Introduction
Breast cancer is a highly prevalent malignancy among
women, which poses a threat to women's health, accounting for
>23.8% of newly diagnosed cancer cases globally and for ~15.4%
of cancer-related mortalities (1).
In recent years, the global incidence and mortality rates of female
breast cancer have shown an increasing trend each year, and its
onset has been noted to occur more frequently in younger
individuals (1,2). Common treatment approaches for breast
cancer include surgery followed by chemotherapy or radiotherapy,
targeted therapy and immunotherapy, as well as endocrine therapy.
Tamoxifen (TAM), as a potent selective estrogen receptor (ER)
modulator, has been reported to competitively bind to the ER on
tumor cells, thereby preventing the action of estrogen on tumor
cell proliferation (3). However,
TAM resistance remains a notable obstacle that can lead to tumor
relapse and metastasis in the majority of patients with breast
cancer. Currently, there is no effective method to reverse TAM
resistance in the clinic (4,5).
Therefore, the identification of novel, non-toxic, broad-spectrum
and cost-effective alternative agents to overcome TAM resistance is
crucial in the prevention and treatment of breast cancer.
Propofol is a short-acting anesthetic used to induce
sedation during a variety of surgical procedures (6). Recently, it has been shown to reduce
cell viability, and inhibit the migratory and invasive abilities of
breast cancer cells (7). In
addition, propofol has been reported to improve the overall
survival (OS) and loco-regional recurrence-free survival (LRRFS) of
patients with non-metastatic breast cancer, compared with
inhalational anesthesia (8).
Furthermore, a previous study demonstrated that propofol can
enhance the sensitivity of patients with breast cancer to
trastuzumab, reducing the local recurrence rate of patients
undergoing breast-conserving surgery (9). These studies have suggested that
propofol may be a promising candidate drug with important
bioactivities that can be used as a therapeutic strategy against
breast cancer. However, the potential regulatory effects of
propofol on TAM-resistant (TR) breast cancer have not yet been
determined.
Based on RNA sequencing (RNAseq) analysis, novel
mechanisms of cancer progression and metastasis, involving numerous
molecular and signaling pathways, have been demonstrated (10). For example, researchers have used
RNAseq analysis to reveal aberrantly activated signaling pathways
in the development of clear cell renal cell carcinoma and to
uncover the oncogenic signaling pathways in patients with acral
melanoma (11,12). Accumulating transcriptome sequencing
data have suggested that the progression of tumorigenesis (such as
cell cycle progression/arrest and suppression of apoptosis), immune
response and metabolic regulation serve essential roles in the
process of drug resistance in breast cancer (13–15).
The progress in transcriptome sequencing has enabled its wider use
in identifying targets of antitumor agents against multiple types
of cancer (16,17).
The present study aimed to explore the potential
regulatory mechanisms underlying the effects of propofol on TR
breast cancer cells. The primary objectives were to detect the
functions of propofol and to uncover the key singling pathways
associated with propofol treatment in MCF7-TR cells. To achieve
these aims, the current study employed a multi-faceted approach
that included in vitro apoptosis and cell cycle analyses,
proliferation and colony formation assays, and RNAseq analysis. By
combining these methods, the study aimed to provide a comprehensive
understanding of the effects of propofol on TR breast cancer, and
to establish a foundation for the development of novel therapeutic
targets for the treatment of TR breast cancer.
Materials and methods
Materials
Propofol (cat. no. HY-B0649) and TAM (cat. no.
HY-13757A) were purchased from MedChemExpress. For the experiments,
propofol and TAM were dissolved in dimethyl sulfoxide (DMSO) and
further diluted in culture medium for storage. Cell Counting Kit-8
(CCK-8) (cat. no. P-CA-001) and phenol red-free high-glucose
Dulbecco's Modified Eagle Medium (DMEM) (cat. no. PM150223) were
purchased from Wuhan Pricella Biotechnology Co., Ltd. Certified
charcoal-stripped fetal bovine serum (FBS; cat. no. 04-201-1A) was
purchased from Biological Industries; Sartorius AG.
Cells
The human breast adenocarcinoma cell line MCF7 was
purchased from American Type Culture Collection (cat. no. HTB-22).
The cells were grown in complete DMEM supplemented with 10%
charcoal-stripped FBS, penicillin (100 U/ml) and streptomycin (100
µg/ml) under standard conditions at 37°C with 5% CO2.
MCF7-TR cells were prepared as described in previous reports with
slight modifications (18,19). Briefly, the induction of TAM
resistance was performed using culture medium containing 10 nM TAM
for 48 h. Subsequently, the drug was removed and the cells
underwent routine culturing, while continuously eliminating
susceptible non-viable cells. Once the cell confluence reached
70–80%, the operation was repeated twice in succession. The
selection process entailed iterative cycles, each time increasing
the drug concentration incrementally. Specifically, the dosage of
the drug was doubled with each iteration. This step-by-step
adjustment continued until the cells could proliferate stably in a
selection culture medium supplemented with 1 µM TAM. The parental
control cells were treated with DMSO and underwent identical
processing procedures as previously described (18,19).
This entire process lasted ~12 months. To maintain TAM resistance,
a concentration of 1 µM TAM was continuously supplemented to the
MCF7-TR cell culture medium. In addition, the expression levels of
two commonly studied TR-associated genes: ER 1 (ESR1) and
ATP binding cassette subfamily B member 1 (ABCB1; also
called P-glycoprotein), were detected in MCF7 and MCF7-TR breast
cancer cells by reverse transcription-quantitative PCR (RT-qPCR).
As shown in Fig. S1A, the
expression of ESR1 was downregulated, whereas ABCB1
was upregulated in MCF7-TR cells as compared with in non-resistant
parental MCF7 controls, which confirmed the successful construction
of TAM resistance (20,21).
Total RNA extraction and RT-qPCR
RT-qPCR was conducted as described in a previous
study (22). Briefly, MCF7-TR cells
and parental non-resistant control cells were lysed in 1 ml
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.) on
ice for 15 min. Subsequently, 200 µl chloroform was added to each
sample to fully dissociate the nucleoprotein complex. Following
centrifugation (12,000 × g, at 4°C for 15 min), the upper aqueous
phase was transferred to a new microcentrifuge tube and mixed with
an additional 500 µl isopropyl alcohol to precipitate the RNA.
After further centrifugation (12,000 × g, at 4°C for 10 min), the
RNA pellet was washed twice with 1 ml 75% ethanol. After
air-drying, the RNA samples were resuspended in 100 µl diethyl
pyrocarbonate-treated deionized water, and their concentrations
were determined using a microspectrophotometer (NanoDrop 2000;
Thermo Fisher Scientific, Inc.). For RT, first-strand cDNA was
synthesized using the RevertAid First Strand cDNA Synthesis Kit
(cat. no. K1622; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Subsequently, qPCR was carried out on
a Bio-Rad CFX-Touch real-time PCR system (Bio-Rad Laboratories,
Inc.) using the PowerTrack™ SYBR Green Master Mix (cat. no. A46012;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. The thermocycling conditions were as follows: 95°C
for 30 sec for cDNA pre-denaturation; 95°C for 10 sec for cDNA
denaturation; 60°C for 30 sec for primer annealing and new strand
extension. With the exception of the pre-denaturation step, the
denaturation, annealing and extension steps were repeated for a
total of 40 cycles. GAPDH was used as a housekeeping gene.
The primer sequences are listed in Table SI.
Cell cycle analysis and apoptosis
detection
Cell cycle progression and the induction of
apoptosis were analyzed using a Gallios flow cytometer (Beckman
Coulter, Inc.) according to the manufacturer's instructions.
For cell cycle analysis, MCF7-TR cells were seeded
in a 12-well plate at a density of 8×104 cells/well and
were incubated overnight. The cells were treated with 20 µM
propofol for 24 h at 37°C in an incubator with 5% CO2,
whereas the control cells were treated with an equal volume of
DMSO. Subsequently, the cells were collected and centrifuged at 300
× g at 4°C for 5 min. The cells were fixed with ice-cold 70%
ethanol for 15 min on ice, washed with PBS, incubated with PI
staining solution containing 50 µg/ml PI, 0.1 mg/ml RNase A and
0.05% Triton X-100 at room temperature for 1 h, and detected using
a flow cytometer. The data were processed using the cell cycle
fitting software ModFit LT 5.0 (Verity Software House, Inc.).
For apoptosis analysis, MCF7-TR cells were seeded in
a 12-well plate at a density of 8×104 cells/well and
were incubated overnight. The cells were treated with 10 or 20 µM
propofol for 24 h at 37°C in an incubator with 5% CO2,
and the control cells were treated with an equal volume of DMSO.
Subsequently, the cells were collected and centrifuged at 300 × g
at 4°C for 5 min. Apoptosis detection was performed using a
fluorescein isothiocyanate Annexin V/PI kit (Becton, Dickinson and
Company) according to the manufacturer's instructions. The samples
were analyzed by fluorescence-activated cell sorting using a flow
cytometer within 1 h of staining. The data were analyzed using
Kaluza v. 1.2 (Beckman Coulter, Inc.), and the percentages of early
apoptotic cells (Annexin V+ and PI−) and late
apoptotic cells (Annexin V+ and PI+) were
calculated.
Cell proliferation assay
Cell proliferation was assessed using the CCK-8
assay according to the manufacturer's instructions. Briefly,
MCF7-TR cells and their parental cells were seeded at a density of
6×103 cells/well and were cultured in 96-well plates
overnight. Subsequently, 2.5, 5 or 10 µM propofol was added to the
cells and incubated for 0, 24, 48, 72 or 96 h. A total of 10 µl
CCK-8 solution was then added to each well and incubated for 1 h.
The absorbance value was detected at 450 nm using a
spectrophotometer (Synergy HT; BioTek; Agilent Technologies,
Inc.).
Colony formation assay
The colony formation assay was performed as
described in a previous study (23). Briefly, MCF7-TR cells were seeded in
6-well plates at a density of 1×103 cells/well and were
cultured overnight. Subsequently, 2.5 µM propofol was added to each
well and the control cells were treated with an equal volume of
DMSO Subsequently, the medium was replaced with fresh complete
medium on day 3. On day 6, the cell colonies were fixed with 4%
paraformaldehyde (cat. no. P0099; Beyotime Institute of
Biotechnology) for 10 min at room temperature. After being washed
twice with distilled water for 2 min each time, the colonies were
stained with 2 ml/well crystal violet solution (cat. no. C0121;
Beyotime Institute of Biotechnology) for 10 min at room
temperature. After washing twice with distilled water (5 min/wash),
the plates were scanned using a scanner (HP ScanJet Pro 2600 f1; HP
Development Company, L.P.) and the colonies were calculated
manually using an inverted light microscope (CKX53; Olympus
Corporation). Colonies with cell numbers >20 were counted.
Total RNA extraction, library
construction and sequencing
A total of 5×106 MCF7-TR cells were
seeded in a 6-cm cell culture dish and cultured overnight.
Subsequently, propofol (10 µM) was added to each plate and the
cells were cultured for 24 h. The control cells were treated with
an equal volume of DMSO. Total RNA was obtained from
propofol-treated or non-treated MCF7-TR cells using TRIzol reagent
according to the manufacturer's instructions. The quality and
concentration of mRNA were detected using a NanoDrop 2000
spectrophotometer (Thermo Fisher Scientific, Inc.). The purified
mRNA from three repeat samples of the solvent control and
propofol-treated MCF7-TR cells was processed to prepare an mRNAseq
library using Illumina Stranded Total RNA Prep with Ribo-Zero Plus
or Ribo-Zero Plus Microbiome kit (Illumina, Inc.) (24). Briefly, ribosomal RNAs were
eliminated by a poly-(A) containing mRNA selection procedure to
minimize their sequencing. Subsequently, the remaining mRNAs were
subjected to cDNA strand synthesis, purification, end-repair,
A-tailing adaptor ligation and PCR amplification. The library
quality was analyzed using Agilent Bioanalyzer 2100 (Agilent
Technologies, Inc.) and the concentrations of the libraries were
detected by qPCR analysis. Subsequently, 20 pM each library was
sequenced using an Illumina NextSeq500 instrument (Illumina, Inc.)
equipped with NextSeq System Suite v2.2.0 software (Illumina,
Inc.). Paired-end reads with a length of 150 bp were harvested
using the NextSeq 550 System High-Output Sequencing Kit (Illumina,
Inc.).
Functional and signaling pathway
enrichment analysis of differentially expressed genes (DEGs)
Differential gene expression analysis and principal
component analysis (PCA) were conducted using the DESeq package
(https://bioconductor.org/packages//2.10/bioc/html/DESeq.html)
and depicted using the FactoMineR package (https://cran.r-project.org/web/packages/FactoMineR/index.html)
in R (version 4.4.2; http://www.r-project.org/). Genes with
|log2FoldChange|>1 and P<0.05 were identified as being
differentially expressed. Two-way cluster analysis on all DEGs was
performed using the pheatmap package (https://cran.r-project.org/web/packages/pheatmap/index.html),
based on expression levels across samples and patterns within
samples, using Euclidean distance and complete linkage hierarchical
clustering. Volcano plots were generated using the ggplot2 package
(https://cran.r-project.org/web/packages/ggplot2/index.html).
Data analysis for gene expression was performed based on the
Database for Annotation, Visualization and Integrated Discovery
(https://davidbioinformatics.nih.gov/). DEGs underwent
GO enrichment analysis to determine the enriched biological
processes, cellular components and molecular functions (http://www.geneontology.org/). The signaling pathways
were investigated using the Kyoto Encyclopedia of Genes and Genomes
(KEGG) pathway (http://www.genome.jp/kegg/). In addition, Gene Set
Enrichment Analysis (GSEA) was performed using GSEA software
(https://www.broadinstitute.org/gsea/)
using the following: Permutation, geneset; metric, Diff_of_classes;
metric, weighted; # permutation, 2,500.
Statistical analysis
Data are presented as the mean ± SD and are
representative of three independent experiments. Data analysis was
performed using GraphPad Prism 7 (Dotmatics). Statistical
significance between two experimental groups was evaluated using an
unpaired two-tailed Student's t-test. Multiple sets of data were
compared using a one- or two-way analysis of variance with Tukey's
post hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Propofol treatment significantly
promotes apoptosis and cell cycle arrest and inhibits proliferation
and colony formation in MCF7-TR cells
Fig. 1A shows the
chemical structure of propofol. To investigate the effect of
propofol on MCF7-TR cells, the cells were incubated with 10 and 20
µM propofol for 24 h. Apoptosis induction was then analyzed by flow
cytometry (Figs. 1B, 1C, S1B
and S1C). Notably, 10 µM propofol
caused only a slight, but significant, increase in both early and
late apoptosis of MCF7-TR cells (Fig.
S1C), whereas 20 µM resulted in a significant increase
(Figs. 1C and S1C). Therefore, 20 µM propofol was
selected for further analysis. Propofol at a concentration of 20 µM
induced a marked increase in the percentage of MCF7-TR cells
present in the G0/G1 phase, and notable
decreases in the percentage of cells present in the S and
G2/M phases of the cell cycle compared with in the
control group, suggesting that it induced significant cell cycle
arrest (Fig. 1D and E). Considering
that 20 µM propofol may induce significant apoptosis and cell cycle
arrest, lower concentrations of propofol, specifically 2.5, 5 and
10 µM, were selected for the cell proliferation assay. Consistent
with these findings, propofol treatment markedly inhibited cell
proliferation in a dose-dependent manner. Notably, when the
concentration of propofol reached 10 µM, it caused a significant
decrease in the proliferation of MCF7-TR cells after 96 h of
incubation (Fig. 1F). MCF7-TR cells
exhibited greater sensitivity to propofol compared with non-TR
control cells, as evidenced by significantly reduced cell
proliferation in MCF7-TR cells following propofol treatment
(Fig. S1D). Since prolonged
exposure to either 5 or 10 µM propofol significantly reduced cell
proliferation (Fig. 1F), a
concentration of 2.5 µM was selected to investigate its effects on
cell colony formation. In the cells treated with 2.5 µM propofol,
both the size and number of colonies were markedly reduced compared
with in the cells treated with a solvent control, indicating that
propofol exerted an inhibitory effect on the colony-forming ability
of MCF7-TR cells (Fig. 1G and H).
These results indicated that propofol exhibited potential antitumor
characteristics in TR breast cancer cells.
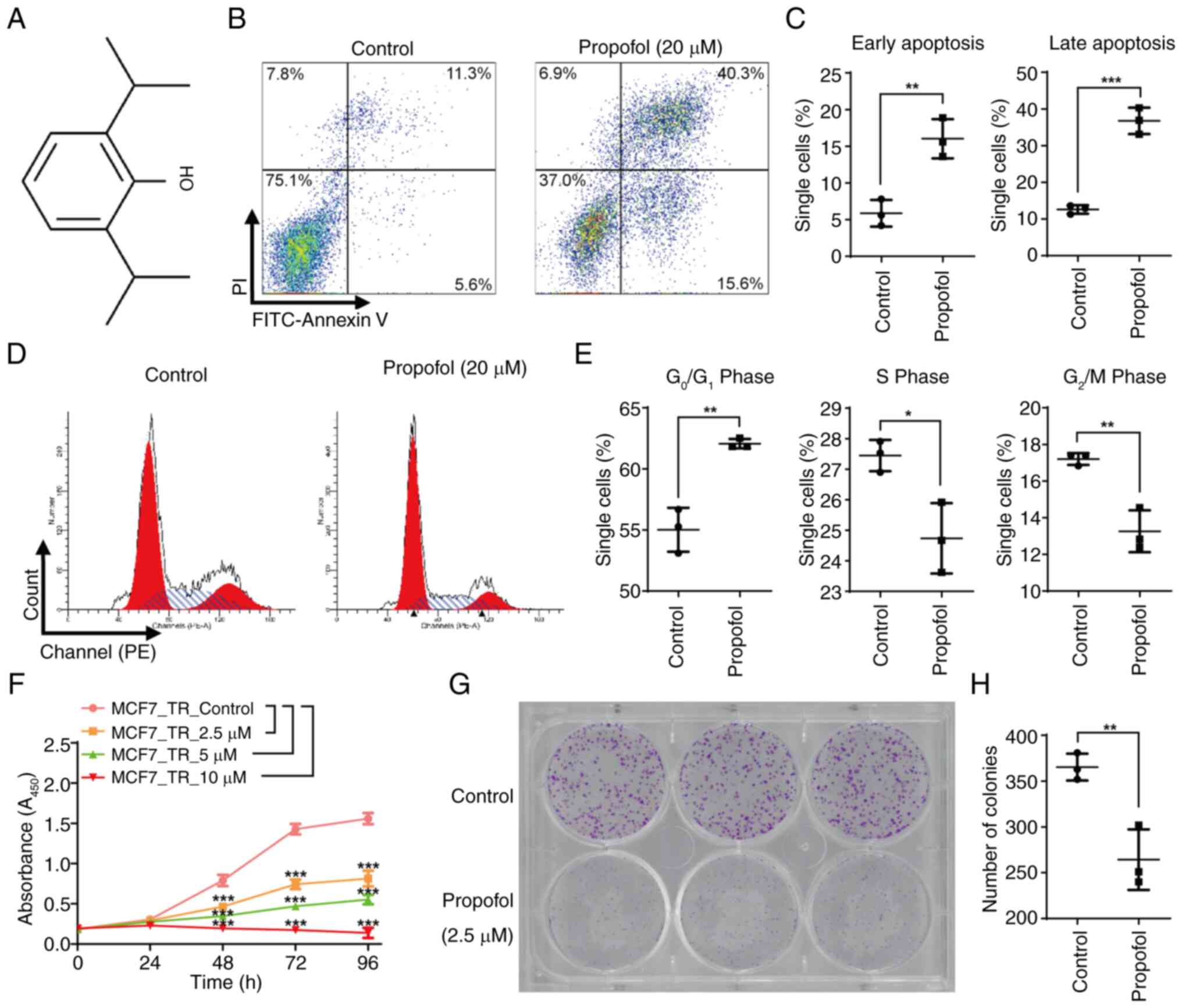 | Figure 1.Propofol significantly promotes
apoptosis and cell cycle arrest, and inhibits proliferation and
colony formation in MCF7-TR cells. (A) Chemical structure of
propofol. (B) Propofol significantly promoted the apoptosis of
MCF7-TR cells. MCF7-TR cells were treated with or without propofol
(20 µM) for 24 h, stained with PI and Annexin V, and apoptosis was
detected using flow cytometry. (C) Percentage of Annexin
V+ and PI− early apoptotic cells, and Annexin
V+ and PI+ late apoptotic cells. (D) Propofol
significantly promoted the cell cycle arrest of MCF7-TR cells.
MCF7-TR cells were treated with or without propofol (20 µM) for 24
h, stained with PI and cell cycle progression was detected using
flow cytometry. (E) Percentage of cells in
G0/G1, S and G2/M phases. (F)
Propofol significantly inhibited the proliferation in MCF7-TR
cells. MCF7-TR cells were treated with 2.5, 5 and 10 µM propofol
for 24, 48, 72 and 96 h, and were incubated with 10 µl Cell
Counting Kit-8 for 1 h before absorbance was measured using a
spectrophotometer. (G) Propofol significantly inhibited colony
formation in MCF7-TR cells. MCF7-TR cells were treated with or
without propofol (2.5 µM) for 6 days, stained with crystal violet
and images were captured using a scanner. (H) Number of colonies in
propofol-treated and untreated MCF7-TR cells. Data are presented as
the mean ± SD. Significant differences were examined using a (C, E
and H) two-tailed Student's t-test or (F) a two-way analysis of
variance. *P<0.05, **P<0.01, ***P<0.001. TR,
tamoxifen-resistant. |
Propofol treatment affects MCF7-TR
gene expression profiles
RNAseq analysis was performed for MCF7-TR cells
treated with or without propofol. Given that a 24-h treatment with
20 µM propofol resulted in a significant increase in the apoptosis
of MCF7-TR cells (Figs. 1B,
1C, S1B and S1C), 10 µM propofol was selected to
investigate its gene regulatory effects on MCF7-TR cells. PCA
indicated optimal intergroup difference and intragroup consistency
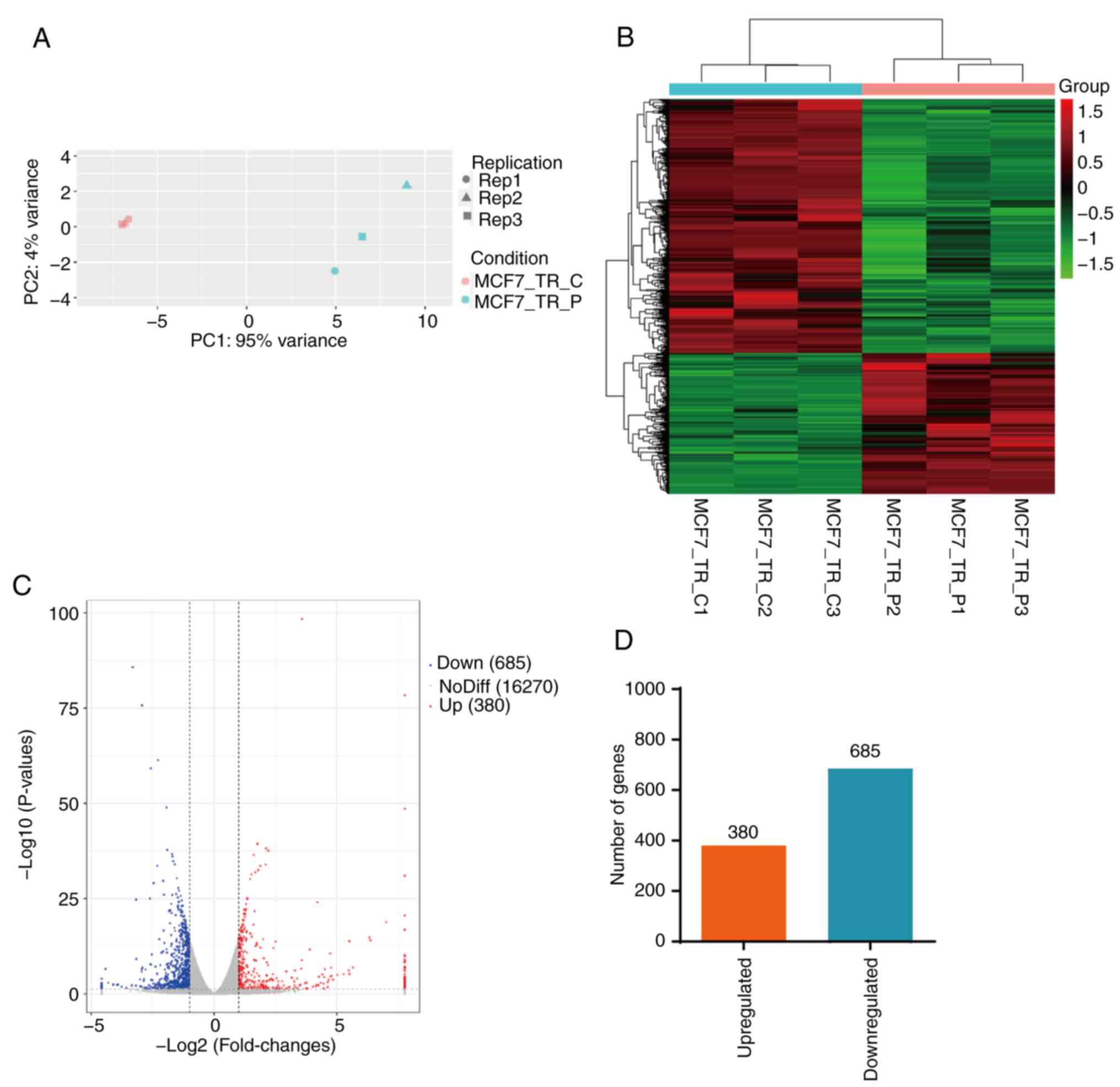
between MCF7-TR cells treated with or without propofol (Fig. 2A). A total of 1,065 DEGs were
identified in MCF7-TR cells following treatment with propofol, in
which the expression levels of 685 genes were markedly
downregulated and those of 380 genes were upregulated compared with
those in the control cells (Fig.
2B-D; Table SII). Overall,
propofol treatment markedly altered the gene expression profiles of
MCF7-TR cells.
GO term classification and KEGG
pathway enrichment analysis
In order to identify the key biological processes in
which the DEGs in propofol-treated MCF7-TR cells were involved, the
molecular functions they exerted and the pivotal signaling pathways
they were involved in, GO term classification and KEGG enrichment
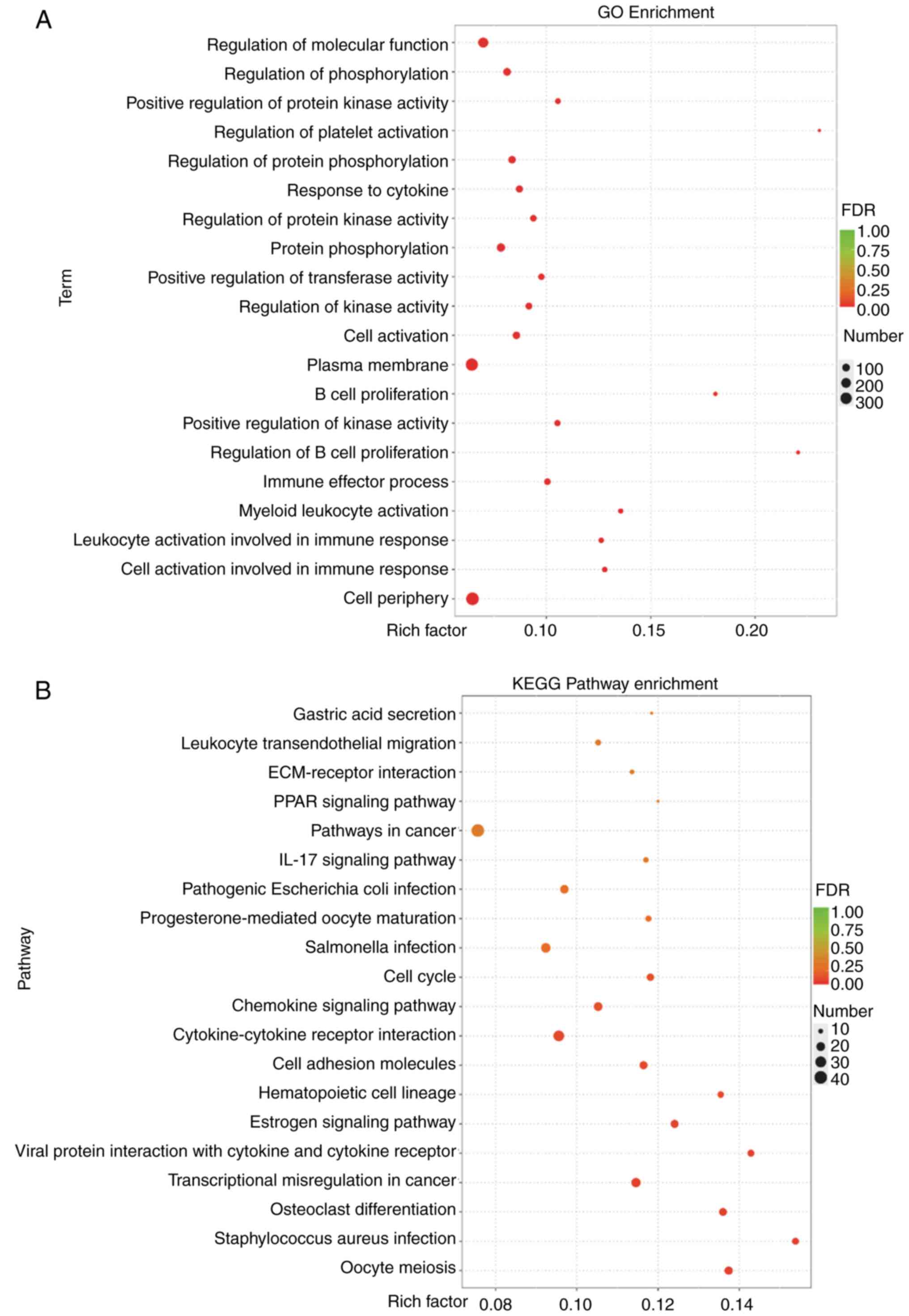
analysis were performed. As shown in Fig. 3A, a high number of DEGs in MCF7-TR
cells treated with propofol were clustered in the ‘cell periphery’
and located in the ‘plasma membrane’, possessing phosphatase
activity, and were involved in the regulation of the immune
response and in the phosphorylation processes. Furthermore, KEGG
enrichment analysis indicated that various enriched pathways in all
treated cells were related to the process of ‘transcriptional
misregulation in cancer’ and ‘cell cycle’, which occur in cancer
biology (Fig. 3B). In addition, the
enriched signaling pathways that were identified were associated
with specific immune response processes, as indicated by
‘cytokine-cytokine receptor interaction’ and ‘chemokine signaling
pathway’, and the endocrine system, as indicated by ‘estrogen
signaling pathway’ and ‘progesterone-mediated oocyte maturation’
according to the rich-factor and false discovery rate (FDR) values
(Fig. 3B). These results indicated
that propofol treatment could markedly regulate tumor
biology-associated processes, immune response and metabolism.
Propofol treatment affects gene
expression profiles in the immune response process in MCF7-TR
cells
Based on the GO term classification and the KEGG
enrichment analysis, the DEGs involved in immune
response-associated signaling pathways were further analyzed. As
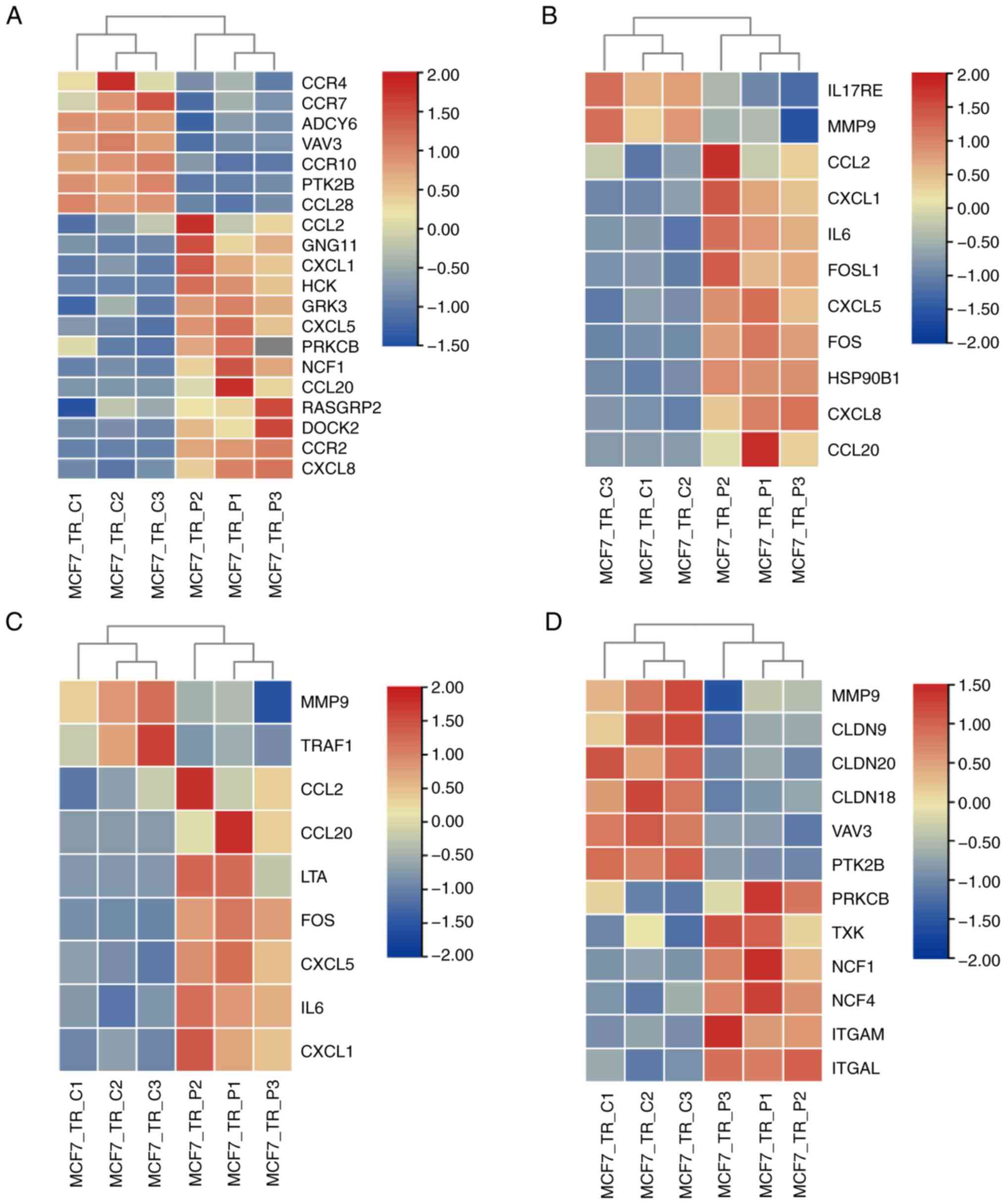
shown in Fig. 4, the top four most
dysregulated signaling pathways, according to the rich factor and
FDR value, in MCF7-TR cells treated with propofol were as follows:
Chemokine signaling pathway (Fig.
4A), IL-17 signaling pathway (Fig.
4B), TNF signaling pathway (Fig.
4C) and leukocyte transendothelial migration (Fig. 4D). Among these pathways, DEGs
affected by propofol treatment were mainly clustered into the
chemokine signaling pathway, in which 13 genes were upregulated and
seven genes were markedly downregulated (Fig. 4A). In addition, for TNF signaling
pathway, two genes (MMP9 and TRAF1) were significantly
downregulated, and seven genes were upregulated upon propofol
administration (Fig. 4C). These
results suggested that propofol administration in MCF7-TR cells
significantly regulated immune response signals, mainly manifested
by the chemokine signaling pathway.
Propofol treatment affects the gene
expression profiles associated with the metabolic process in
MCF7-TR cells
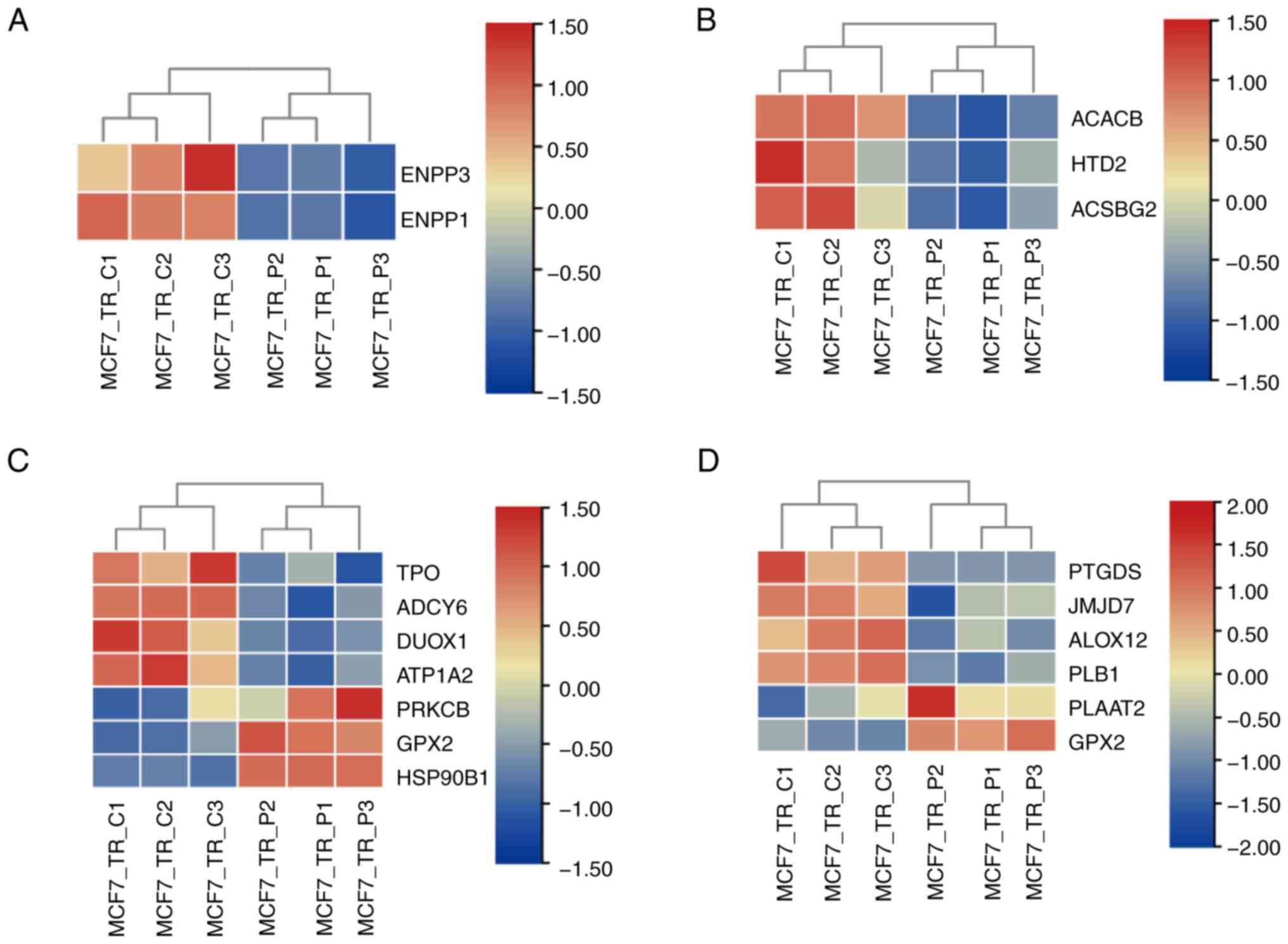
The enriched DEGs involved in the metabolic process,
according to the rich factor and FDR value, were analyzed and are
shown in Fig. 5. The impact of
propofol on metabolism was characterized by riboflavin metabolism
(Fig. 5A), fatty acid biosynthesis
(Fig. 5B), thyroid hormone
synthesis (Fig. 5C) and arachidonic
acid metabolism (Fig. 5D). Among
these processes, the effect of propofol on metabolic regulation was
mainly characterized by inhibition of the expression levels of
metabolism-related genes. No upregulated genes were noted in the
processes of riboflavin metabolism and fatty acid biosynthesis, and
higher numbers of downregulated genes were noted in the thyroid
hormone synthesis and arachidonic acid metabolism processes in
MCF7-TR cells following treatment with propofol compared with in
the control cells. These findings indicated that propofol was
involved in regulation of the metabolic process, which was mainly
dependent on downregulation of the expression levels of metabolic
genes.
Signaling pathways involved in the
sensitivity of MCF7-TR cells to propofol
To further characterize the molecular functions
involved in the immune response and metabolism, based on the
findings from the GO and KEGG analyses in propofol-treated MCF7-TR
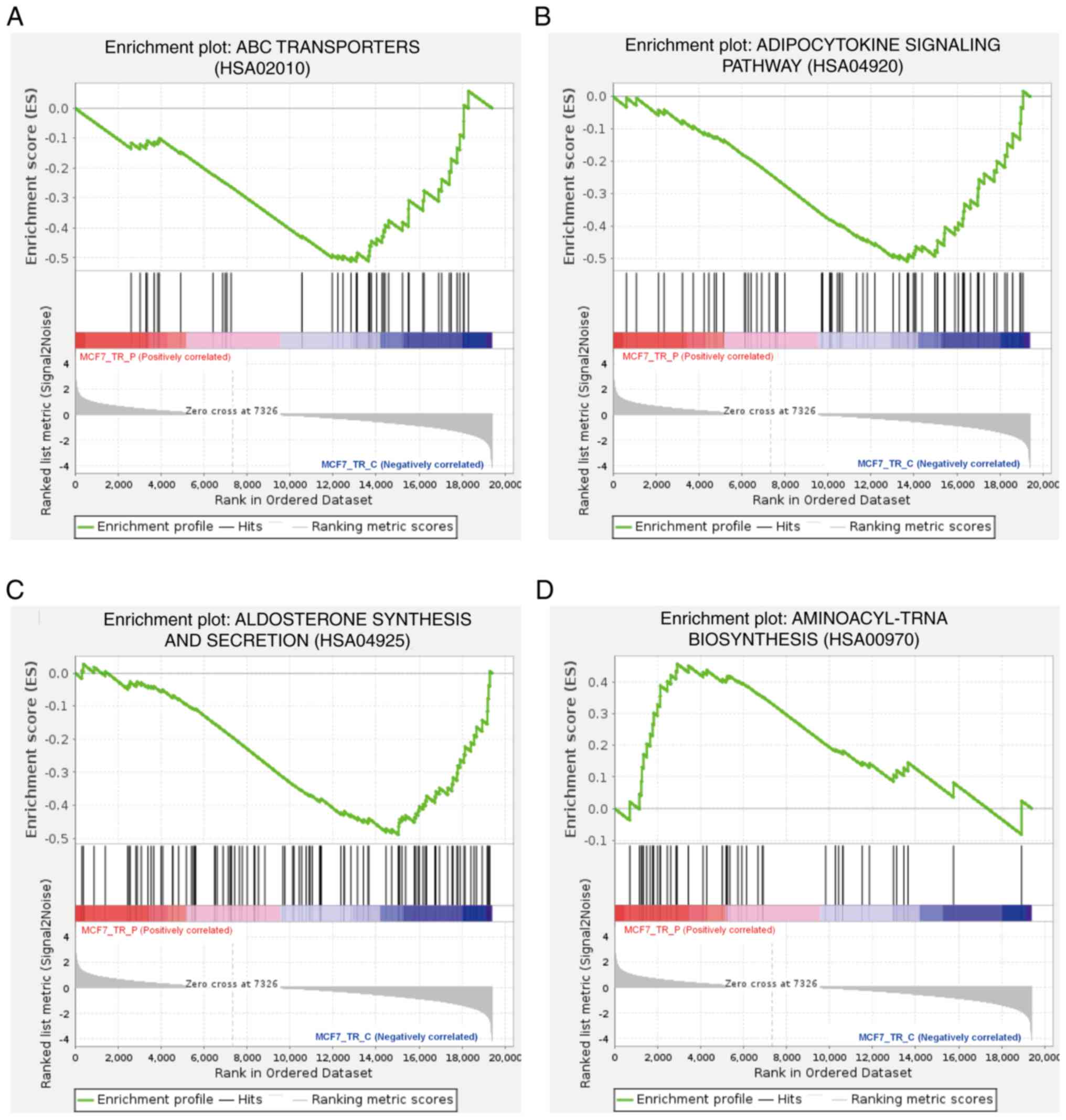
cells, GSEA was performed on the RNAseq data (Fig. 6). The negatively enriched gene sets
were involved in ABC transporters (Fig.
6A), the adipocytokine signaling pathway (Fig. 6B), and aldosterone synthesis and
secretion (Fig. 6C), while the
positively enriched gene set was involved in the aminoacyl-tRNA
biosynthesis (Fig. 6D). These gene
sets were composed of genes associated with certain metabolites,
which indicated that propofol functions partially by activating and
inhibiting metabolism-related signaling pathways.
Discussion
Currently, breast cancer is the leading cause of
morbidity and mortality among women. Among all types of breast
cancer, ER+ breast cancer is the most common, and the ER
signaling pathway serves a key role in its occurrence and
development (25). Therefore,
endocrine therapy that blocks the effects of estrogen has become
the first-line treatment for patients with ER+ breast
cancer (26). However, the primary
and acquired resistance encountered by patients receiving endocrine
drugs, such as TAM, remains an unsolved clinical challenge. The
present study demonstrated that propofol can affect cell cycle
progression and induce the apoptosis of TR breast cancer cell lines
by altering gene expression profiles involved in the regulation of
tumorigenesis, immune response and metabolism.
Numerous studies have suggested that the
development, metastasis and recurrence of tumors are closely
related to the cell cycle, apoptosis and proliferation, and
disruption of these events have been confirmed to be the
fundamental hallmarks of human malignancies (27–29).
Notably, misregulation in the cell cycle-related signaling pathways
can result in the unlimited proliferation of tumor cells. Moreover,
it has been reported that one approach to enhance the sensitivity
of cancer cells to chemotherapeutics can be achieved by combining
them with cell cycle regulators (30). Preclinical studies have shown that
propofol can significantly inhibit the development of breast cancer
tumors by promoting apoptosis and arresting the cell cycle
(7,31–33).
Furthermore, TR breast cancer cells exhibit greater sensitivity to
propofol compared with non-TR breast cancer cells. For non-TR
cells, a minimum of 25 µg/ml propofol (equivalent to 140 µM) is
required to induce significant apoptosis (7), whereas in the present study, only 20
µM propofol was sufficient to achieve similar effects in TR breast
cancer cells. Additionally, the study revealed that concentrations
as low as 2.5 µM propofol could inhibit cell proliferation and
colony formation in TR breast cancer cells. These concentrations
are much lower than those used for clinical anesthesia induction
(1.5–2.5 mg/kg body weight) and maintenance (4–12 mg/kg body
weight/h) (34). This lower
effective concentration is particularly important for patients with
TR breast cancer, as it suggests a broader therapeutic window for
propofol (35). In the clinical
setting, the effects of propofol have been perplexing. A previous
study has shown a lack of association between propofol-based total
intravenous anesthesia and inhalation anesthesia and long-term
survival following cancer surgery in Korean individuals (36). However, a broader meta-analysis
(including four randomized clinical trials and 13 retrospective
cohort studies) demonstrated that propofol-based anesthesia can
significantly improve OS and LRRFS in patients with non-metastatic
breast cancer, compared with inhalational anesthesia (8). In the present study, the antitumor
activity of propofol was characterized by inhibited cell cycle
progression, increased cell apoptosis and inhibited proliferation.
These findings suggested that propofol may act as a cell cycle
regulator in TR breast cancer cells and could be a potential
effective drug for patients who are resistant to anti-endocrine
therapy.
In accordance with the in vitro results, the
data obtained from transcriptome sequencing analysis indicated that
a large number of DEGs in MCF7-TR cells treated with propofol were
enriched in the process of cell cycle and transcriptional
misregulation. The cell cycle mainly consists of the following two
key events: Interphase, and the mitotic or M phase. The transitions
between the cell cycle phases are triggered by the cyclin-dependent
kinases (CDKs) and their binding ligands in the cyclin protein
family (37,38). CDK4/6 inhibitors have been employed
as potent, selective and orally bioavailable treatments for hormone
receptor-positive, HER2− breast cancer (39,40).
In the present study, propofol administration resulted in a marked
inhibition in the number of MCF7-TR cells in S phase of the cell
cycle, implying a potential association with CDK4/6 signals.
Preclinical studies have indicated that various cell
cycle-associated proteins are implicated in the resistance of
tumors to CDK4/6 inhibitors. For example, the dysregulation of the
cell cycle specific proteins INK4, p21 and P27 has been reported to
mediate the resistance to CDK4/6 inhibitors in breast cancer
(41–43). CDK4/6 inhibition and cytostasis
evasion are two critical events that occur in ER+ breast
cancer cells, and combined targeting of both CDK4/6 and PI3K can
induce cancer cell apoptosis in vitro and in patient-derived
tumor xenograft models, and prevent resistance to CDK4/6 inhibitors
by reducing the levels of cyclin D1 and other G1-S
cyclins, which ultimately results in tumor regression (44). The aforementioned studies indicate
that the targets of propofol in TR breast cancer cells may also
function as components of G1-S cyclins, which require
further exploration in future studies.
In addition to the DEGs enriched in the cell cycle
involved in the regulation of tumor occurrence and recurrence,
those affected by propofol treatment were also clustered into
immune and metabolic signaling pathways. The results from KEGG
pathway analysis indicated that certain DEGs were significantly
enriched into the chemokine signaling pathway. A previous study has
indicated that C-X-C motif ligand (CXCL)10, which is a
pro-inflammatory cytokine secreted by tumor cells, serves a vital
role in TR in breast cancer (45).
CXCL10 can promote breast cancer proliferation and growth via both
estrogen-dependent and -independent pathways, whereas CXCL10
inhibition has been shown to reverse the resistance of cells to TAM
(46). In addition, chemokines and
their receptors, such as C-X3-C motif chemokine ligand 1, CXCL12
and C-X-C chemokine receptor type 4 have been identified as
biomarkers for TR breast cancer therapy (47,48).
Moreover, the present study indicated that ~40 DEGs contributed to
regulation of the cytokine-cytokine receptor interaction,
manifested by the activation of IL-17 and the TNF signaling
pathway. TNF receptor-associated factor 1 (TRAF1) is a signaling
adaptor known for its role in TNF receptor-induced cell survival,
and serves a pivotal role in tumorigenesis and metastasis (49). The expression levels of TRAF1 have
been reported to be significantly associated with a longer
disease-free survival rate of breast cancer (50), and knockdown of TRAF1 expression may
increase TAM sensitivity in breast cancer cells (51). In the present study, propofol
downregulated TRAF1 levels, suggesting its potential for reversing
TAM resistance in breast cancer, which requires further
elucidation. In addition, the present study demonstrated that
metabolic processes, such as riboflavin metabolism, fatty acid
biosynthesis, thyroid hormone synthesis and arachidonic acid
metabolism, were processes involved in the response of MCF7-TR
cells to propofol; within these processes, the expression levels of
the majority of the genes were downregulated. The results of the
present study are supported by previous findings reported by Jiang
et al (52); this previous
study demonstrated that targeting fatty acid metabolism inhibition
could overcome TAM resistance in ER+ breast cancer.
Furthermore, the results from the GSEA indicated that significant
metabolites, including adipocytokines, aldosterone and
aminoacyl-tRNA, were strongly associated with propofol sensitivity.
These findings suggested that the activation and/or inhibition of
the signaling pathways in which these metabolites are involved may
have an important role in counteracting TAM resistance in breast
cancer cells. Collectively, the present study elucidated a
potential mechanism by which propofol regulates functions in TR
breast cancer cells at the transcriptional level, providing
valuable insights for the clinical application of this anesthetic
drug, particularly in individuals who are resistant to TAM. Future
investigations employing gene knockout and transgene experiments,
as well as in vivo and in vitro studies, will be
conducted to further explore the immune response and metabolic
pathways implicated in the present study, aiming to identify the
underlying mechanism for propofol-mediated regulation of TR breast
cancer.
The present study revealed that a 24-h incubation
with 20 µM propofol was sufficient to promote cell cycle arrest and
trigger apoptosis, demonstrating its potential as a cell cycle
regulator for the treatment of breast cancer. Although the impact
of different propofol concentrations on the gene expression profile
of cells remains unclear, cell phenotype assays suggested that a
lower concentration with a longer duration (2.5 or 5 µM for 3–6
days) may regulate the expression of genes associated with cell
proliferation. Conversely, a shorter culturing time with a higher
concentration of propofol (10 or 20 µM for 24 h) may induce the
expression of genes related to apoptosis, cell cycle arrest,
inflammatory response and metabolic changes. As the results of the
present study indicated lower effective concentrations of propofol
on TR breast cancer cells, and identified several therapeutic genes
and pathway targets of propofol in TR breast cancer cells, this
work is anticipated to make notable contributions to the field of
breast cancer treatment and may lead to more effective therapies
for patients with TAM resistance. However, the present study was
constrained by its exclusive focus on gene transcription in cell
lines and the exclusion of potential regulatory mechanisms
associated with protein expression levels. Future experiments will
focus on investigating the DEGs and their associated signaling
pathways. Gene knockout and transgenic mouse models, as well as
cell lines, will be utilized to elucidate the mechanisms by which
these DEGs and pathways confer resistance to TAM. Additionally, the
synergistic effects of combining small molecule inhibitors
targeting these DEGs and pathways with propofol will be explored to
develop more effective therapeutic strategies. Clinically, it is
also crucial to evaluate the potential benefits of the use of
propofol for patients with TR breast cancer, which can
significantly guide the selection of anesthetics in breast cancer
surgery.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by a grant from the Scientific Research
Project of Higher Education Institutions of Inner Mongolia
Autonomous Region (grant no. NJZZ23016).
Availability of data and materials
The RNA sequencing data generated in the present
study may be found in the NCBI BioProject database under accession
number PRJNA1021154 or at the following URL: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1021154.
The other data generated in the present study may be requested from
the corresponding author.
Authors' contributions
RY, CG and YL conceptualized the study. RY and JG
designed the study, and collected and analyzed the data. RY, JG, YL
and CG wrote and edited the manuscript. CG provided the funding. CG
and YL supervised the research. RY, YL and CG confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CDK
|
cyclin-dependent kinase
|
|
DEG
|
differentially expressed gene
|
|
ER
|
estrogen receptor
|
|
GO
|
Gene Ontology
|
|
GSEA
|
Gene Set Enrichment Analysis
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
PCA
|
principal component analysis
|
References
|
1
|
Bray F, Laversanne M, Sung H, Ferlay J,
Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 74:229–263. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD, Wagle NS and Jemal
A: Cancer statistics, 2023. CA Cancer J Clin. 73:17–48. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim J and Munster PN: Estrogens and breast
cancer. Ann Oncol. 36:134–148. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen F, Wang L, Feng Y, Ma W, Liu J, Bi Q,
Song Y, Gao R and Jia Y: F-box and leucine-rich repeat protein 16
controls tamoxifen sensitivity via regulation of mitochondrial
respiration in estrogen receptor-positive breast cancer cells. Hum
Cell. 36:2087–2098. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hanker AB, Sudhan DR and Arteaga CL:
Overcoming endocrine resistance in breast cancer. Cancer Cell.
37:496–513. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kazi M, Alqahtani A, Alharbi M, Ahmad A,
Hussain MD, Alothaid H and Aldughaim MS: The development and
optimization of lipid-based self-nanoemulsifying drug delivery
systems for the intravenous delivery of propofol. Molecules.
28:14922023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sun P, Huang H, Ma JC, Feng B, Zhang Y,
Qin G, Zeng W and Cui ZK: Repurposing propofol for breast cancer
therapy through promoting apoptosis and arresting cell cycle. Oncol
Rep. 52:1552024. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang Y, Yu P, Bian L, Huang W, Li N and
Ye F: Survival benefits of propofol-based versus inhalational
anesthesia in non-metastatic breast cancer patients: a
comprehensive meta-analysis. Sci Rep. 14:163542024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tian D, Tian M, Ma ZM, Zhang LL, Cui YF
and Li JL: Anesthetic propofol epigenetically regulates breast
cancer trastuzumab resistance through IL-6/miR-149-5p axis. Sci
Rep. 10:88582020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Müller MR, Burmeister A, Skowron MA,
Stephan A, Bremmer F, Wakileh GA, Petzsch P, Köhrer K, Albers P and
Nettersheim D: Therapeutical interference with the epigenetic
landscape of germ cell tumors: A comparative drug study and new
mechanistical insights. Clin Epigenetics. 14:52022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Moon S, Kim HJ, Lee Y, Lee YJ, Jung S, Lee
JS, Hahn SH, Kim K, Roh JY and Nam S: Oncogenic signaling pathways
and hallmarks of cancer in Korean patients with acral melanoma.
Comput Biol Med. 154:1066022023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang J, Liu F, Guo W, Bi X, Yuan S,
Shayiti F, Pan T, Li K and Chen P: Single-cell transcriptome
sequencing reveals aberrantly activated inter-tumor cell signaling
pathways in the development of clear cell renal cell carcinoma. J
Transl Med. 22:372024. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Salvati A, Melone V, Sellitto A, Rizzo F,
Tarallo R, Nyman TA, Giurato G, Nassa G and Weisz A: Combinatorial
targeting of a chromatin complex comprising Dot1L, menin and the
tyrosine kinase BAZ1B reveals a new therapeutic vulnerability of
endocrine therapy-resistant breast cancer. Breast Cancer Res.
24:522022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang S, Bei Y, Tian Q, He J, Wang R, Wang
Q, Sun L, Ke J, Xie C and Shen P: PFKFB4 facilitates palbociclib
resistance in oestrogen receptor-positive breast cancer by
enhancing stemness. Cell Prolif. 56:e133372023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhu Y, Zhang H, Pan C, He G, Cui X, Yu X,
Zhang X, Wu D, Yang J, Wu X, et al: Integrated tumor genomic and
immune microenvironment analysis identifies predictive biomarkers
associated with the efficacy of neoadjuvant therapy for
triple-negative breast cancer. Cancer Med. 12:5846–5858. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li R, Ferdinand JR, Loudon KW, Bowyer GS,
Laidlaw S, Muyas F, Mamanova L, Neves JB, Bolt L, Fasouli ES, et
al: Mapping single-cell transcriptomes in the intra-tumoral and
associated territories of kidney cancer. Cancer Cell.
40:1583–1599.e1510. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shimizu H and Nakayama KI: A 23 gene-based
molecular prognostic score precisely predicts overall survival of
breast cancer patients. EBioMedicine. 46:150–159. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xue Y, Lian W, Zhi J, Yang W, Li Q, Guo X,
Gao J, Qu H, Lin W, Li Z, et al: HDAC5-mediated deacetylation and
nuclear localisation of SOX9 is critical for tamoxifen resistance
in breast cancer. Br J Cancer. 121:1039–1049. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lü M, Ding K, Zhang G, Yin M, Yao G, Tian
H, Lian J, Liu L, Liang M, Zhu T and Sun F: MicroRNA-320a
sensitizes tamoxifen-resistant breast cancer cells to tamoxifen by
targeting ARPP-19 and ERRγ. Sci Rep. 5:87352015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yao J, Deng K, Huang J, Zeng R and Zuo J:
Progress in the understanding of the mechanism of tamoxifen
resistance in breast cancer. Front Pharmacol. 11:5929122020.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Leslie EM, Deeley RG and Cole SP:
Multidrug resistance proteins: Role of P-glycoprotein, MRP1, MRP2,
and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol.
204:216–237. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu H, Wang N, Yang R, Luan J, Cao M, Zhai
C, Wang S, Wei M, Wang D, Qiao J, et al: E3 ubiquitin ligase NEDD4L
negatively regulates skin tumorigenesis by inhibiting IL-6/GP130
signaling pathway. J Invest Dermatol. 144:2453–2464.e2411. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yin R, Gao J and Liu Y: Mechanisms
analysis for formononetin counteracted-osimertinib resistance in
non-small cell lung cancer cells: From the insight into the gene
transcriptional level. Chem Biol Drug Des. 103:e144352024.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang Z, Gerstein M and Snyder M: RNA-Seq:
A revolutionary tool for transcriptomics. Nat Rev Genet. 10:57–63.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Angus L, Smid M, Wilting SM, Bos MK,
Steeghs N, Konings I, Tjan-Heijnen VCG, van Riel J, van de Wouw AJ,
Cpct C, et al: Genomic alterations associated with estrogen
receptor pathway activity in metastatic breast cancer have a
differential impact on downstream ER signaling. Cancers (Basel).
15:44162023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sasada S, Kondo N, Hashimoto H, Takahashi
Y, Terata K, Kida K, Sagara Y, Ueno T, Anan K, Suto A, et al:
Prognostic impact of adjuvant endocrine therapy for estrogen
receptor-positive and HER2-negative T1a/bN0M0 breast cancer. Breast
Cancer Res Treat. 202:473–483. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Barathan M, Vellasamy KM, Ibrahim ZA,
Mariappan V, Hoong SM and Vadivelu J: Zerumbone mediates apoptosis
and induces secretion of proinflammatory cytokines in breast
carcinoma cell culture. Iran J Basic Med Sci. 24:1538–1545.
2021.PubMed/NCBI
|
|
28
|
Icard P, Shulman S, Farhat D, Steyaert JM,
Alifano M and Lincet H: How the Warburg effect supports
aggressiveness and drug resistance of cancer cells? Drug Resist
Updat. 38:1–11. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kowsari H, Davoodvandi A, Dashti F,
Mirazimi SMA, Bahabadi ZR, Aschner M, Sahebkar A, Gilasi HR,
Hamblin MR and Mirzaei H: Resveratrol in cancer treatment with a
focus on breast cancer. Curr Mol Pharmacol. 16:346–361. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rasoolnezhad M, Safaralizadeh R,
Hosseinpour Feizi MA, Banan-Khojasteh SM, Roshani Asl E, Lotfinejad
P and Baradaran B: MiR-138-5p improves the chemosensitivity of
MDA-MB-231 breast cancer cell line to paclitaxel. Mol Biol Rep.
50:8407–8420. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sun C, Liu P, Pei L, Zhao M and Huang Y:
Propofol inhibits proliferation and augments the anti-tumor effect
of doxorubicin and paclitaxel partly through promoting ferroptosis
in triple-negative breast cancer cells. Front Oncol. 12:8379742022.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang H, Zhao L, Wu J, Hong J and Wang S:
Propofol induces ROS-mediated intrinsic apoptosis and migration in
triple-negative breast cancer cells. Oncol Lett. 20:810–816. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang X, Li F, Zheng Y, Wang X, Wang K, Yu
Y and Zhao H: Propofol reduced mammosphere formation of breast
cancer stem cells via pd-l1/nanog in vitro. Oxid Med Cell Longev.
2019:90782092019.PubMed/NCBI
|
|
34
|
Malekmohammadi M, Price CM, Hudson AE,
DiCesare JAT and Pouratian N: Propofol-induced loss of
consciousness is associated with a decrease in thalamocortical
connectivity in humans. Brain. 142:2288–2302. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wakai A, Blackburn C, McCabe A, Reece E,
O'Connor G, Glasheen J, Staunton P, Cronin J, Sampson C, McCoy SC,
et al: The use of propofol for procedural sedation in emergency
departments. Cochrane Database Syst Rev.
2015:CD0073992015.PubMed/NCBI
|
|
36
|
Yoon S, Jung SY, Kim MS, Yoon D, Cho Y and
Jeon Y: Impact of propofol-based total intravenous anesthesia
versus inhalation anesthesia on long-term survival after cancer
surgery in a nationwide cohort. Ann Surg. 278:1024–1031. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Faienza F, Polverino F, Rajendraprasad G,
Milletti G, Hu Z, Colella B, Gargano D, Strappazzon F, Rizza S,
Vistesen MV, et al: AMBRA1 phosphorylation by CDK1 and PLK1
regulates mitotic spindle orientation. Cell Mol Life Sci.
80:2512023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Milletti G, Colicchia V and Cecconi F:
Cyclers' kinases in cell division: From molecules to cancer
therapy. Cell Death Differ. 30:2035–2052. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Decker T, Lüdtke-Heckenkamp K, Melnichuk
L, Hirmas N, Lübbe K, Zahn MO, Schmidt M, Denkert C, Lorenz R,
Müller V, et al: Anti-hormonal maintenance treatment with the
CDK4/6 inhibitor ribociclib after 1st line chemotherapy in hormone
receptor positive / HER2 negative metastatic breast cancer: A phase
II trial (AMICA). Breast. 72:1035752023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ergun Y, Dogan M, Ucar G, Karacin P and
Karacin C: Efficacy of adjuvant CDK4/6 inhibitors in hormone
receptor-positive breast cancer: A systematic review and
meta-analysis. Expert Opin Pharmacother. 24:1901–1909. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li Q, Jiang B, Guo J, Shao H, Del Priore
IS, Chang Q, Kudo R, Li Z, Razavi P, Liu B, et al: INK4 tumor
suppressor proteins mediate resistance to CDK4/6 kinase inhibitors.
Cancer Discov. 12:356–371. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pandey K, An HJ, Kim SK, Lee SA, Kim S,
Lim SM, Kim GM, Sohn J and Moon YW: Molecular mechanisms of
resistance to CDK4/6 inhibitors in breast cancer: A review. Int J
Cancer. 145:1179–1188. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Paternot S, Bockstaele L, Bisteau X,
Kooken H, Coulonval K and Roger PP: Rb inactivation in cell cycle
and cancer: The puzzle of highly regulated activating
phosphorylation of CDK4 versus constitutively active CDK-activating
kinase. Cell Cycle. 9:689–699. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Herrera-Abreu MT, Palafox M, Asghar U,
Rivas MA, Cutts RJ, Garcia-Murillas I, Pearson A, Guzman M,
Rodriguez O, Grueso J, et al: Early adaptation and acquired
resistance to CDK4/6 inhibition in estrogen receptor-positive
breast cancer. Cancer Res. 76:2301–2013. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mihály Z, Kormos M, Lánczky A, Dank M,
Budczies J, Szász MA and Győrffy B: A meta-analysis of gene
expression-based biomarkers predicting outcome after tamoxifen
treatment in breast cancer. Breast Cancer Res Treat. 140:219–232.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wu X, Sun A, Yu W, Hong C and Liu Z:
CXCL10 mediates breast cancer tamoxifen resistance and promotes
estrogen-dependent and independent proliferation. Mol Cell
Endocrinol. 512:1108662020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cao Z, Jin Z, Zeng L, He H, Chen Q, Zou Q,
Ouyang D, Luo N, Zhang Y, Yuan Y and Yi W: Prognostic and
tumor-immune infiltration cell signatures in tamoxifen-resistant
breast cancers. Gland Surg. 10:2766–2779. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gonçalves TL, de Araújo LP and Pereira
Ferrer V: Tamoxifen as a modulator of CXCL12-CXCR4-CXCR7 chemokine
axis: A breast cancer and glioblastoma view. Cytokine.
170:1563442023. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Abdul-Sater AA, Edilova MI, Clouthier DL,
Mbanwi A, Kremmer E and Watts TH: The signaling adaptor TRAF1
negatively regulates toll-like receptor signaling and this
underlies its role in rheumatic disease. Nat Immunol. 18:26–35.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lee HJ, Lee JJ, Song IH, Park IA, Kang J,
Yu JH, Ahn JH and Gong G: Prognostic and predictive value of
NanoString-based immune-related gene signatures in a neoadjuvant
setting of triple-negative breast cancer: Relationship to
tumor-infiltrating lymphocytes. Breast Cancer Res Treat.
151:619–627. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Weng L, Ziliak D, Lacroix B, Geeleher P
and Huang RS: Integrative ‘omic’ analysis for tamoxifen sensitivity
through cell based models. PLoS One. 9:e934202014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Jiang C, Zhu Y, Chen H, Lin J, Xie R, Li
W, Xue J, Chen L, Chen X and Xu S: Targeting c-Jun inhibits fatty
acid oxidation to overcome tamoxifen resistance in estrogen
receptor-positive breast cancer. Cell Death Dis. 14:6532023.
View Article : Google Scholar : PubMed/NCBI
|