 min
and stained with 0.1% crystal violet at room temperature for 30
min. Cells on the surface of the upper membrane were removed, while
those on the lower surface were analyzed. Images were acquired
under an inverted fluorescence microscopy (Leica, DMi8, Germany)
and analyzed using image J (1.48v).
Gene set enrichment analysis
To elucidate the immunological impact of FANCE
expression in OSCC, Gene Set Enrichment Analysis (GSEA) was
performed. This analysis was executed using the clusterProfiler
package within RStudio v. 3.5.3 (RStudio, Inc.), where gene sets
were analyzed for differential expression between high and low
FANCE-expressing OSCC samples. Significance was determined via
phenotype-based permutation tests, with an FDR threshold of 0.25
for identifying notable gene sets. Visual representations were
constructed with enrichplot and patchwork, offering a clear
depiction of the enrichment results.
FANCE-immune checkpoint correlation
analysis
To assess the association between FANCE expression
and immune checkpoint genes, RNA sequencing data were assessed from
the TCGA OSCC cohort. The expression levels of FANCE and immune
checkpoint genes (PD1, PDL1, CTLA4, LAG3, HAVCR2 and TIGIT) were
extracted and subjected to statistical analysis using Pearson
correlation coefficient. Scatter plots were generated using ggplot2
in RStudio v. 3.5.3 (RStudio, Inc.) to visualize the association
between FANCE and the immune checkpoint markers. Statistical
significance for all correlations was evaluated with P-values
calculated, with P<0.01 considered significant.
Immunohistochemical (IHC)
staining
The present study involved pathological sections
from 13 patients (9 male and 4 female; mean age, 55.62 years; age
range, 36–80 years) obtained from the Department of Pathology at
Peking University Shenzhen Hospital (Shenzhen, China) between
January 2020 and December 2023. The specimens were selected
(inclusion criteria of age between 18 and 85 years and
pathologically diagnosed as oral squamous cell carcinoma to
represent a diverse patient demographic and ensure the relevance of
the present findings to the broader OSCC population, while
excluding those with severe sample quality problems affecting
subsequent staining and patients having other major interfering
diseases or a history of special treatments and other unstated
factors. Human sample collection was approved by The Ethical
Committee of The Peking University Shenzhen Hospital (Shenzhen,
China; approval no. 2023-177) and written informed consent was
obtained from all participants. Fresh OSCC samples were obtained
from the operating room and fixed in 10% neutral buffered formalin
for 72 h at room temperature, dehydrated in gradient alcohol
solution (50, 70, 80, 95, 95 and 100 alcohol, each for 1 h) and
paraffin embedded. Paraffin embedded tumor sections with a
thickness of 4 µm were deparaffinized with xylene I for 15 and
xylene II for 10 min, and rehydrated with 100, 100, 95, 95 and 80%
ethanol for 5 min each. Sections were then washed twice for 5 min
each with dH2O. Slides were heated in a microwave in citrate
antigen retrieval solution (cat. no. MVS-0066; MBX Bioscience) or
TRIS-EDTA antigen retrieval solution (cat. no. BL617A; Biosharp
Biotechnology) until boiling (100°C), followed by incubation at the
boiling state for 20 min. After cooling, sections were washed in
PBS three times for 5 min each. Antigen retrieval was achieved by
blocking with 3% H2O2 for 30 min at room
temperature. Blocking was performed using 10% goat serum (cat. no.
ZLI-9022; ZSGB-BIO) at room temperature for 20 min. The slices were
incubated with primary antibody at 4°C overnight. The following
primary antibodies: Anti-FANCE (1:400; cat. no. GTX110184; GeneTex,
Inc.) anti-CD68 [1:200; cat. no. 76437; Cell Signaling Technology,
Inc. (CST)], anti-CD14 (cat. no. ZA-0532; ZSGB Biotech, Beijing,
China) and anti-CD206 (1:400; CST; cat. no. 91992). The slices were
incubated with (cat. no. KIT-9710; MBX Biosciences) biotin-labeled
goat anti-mouse/rabbit IgG at room temperature for 10 min, and then
incubated with streptomyces peroxidase at room temperature for 10
min. The washing step was repeated, the slices were stained using
DAB (cat. no. DAB-1031; MBX Biosciences), and the reaction was
aborted with dH2O. The slices were counterstain with hematoxylin at
room temperature for 2 min. Stained sections were visualized using
a BX53 fluorescence microscope (Olympus Corporation) and analyzed
using Image J (1.48v; National Institutes of Health).
Statistical analysis
All data were analyzed using GraphPad Prism 5.0
(Dotmatics) and RStudio software (v. 3.5.3). The R packages used
included ‘DESeq2’, ‘survival’, ‘kmeans’, ‘factoextra’,
‘MCPcounter’, ‘xCell’, ‘CIBERSORT’, ‘TIMER’, ‘ggplot2’, ‘dplyr’,
‘tidyr’, ‘purrr’, ‘stringr’ and ‘reshape2’. Data were presented as
the mean ± standard deviation. All assays were repeated in at least
three independent experiments. P<0.05 were considered to
indicate a statistically significant difference. Significance was
assessed using an unpaired two-tailed Student's t-test for
two-group comparisons, Kaplan-Meier method with log-rank tests for
survival analysis and Pearson correlation coefficient for
correlation analyses.
Results
FANCE expression is upregulated in
patients with OSCC
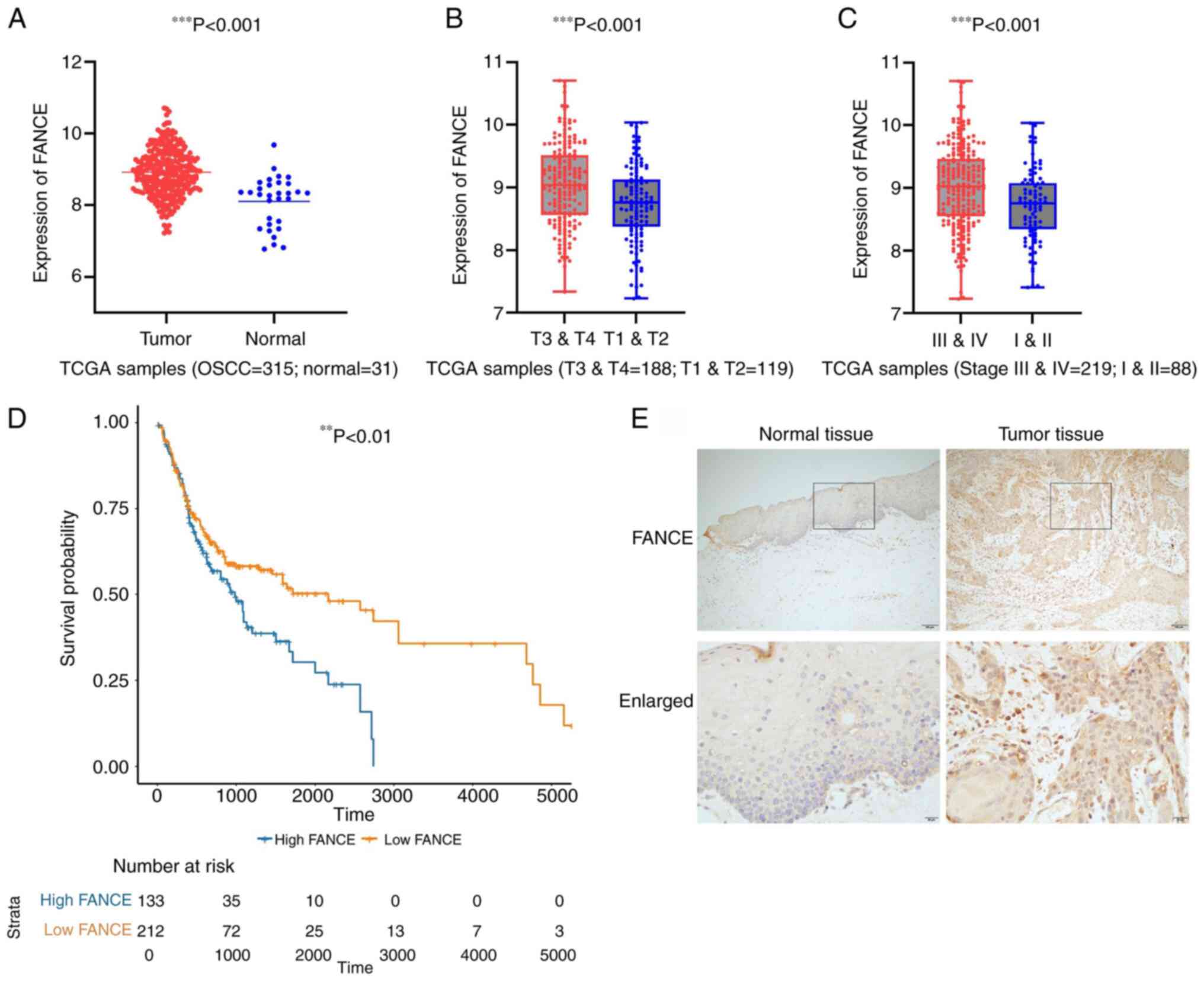
RNA expression (fold-change >2.0; P<0.05) and
the clinical features of 315 patients with OSCC derived from TCGA
database were analyzed to determine the function of FANCE. Table I summarized the clinical features of
patients with OSCC. The expression of FANCE was significantly
higher in the tumor tissues compared with the normal tissues
(Fig. 1A), and was positively
associated with T classification and clinical stage (Fig. 1B and C). However, no significant
statistical differences in FANCE expression were found in other
clinical parameters, such as N classification, perineural invasion
or lymphovascular invasion, as detailed in Table II. The results of the Kaplan-Meier
curves revealed a correlation between enhanced FANCE expression and
poor survival among the patients with OSCC (Fig. 1D). The upregulation of FANCE
expression in tumor tissues was also validated by the IHC staining
(Fig. 1E). Thus, enhanced FANCE
expression was associated with larger tumors, advanced clinical
stage and poor survival in patients with OSCC. The expression data
of FANCE and the clinical information of the cases are provided in
Tables SI and II, respectively.
 | Table I.Basic information of patients with
oral squamous cell carcinoma in the head and neck squamous cell
carcinoma dataset. |
Table I.
Basic information of patients with
oral squamous cell carcinoma in the head and neck squamous cell
carcinoma dataset.
| Clinical
parameter | n |
|---|
| Location | 315 |
|
Alveolar ridge | 18 |
| Buccal
mucosa | 22 |
| Floor
of mouth | 62 |
| Hard
palate | 7 |
|
Lip | 3 |
| Oral
cavity | 73 |
| Oral
tongue | 130 |
| Clinical T
stage | 307 |
| T1 | 19 |
| T2 | 100 |
| T3 | 76 |
| T4 | 112 |
| Clinical N
stage | 303 |
| N0 | 165 |
| N1 | 56 |
| N2 | 80 |
| N3 | 2 |
| Clinical M
stage | 300 |
| M0 | 298 |
| M1 | 2 |
| Clinical stage | 307 |
| I | 12 |
| II | 76 |
|
III | 65 |
|
IVA | 154 |
| Lymphovascular
invasion | 234 |
| No | 164 |
|
Yes | 70 |
| Margin status | 302 |
|
Close | 35 |
|
Negative | 230 |
|
Positive | 37 |
| Histological
grade | 311 |
| G1 | 49 |
| G2 | 196 |
| G3 | 66 |
| Perineural invasion
present | 247 |
| No | 112 |
|
Yes | 135 |
| Recurrence | 277 |
| No | 199 |
|
Yes | 78 |
| Table II.Comparative analysis of FANCE
expression in patients with oral squamous cell carcinoma. |
Table II.
Comparative analysis of FANCE
expression in patients with oral squamous cell carcinoma.
| Clinical
characteristic | na | Median
differenceb | P-value |
|---|
| Alcohol use (no vs.
yes) | 308 | −0.113 | 0.403 |
| Smoking (no vs.
yes) | 315 | 0.029 | 0.633 |
| N classification
(N0 vs. N+) | 271 | 0.061 | 0.301 |
| T classification
(T1 & T2 vs. T3 & T4) | 297 | −0.303 | 0.001 |
| Stage (I & II
vs. III & IV) | 293 | −0.239 | 0.010 |
| Histological grade
(G1 and G2 vs. G3) | 311 | 0.212 | 0.161 |
| Perineural invasion
(negative vs. positive) | 247 | 0.022 | 0.502 |
| Lymphovascular
invasion (negative vs. positive) | 234 | 0.049 | 0.977 |
| Recurrence (no vs.
yes) | 277 | 0.006 | 0.691 |
| Outcome (alive vs.
dead) | 315 | −0.208 | 0.132 |
Integrated cluster analysis of
patients with OSCC
The flowchart of bioinformatics analysis of the OSCC
dataset are shown in Fig. 2.
Integrated cluster analysis of the samples of patients with OSCC
revealed distinct gene expression profiles within the dataset.
Employing the elbow method, the optimal number of clusters was
determined to be two (k=2), as visualized in Fig. 3A, where the total within-cluster sum
of squares reached an inflection point. K-means clustering analysis
further segregated the OSCC samples into two subgroups: Cluster 1
(n=174) and Cluster 2 (n=141). Principal component analysis (PCA)
demonstrated the first two components accounted for 11.3 and 8.3%
of the total observed variance, respectively, with the PCA plot
exhibiting distinct yet slightly overlapping gene expression
profiles for the two clusters as depicted in Fig. 3B. The expression level of the FANCE
gene was found to be significantly different between the two
clusters. As shown in the box plot (Fig. 3C), Cluster 1 had a higher mean FANCE
gene expression (9.038±0.595) compared with Cluster 2
(8.787±0.690), with the differences being statistically significant
(P<0.05). For detailed FANCE expression data by cluster, see
Table SIII. Further comparative
analysis of the TME using the ESTIMATE algorithm revealed notable
disparities between the clusters. Cluster 1 presented with a
universally lower mean (± SD) stroma, immune and ESTIMATE scores
(−675.062±534.185, 307.954±701.403, and −367.108±1117.081,
respectively) compared with Cluster 2 (233.293±695.683,
623.624±802.598 and 856.917±1353.531, respectively), with these
differences reaching statistical significance (P<0.05; Fig. 3D). Furthermore, the macrophage
profile of each cluster was assessed using multiple algorithms,
including MCP-counter, xCell, CIBERSORT and TIMER. Macrophages were
significantly more abundant in Cluster 2, as consistently indicated
by all applied algorithms. This is depicted in Fig. 3E-H, where the macrophage abundance
scores of the TIMER algorithm were significantly higher for Cluster
2 compared with Cluster 1. Both M0 and M2 macrophages were
substantially more prevalent in Cluster 2, while M1 macrophages
were observed to be more abundant in Cluster 1, as detailed in the
CIBERSORT analysis (Fig. 3F). The
MCP-counter analysis also demonstrated a significantly higher
monocyte lineage score in Cluster 2 (Fig. 3G). Moreover, xCell algorithm-based
analysis revealed a significantly increased abundance of
macrophages, including both M1 and M2 subsets, in Cluster 2
(Fig. 3H), highlighting a distinct
profile of macrophage infiltration in this subgroup.
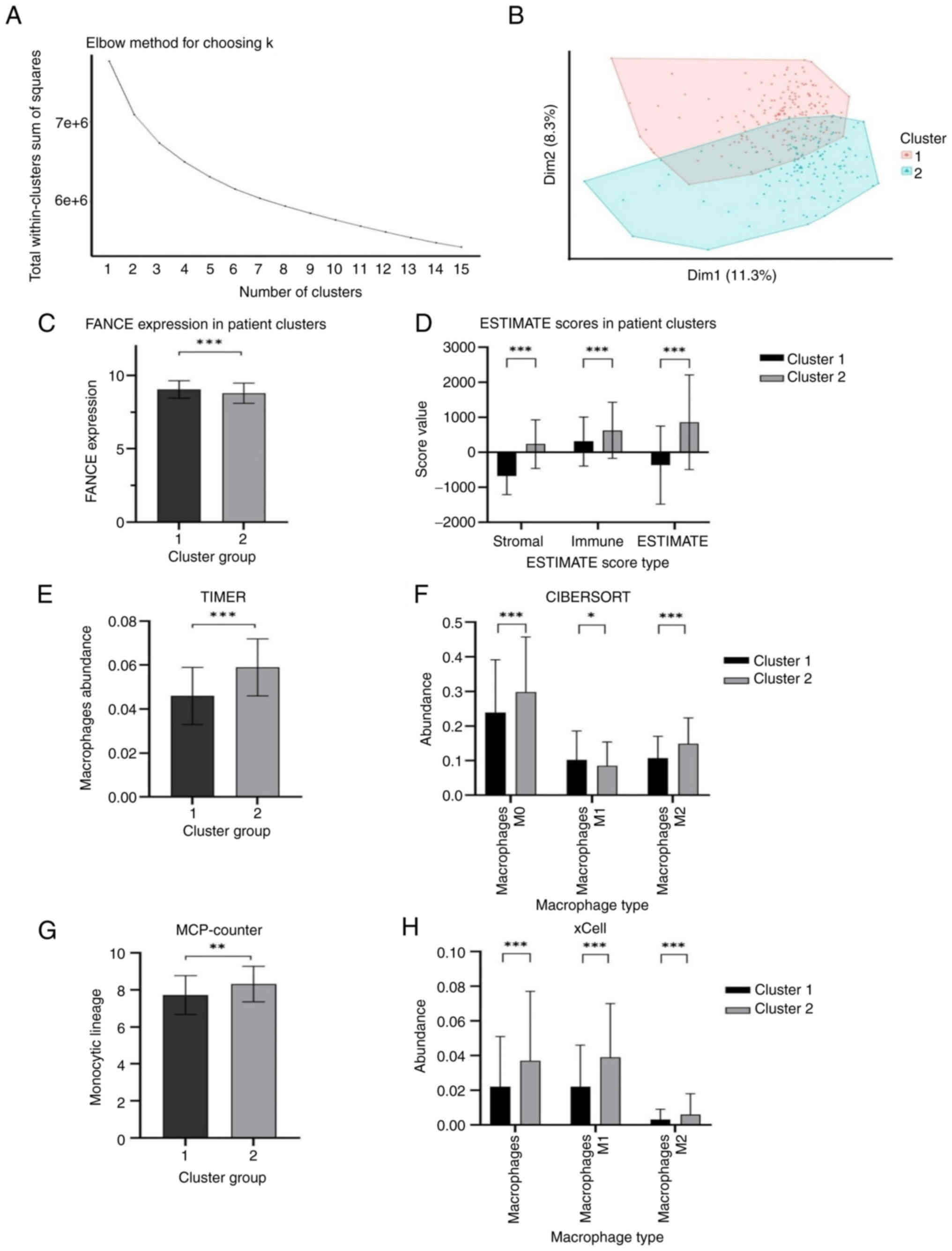 | Figure 3.Integrated cluster analysis of
samples from patients with OSCC. Statistical analysis was performed
using the two-tailed unpaired student's t-tests, and data are
presented as the mean ± SD. (A) Elbow method identifying k=2 as the
optimal cluster number. (B) k-means clustering on the first two
principal components. (C) Box plot of FANCE expression levels in
two OSCC subgroups (***P<0.001). (D) Bar chart of TME scores
comparing Cluster 1 and 2, (***P<0.001). (E) TIMER analysis
shows macrophage abundances for Cluster 1 and Cluster 2. (F)
CIBERSORT analysis presents macrophage abundances for Cluster 1 and
Cluster 2 across Macrophages M0, M1, and M2 types, with indicated
statistical significance (*P<0.05, ***P<0.001). (G)
MCP-counter analysis displays monocyte lineage scores for
macrophages in Cluster 1 and Cluster 2, with indicated statistical
significance (**P<0.01). (H) xCell analysis depicts macrophage
abundances for Cluster 1 and Cluster 2 across Macrophages, M1, and
M2 types. ***P<0.001). FANCE, Fanconi anemia complementation
group E; OSCC, oral squamous cell carcinoma; TME, tumor
microenvironment; MCP, Microenvironment Cell Populations. |
Knockdown of FANCE inhibits cell
proliferation and migration in OSCC cells
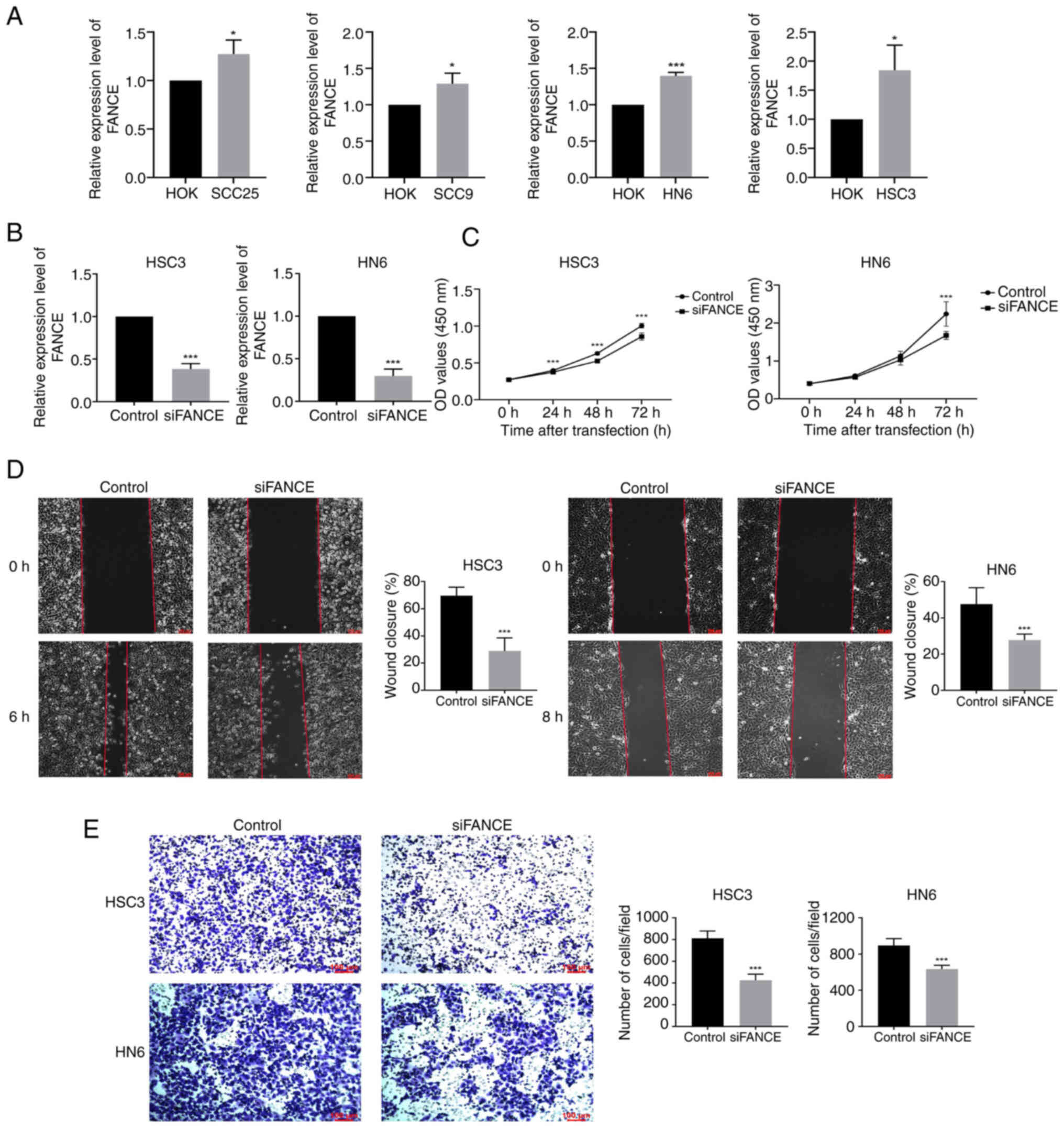
The expression of FANCE in OSCC cells was validated
by RT-qPCR. The mRNA expression of FANCE was significantly enhanced
in SCC25, SCC9, HN6 and HSC3 cells compared with HOK cells; FANCE
expression was enhanced by 1.3- and 1.8-fold in HN6 and HSC3 cells,
respectively, compared with HOK cells (Fig. 4A). The elevated expression of FANCE
in OSCC cells indicated its potential oncogenic role in oral
carcinogenesis. FANCE expression was knocked down in HSC3 and HN6
cells by siRNA transfection and its efficiency was confirmed by
RT-qPCR to further analyze FANCE function in OSCC. Knockdown of
FANCE significantly inhibited its mRNA expression in HSC3 and HN6
cells by ~65% compared with the control. (Fig. 4B).
The effects of FANCE knockdown on cell proliferation
were determined using CCK-8 assays and revealed significantly
inhibited proliferative ability compared with controls, and the
wound healing rates of knockdown HSC3 and HN6 cells were also
decreased (Fig. 4C and D). Knocking
down FANCE inhibited HSC3 and HN6 cell migration in Transwell
assays by 50 and 30%, respectively, compared with controls
(Fig. 4E). Overall, these findings
indicate that FANCE expression is upregulated in OSCC cells and
that its knockdown inhibits their proliferation and migration.
FANCE expression modulates the OSCC
microenvironment
GSEA analysis revealed that FANCE expression is
intricately linked to the OSCC TME, particularly in its regulatory
effects on immune pathways (Table
III). FANCE may act to suppress certain immune pathways under
normal conditions, with its reduced expression potentially leading
to the activation of these pathways (14), as indicated by negative enrichment
scores in gene sets related to systemic lupus erythematosus and
asthma. This indicates a dynamic role for FANCE in modulating the
immune response within the TME.
| Table III.Gene set enrichment analysis results
of immune-related gene sets in oral squamous cell carcinoma. |
Table III.
Gene set enrichment analysis results
of immune-related gene sets in oral squamous cell carcinoma.
| Gene sets | Normalized
enrichment score | Adjusted
P-value |
|---|
| Malaria | −0.806 | <0.001 |
| Primary
immunodeficiency | −0.794 | <0.001 |
| Systemic lupus
erythematosus | −0.794 | <0.001 |
| Asthma | −0.793 | <0.001 |
| Allograft
rejection | −0.765 | <0.001 |
| Viral protein
interaction with cytokine and cytokine receptor | −0.761 | <0.001 |
| Graft-vs.-host
disease | −0.759 | <0.001 |
| African
trypanosomiasis | −0.753 | <0.001 |
| Intestinal immune
network for IgA production | −0.751 | <0.001 |
| Hematopoietic cell
lineage | −0.730 | <0.001 |
| Type I diabetes
mellitus | −0.728 | <0.001 |
| Autoimmune thyroid
disease | −0.722 | <0.001 |
| Rheumatoid
arthritis | −0.722 | <0.001 |
| Inflammatory bowel
disease | −0.698 | <0.001 |
| Cardiac muscle
contraction | −0.695 | <0.001 |
| Renin-angiotensin
system | −0.682 | 0.002 |
| Hypertrophic
cardiomyopathy | −0.679 | <0.001 |
| Leishmaniasis | −0.678 | <0.001 |
| Antigen processing
and presentation | −0.677 | <0.001 |
| Dilated
cardiomyopathy | −0.675 | <0.001 |
FANCE expression showed significant negative
correlations with multiple immune checkpoint genes in OSCC,
including programmed Cell Death 1), CD274 (Programmed Death-Ligand
1), CTLA4, LAG3 (Lymphocyte-Activation Gene 3), HAVCR2 (Hepatitis A
Virus Cellular Receptor 2) and TIGIT (T cell immunoglobulin and
ITIM domain). Scatter plots demonstrated these negative
correlations, with Pearson correlation coefficients and
corresponding P-values indicating statistical significance for each
association (Fig. 5A). Further
analysis of TCGA data revealed a negative correlation between FANCE
expression and the macrophage markers CD68, CD14,and CD206
(Fig. 5B). This correlation was
mirrored in the IHC staining of OSCC tumor tissues, where decreased
FANCE expression was associated with increased expression of
macrophage markers (Fig. 5C). These
findings indicate that FANCE expression plays a crucial role in
modulating the TME in OSCC, influencing both the infiltration of
immune cells and the overall immunological landscape of the
tumor.
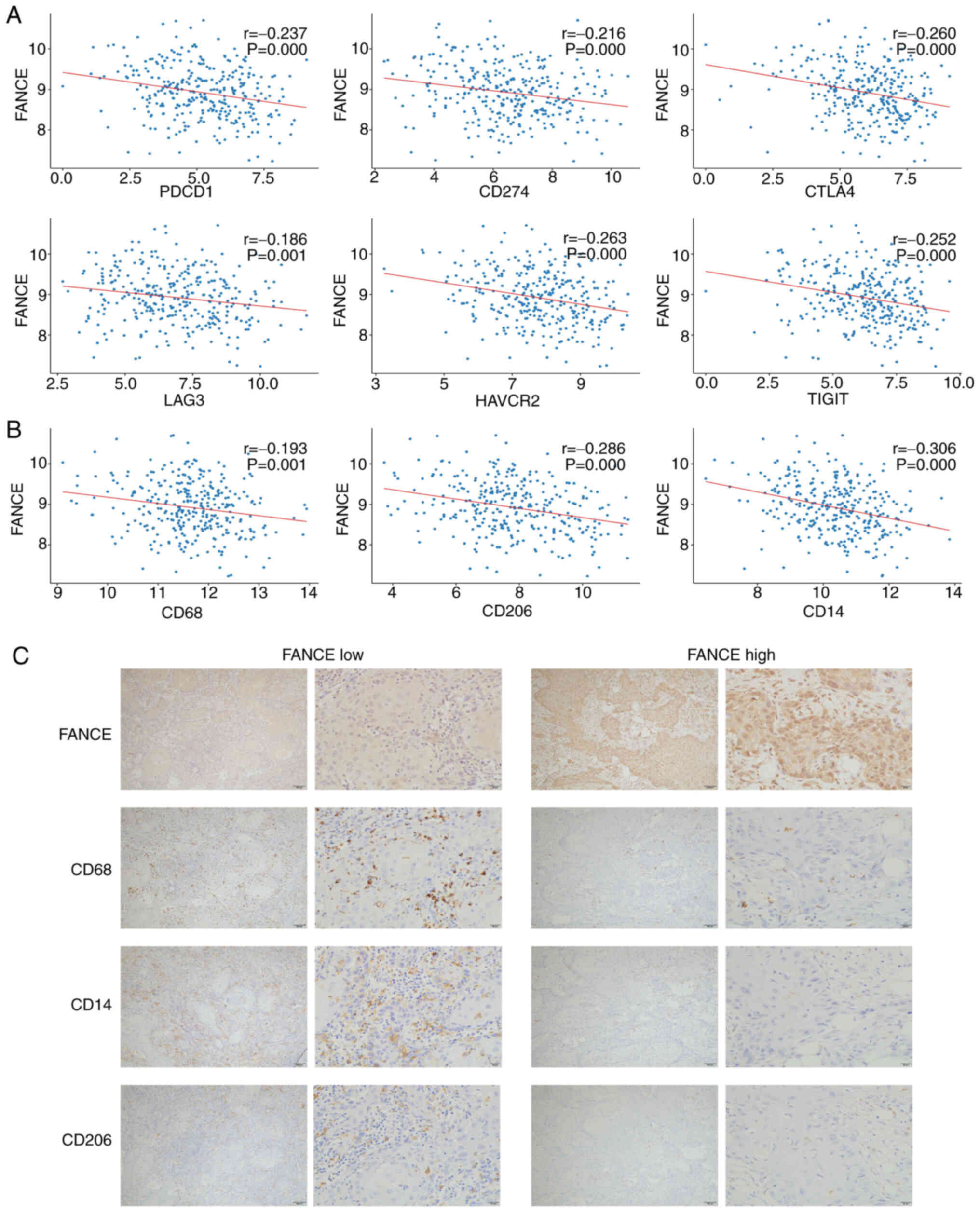 | Figure 5.FANCE expression modulates the OSCC
tumor microenvironment. (A) Scatter plots demonstrating the
negative correlation between FANCE expression and the expression of
immune checkpoint genes (PDCD1, CD274, CTLA4, LAG3, HAVCR2 and
TIGIT) in OSCC. Each plot includes the Pearson correlation
coefficient (r) and P-value indicating statistical significance,
with all P≤0.001. (B) Correlation between FANCE and OSCC macrophage
markers (CD68, CD14 and CD206) based on TCGA data analysis. Pearson
correlation coefficients are shown for each association. All
P≤0.001. (C) Immunohistochemical staining of FANCE, CD68, CD14 and
CD206 in OSCC tissues. Representative images are shown
demonstrating the inverse association between FANCE expression and
macrophage markers (scale bars, 100 and 20 µm, respectively).
FANCE, Fanconi anemia complementation group E; OSCC, oral squamous
cell carcinoma; TCGA, The Cancer Genome Atlas; PDCD1, programmed
Cell Death 1; CD274, Cluster of Differentiation 274; CTLA4,
Cytotoxic T-Lymphocyte-Associated Protein 4; LAG3, Lymphocyte
Activation Gene 3; HAVCR2, Hepatitis A Virus Cellular Receptor 2;
TIGIT, T Cell Immunoglobulin and ITIM Domain. |
Integrated analyses highlight the complex role of
FANCE in the OSCC TME, influencing not only immune cell
infiltration but also the balance between immunosuppressive and
antitumor immune responses. The findings suggest that FANCE may
serve as a potential therapeutic target for modulating the TME,
potentially enhancing the efficacy of immunotherapies in OSCC.
Discussion
In the present study, the role of FANCE in the
pathogenesis of OSCC was investigated, and insights into its
function in tumor biology and potential as a diagnostic marker were
revealed. Bioinformatics analysis of a TCGA dataset revealed an
association between enhanced expression of FANCE in tumor tissues
and poor survival in patients with OSCC. Machine learning and
immune microenvironment analyses enriched comprehension of the role
of FANCE in tumor biology. Functional assays indicated that FANCE
knockdown inhibited OSCC cell proliferation and migration,
confirming its role in oncogenic processes. GSEA showed negative
enrichment scores for FANCE, linking it to immune pathways.
Correlation analysis identified significant negative associations
between FANCE expression and immune checkpoint genes. IHC staining
revealed a negative association between FANCE and the immune
markers CD14, CD206 and CD68. Collectively, these findings enhanced
understanding of the pathophysiology of OSCC and emphasized the
multifaceted involvement of FANCE in tumor progression and its
interaction with the immune environment.
The findings of the present study demonstrated the
enhanced expression of FANCE in OSCC tissues in the TCGA dataset.
This convergence underscores the potential oncogenic role of FANCE
in OSCC and suggests that its enhanced expression could serve as a
reliable and reproducible biomarker. These data contribute to
accumulating evidence implicating FANCE in the development of OSCC.
Previous in-depth evaluations of robust datasets have provided a
solid foundation for understanding of the function of FANCE in
diseases including esophageal cancer, hepatocellular carcinoma,
breast/ovarian and lung cancer, uterine sarcoma, primary ovarian
insufficiency, and common variable immunodeficiency, which
mitigates concerns arising from the limitations of the analyzed
dataset. Acknowledging the need for integrated bioinformatics and
clinical studies, ongoing investigation will further elucidate the
clinical application of FANCE in the prognosis and therapy of OSCC.
The correlation between FANCE expression and immune cell markers
revealed by IHC staining is a qualitative assessment supported by
similar methodologies used in other studies, following a
well-established approach (15,16).
This method is recognized for its effectiveness in illustrating the
complex dynamics within the tumor microenvironment (7). Notably, across multiple OSCC tumor
samples, an association between decreased FANCE expression and
increased expression of macrophage markers (CD68, CD14, CD206) was
consistently observed. Although these observations were made
through microscopic examination rather than statistical analysis,
the consistency of this pattern across the samples in the present
study adds robustness to the conclusions.
Unsupervised learning was used to segment the OSCC
dataset, initially based on overall gene expression profiles rather
than FANCE expression alone. This approach allowed the discovery of
significant differences in FANCE expression between clusters, which
became a key entry point for exploring the complex roles of FANCE
across molecular subtypes. Given the high heterogeneity of OSCC,
this method provided a broader perspective on the role of FANCE,
beyond just its expression levels, and helped avoid confirmation
bias. The k-means clustering (11,12)
revealed distinct molecular subtypes, enabling the detection of
subtle but meaningful FANCE expression differences. These findings
not only highlight the intricate interplay between FANCE and the
OSCC microenvironment but also lay the groundwork for subsequent
functional analyses, allowing for a more nuanced understanding of
the role of FANCE in the molecular landscape of OSCC. Although the
differences in FANCE expression between the two groups may not
appear striking, they are statistically significant, attributed to
the large sample size that allows subtle variations to reach
significance. This approach reduces the risk of confirmation bias
and provides a broader perspective on the complex interplay between
FANCE expression and the OSCC microenvironment.
Immune cell infiltration was analyzed using
MCP-counter (12), xCell (17), CIBERSORT (18) and TIMER (17) algorithms, each offering a
comprehensive perspective on macrophage infiltration. MCP-counter
and xCell assessments are based on marker genes of specific cell
types, thus accurately mapping distribution and abundance of
different immune cell types within samples (17). The CIBERSORT and TIMER methods used
a deconvolution-based approach that evaluates samples as mixtures
of various immune cells. They can precisely deduce the proportion
and expression of each immune cell type via complex
computational models (19).
Although the present analysis reveals some discrepancies,
particularly in M1 macrophage abundance between CIBERSORT and
xCell, these differences underscore the importance of
cross-validating results to ensure robust conclusions. This
discrepancy likely arises from inherent differences in the
reference gene sets and statistical models used by the two
algorithms, which could affect their sensitivity and specificity in
detecting certain immune cell types. Notably, the observed
differences in macrophage abundance between the molecular subtypes
identified through clustering suggest that FANCE could be involved
in modulating the immune microenvironment across different OSCC
subtypes. While these findings are preliminary, they provide a
foundation for further investigation into the role of FANCE in
OSCC. Additionally, these insights may guide the selection of
appropriate markers for subsequent IHC experiments, thereby helping
to clarify the role of FANCE within the broader immunological
landscape of OSCC.
The GSEA highlights the multifaceted role of FANCE
in OSCC, linking its influence on immune pathways with broader
biological processes, including cell growth and migration. While
the exploration of the effects of FANCE on cell proliferation and
migration and its impact on the TME initially seemed like separate
investigations, the integrated approach reveals that these
processes may be interconnected. Specifically, the negative
enrichment of gene sets associated with immune regulation, such as
systemic lupus erythematosus and asthma, suggests that FANCE may
normally suppress these pathways. Upon downregulation of FANCE,
this suppression could be lifted, potentially activating immune
responses that influence tumor behavior, including proliferation
and migration. Furthermore, the observed negative correlation
between FANCE expression and immune checkpoint genes indicates that
FANCE might play a role in modulating the tumor microenvironment,
which could affect immune evasion and tumor aggressiveness. These
findings underscore the complex role of FANCE in OSCC, not only in
regulating cellular behaviors but also in shaping the immune
landscape, reinforcing the value of this integrated approach.
The functional assays of the present study provided
a compelling account of the role of FANCE in OSCC, as knockdown of
FANCE significantly inhibited cell proliferation and migration.
These findings support the canonical role of FANCE in DNA repair
and cellular stability, suggesting that its deregulation could
promote oncogenesis (20,21). While the results of the present
study demonstrate a clear impact on cell viability and behavior,
the specific mechanisms through which FANCE contributes to these
phenotypical changes in OSCC remain unclear. Further investigation
is warranted to explore the interplay between FANCE activity and
other cellular pathways such as p53 signaling and apoptosis.
Additionally, the present bioinformatics analyses, including GSEA
and immune checkpoint analysis, highlight the broader impact of
FANCE on the tumor microenvironment, suggesting a link between
FANCE expression and immune evasion mechanisms. These findings also
align with previous literature that implicates FANCE in cancer
progression (3,4,6,7,22–24),
supporting its consideration as a potential therapeutic target.
However, the varying results across different studies reflect the
complexity of the role of FANCE in cancer biology, underscoring the
importance of a nuanced and carefully tailored approach when
evaluating FANCE as a target in clinical settings (25,26).
The significant negative correlation between the
expression of FANCE and CD68 in OSCC demonstrates its potential
immunomodulatory role, particularly in influencing the infiltration
of proinflammatory M1 macrophages within the TME (27). Antitumorigenic M1 macrophages are
typically associated with an improved prognosis of cancer (28), and their negative correlation with
enhanced FANCE expression suggests a shift towards an
immunosuppressive and tumor-promoting microenvironment (6). A negative correlation between FANCE
and CD14 and CD206 also indicated initial macrophage activation and
an M2 phenotype transition (29,30),
respectively. Nevertheless, these preliminary findings align with
TCGA analyses of patients in the cBioPortal database (6,7),
suggesting a consistent negative correlation between FANCE and
macrophage markers. Cross-validation using a public genomic
database supports the relevance of the present findings and
suggests their applicability to a broader range of OSCC.
Elucidating the mechanistic link between FANCE and macrophage
polarization is crucial for developing strategies to manipulate the
tumor immune microenvironment and enhance the efficacy of
immunotherapy in patients with OSCC. Further studies are required
to elucidate the precise regulatory function of FANCE and its
potential as a therapeutic target.
The present study contributes significantly to the
understanding of the functional role of FANCE in OSCC. The results
derived from cell lines and retrospective data may not completely
mimic the complexity of tumor biology in vivo (31). Thus, further investigation is
required to validate these findings through clinical trials and
models in vivo to assess the therapeutic viability of
targeting FANCE. Future studies to elucidate details of the
molecular mechanisms of FANCE in tumorigenesis and tumor immunity
are warranted. Investigating FANCE interactions with other DNA
repair pathways and its impact on OSCC immunogenicity might lead to
novel therapeutic targets and the refinement of current treatment
paradigms. Furthermore, the integration of FANCE-associated
research into advancements in precision medicine may enhance
patient management and improve OSCC outcomes.
The present study advances the understanding of the
role of FANCE in OSCC, but several limitations must be
acknowledged. Firstly, the present research primarily relies on
retrospective clinical data and in vitro experiments, which,
while providing valuable insights, do not fully capture the
complexity of OSCC in vivo. Although the present study
focused on extensive clinical data from a public database, offering
a direct reflection of the human condition, animal studies are
crucial for validating these findings in a more physiologically
relevant context. Additionally, while the present study
concentrated on the role of FANCE within the tumor immune
microenvironment, its involvement in DNA repair pathways and
potential impact on therapy resistance require further exploration.
Future endeavors will aim plan to conduct animal studies that
complement the current findings and deepen the understanding of the
role of FANCE in OSCC. These future directions will help refine
therapeutic strategies and contribute to more personalized
treatment approaches, ultimately improving patient outcomes.
In conclusion, the results of the present study
demonstrated that FANCE is upregulated in OSCC and is correlated
with poor survival. Furthermore, FANCE participates in oncogenesis
by influencing OSCC cell proliferation and migration. With the help
of machine learning clustering and immune infiltration analysis, it
was demonstrated that FANCE also plays crucial roles in modulating
the immune microenvironment. Overall, this multi-dimensional
evidence underlines the potential of FANCE as a therapeutic target
for treating OSCC.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present research was supported by The Basic Research Program
of Shenzhen Innovation Council (grant no. JCYJ20210324110014037),
Shenzhen Clinical Medical Research Center for Oral Diseases (grant
no. 20210617170745001-SCRC202201001) and Shenzhen High-level
Hospital Construction Fund, Peking University Shenzhen Hospital
Scientific Research Fund (grant no. KYQD2023253).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
BL designed the experiments and drafted the
manuscript. YC, YL, XL and JZ conducted the experiments. BL, HY, YS
and YC conducted statistical analysis. HY and YS contributed to the
study conception, design and interpretation. BL, HY and YS confirm
the authenticity of all the raw data. All authors have read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
The study was conducted according to the Declaration
of Helsinki. Human sample collection was approved by the Ethical
Committee of the Peking University Shenzhen Hospital (Shenzhen,
China; approval no. 2023-177). Written informed consent was
obtained from all participants, and the consent forms were
collected and stored in accordance with the ethical standards of
the institution.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Alkire BC, Bergmark RW, Chambers K, Cheney
ML and Meara JG: Head and neck cancer in South Asia: Macroeconomic
consequences and the role of surgery. Lancet. 385 Suppl 2:S562015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chi AC, Day TA and Neville BW: Oral cavity
and oropharyngeal squamous cell carcinoma-an update. CA Cancer J
Clin. 65:401–421. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gordon SM, Alon N and Buchwald M: FANCC,
FANCE, and FANCD2 form a ternary complex essential to the integrity
of the Fanconi anemia DNA damage response pathway. J Biol Chem.
280:36118–36125. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zheng C, Ren Z, Chen H, Yuan X, Suye S,
Yin H and Fu C: Reduced FANCE confers genomic instability and
malignant behavior by regulating cell cycle progression in
endometrial cancer. J Cancer. 14:2670–2685. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Taniguchi T, Tischkowitz M, Ameziane N,
Hodgson SV, Mathew CG, Joenje H, Mok SC and D'Andrea AD: Disruption
of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian
tumors. Nat Med. 9:568–574. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lin B, Li H, Zhang T, Ye X, Yang H and
Shen Y: Comprehensive analysis of macrophage-related multigene
signature in the tumor microenvironment of head and neck squamous
cancer. Aging (Albany NY). 13:5718–5747. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou Z, Yin H, Suye S, He J and Fu C:
Pan-cancer analysis of the prognostic and immunological role of
Fanconi anemia complementation group E. Front Genet.
13:10249892023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Del Valle J, Rofes P, Moreno-Cabrera JM,
López-Dóriga A, Belhadj S, Vargas-Parra G, Teulé À, Cuesta R, Muñoz
X, Campos O, et al: Exploring the role of mutations in fanconi
anemia genes in hereditary cancer patients. Cancers (Basel).
12:8292020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
George M, Solanki A, Chavan N, Rajendran
A, Raj R, Mohan S, Nemani S, Kanvinde S, Munirathnam D, Rao S, et
al: A comprehensive molecular study identified 12 complementation
groups with 56 novel FANC gene variants in Indian Fanconi anemia
subjects. Hum Mutat. 42:1648–1665. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Snyder ER, Ricker JL, Chen Z and Waes CV:
Variation in cisplatinum sensitivity is not associated with Fanconi
Anemia/BRCA pathway inactivation in head and neck squamous cell
carcinoma cell lines. Cancer Lett. 245:75–80. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bello-Orgaz G, Menendez HD and Camacho D:
Adaptive k-means algorithm for overlapped graph clustering. Int J
Neural Syst. 22:12500182012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang Z, Pan Q, Ge H, Xing L, Hong Y and
Chen P: Deep learning-based clustering robustly identified two
classes of sepsis with both prognostic and predictive values.
EBioMedicine. 62:1030812020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Song L: A possible approach for stem cell
gene therapy of Fanconi anemia. Curr Gene Ther. 9:26–32. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zaki MYW, Alhasan SF, Shukla R, McCain M,
Laszczewska M, Geh D, Patman GL, Televantou D, Whitehead A,
Maurício JP, et al: Sulfatase-2 from cancer associated fibroblasts:
An environmental target for hepatocellular carcinoma? Liver Cancer.
11:540–557. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang X, Wang J, Zhao J, Wang H, Chen J and
Wu J: HMGA2 facilitates colorectal cancer progression via
STAT3-mediated tumor-associated macrophage recruitment.
Theranostics. 12:963–975. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Aran D, Hu Z and Butte AJ: xCell:
Digitally portraying the tissue cellular heterogeneity landscape.
Genome Biol. 18:2202017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Newman AM, Liu CL, Green MR, Gentles AJ,
Feng W, Xu Y, Hoang CD, Diehn M and Alizadeh AA: Robust enumeration
of cell subsets from tissue expression profiles. Nat Methods.
12:453–457. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen B, Khodadoust MS, Liu CL, Newman AM
and Alizadeh AA: Profiling tumor infiltrating immune cells with
CIBERSORT. Methods Mol Biol. 1711:243–259. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Takahashi J, Masuda T, Kitagawa A, Tobo T,
Nakano Y, Abe T, Ando Y, Kosai K, Kobayashi Y, Matsumoto Y, et al:
Fanconi anemia complementation group E, a DNA repair-related gene,
is a potential marker of poor prognosis in hepatocellular
carcinoma. Oncology. 100:101–113. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Polito D, Cukras S, Wang X, Spence P,
Moreau L, D'Andrea AD and Kee Y: The carboxyl terminus of FANCE
recruits FANCD2 to the Fanconi Anemia (FA) E3 ligase complex to
promote the FA DNA repair pathway. J Biol Chem. 289:7003–7010.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gille JJ, Floor K, Kerkhoven L, Ameziane
N, Joenje H and de Winter JP: Diagnosis of fanconi anemia: Mutation
analysis by multiplex ligation-dependent probe amplification and
PCR-based sanger sequencing. Anemia. 2012:6032532012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Philip PA, Azar I, Xiu J, Hall MJ,
Hendifar AE, Lou E, Hwang JJ, Gong J, Feldman R, Ellis M, et al:
Molecular characterization of KRAS wild-type tumors in patients
with pancreatic adenocarcinoma. Clin Cancer Res. 28:2704–2714.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pillonetto DV, Piovezan BZ, Nichele S,
Lima ACM, Pasquini R, Pereira NF and Bonfim C: Investigation of
mutations in Fanconi anemia genes and malignancy predisposition in
Brazilian patients. Int J Lab Hematol. 45:82–89. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fu C, Begum K, Jordan PW, He Y and
Overbeek PA: Dearth and delayed maturation of testicular germ cells
in fanconi anemia E mutant male mice. PLoS One. 11:e01598002016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fu C, Begum K and Overbeek PA: Primary
ovarian insufficiency induced by fanconi anemia E mutation in a
mouse model. PLoS One. 11:e01442852016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ferrisse TM, de Oliveira AB, Palacon MP,
Silva EV, Massucato EMS, de Almeida LY, Léon JE and Bufalino A: The
role of CD68+ and CD163+ macrophages in immunopathogenesis of oral
lichen planus and oral lichenoid lesions. Immunobiology.
226:1520722021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mantovani A, Marchesi F, Malesci A, Laghi
L and Allavena P: Tumour-associated macrophages as treatment
targets in oncology. Nat Rev Clin Oncol. 14:399–416. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jayasingam SD, Citartan M, Thang TH, Mat
Zin AA, Ang KC and Ch'ng ES: Evaluating the polarization of
tumor-associated macrophages into M1 and M2 phenotypes in human
cancer tissue: Technicalities and challenges in routine clinical
practice. Front Oncol. 9:15122020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schraufstatter IU, Zhao M, Khaldoyanidi SK
and Discipio RG: The chemokine CCL18 causes maturation of cultured
monocytes to macrophages in the M2 spectrum. Immunology.
135:287–298. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kimura Y: New anticancer agents: In vitro
and in vivo evaluation of the antitumor and antimetastatic actions
of various compounds isolated from medicinal plants. In Vivo.
19:37–60. 2005.PubMed/NCBI
|