According to data from the 2024 National Cancer
Institute and the Centers for Disease Control and Prevention, the
age-standardized incidence rate of breast cancer for women during
the 2017–2021 period was 131.8 per 100,000 women. Triple-negative
breast cancer (TNBC) is a highly heterogeneous subtype of breast
cancer with poor prognosis, accounting for 10–20% of all breast
cancer cases and having the lowest survival rate (78%) among all
subtypes (1). Due to the lack of
expression of estrogen receptor, progesterone receptor and human
epidermal growth factor receptor 2 (HER2), TNBC is insensitive to
traditional hormone therapy and targeted treatments, leaving
chemotherapy as the primary treatment option for TNBC. However, the
high rates of recurrence and metastasis in TNBC highlight the
urgent need to identify novel therapeutic strategies and molecular
targets to improve patient survival (2).
The primary risk factor for breast cancer is a
mutation in either of the breast cancer susceptibility genes breast
cancer susceptibility gene (BRCA)1 or BRCA2. A considerable
proportion of patients with TNBC carry germline BRCA1/2 mutations.
Tumors harboring BRCA1/2 mutations are highly dependent on poly
(ADP-ribose) polymerase (PARP) proteins, which are essential for
repairing single-strand DNA damage through the base excision repair
pathway (3). Consequently, PARP
inhibitors, due to their unique mechanism of blocking DNA damage
repair, are particularly suited for treating tumors with DNA repair
deficiencies (4). Over the past
decades, PARP inhibitors have emerged as promising drugs in
clinical oncology (5,6).
To date, numerous clinical research articles have
been published on PARP inhibitors in the treatment of breast
cancer, ovarian cancer and other diseases. At present, there are
eight PARP inhibitors, of which only six have been approved for
global marketing, excluding Iniparib and Veliparib. These approved
drugs include Olaparib (NCT02734004), Rucaparib (NCT02042378),
Niraparib (NCT05734911), Talazoparib (NCT04987931), Fuzuloparib
(NCT05753826) and Pamiparib (NCT03333915) (7–9), and
are used for the treatment of ovarian cancer (such as NCT03333915),
pancreatic cancer (such as NCT02677038), breast cancer (such as
NCT01074970), prostate cancer (such as NCT03516812) and other solid
tumors (10).
It is worth noting that not all PARP inhibitors have
achieved highly satisfactory results in the treatment of breast
cancer due to the emergence of drug resistance, toxic side effects,
such as bone marrow suppression and gastrointestinal discomfort, as
well as limitations in the target population (e.g., the efficacy of
the drug depends on the BRCA gene mutation status) (11). Clinical research on certain PARP
inhibitors is still exploring more suitable dosing regimens and
combination therapy strategies (12–15).
Numerous studies have focused on PARP and its
inhibitors as therapeutic targets (16–18).
However, in the actual clinical treatment of patients with TNBC,
the efficacy of PARP inhibitors does not always mirror the
promising results observed in experimental models, thus warranting
reflection from researchers and clinicians on the outcomes. The
main objective of the present study was to explore potential
directions for advancing the use of PARP inhibitors in TNBC
treatment by integrating feasible therapeutic strategies with the
current clinical trial performance of PARP inhibitors in TNBC. The
present review focuses on the molecular mechanisms of PARP and its
inhibitors, aiming to provide insights into optimizing the use of
these agents in future TNBC therapies.
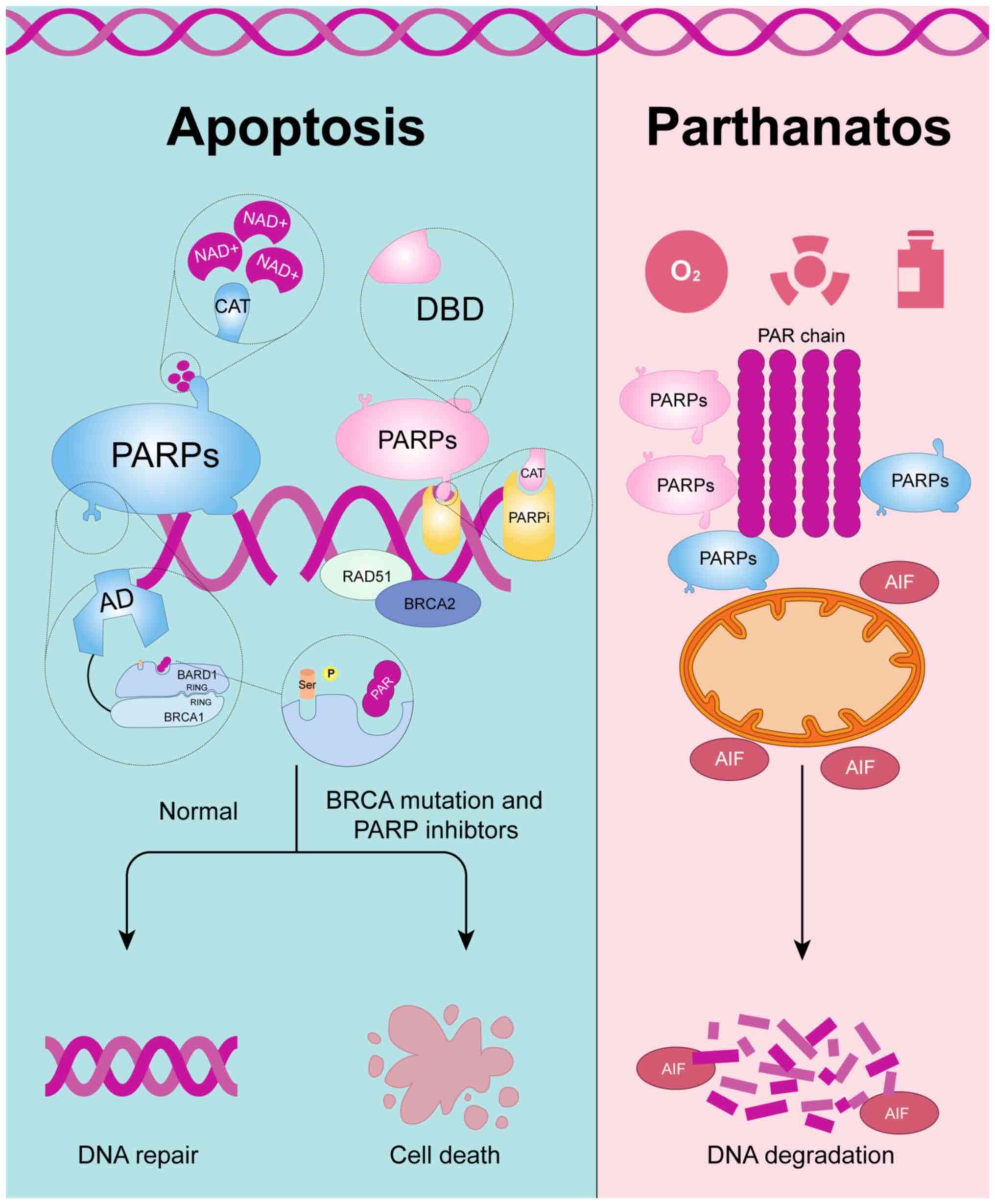
PARPs are a family of DNA repair enzymes that were
first identified in the 1960s (19). To date, 17 members of the PARP
family have been identified, with PARP-1, PARP-2 and PARP-3 being
the most extensively studied. PARP-1 is a protease characterized by
highly conserved domains, consisting of three main functional
regions: The DNA-binding domain (DBD), the auto-modification domain
(AD) and the catalytic (CAT) domain. The DBD is responsible for
recognizing and binding to damaged DNA fragments, particularly
single-strand breaks (SSBs). Once PARP-1 detects DNA damage, it
binds to the damaged site via its DBD and catalyzes the conversion
of NAD+ through its CAT, transferring ADP-ribose units from NAD+ to
PARP itself or other acceptor proteins, forming PAR chains. This
modification, known as poly(ADP-ribosyl)ation (PARylation), rapidly
initiates a series of downstream signals, coordinating the protein
network involved in DNA repair (20). In this process, the AD, primarily
associated with BRCA1 C-terminal (BRCT) regions, plays a key role
in facilitating protein-protein interactions and regulating PARP's
dissociation from DNA after it recognizes and binds to DNA-damage
sites. During DNA repair, PARP identifies and binds to SSBs through
its zinc finger domain, which subsequently activates its CAT
domain. The CAT domain is responsible for enzymatic activity,
including the cleavage of NAD+ and the formation of PAR chains
(Fig. 1). This process alters the
spatial conformation of PARP, weakening its binding affinity to DNA
and allowing it to dissociate from the site of the DNA break
(21,22).
PARP inhibitors have shown promise for treating
patients with cancer carrying BRCA1/2 mutations, particularly
demonstrating significant clinical efficacy in ovarian and breast
cancer (23,24). Based on the strategy of targeting
homologous recombination repair deficiency (HRD), PARP inhibitors
block DNA repair, leading to cancer cell death and effectively
suppressing tumor growth. However, as their clinical applications
have expanded, the issue of drug resistance has gradually emerged
(25). Overcoming resistance to
PARP inhibitors and extend their use to patients without BRCA
mutations have become key areas of current research.
Increasing evidence suggests that PARP-1 activation
can be triggered by a range of other stimuli, such as
protein-protein interactions or changes in redox states. These
findings underscore its role as a regulator involved in chromatin
remodeling and histone PARylation, or as a transcriptional
co-factor in gene expression regulation (26–28).
Furthermore, excessive activation of PARP can be
induced by various stressors, including oxidative stress, radiation
and chemotherapeutic agents, leading to an energy-depleting form of
cell death known as parthanatos, along with apoptosis-inducing
factor release and widespread DNA degradation (Fig. 1). Parthanatos has been applied in
neuroscience (29,30); however, it remains largely
unexplored in oncology. Therefore, there is considerable potential
for further investigation in this area.
In the field of DNA damage repair and breast cancer,
particularly TNBC, the key molecular players are most notably
p53 and BRCA1/2. Mutations in p53 can
influence the expression of key genes associated with cell
proliferation, including MYC proto-oncogene, bHLH transcription
factor, C-X-C motif chemokine ligand 1 and cyclin E2, through
direct or indirect pathways (31–33).
Although TP53 is the most frequently altered gene in breast cancer,
p53 remains in its wild-type form >2/3 of cases. However,
in basal-like breast cancer cases, most of which are triple
negative, the mutation frequency of p53 reaches 88%
(34). BRCA1 and
BRCA2 are tumor suppressor genes linked to breast cancer,
playing essential roles in homologous recombination repair (HRR).
Mutations in BRCA1 are frequently observed in patients with
breast cancer, resulting in defects in DNA repair, abnormal
centrosome replication, and impaired cell cycle and spindle
checkpoint functions. This has led to the hypothesis that
BRCA1 mutations contribute to tumorigenesis through genomic
instability (35). These mutations
not only endow cells with malignant potential but also allow the
use of targeted therapies.
The concept of synthetic lethality refers to the
phenomenon where two different metabolic pathways or gene functions
compensate for one another; inhibiting either pathway alone does
not result in cell death, but simultaneous inhibition of both leads
to cell death, which also applies to tumor cells (36). Mutations in the p53 and
BRCA1/2 genes in patients disable one of the primary DNA
repair pathways, and further inhibition of the DNA repair function
of PARP leads to pronounced cell death (37). This theory has been validated in
numerous experiments. For instance, Bryant et al (38) demonstrated that BRCA2
deficiency (either BRCA2+/− or BRCA2−/−)
resulted in HRR defects, and mice with BRCA2 mutations showed a
high sensitivity to PARP inhibitors, which induced replication fork
collapse and selectively killed the defective cells, while cells
with normal BRCA function remained largely unaffected. Farmer et
al (39) elucidated the
mechanism involving the interaction between BRCA2 and RAD51,
highlighting that the rapid recruitment of RAD51 to DNA damage
sites was fundamental to the HRR pathway. While PARP1 does not play
a direct role in HRR, its absence increases the need for an
efficient HRR pathway, possibly due to the conversion of unrepaired
SSBs into double-strand breaks. Normally, BRCA2 recruits RAD51 to
these sites for HRR, but without functional BRCA2, HRR does not
occur (39).
BRCA1-associated ring domain 1 (BARD1) is a protein
that forms a heterodimer with BRCA1 and plays a crucial role in DNA
damage repair. Regarding the mechanism by which BRCA molecules are
recruited to DNA damage sites, the role of BARD1 has been widely
recognized. BARD1, the primary partner of BRCA1, contains tandem
BRCT motifs. The heterodimer of BARD1 and BRCA1 is targeted
to sites of DNA damage mainly through their ring-like structures,
while the BRCT domains, which recognize phosphorylated serine
motifs, influence the translocation of the dimer at DNA sites
(40,41). Additionally, the interaction between
BRCT and PAR occurs through the binding of each ADP-ribose unit in
PAR, thereby ensuring the adequate progression of HRR (42,43).
Protein ubiquitination not only plays a key role in
protein degradation but also has crucial functions in DNA damage
repair and maintaining genomic stability. A study has shown that
checkpoint protein with FHA and RING domains (CHFR), through its
interaction with PARP1, regulates the crosstalk between PARylation
and ubiquitination. PARP1 recruits downstream DNA repair factors
via PAR modification, while CHFR may regulate the ubiquitination of
PAR and repair proteins, thereby modulating the duration and
intensity of PARP1 activity during the DNA damage repair process
(44). This coordinated regulation
is critical to ensure the accuracy and timeliness of DNA damage
repair, and it may also confer sensitivity to PARP inhibitors in
TNBC cells (45). Furthermore, a
diverse range of ubiquitin ligases have been identified, including
deltex E3 ubiquitin ligase (DTX), ring finger protein 8(RNF8) and
mediator of DNA damage checkpoint 1(MDC1) (46–49).
The combined use of drugs targeting ubiquitin ligases exacerbates
the accumulation of DNA damage, leading to unrepaired DNA breaks in
cancer cells, ultimately triggering cell death.
Epigenetic modifications refer to alterations in
gene expression that do not involve changes to the DNA sequence
itself. Common epigenetic modifications include DNA methylation and
histone modifications (such as acetylation, methylation and
phosphorylation), which regulate gene expression by altering
chromatin structure (50). One of
the central mechanisms of epigenetic regulation involves modifying
chromatin conformation to control gene accessibility. PARP-1 and
other members of the PARP family modulate chromatin plasticity
through the PARylation of histones (51,52).
For example, previous research has shown that histones such as H2A
at K13, H2B at K30, H3 at K27 and K37, and H4 at K16 can serve as
ADP-ribose acceptor sites (53).
The PARylation of H2 introduces a considerable negative charge
around chromatin proteins, leading to chromatin decondensation,
thereby facilitating the access of DNA repair factors to sites of
damage (54). Zhou et al
(55) treated trophoblast stem
cells with a DNA-modifying agent in PAR glycohydrolase-deficient
cells and found that excessive PARylation of histones, due to
inhibition of PAR chain hydrolysis, increased the sensitivity of
the cells to DNA damage-induced cell death.
Activation of PARP-1 can also alter chromatin
structure by recruiting chromatin remodeling complexes such as the
Switch/sucrose non-fermentable complex, which plays a critical role
in regulating chromatin structure and gene expression by altering
chromatin accessibility (56).
However, in tumor cells, this can more easily induce synthetic
lethality (57). It is worth noting
that PARylation can activate gene expression but can also regulate
gene silencing through chromatin compaction. For instance, SIRT1, a
deacetylase that promotes cell survival, competes with PARP-1 as
both rely on NAD+ as a substrate. Overactivation of PARP-1 can
reduce SIRT1 activity, thereby contributing to gene silencing
(58). Previous research has
suggested that certain viruses can exploit PARP-1 to modify viral
episomes or host genes, aiding long-term viral infections of DNA
viruses, which can lead to tumorigenesis (59).
Long non-coding RNAs (lncRNAs) are also involved in
the epigenetic regulation of gene expression by interacting with
epigenetic regulatory protein complexes, such as histone-modifying
enzymes and DNA methyltransferases, and are closely associated with
apoptosis, autophagy and transformation. Increasing evidence has
revealed their role in promoting tumor initiation, invasion and
malignancy (60–62). A PAR-related study has reported that
a human lncRNA-encoded micropeptide called PACMP not only inhibits
the ubiquitination of C-terminal binding protein-interacting
protein by blocking its interaction with KLHL15 but also
directly binds to DNA damage-induced PAR chains, thus enhancing
PARP1-dependent PARylation. Targeting lnc15.2/PACMP has shown
reversal of resistance to several chemotherapy drugs, including
PARP inhibitors, ataxia telangiectasia and Rad3-related inhibitors
and cyclin-dependent kinase-4/6 inhibitors (63). However, there is limited research on
targeted therapy of lncRNAs related to breast cancer in combination
with PARP inhibitors, thus offering a broad scope for exploration
in the future.
Both the inhibition and activation of PARP can
induce cell death, with the former primarily attributed to
apoptosis caused by DNA damage within a controllable range for the
cell, while the latter triggers energy depletion and widespread
degradation of genetic material by exhausting the precursors of
PAR, such as NAD+ and ATP (64).
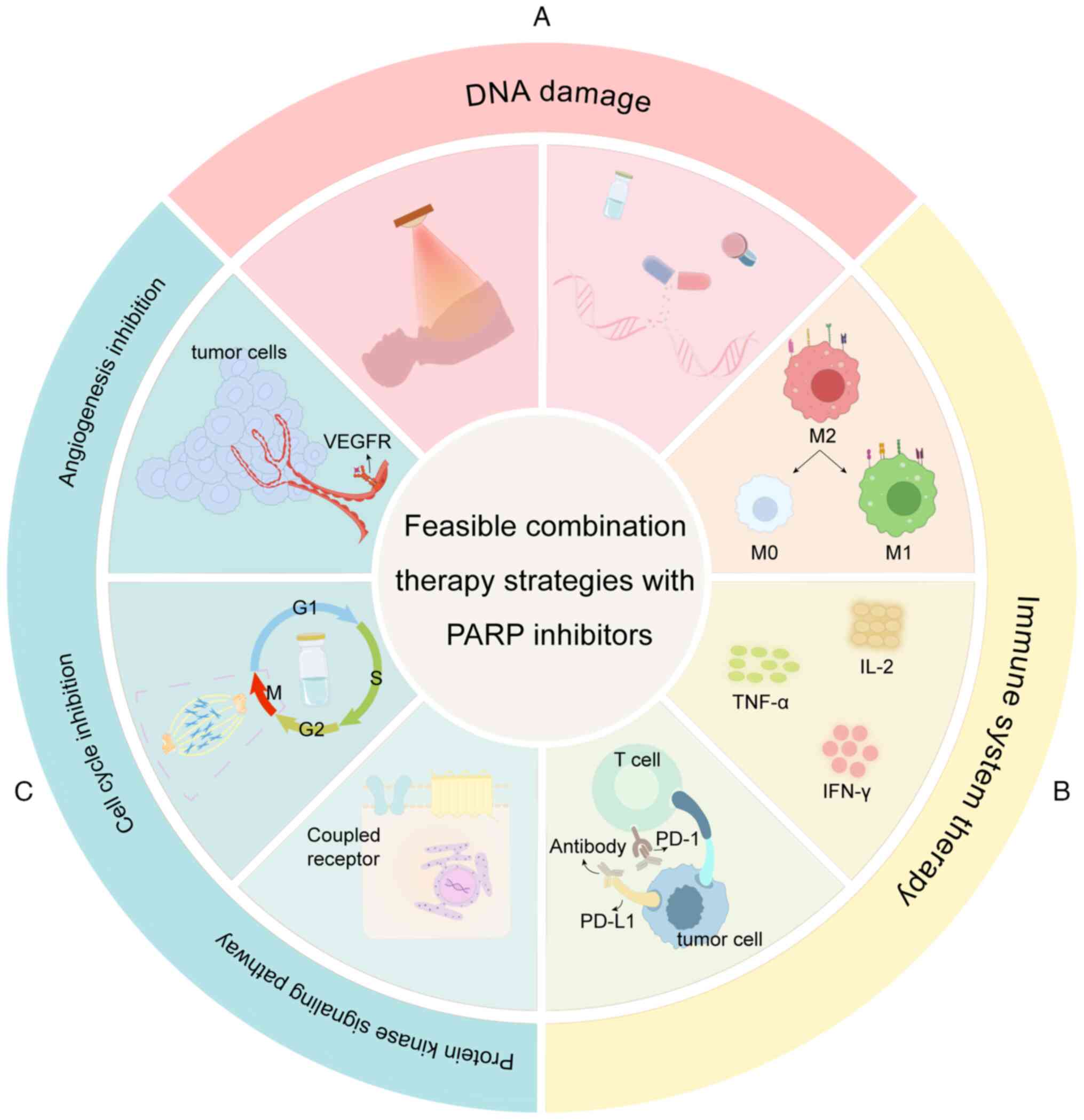
For TNBC, the mainstream treatment still focuses on
PARP inhibitors, mainly in combination with other clinical
chemotherapy agents. For example, combining PARP inhibitors with
DNA cross-linking agents such as carboplatin and cisplatin
exacerbates DNA damage, while combining them with microtubule
inhibitors such as paclitaxel increases replication stress in tumor
cells. Additionally, targeting signaling pathways such as PI3K/Akt
inhibition or anti-angiogenesis agents can further prevent tumor
growth and metastasis (Fig. 2)
(65).
There is a close association between immunotherapy
and PARP inhibitors. From a metabolomics perspective, Ding et
al (66) reported that
guanosine diphosphate-mannose (GDP-M), a metabolite, accumulates in
cells and reduces the interaction between BRCA2 and
ubiquitin-specific peptidase 21. This promotes the
ubiquitin-mediated degradation of BRCA2, thereby inhibiting HRR.
Furthermore, the authors found that the combination of GDP-M and
DNA-damaging agents activates STING-dependent antitumor immunity in
immunocompetent mouse models. Using whole-genome clustered
regularly interspaced short palindromic repeats screening, Zhong
et al (67) identified PARP1
as a restriction factor for herpes simplex virus 1 replication. By
combining their custom-designed oncolytic virus with a PARP
inhibitor, the authors were able to render TNBC highly sensitive to
immune checkpoint inhibitors. Seaweed polysaccharides have been
recommended as anticancer supplements and for boosting human
health. Chen et al (68)
observed that oligo-fucoidan combined with Olaparib more
effectively inhibited the formation of TNBC stem cell mammospheres,
while also suppressing the oncogenic IL-6/phosphorylated EGFR/PD-L1
pathway. This combination promoted the repolarization of M2
macrophages into M0-like and M1-like macrophages, thereby inducing
immune activation. In the same study, this effect has been
confirmed in mice, where oral administration of oligo-fucoidan
alongside Olaparib inhibited postoperative TNBC recurrence and
metastasis (68).
Before evaluating the clinical efficacy of PARP
inhibitors, it is essential to understand their mechanisms of
action. The catalytic function of PARP enzymes stems from the CAT
domain, which includes a binding site for NAD+. PARP inhibitors
competitively bind to the catalytic active site of PARP, thereby
blocking the entry of NAD+. Simultaneously, when PARP inhibitors
bind to the catalytic site, PARP is unable to complete PARylation
and dissociates from DNA damage sites. This trapping effect
prevents other DNA repair factors from reaching the damage site,
further exacerbating DNA damage. Therefore, the efficacy of a PARP
inhibitor largely depends on its binding strength to the CAT domain
and its ability to trap PARP at DNA damage sites (22,69).
Over the past two decades, numerous PARP inhibitors
have entered clinical trials. These drugs include Iniparib
(NCT01173497) (70) and Veliparib
(NCT02163694) (71) Olaparib
(NCT02734004) (72), Rucaparib
(NCT02042378) (73), Niraparib
(NCT05734911) (74), Talazoparib
(NCT04987931) (75), Fuzuloparib
(NCT05753826, Recruiting) and Pamiparib (NCT03333915) (76). Understanding their efficacy in these
trials provides valuable insights for developing effective clinical
treatment strategies for patients in the future.
Iniparib (chemical name: BSI-201) was initially
considered one of the promising candidate drugs in the category of
PARP inhibitors. In 2011, it entered phase II clinical trials and
demonstrated significant efficacy in treating TNBC (clinical trial
no. NCT00540358) (77). However,
subsequent phase III randomized controlled trials (clinical trial
no. NCT00938652) (78) delivered
disappointing results. In the trial, 261 patients treated with the
combination of gemcitabine + carboplatin + Iniparib were compared
with 258 patients receiving gemcitabine + carboplatin alone, and no
statistically significant difference was observed in overall
survival (OS) [hazard ratio (HR)=0.88; 95% confidence interval
(CI), 0.69–1.12; P=0.28] or progression-free survival (PFS)
(HR=0.79; 95% CI, 0.65–0.98; P=0.027). These results indicated that
the potential benefits of Iniparib required further evaluation
(78).
Olaparib (chemical name: AZD2281), developed by
AstraZeneca, is the first widely used PARP inhibitor in clinical
settings. Its earliest clinical trials began in 2005, focusing on
breast and ovarian cancer cases with BRCA1/2 mutations, and it also
showed efficacy in HER2-negative metastatic breast cancer (81). In 2009, the results of a phase I
clinical trial (clinical trial no. NCT00516373) demonstrated the
effectiveness of Olaparib in BRCA-mutated ovarian and breast cancer
(23). Subsequently, a phase II,
multicenter, open-label, non-randomized study (clinical trial no.
NCT00679783) was conducted to evaluate Olaparib treatment in women
with ovarian cancer and TNBC. The results showed that 18 out of 64
patients with ovarian cancer had a confirmed objective response,
while no responses were observed in any of the 26 patients with
TNBC (15). Similarly, a
multicenter phase II clinical study in 2015 reported the
therapeutic response in germline BRCA1/2-mutated tumors, with a
relatively low response rate of 12.9% (8/62; 95% CI, 5.7–23.9) in
breast cancer. Additionally, 47% of the patients experienced stable
disease for >8 weeks (95% CI, 34.0–59.9) (82).
Whether used as a monotherapy or in combination with
other drugs, the majority of the early clinical studies reported
that ≥50% of patients experienced grade ≥3 adverse events (82–86).
Previous studies have also evaluated the safety and tolerability of
combining Olaparib with radiotherapy. For instance, Loap et
al (87–89) conducted the long-term RADIOPARP
phase I trial in 24 patients with TNBC who exhibited residual
disease after neoadjuvant chemotherapy. These patients were treated
with four different doses of Olaparib (50, 100, 150 and 200 mg,
twice daily). Initially, no dose-limiting toxicities were observed,
and after 1 year of follow-up, no treatment-related grade ≥3
toxicities were noted. Additionally, the 3-year OS and event-free
survival rates were 83% (95% CI, 70–100%) and 65% (95% CI, 48–88%),
respectively.
In recent years, the majority of clinical trials
involving Olaparib have progressed to phase II or III (Table I). Based on the trial endpoints, it
can be observed that Olaparib has improved pathological complete
response and OS in patients, although the improvements are not
always statistically significant. Additionally, certain studies
have not only focused on efficacy but also on quality of life
(QoL). For example, a patient-reported outcomes study indicated
that patients receiving Olaparib experienced statistically
significantly higher levels of fatigue at 6 and 12 months during
the treatment and follow-up periods compared to those receiving
placebo. However, this did not reach the predetermined clinical
significance threshold of a 3-point difference. At 18 and 24
months, the Olaparib and placebo scores were similar (90). In conclusion, the importance of
these clinical studies lies in helping physicians make more precise
decisions about treatment options by considering both QoL and
efficacy, ensuring that therapeutic effectiveness is maximized
while minimizing the burden on the QoL of patients.
The earliest clinical study on Rucaparib (chemical
name: AG-014699) was reported in 2008, when Plummer et al
(91) evaluated the safety,
efficacy, pharmacokinetics and pharmacodynamics of AG-014699 in
combination with temozolomide in adult patients with advanced
malignant tumors. The study observed encouraging evidence of
activity. In 2016, clinical research focusing on breast
cancer-related treatments indicated that Rucaparib was well
tolerated at doses ≤480 mg/day and was a potent PARP inhibitor,
providing sustained inhibition for ≥24 h after a single dose,
although specific efficacy data for patients with breast cancer
were not provided (92).
Another phase II clinical trial in patients with
TNBC found that Rucaparib significantly reduced circulating tumor
DNA levels in homologous recombination repair-deficient cancer and
induced the expression of interferon response genes, suggesting
that Rucaparib may enhance antitumor effects not only through
inhibition of DNA repair pathways but also by triggering DNA
damage-induced immune responses (93). In another study, Kristeleit et
al (94) evaluated the
combination of Rucaparib with the immune checkpoint inhibitor
Atezolizumab in advanced gynecological cancer or TNBC. Patients
with higher PD-L1 levels and BRCA mutations prior to treatment were
more likely to experience adverse reactions such as
gastrointestinal symptoms, elevated levels of liver enzymes or
anemia. Markers of DNA damage repair (such as RAD51 foci)
and apoptosis markers were also reduced. This suggests that there
may be a synergistic effect between the immune response and PARP
inhibitors. Rucaparib may enhance tumor antigen production by
promoting DNA damage, thereby activating the immune system.
Niraparib (chemical name: MK-4827) is an oral,
potent and selective inhibitor of PARP-1 and PARP-2, capable of
inducing functional loss in BRCA and phosphatase and tensin
homolog. In 2013, Sandhu et al (95) clinically evaluated its safety and
tolerability, establishing 300 mg/day as the maximum tolerated
dose. The oral bioavailability of Niraparib in humans was measured
at 72.7% (96). In the
multi-country, phase III Niraparib in Patients with
Platinum-Sensitive Recurrent Ovarian Cancer trial (NCT01847274)
conducted in adult patients with platinum-sensitive recurrent
ovarian cancer (97), Niraparib
significantly prolonged the median PFS, regardless of the presence
of germline BRCA mutations or HRD, highlighting its distinct
advantage (98).
Although clinical research on the treatment of TNBC
with Niraparib remains limited, a combination therapy study
involving Niraparib (MK-4827) and fractionated radiotherapy in the
MDA-MB-231 TNBC cell line has demonstrated promising results.
Regardless of p53 status, MK-4827 reduced tumor PAR levels within 1
h of administration, with the effect lasting ≤24 h (99). These findings provide a foundation
for future research in this area.
Talazoparib (chemical name: BMN673), developed by
BioMarin Pharmaceutical Inc., is a second-generation PARP
inhibitor. Previous research has shown that BMN673 exhibits
selective cytotoxicity against tumor cells and induces DNA repair
biomarkers at notably lower concentrations compared to
first-generation PARP1/2 inhibitors such as Olaparib, Rucaparib and
Veliparib (PARP1 half-maximal inhibitory concentration, 0.57
nmol/l) (100). In a 2017 phase I
clinical trial (clinical trial no. NCT01286987), Talazoparib
demonstrated notable antitumor activity and safety as a
monotherapy, with 7 out of 14 patients with BRCA-mutated breast
cancer showing confirmed responses, resulting in a response rate of
50%. Grade 3–4 adverse events included anemia (17 out of 71
patients; 24%) and thrombocytopenia (13 out of 71 patients; 18%)
(12).
Talazoparib has been widely applied in clinical
treatments for breast cancer, primarily as a monotherapy to assess
efficacy (Table II). For instance,
a phase II single-arm, open-label study (clinical trial no.
NCT03499353) evaluated the efficacy of Talazoparib as a neoadjuvant
therapy in patients with TNBC, and achieved a pathological complete
response rate of 45.8% (95% CI, 29.4–63.2%) in the evaluable
population, demonstrating considerable activity (101).
It is important to note that, when combining
Talazoparib with other potent anticancer drugs, careful attention
must be paid to dosing and administration sequence. Previous
research has indicated a potential synergistic effect between
antibody-drug conjugates (ADCs) and PARP inhibitors. ADCs can
effectively increase DNA damage in tumor cells, and subsequent use
of PARP inhibitors can further block DNA repair, leading to cancer
cell apoptosis (102). However,
simultaneous administration in clinical settings has shown
significant toxicity, particularly myelosuppression. Sequential
administration, however, creates a time window that allows PARP
inhibitors to act more effectively within tumor cells without
increasing toxicity (13).
Veliparib, a PARP inhibitor, has been evaluated in
combination with other drugs in clinical trials for various cancer
types. In the treatment of TNBC, its combination with cisplatin or
carboplatin has shown good tolerability and response rates
(14). In addition to its use in
classic gynecological tumors such as breast and ovarian cancer,
Veliparib has also been investigated in the treatment of non-small
cell lung cancer, pancreatic cancer, prostate cancer and acute
myeloid leukemia (103–106). Although Veliparib has not yet
received broad clinical approval, its extensive research and
application in various cancer types provides promising insights for
future therapeutic strategies.
Recently developed in China, Fuzuloparib and
Pamiparib are novel PARP inhibitors. In a phase I clinical trial
(clinical trial no. NCT03945604) of Fuzuloparib for patients with
recurrent or metastatic TNBC, 29 patients received camrelizumab
(200 mg, every 2 weeks), Apatinib (375 or 500 mg, daily) and
Fuzuloparib (starting dose of 100 mg, twice daily). In total, 2
patients (6.9%; 95% CI, 0.9–22.8) had an objective response, while
the disease control rate was 62.1% (95% CI, 42.3–79.3). The median
PFS was 5.2 months (95% CI, 3.6–7.3), while the 12-month OS rate
was 64.2% (95% CI, 19.0–88.8), indicating controllable safety and
preliminary antitumor activity (107). Similarly, another phase I trial
(clinical trial no. NCT03075462) confirmed the acceptable safety of
Fuzuloparib combined with Apatinib in patients with advanced
ovarian cancer or TNBC (108).
In addition, Pamiparib in combination with the
immune checkpoint inhibitor Tislelizumab demonstrated efficacy in a
dose-escalation study for treating various advanced solid tumors
(109). In monotherapy for
patients with non-mucinous high-grade ovarian cancer and TNBC in
China, the most common adverse effects were fatigue, nausea and
hemoglobin reduction. Among all patients with TNBC who were
evaluable under the Response Evaluation Criteria in Solid Tumors
criteria (n=5), disease progression was reported, indicating the
need for further evidence to support its efficacy in breast cancer
(76).
Although PARP inhibitors have shown significant
efficacy in cancer cases with BRCA mutations, resistance remains
one of the most challenging clinical issues. Resistance can arise
through various mechanisms, and previous studies have uncovered
some key pathways. For example, certain resistant cancer cells can
overcome the effects of PARP inhibitors by reversing BRCA gene
mutations or restoring BRCA1/2 function (11,110).
Additionally, PARP inhibitors act not only by inhibiting PARP
activity but also by inducing PARP trapping, which increases DNA
damage. Resistant cancer cells can reduce PARP trapping by altering
the expression or function of PARP-related proteins, thereby
diminishing the efficacy of PARP inhibitors (111). Moreover, cancer cells may repair
DNA through alternative pathways, using different enzymes or repair
mechanisms. For instance, tankyrase, which has a catalytic activity
similar to PARP, regulates other proteins through ADP-ribosylation.
This suggests that tankyrase may serve as an alternative mechanism
during PARP inhibitor therapy, and several studies have summarized
its importance in cancer treatment (112–114). It is also noteworthy that
upregulation of the ABCB1 gene, which encodes the
P-glycoprotein efflux pump, may contribute to drug efflux and
resistance to PARP inhibitors (115).
As a result, numerous studies have evaluated the
efficacy of PARP inhibitors in tumors without BRCA mutations,
expanding their potential application beyond BRCA-mutated tumors.
HRD testing tools have been developed to assess genomic instability
(for example, copy number alterations, gene rearrangements and
microsatellite instability) in tumors. This enables the
identification of HRD-positive patients who are more likely to
benefit from PARP therapy (116).
Gasdermin-C (GSDMC) is a member of the gasdermin family protein,
which contains a gasdermin domain, and GSDMC/caspase-8 are key
molecules in pyroptosis. Wang et al (117) found that treatment with PARP
inhibitors triggered GSDMC/caspase-8-mediated pyroptosis in cancer
cells. Additionally, GSDMC-mediated pyroptosis increased the
population of memory CD8+ T cells in the lymph nodes,
spleen and tumors, thereby promoting the infiltration of cytotoxic
CD8+ T cells into the tumor microenvironment. Notably,
this process was independent of BRCA mutations.
Furthermore, the efficacy of PARP inhibitors in
non-BRCA-mutated tumors has being explored. For instance, a phase
II trial evaluating Olaparib in patients with advanced TNBC or
high-grade serous and/or undifferentiated ovarian cancer without
BRCA1 or BRCA2 mutations found that, among 63 patients with ovarian
cancer, the objective response rate was 41% in the mutation group
(n=17) and 24% in the non-mutation group (n=46). No responses were
reported in any of the 26 patients with breast cancer (15). Another similar study also explored
the use of PARP inhibitors in patients with BRCA wild-type TNBC
(118).
Replication fork stability can endow BRCA-deficient
cells with resistance. A defect in PTIP, a paired box transcription
activation domain interaction protein, does not restore homologous
recombination but inhibits the recruitment of the MRE11 nuclease to
stalled replication forks. This, in turn, protects nascent DNA
strands from extensive degradation (119). Tan and Xu (120) found that ubiquitin-fold modifier
modification (UFMylation) played a critical role in replication
fork dynamics in BRCA1-deficient cells, where PTIP UFMylation
promoted the resection of nascent DNA, conferring resistance to
BRCA1-deficient cells. Furthermore, Tian et al (121) demonstrated that, during
replication stress, UFL1, a UFM1-specific E3 ligase,
localized to stalled forks and catalyzed PTIP UFMylation, thus
providing a mechanism for UFMylation in PTIP regulation. These
studies offer potential avenues for overcoming resistance to PARP
inhibitors in patients with BRCA-deficient TNBC.
In conclusion, identifying more precise molecular
biomarkers, optimizing drug sequencing and dosing in resistant
patients, and improving the use of PARP-related cell death
mechanisms (such as energy depletion) are potential strategies for
the future treatment of breast cancer, particularly TNBC.
The present review focuses on the role of PARP
inhibitors in the treatment of TNBC, particularly highlighting
their efficacy in BRCA mutation-associated tumors. By analyzing key
mechanisms of DNA damage repair, it was observed that, in multiple
clinical trials, PARP inhibitors have demonstrated not only strong
efficacy in BRCA-mutated TNBC but also potential in
non-BRCA-mutated tumors. Despite the notable efficacy of PARP
inhibitors in patients with BRCA-mutated TNBC, the emergence of
resistance remains a major challenge in clinical applications, and
identifying additional biomarkers to combat this resistance
requires further exploration.
Future research should focus on overcoming
resistance to PARP inhibitors through combination therapy
strategies while refining patient selection by integrating genomic
data and biomarker analysis. This should help ensure optimal
efficacy with the most appropriate dosing for the suitable
patients. Additionally, further investigation of combination
treatment regimens and sequencing should provide improved survival
benefits to patients.
The authors would like to thank Ms. Sihan Lin (First
Clinical College, Wuhan University, China) for providing important
contributions to Figure 2 during
the revision of the manuscript.
Funding: No funding was received.
Not applicable.
YH and LW contributed to the study conception and
design. YH proposed the concept of the study and drafted for the
article, and performed literature search and data analysis. LW
determined the writing framework of the article, put forward
valuable revision suggestions for the first draft and also provided
assistance in the detailed revisions of the article. Both authors
read and approved the final version of the manuscript. Data
authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Giaquinto AN, Sung H, Newman LA, Freedman
RA, Smith RA, Star J, Jemal A and Siegel RL: Breast cancer
statistics 2024. CA Cancer J Clin. 74:477–495. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wu SY, Wang H, Shao ZM and Jiang YZ:
Triple-negative breast cancer: New treatment strategies in the era
of precision medicine. Sci China Life Sci. 64:372–388. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yuan P, Ma N and Xu B: Poly (adenosine
diphosphate-ribose) polymerase inhibitors in the treatment of
triple-negative breast cancer with homologous repair deficiency.
Med Res Rev. 44:2774–2792. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Konstantinopoulos PA, Ceccaldi R, Shapiro
GI and D'Andrea AD: Homologous recombination deficiency: Exploiting
the fundamental vulnerability of ovarian cancer. Cancer Discov.
5:1137–1154. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
de Bono J, Mateo J, Fizazi K, Saad F,
Shore N, Sandhu S, Chi KN, Sartor O, Agarwal N, Olmos D, et al:
Olaparib for metastatic castration-resistant prostate cancer. N
Engl J Med. 382:2091–2102. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mateo J, Porta N, Bianchini D, McGovern U,
Elliott T, Jones R, Syndikus I, Ralph C, Jain S, Varughese M, et
al: Olaparib in patients with metastatic castration-resistant
prostate cancer with DNA repair gene aberrations (TOPARP-B): A
multicentre, open-label, randomised, phase 2 trial. Lancet Oncol.
21:162–174. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Markham A: Pamiparib: First Approval.
Drugs. 81:1343–1348. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee A: Fuzuloparib: First Approval. Drugs.
81:1221–1226. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xiao F, Wang Z, Qiao L, Zhang X, Wu N,
Wang J and Yu X: Application of PARP inhibitors combined with
immune checkpoint inhibitors in ovarian cancer. J Transl Med.
22:7782024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Q, Qian W, Zhang Y, Hu L, Chen S and
Xia Y: A new wave of innovations within the DNA damage response.
Signal Transduct Target Ther. 8:3382023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Afghahi A, Timms KM, Vinayak S, Jensen KC,
Kurian AW, Carlson RW, Chang PJ, Schackmann E, Hartman AR, Ford JM
and Telli ML: Tumor BRCA1 reversion mutation arising during
neoadjuvant platinum-based chemotherapy in triple-negative breast
cancer is associated with therapy resistance. Clin Cancer Res.
23:3365–3370. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
de Bono J, Ramanathan RK, Mina L, Chugh R,
Glaspy J, Rafii S, Kaye S, Sachdev J, Heymach J, Smith DC, et al:
Phase I, dose-escalation, two-part trial of the PARP inhibitor
talazoparib in patients with advanced germline BRCA1/2 mutations
and selected sporadic cancers. Cancer Discov. 7:620–629. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bardia A, Sun S, Thimmiah N, Coates JT, Wu
B, Abelman RO, Spring L, Moy B, Ryan P, Melkonyan MN, et al:
Antibody-drug conjugate sacituzumab govitecan enables a sequential
TOP1/PARP inhibitor therapy strategy in patients with breast
cancer. Clin Cancer Res. 30:2917–2924. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rodler ET, Kurland BF, Griffin M, Gralow
JR, Porter P, Yeh RF, Gadi VK, Guenthoer J, Beumer JH, Korde L, et
al: Phase I study of veliparib (ABT-888) combined with cisplatin
and vinorelbine in advanced triple-negative breast cancer and/or
BRCA mutation-associated breast cancer. Clin Cancer Res.
22:2855–2864. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gelmon KA, Tischkowitz M, Mackay H,
Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M,
Gilks B, et al: Olaparib in patients with recurrent high-grade
serous or poorly differentiated ovarian carcinoma or
triple-negative breast cancer: A phase 2, multicentre, open-label,
non-randomised study. Lancet Oncol. 12:852–861. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhu S, Wu Y, Song B, Yi M, Yan Y, Mei Q
and Wu K: Recent advances in targeted strategies for
triple-negative breast cancer. J Hematol Oncol. 16:1002023.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Subhan MA, Parveen F, Shah H, Yalamarty
SSK, Ataide JA and Torchilin VP: Recent advances with precision
medicine treatment for breast cancer including triple-negative
sub-type. Cancers (Basel). 15:22042023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Won KA and Spruck C: Triple-negative
breast cancer therapy: Current and future perspectives (Review).
Int J Oncol. 57:1245–1261. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kraus WL: PARPs and ADP-ribosylation: 50
Years … and counting. Mol Cell. 58:902–910. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schreiber V, Dantzer F, Ame JC and de
Murcia G: Poly(ADP-ribose): Novel functions for an old molecule.
Nat Rev Mol Cell Biol. 7:517–528. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Duma L and Ahel I: The function and
regulation of ADP-ribosylation in the DNA damage response. Biochem
Soc Trans. 51:995–1008. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rouleau-Turcotte É and Pascal JM:
ADP-ribose contributions to genome stability and PARP enzyme
trapping on sites of DNA damage; paradigm shifts for a
coming-of-age modification. J Biol Chem. 299:1053972023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fong PC, Boss DS, Yap TA, Tutt A, Wu P,
Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, et
al: Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA
mutation carriers. N Engl J Med. 361:123–134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tutt A, Robson M, Garber JE, Domchek SM,
Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler
RK, et al: Oral poly(ADP-ribose) polymerase inhibitor olaparib in
patients with BRCA1 or BRCA2 mutations and advanced breast cancer:
A proof-of-concept trial. Lancet. 376:235–244. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim D and Nam HJ; PARP inhibitors, :
Clinical limitations and recent attempts to overcome them. Int J
Mol Sci. 23:84122022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim MY, Mauro S, Gévry N, Lis JT and Kraus
WL: NAD+-dependent modulation of chromatin structure and
transcription by nucleosome binding properties of PARP-1. Cell.
119:803–814. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kun E, Kirsten E, Mendeleyev J and Ordahl
CP: Regulation of the enzymatic catalysis of poly(ADP-ribose)
polymerase by dsDNA, polyamines, Mg2+, Ca2+, histones H1 and H3,
and ATP. Biochemistry. 43:210–216. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kraus WL: Transcriptional control by
PARP-1: Chromatin modulation, enhancer-binding, coregulation, and
insulation. Curr Opin Cell Biol. 20:294–302. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Baek SH, Bae ON, Kim EK and Yu SW:
Induction of mitochondrial dysfunction by poly(ADP-ribose) polymer:
Implication for neuronal cell death. Mol Cells. 36:258–266. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tuo QZ, Zhang ST and Lei P: Mechanisms of
neuronal cell death in ischemic stroke and their therapeutic
implications. Med Res Rev. 42:259–305. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang Z, Li Y, Yang J, Sun Y, He Y, Wang Y,
Liang Y, Chen X, Chen T, Han D, et al: CircCFL1 promotes TNBC
stemness and immunoescape via deacetylation-mediated c-Myc
deubiquitylation to facilitate mutant TP53 transcription. Adv Sci
(Weinh). 11:e24046282024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
De Souza C, Madden JA, Minn D, Kumar VE,
Montoya DJ, Nambiar R, Zhu Z, Xiao WW, Tahmassebi N, Kathi H, et
al: The P72R polymorphism in R248Q/W p53 mutants modifies the
mutant effect on epithelial to mesenchymal transition phenotype and
cell invasion via CXCL1 expression. Int J Mol Sci. 21:80252020.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Macdonald FH, Yao D, Quinn JA and
Greenhalgh DA: PTEN ablation in Ras(Ha)/Fos skin carcinogenesis
invokes p53-dependent p21 to delay conversion while p53-independent
p21 limits progression via cyclin D1/E2 inhibition. Oncogene.
33:4132–4143. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Marvalim C, Datta A and Lee SC: Role of
p53 in breast cancer progression: An insight into p53 targeted
therapy. Theranostics. 13:1421–1442. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Deng CX: Tumorigenesis as a consequence of
genetic instability in Brca1 mutant mice. Mutat Res. 477:183–189.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Du Y, Luo L, Xu X, Yang X, Yang X, Xiong
S, Yu J, Liang T and Guo L: Unleashing the power of synthetic
lethality: Augmenting treatment efficacy through synergistic
integration with chemotherapy drugs. Pharmaceutics. 15:24332023.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Anders CK, Winer EP, Ford JM, Dent R,
Silver DP, Sledge GW and Carey LA: Poly(ADP-Ribose) polymerase
inhibition: ‘Targeted’ therapy for triple-negative breast cancer.
Clin Cancer Res. 16:4702–4710. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bryant HE, Schultz N, Thomas HD, Parker
KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ and Helleday T:
Specific killing of BRCA2-deficient tumours with inhibitors of
poly(ADP-ribose) polymerase. Nature. 434:913–917. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Farmer H, McCabe N, Lord CJ, Tutt AN,
Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I,
Knights C, et al: Targeting the DNA repair defect in BRCA mutant
cells as a therapeutic strategy. Nature. 434:917–921. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tarsounas M and Sung P: The
antitumorigenic roles of BRCA1-BARD1 in DNA repair and replication.
Nat Rev Mol Cell Biol. 21:284–299. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Peña-Guerrero J, Fernández-Rubio C,
García-Sosa AT and Nguewa PA: BRCT domains: Structure, functions,
and implications in disease-new therapeutic targets for innovative
drug discovery against infections. Pharmaceutics. 15:18392023.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li M and Yu X: Function of BRCA1 in the
DNA damage response is mediated by ADP-ribosylation. Cancer Cell.
23:693–704. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Becker JR, Clifford G, Bonnet C, Groth A,
Wilson MD and Chapman JR: BARD1 reads H2A lysine 15 ubiquitination
to direct homologous recombination. Nature. 596:433–437. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu C, Wu J, Paudyal SC, You Z and Yu X:
CHFR is important for the first wave of ubiquitination at DNA
damage sites. Nucleic Acids Res. 41:1698–1710. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wu W, Zhao J, Xiao J, Wu W, Xie L, Xie X,
Yang C, Yin D and Hu K: CHFR-mediated degradation of RNF126 confers
sensitivity to PARP inhibitors in triple-negative breast cancer
cells. Biochem Biophys Res Commun. 573:62–68. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Djerir B, Marois I, Dubois JC, Findlay S,
Morin T, Senoussi I, Cappadocia L, Orthwein A and Maréchal A: An E3
ubiquitin ligase localization screen uncovers DTX2 as a novel
ADP-ribosylation-dependent regulator of DNA double-strand break
repair. J Biol Chem. 300:1075452024. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yan Q, Xu R, Zhu L, Cheng X, Wang Z, Manis
J and Shipp MA: BAL1 and its partner E3 ligase, BBAP, link
Poly(ADP-ribose) activation, ubiquitylation, and double-strand DNA
repair independent of ATM, MDC1, and RNF8. Mol Cell Biol.
33:845–857. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kolas NK, Chapman JR, Nakada S, Ylanko J,
Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson
TM, et al: Orchestration of the DNA-damage response by the RNF8
ubiquitin ligase. Science. 318:1637–1640. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Marteijn JA, Bekker-Jensen S, Mailand N,
Lans H, Schwertman P, Gourdin AM, Dantuma NP, Lukas J and Vermeulen
W: Nucleotide excision repair-induced H2A ubiquitination is
dependent on MDC1 and RNF8 and reveals a universal DNA damage
response. J Cell Biol. 186:835–847. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tian H, Luan P, Liu Y and Li G:
Tet-mediated DNA methylation dynamics affect chromosome
organization. Nucleic Acids Res. 52:3654–3666. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Messner S and Hottiger MO: Histone
ADP-ribosylation in DNA repair, replication and transcription.
Trends Cell Biol. 21:534–542. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hottiger MO: ADP-ribosylation of histones
by ARTD1: An additional module of the histone code? FEBS Lett.
585:1595–1599. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Messner S, Altmeyer M, Zhao H, Pozivil A,
Roschitzki B, Gehrig P, Rutishauser D, Huang D, Caflisch A and
Hottiger MO: PARP1 ADP-ribosylates lysine residues of the core
histone tails. Nucleic Acids Res. 38:6350–6362. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Rouleau M, Patel A, Hendzel MJ, Kaufmann
SH and Poirier GG: PARP inhibition: PARP1 and beyond. Nat Rev
Cancer. 10:293–301. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhou Y, Feng X and Koh DW: Enhanced DNA
accessibility and increased DNA damage induced by the absence of
poly(ADP-ribose) hydrolysis. Biochemistry. 49:7360–7366. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Martin BJE, Ablondi EF, Goglia C, Mimoso
CA, Espinel-Cabrera PR and Adelman K: Global identification of
SWI/SNF targets reveals compensation by EP400. Cell.
186:5290–5307.e26. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wanior M, Krämer A, Knapp S and Joerger
AC: Exploiting vulnerabilities of SWI/SNF chromatin remodelling
complexes for cancer therapy. Oncogene. 40:3637–3654. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kolthur-Seetharam U, Dantzer F, McBurney
MW, de Murcia G and Sassone-Corsi P: Control of AIF-mediated cell
death by the functional interplay of SIRT1 and PARP-1 in response
to DNA damage. Cell Cycle. 5:873–877. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Sobotka AA and Tempera I: PARP1 as an
epigenetic modulator: Implications for the regulation of host-viral
dynamics. Pathogens. 13:1312024. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ahmad M, Weiswald LB, Poulain L, Denoyelle
C and Meryet-Figuiere M: Involvement of lncRNAs in cancer cells
migration, invasion and metastasis: Cytoskeleton and ECM crosstalk.
J Exp Clin Cancer Res. 42:1732023. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Bartonicek N, Maag JL and Dinger ME: Long
noncoding RNAs in cancer: Mechanisms of action and technological
advancements. Mol Cancer. 15:432016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Jiang J, Lu Y, Zhang F, Huang J, Ren XL
and Zhang R: The emerging roles of long noncoding RNAs as hallmarks
of lung cancer. Front Oncol. 11:7615822021. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhang C, Zhou B, Gu F, Liu H, Wu H, Yao F,
Zheng H, Fu H, Chong W, Cai S, et al: Micropeptide PACMP inhibition
elicits synthetic lethal effects by decreasing CtIP and
poly(ADP-ribosyl)ation. Mol Cell. 82:1297–1312.e8. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Siegel C and McCullough LD: NAD+ depletion
or PAR polymer formation: Which plays the role of executioner in
ischaemic cell death? Acta Physiol (Oxf). 203:225–234. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
De P, Sun Y, Carlson JH, Friedman LS,
Leyland-Jones BR and Dey N: Doubling down on the PI3K-AKT-mTOR
pathway enhances the antitumor efficacy of PARP inhibitor in triple
negative breast cancer model beyond BRCA-ness. Neoplasia. 16:43–72.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ding JH, Xiao Y, Yang F, Song XQ, Xu Y,
Ding XH, Ding R, Shao ZM, Di GH and Jiang YZ: Guanosine
diphosphate-mannose suppresses homologous recombination repair and
potentiates antitumor immunity in triple-negative breast cancer.
Sci Transl Med. 16:eadg77402024. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Zhong Y, Le H, Zhang X, Dai Y, Guo F, Ran
X, Hu G, Xie Q, Wang D and Cai Y: Identification of restrictive
molecules involved in oncolytic virotherapy using genome-wide
CRISPR screening. J Hematol Oncol. 17:362024. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Chen LM, Yang PP, Al Haq AT, Hwang PA, Lai
YC, Weng YS, Chen MA and Hsu HL: Oligo-Fucoidan supplementation
enhances the effect of Olaparib on preventing metastasis and
recurrence of triple-negative breast cancer in mice. J Biomed Sci.
29:702022. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Rudolph J, Jung K and Luger K: Inhibitors
of PARP: Number crunching and structure gazing. Proc Natl Acad Sci
USA. 119:e21219791192022. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Anders C, Deal AM, Abramson V, Liu MC,
Storniolo AM, Carpenter JT, Puhalla S, Nanda R, Melhem-Bertrandt A,
Lin NU, et al: TBCRC 018: phase II study of iniparib in combination
with irinotecan to treat progressive triple negative breast cancer
brain metastases. Breast Cancer Res Treat. 146:557–566. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Diéras V, Han HS, Kaufman B, Wildiers H,
Friedlander M, Ayoub JP, Puhalla SL, Bondarenko I, Campone M,
Jakobsen EH, et al: Veliparib with carboplatin and paclitaxel in
BRCA-mutated advanced breast cancer (BROCADE3): A randomised,
double-blind, placebo-controlled, phase 3 trial. Lancet Oncol.
21:1269–1282. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Domchek SM, Postel-Vinay S, Im SA, Park
YH, Delord JP, Italiano A, Alexandre J, You B, Bastian S, Krebs MG,
et al: Olaparib and durvalumab in patients with germline
BRCA-mutated metastatic breast cancer (MEDIOLA): An open-label,
multicentre, phase 1/2, basket study. Lancet Oncol. 21:1155–1164.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Shroff RT, Hendifar A, McWilliams RR, Geva
R, Epelbaum R, Rolfe L, Goble S, Lin KK, Biankin AV, Giordano H, et
al: Rucaparib monotherapy in patients with pancreatic cancer and a
known deleterious BRCA mutation. JCO Precis Oncol.
2018.PO.17.00316. 2018. View Article : Google Scholar
|
|
74
|
Zhao M, Qiu S, Wu X, Miao P, Jiang Z, Zhu
T, Xu X, Zhu Y, Zhang B, Yuan D, et al: Efficacy and safety of
niraparib as first-line maintenance treatment for patients with
advanced ovarian cancer: Real-world data from a multicenter study
in China. Target Oncol. 18:869–883. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Savill KMZ, Ivanova J, Asgarisabet P,
Falkenstein A, Balanean A, Niyazov A, Ryan JC, Kish J, Gajra A and
Mahtani RL: Characteristics, treatment, and outcomes of real-world
talazoparib-treated patients with germline BRCA-mutated advanced
HER2-negative breast cancer. Oncologist. 28:414–424. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Xu B, Yin Y, Dong M, Song Y, Li W, Huang
X, Wang T, He J, Mu X, Li L, et al: Pamiparib dose escalation in
Chinese patients with non-mucinous high-grade ovarian cancer or
advanced triple-negative breast cancer. Cancer Med. 10:109–118.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
O'Shaughnessy J, Osborne C, Pippen JE,
Yoffe M, Patt D, Rocha C, Koo IC, Sherman BM and Bradley C:
Iniparib plus chemotherapy in metastatic triple-negative breast
cancer. N Engl J Med. 364:205–214. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
O'Shaughnessy J, Schwartzberg L, Danso MA,
Miller KD, Rugo HS, Neubauer M, Robert N, Hellerstedt B, Saleh M,
Richards P, et al: Phase III study of iniparib plus gemcitabine and
carboplatin versus gemcitabine and carboplatin in patients with
metastatic triple-negative breast cancer. J Clin Oncol.
32:3840–3847. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Mateo J, Ong M, Tan DS, Gonzalez MA and de
Bono JS: Appraising iniparib, the PARP inhibitor that never
was-what must we learn? Nat Rev Clin Oncol. 10:688–696. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Liu X, Shi Y, Maag DX, Palma JP, Patterson
MJ, Ellis PA, Surber BW, Ready DB, Soni NB, Ladror US, et al:
Iniparib nonselectively modifies cysteine-containing proteins in
tumor cells and is not a bona fide PARP inhibitor. Clin Cancer Res.
18:510–523. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Wu HL, Luo ZY, He ZL, Gong Y, Mo M, Ming
WK and Liu GY: All HER2-negative breast cancer patients need gBRCA
testing: Cost-effectiveness and clinical benefits. Br J Cancer.
128:638–646. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Kaufman B, Shapira-Frommer R, Schmutzler
RK, Audeh MW, Friedlander M, Balmaña J, Mitchell G, Fried G,
Stemmer SM, Hubert A, et al: Olaparib monotherapy in patients with
advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol.
33:244–250. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Dent RA, Lindeman GJ, Clemons M, Wildiers
H, Chan A, McCarthy NJ, Singer CF, Lowe ES, Watkins CL and
Carmichael J: Phase I trial of the oral PARP inhibitor olaparib in
combination with paclitaxel for first- or second-line treatment of
patients with metastatic triple-negative breast cancer. Breast
Cancer Res. 15:R882013. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Liu JF, Tolaney SM, Birrer M, Fleming GF,
Buss MK, Dahlberg SE, Lee H, Whalen C, Tyburski K, Winer E, et al:
A Phase 1 trial of the poly(ADP-ribose) polymerase inhibitor
olaparib (AZD2281) in combination with the anti-angiogenic
cediranib (AZD2171) in recurrent epithelial ovarian or
triple-negative breast cancer. Eur J Cancer. 49:2972–2978. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Lee JM, Hays JL, Annunziata CM, Noonan AM,
Minasian L, Zujewski JA, Yu M, Gordon N, Ji J, Sissung TM, et al:
Phase I/Ib study of olaparib and carboplatin in BRCA1 or BRCA2
mutation-associated breast or ovarian cancer with biomarker
analyses. J Natl Cancer Inst. 106:dju0892014. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Del Conte G, Sessa C, von Moos R, Viganò
L, Digena T, Locatelli A, Gallerani E, Fasolo A, Tessari A,
Cathomas R and Gianni L: Phase I study of olaparib in combination
with liposomal doxorubicin in patients with advanced solid tumours.
Br J Cancer. 111:651–659. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Loap P, Loirat D, Berger F, Ricci F,
Vincent-Salomon A, Ezzili C, Mosseri V, Fourquet A, Ezzalfani M and
Kirova Y: Combination of olaparib and radiation therapy for triple
negative breast cancer: Preliminary results of the RADIOPARP phase
1 trial. Int J Radiat Oncol Biol Phys. 109:436–440. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Loap P, Loirat D, Berger F, Cao K, Ricci
F, Jochem A, Raizonville L, Mosseri V, Fourquet A and Kirova Y:
Combination of Olaparib with radiotherapy for triple-negative
breast cancers: One-year toxicity report of the RADIOPARP Phase I
trial. Int J Cancer. 149:1828–1832. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Loap P, Loirat D, Berger F, Rodrigues M,
Bazire L, Pierga JY, Vincent-Salomon A, Laki F, Boudali L,
Raizonville L, et al: Concurrent Olaparib and radiotherapy in
patients with triple-negative breast cancer: The phase 1 Olaparib
and radiation therapy for triple-negative breast cancer trial. JAMA
Oncol. 8:1802–1808. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Ganz PA, Bandos H, Španić T, Friedman S,
Müller V, Kuemmel S, Delaloge S, Brain E, Toi M, Yamauchi H, et al:
Patient-reported outcomes in OlympiA: A phase III, randomized,
placebo-controlled trial of adjuvant Olaparib in gBRCA1/2 mutations
and high-risk human epidermal growth factor receptor 2-negative
early breast cancer. J Clin Oncol. 42:1288–1300. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Plummer R, Jones C, Middleton M, Wilson R,
Evans J, Olsen A, Curtin N, Boddy A, McHugh P, Newell D, et al:
Phase I study of the poly(ADP-ribose) polymerase inhibitor,
AG014699, in combination with temozolomide in patients with
advanced solid tumors. Clin Cancer Res. 14:7917–7923. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Drew Y, Ledermann J, Hall G, Rea D,
Glasspool R, Highley M, Jayson G, Sludden J, Murray J, Jamieson D,
et al: Phase 2 multicentre trial investigating intermittent and
continuous dosing schedules of the poly(ADP-ribose) polymerase
inhibitor rucaparib in germline BRCA mutation carriers with
advanced ovarian and breast cancer. Br J Cancer. 114:723–730. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Chopra N, Tovey H, Pearson A, Cutts R,
Toms C, Proszek P, Hubank M, Dowsett M, Dodson A, Daley F, et al:
Homologous recombination DNA repair deficiency and PARP inhibition
activity in primary triple negative breast cancer. Nat Commun.
11:26622020. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Kristeleit R, Leary A, Oaknin A, Redondo
A, George A, Chui S, Seiller A, Liste-Hermoso M, Willis J, Shemesh
CS, et al: PARP inhibition with rucaparib alone followed by
combination with atezolizumab: Phase Ib COUPLET clinical study in
advanced gynaecological and triple-negative breast cancers. Br J
Cancer. 131:820–831. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Sandhu SK, Schelman WR, Wilding G, Moreno
V, Baird RD, Miranda S, Hylands L, Riisnaes R, Forster M, Omlin A,
et al: The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827)
in BRCA mutation carriers and patients with sporadic cancer: A
phase 1 dose-escalation trial. Lancet Oncol. 14:882–892. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
van Andel L, Rosing H, Zhang Z, Hughes L,
Kansra V, Sanghvi M, Tibben MM, Gebretensae A, Schellens JHM and
Beijnen JH: Determination of the absolute oral bioavailability of
niraparib by simultaneous administration of a (14)C-microtracer and
therapeutic dose in cancer patients. Cancer Chemother Pharmacol.
81:39–46. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Mirza MR, Monk BJ, Herrstedt J, Oza AM,
Mahner S, Redondo A, Fabbro M, Ledermann JA, Lorusso D, Vergote I,
et al: Niraparib maintenance therapy in platinum-sensitive,
recurrent ovarian cancer. N Engl J Med. 375:2154–2164. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Scott LJ: Niraparib: First global
approval. Drugs. 77:1029–1034. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Wang L, Mason KA, Ang KK, Buchholz T,
Valdecanas D, Mathur A, Buser-Doepner C, Toniatti C and Milas L:
MK-4827, a PARP-1/-2 inhibitor, strongly enhances response of human
lung and breast cancer xenografts to radiation. Invest New Drugs.
30:2113–2120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Shen Y, Rehman FL, Feng Y, Boshuizen J,
Bajrami I, Elliott R, Wang B, Lord CJ, Post LE and Ashworth A: BMN
673, a novel and highly potent PARP1/2 inhibitor for the treatment
of human cancers with DNA repair deficiency. Clin Cancer Res.
19:5003–5015. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Litton JK, Beck JT, Jones JM, Andersen J,
Blum JL, Mina LA, Brig R, Danso M, Yuan Y, Abbattista A, et al:
Neoadjuvant talazoparib in patients with germline BRCA1/2
mutation-positive, early-stage triple-negative breast cancer:
Results of a phase II study. Oncologist. 28:845–855. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Li Y, Li L, Fu H, Yao Q, Wang L and Lou L:
Combined inhibition of PARP and ATR synergistically potentiates the
antitumor activity of HER2-targeting antibody-drug conjugate in
HER2-positive cancers. Am J Cancer Res. 13:161–175. 2023.PubMed/NCBI
|
|
103
|
Kozono DE, Stinchcombe TE, Salama JK,
Bogart J, Petty WJ, Guarino MJ, Bazhenova L, Larner JM, Weiss J,
DiPetrillo TA, et al: Veliparib in combination with
carboplatin/paclitaxel-based chemoradiotherapy in patients with
stage III non-small cell lung cancer. Lung Cancer. 159:56–65. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
O'Reilly EM, Lee JW, Zalupski M, Capanu M,
Park J, Golan T, Tahover E, Lowery MA, Chou JF, Sahai V, et al:
Randomized, multicenter, phase II trial of gemcitabine and
cisplatin with or without veliparib in patients with pancreas
adenocarcinoma and a germline BRCA/PALB2 mutation. J Clin Oncol.
38:1378–1388. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Hussain M, Carducci MA, Slovin S, Cetnar
J, Qian J, McKeegan EM, Refici-Buhr M, Chyla B, Shepherd SP,
Giranda VL and Alumkal JJ: Targeting DNA repair with combination
veliparib (ABT-888) and temozolomide in patients with metastatic
castration-resistant prostate cancer. Invest New Drugs. 32:904–912.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Gojo I, Beumer JH, Pratz KW, McDevitt MA,
Baer MR, Blackford AL, Smith BD, Gore SD, Carraway HE, Showel MM,
et al: A phase 1 study of the PARP inhibitor veliparib in
combination with temozolomide in acute myeloid leukemia. Clin
Cancer Res. 23:697–706. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Zhang Q, Shao B, Tong Z, Ouyang Q, Wang Y,
Xu G, Li S and Li H: A phase Ib study of camrelizumab in
combination with apatinib and fuzuloparib in patients with
recurrent or metastatic triple-negative breast cancer. BMC Med.
20:3212022. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Liu Y, Wang W, Yin R, Zhang Y, Zhang Y,
Zhang K, Pan H, Wang K, Lou G, Li G, et al: A phase 1 trial of
fuzuloparib in combination with apatinib for advanced ovarian and
triple-negative breast cancer: Efficacy, safety, pharmacokinetics
and germline BRCA mutation analysis. BMC Med. 21:3762023.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Friedlander M, Meniawy T, Markman B,
Mileshkin L, Harnett P, Millward M, Lundy J, Freimund A, Norris C,
Mu S, et al: Pamiparib in combination with tislelizumab in patients
with advanced solid tumours: Results from the dose-escalation stage
of a multicentre, open-label, phase 1a/b trial. Lancet Oncol.
20:1306–1315. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Lord CJ and Ashworth A: Mechanisms of
resistance to therapies targeting BRCA-mutant cancers. Nat Med.
19:1381–1388. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Murai J, Huang SY, Das BB, Renaud A, Zhang
Y, Doroshow JH, Ji J, Takeda S and Pommier Y: Trapping of PARP1 and
PARP2 by clinical PARP inhibitors. Cancer Res. 72:5588–5599. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Damale MG, Pathan SK, Shinde DB, Patil RH,
Arote RB and Sangshetti JN: Insights of tankyrases: A novel target
for drug discovery. Eur J Med Chem. 207:1127122020. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Yu M, Yang Y, Sykes M and Wang S:
Small-molecule inhibitors of tankyrases as prospective therapeutics
for cancer. J Med Chem. 65:5244–5273. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Smith S, Giriat I, Schmitt A and de Lange
T: Tankyrase, a poly(ADP-ribose) polymerase at human telomeres.
Science. 282:1484–1487. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Rottenberg S, Jaspers JE, Kersbergen A,
van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M,
Zevenhoven J, Lau A, et al: High sensitivity of BRCA1-deficient
mammary tumors to the PARP inhibitor AZD2281 alone and in
combination with platinum drugs. Proc Natl Acad Sci USA.
105:17079–17084. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Abkevich V, Timms KM, Hennessy BT, Potter
J, Carey MS, Meyer LA, Smith-McCune K, Broaddus R, Lu KH, Chen J,
et al: Patterns of genomic loss of heterozygosity predict
homologous recombination repair defects in epithelial ovarian
cancer. Br J Cancer. 107:1776–1782. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Wang S, Chang CW, Huang J, Zeng S, Zhang
X, Hung MC and Hou J: Gasdermin C sensitizes tumor cells to PARP
inhibitor therapy in cancer models. J Clin Invest. 134:e1668412024.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Stringer-Reasor EM, May JE, Olariu E,
Caterinicchia V, Li Y, Chen D, Della Manna DL, Rocque GB, Vaklavas
C, Falkson CI, et al: An open-label, pilot study of veliparib and
lapatinib in patients with metastatic, triple-negative breast
cancer. Breast Cancer Res. 23:302021. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Ray Chaudhuri A, Callen E, Ding X, Gogola
E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et
al: Replication fork stability confers chemoresistance in
BRCA-deficient cells. Nature. 535:382–387. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Tan Q and Xu X: PTIP UFMylation promotes
replication fork degradation in BRCA1-deficient cells. J Biol Chem.
300:1073122024. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Tian T, Chen J, Zhao H, Li Y, Xia F, Huang
J, Han J and Liu T: UFL1 triggers replication fork degradation by
MRE11 in BRCA1/2-deficient cells. Nat Chem Biol. 20:1650–1661.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Tutt ANJ, Garber JE, Kaufman B, Viale G,
Fumagalli D, Rastogi P, Gelber RD, de Azambuja E, Fielding A,
Balmaña J, et al: Adjuvant olaparib for patients with BRCA1- or
BRCA2-mutated breast cancer. N Engl J Med. 384:2394–2405. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Gelmon KA, Fasching PA, Couch FJ, Balmaña
J, Delaloge S, Labidi-Galy I, Bennett J, McCutcheon S, Walker G and
O'Shaughnessy J; Collaborating Investigator, : Clinical
effectiveness of olaparib monotherapy in germline BRCA-mutated,
HER2-negative metastatic breast cancer in a real-world setting:
Phase IIIb LUCY interim analysis. Eur J Cancer. 152:68–77. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Pusztai L, Yau C, Wolf DM, Han HS, Du L,
Wallace AM, String-Reasor E, Boughey JC, Chien AJ, Elias AD, et al:
Durvalumab with olaparib and paclitaxel for high-risk HER2-negative
stage II/III breast cancer: Results from the adaptively randomized
I-SPY2 trial. Cancer Cell. 39:989–998.e5. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Batalini F, Xiong N, Tayob N, Polak M,
Eismann J, Cantley LC, Shapiro GI, Adalsteinsson V, Winer EP,
Konstantinopoulos PA, et al: Phase 1b clinical trial with alpelisib
plus olaparib for patients with advanced triple-negative breast
cancer. Clin Cancer Res. 28:1493–1499. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Geyer CE Jr, Garber JE, Gelber RD, Yothers
G, Taboada M, Ross L, Rastogi P, Cui K, Arahmani A, Aktan G, et al:
Overall survival in the OlympiA phase III trial of adjuvant
olaparib in patients with germline pathogenic variants in BRCA1/2
and high-risk, early breast cancer. Ann Oncol. 33:1250–1268. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Robson ME, Im SA, Senkus E, Xu B, Domchek
SM, Masuda N, Delaloge S, Tung N, Armstrong A, Dymond M, et al:
OlympiAD extended follow-up for overall survival and safety:
Olaparib versus chemotherapy treatment of physician's choice in
patients with a germline BRCA mutation and HER2-negative metastatic
breast cancer. Eur J Cancer. 184:39–47. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Yamauchi H, Toi M, Takayama S, Nakamura S,
Takano T, Cui K, Campbell C, De Vos L, Geyer C Jr and Tutt A:
Adjuvant olaparib in the subset of patients from Japan with BRCA1-
or BRCA2-mutated high-risk early breast cancer from the phase 3
OlympiA trial. Breast Cancer. 30:596–605. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Senkus E, Delaloge S, Domchek SM, Conte P,
Im SA, Xu B, Armstrong A, Masuda N, Fielding A, Robson M and Tung
N: Olaparib efficacy in patients with germline BRCA-mutated,
HER2-negative metastatic breast cancer: Subgroup analyses from the
phase III OlympiAD trial. Int J Cancer. 153:803–814. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Ring A, Kilburn LS, Pearson A, Moretti L,
Afshari-Mehr A, Wardley AM, Gurel B, Macpherson IR, Riisnaes R,
Baird RD, et al: Olaparib and ceralasertib (AZD6738) in patients
with triple-negative advanced breast cancer: Results from cohort E
of the plasmaMATCH trial (CRUK/15/010). Clin Cancer Res.
29:4751–4759. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Balmaña J, Fasching PA, Couch FJ, Delaloge
S, Labidi-Galy I, O'Shaughnessy J, Park YH, Eisen AF, You B,
Bourgeois H, et al: Clinical effectiveness and safety of olaparib
in BRCA-mutated, HER2-negative metastatic breast cancer in a
real-world setting: Final analysis of LUCY. Breast Cancer Res
Treat. 204:237–248. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Tan TJ, Sammons S, Im YH, She L, Mundy K,
Bigelow R, Traina TA, Anders C, Yeong J, Renzulli E, et al: Phase
II DORA study of olaparib with or without durvalumab as a
chemotherapy-free maintenance strategy in platinum-pretreated
advanced triple-negative breast cancer. Clin Cancer Res.
30:1240–1247. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Abraham JE, Pinilla K, Dayimu A, Grybowicz
L, Demiris N, Harvey C, Drewett LM, Lucey R, Fulton A, Roberts AN,
et al: The PARTNER trial of neoadjuvant olaparib with chemotherapy
in triple-negative breast cancer. Nature. 629:1142–1148. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Litton JK, Rugo HS, Ettl J, Hurvitz SA,
Gonçalves A, Lee KH, Fehrenbacher L, Yerushalmi R, Mina LA, Martin
M, et al: Talazoparib in patients with advanced breast cancer and a
germline BRCA mutation. N Engl J Med. 379:753–763. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Ettl J, Quek RGW, Lee KH, Rugo HS, Hurvitz
S, Gonçalves A, Fehrenbacher L, Yerushalmi R, Mina LA, Martin M, et
al: Quality of life with talazoparib versus physician's choice of
chemotherapy in patients with advanced breast cancer and germline
BRCA1/2 mutation: Patient-reported outcomes from the EMBRACA phase
III trial. Ann Oncol. 29:1939–1947. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Litton JK, Scoggins ME, Hess KR, Adrada
BE, Murthy RK, Damodaran S, DeSnyder SM, Brewster AM, Barcenas CH,
Valero V, et al: Neoadjuvant talazoparib for patients with operable
breast cancer with a germline BRCA pathogenic variant. J Clin
Oncol. 38:388–394. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Litton JK, Hurvitz SA, Mina LA, Rugo HS,
Lee KH, Gonçalves A, Diab S, Woodward N, Goodwin A, Yerushalmi R,
et al: Talazoparib versus chemotherapy in patients with germline
BRCA1/2-mutated HER2-negative advanced breast cancer: Final overall
survival results from the EMBRACA trial. Ann Oncol. 31:1526–1535.
2020. View Article : Google Scholar : PubMed/NCBI
|