Introduction
Given that cancer is the second most prevalent cause
of death worldwide after cardiovascular disease, the investigation
and development of new and safe anticancer medications remains a
top priority (1,2). For example, the National Center for
Health Statistics predicted that in the United States in 2024 there
would be ~2,001,140 new cases of cancer and ~611,720 cancer-related
deaths (3).
The chemotherapeutic drug paclitaxel (PTX, brand
name Taxol®) is a secondary metabolite that has been
used for years to treat a variety of cancers (4–6). It is
a diterpenoid pseudo-alkaloid, consisting of a taxane ring and an
N-benzoylphenylisoserine group, that was first isolated from the
bark of the Pacific yew tree (Taxus brevifolia) in 1963
(7). It belongs to the taxane
family of medications and is known to prevent cancer cells from
growing and dividing abnormally. PTX is used to treat a variety of
malignancies, including breast (8–10),
ovarian (11,12) and lung cancers (13). PTX has a mechanism of action (MoA)
distinct from those of typical anticancer medications, which
usually target DNA and RNA. Instead of attacking these genetic
materials, PTX intervenes during the mitotic phase of cell
division. It promotes the polymerization of tubulin and facilitates
the assembly of microtubules, thereby stabilizing them. This can
render the microtubules dysfunctional, leading to the cessation of
cell growth (14,15).
While PTX has been shown to have clear benefits for
the treatment of cancer, it also has several undesirable side
effects, including hair loss, peripheral neuropathy, nausea and
vomiting (16,17). Over time, cancer cells may become
resistant to PTX, which compromises its effectiveness (18–21).
Researchers are actively looking for strategies to address this
issue by combining PTX with other drugs (22–24).
In addition, there are two main strategies for increasing the
effectiveness of PTX and overcoming its side effects. One is to
synthesize analogs with more potent anticancer action. The other is
to overcome the limitations of PTX by creating a prodrug. A variety
of prodrugs can be produced by attaching small or large molecules
to PTX.
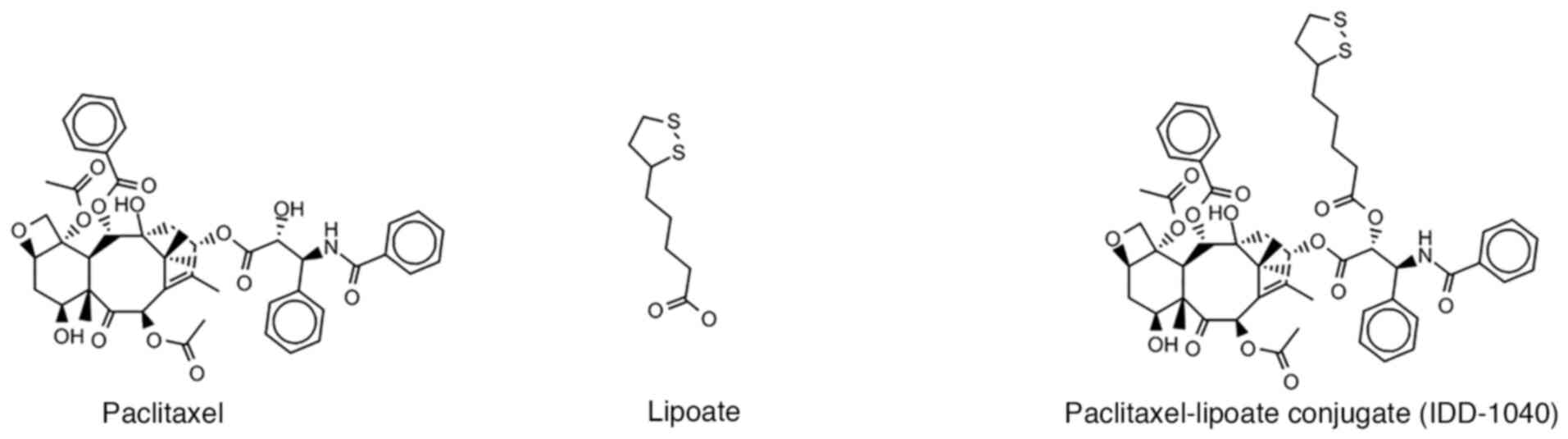
In the present study, a small molecule prodrug,
PTX-lipoate (IDD-1040), obtained by condensing a lipoic acid moiety
with the PTX hydroxyl group at C2′ position to form an esterified
PTX prodrug, was investigated (Fig.
1). Lipoate, also known as a-lipoic acid, possesses antioxidant
properties (25). Our previous
study on IDD-1040 showed that it exhibits superior inhibition of
tumor growth and more powerful antitumor effects compared with PTX
alone. Its effectiveness for slowing tumor growth is evident long
after treatment has stopped, and its administration is associated
with a low level of toxicity and a decreased mortality rate in mice
(25). Collectively, these results
clearly indicate the potential of IDD-1040 as a cancer treatment
candidate and support its advancement to clinical testing. However,
to use IDD-4010 most effectively in patients and support its
development as a medication, it is crucial to have a complete
understanding of its pharmacokinetic features. Pharmacokinetic
studies have become the cornerstone of a strong drug development
program. In the present case, they are necessary to increase the
likelihood of this drug being approved for clinical use. Due to its
structure (Fig. 1) and
physicochemical properties, IDD-1040, like PTX, has very low water
solubility (25). Both compounds
are lipophilic and neutral, lacking any basic or acidic
ionizability, which means that pH manipulation is not able to
increase their solubility. Given that the poor aqueous solubility
of PTX has presented an obstacle to its clinical application, the
development a delivery system for IDD-1040 may also be expected to
present considerable challenges.
The aim of the current study was to investigate the
pharmacokinetic characteristics of the PTX-lipoate prodrug. It is
hypothesized that conjugation with lipoic acid would increase the
therapeutic effectiveness of PTX while reducing its side effects,
thereby expanding the range of therapy options for patients.
Materials and methods
Chemicals and reagents
The synthesis of IDD-1040 was performed as detailed
in our previous study (25).
Following synthesis, IDD-1040 (100 µg) was stored under a nitrogen
atmosphere at −18°C until subjected to analysis. Liquid
chromatography-mass spectrometry (LC/MS)-grade acetonitrile (J.T.
Baker; Thermo Fisher Scientific, Inc.) and LC/MS-grade methanol and
water (Bio-Lab Ltd.) were utilized for the analytical experiments.
The high-performance liquid chromatography (HPLC) cartridge
employed was LiChroCART® 250-4 LiChrospher®
100 RP-18 (5 µm), while a LiChroCART® 4-4
LiChrospher® 100 RP-18 (5 µm) guard column was used to
safeguard the integrity of the analytical column (both from Merck
KGaA).
Instrumentation
High Performance Liquid Chromatography (HPLC) system
(Thermo Fisher Scientific, Inc.) included an Accela pump with a
degasser module, an Accela autosampler and an Accela
photodiode-array (PDA) detector. To record the mass spectrum, the
HPLC system was connected to a TSQ Quantum Access Max triple-stage
quadrupole mass spectrometer (Thermo Fisher Scientific, Inc.) via a
heated electrospray ionization (H-ESI) interface. Data acquisition
and processing were performed using Xcalibur™ software
version 4.3 (Thermo Fisher Scientific, Inc.).
Chromatographic conditions
Optimal chromatographic separation was achieved via
the use of an isocratic mobile phase consisting of a binary solvent
mixture of methanol:water, 82:18, v/v, at a constant flow rate of
1.0 ml/min, with a total run time of 7 min. The chromatographic
oven temperature was maintained at 40°C, while the autosampler tray
temperature was carefully controlled at 5°C. Samples with a volume
of 5 µl were introduced into the column via a partial loop
injection system, and the injection needle was thoroughly cleansed
with 1 ml methanol:water, 80:20, v/v, between successive injections
to prevent cross-contamination. The initial investigation of the
optimal UV detection wavelength was conducted using the PDA
detector within a spectral range of 200–300 nm. Final
quantification was carried out at a fixed wavelength of 225 nm.
Mass spectrometric conditions
The mass spectrometer was configured to operate in
positive ionization mode. The key operational parameters included
an ionization spray voltage of 3.0 kV, a skimmer offset of 0 V and
a capillary transfer tube temperature of 350°C. The nitrogen sheath
gas and auxiliary gas flow rates were set at 35 and 30 l/min,
respectively. The vaporizing temperature in the H-ESI source was
maintained at a consistent 250°C, and a rapid scan time of 0.1 sec
was employed.
Preparation of the standard and stock
solutions
Stock solution A (1.0 mg/ml) was prepared by
dissolving 10.0 mg IDD-1040 in acetonitrile in a 10.0-ml volumetric
flask. Following this, stock solution B (0.1 mg/ml), was generated
by diluting 1.0 ml stock solution A with acetonitrile in a 10.0-ml
volumetric flask. These stock solutions were stored in a freezer at
−20°C. To construct the linear calibration curve, eight standards
were created, at concentrations of 10, 20, 50, 100, 200, 500, 750
and 1,000 µg/ml. To ensure accuracy, the calibration samples were
meticulously prepared in duplicate and analyzed in duplicate as
well.
MoA assessment
In vitro tubulin polymerization assay
To elucidate the MoA of IDD-1040, an in vitro
tubulin polymerization assay was conducted. This MoA was
investigated because IDD-1040 is a prodrug of PTX, which binds
tubulin and stabilizes microtubules during cell division. The
Tubulin Polymerization Assay Kit (cat. no. BK006P; Cytoskeleton,
Inc.) was used. This assay is based on the original methodologies
developed by Shelanski et al (26) and Lee and Timasheff (27), which demonstrated that the amount of
light scattering by microtubules is directly proportional to the
concentration of microtubule polymer.
Preparation of the cold tubulin
polymerization buffer
To initiate the experiment, cold tubulin
polymerization buffer was prepared as follows. A 750-µl volume of
general tubulin buffer was combined with a 250-µl volume of tubulin
glycerol buffer, and 10 µl GTP stock solution (100 mM) was added to
the mixture. The general tubulin buffer was warmed to room
temperature to facilitate tubulin ligand dilution in the next
step.
Tubulin dilution
Tubulin (100 µl) was rapidly thawed and diluted with
210 µl tubulin polymerization buffer to achieve a final
concentration of 3 mg/ml.
Sample preparation
The following samples were prepared: General tubulin
buffer (negative control); PTX at a final concentration of 100 µM
(positive control), as specified in the kit; IDD-1040 at a final
concentration of 100 µM; and IDD-1040 at a final concentration of
500 µM. Then, 30 µl of each sample was added to 270 µl diluted
tubulin.
Spectrophotometric analysis
The spectrophotometric analysis was conducted with
an Evolution 300 spectrophotometer (Thermo Fisher Scientific, Inc.)
equipped with a controlled heating system. Cuvettes with a volume
of 750 µl were used. The samples were incubated at 37°C, and
absorbance readings were taken at 340 nm for a total duration of 10
min, for 60 cycles of 10 sec each.
In vivo pharmacokinetic study
The in vivo experiment was performed by
Pharmaseed Ltd., a good laboratory practice (GLP)-certified
laboratory, following the guidelines of the Organisation for
Economic Co-operation and Development Principles of GLP
[C(97)186/final]. Animal handling was performed in accordance with
the guidelines of the National Institutes of Health and the
Association for Assessment and Accreditation of Laboratory Animal
Care. The study was performed after approval by the Israel Board
for Animal Experiments (approval no. GB06/68708), in compliance
with the Israel Animal Welfare Act. The formulation used for in
vivo studies consisted of 6 mg/ml of the active compound in
solution containing 527 mg Cremophor EL (polyethoxylated castor oil
surfactant) and 49.7% (v/v) dehydrated alcohol. Before
administration, the solution was diluted to 0.3–1.2 mg/ml.
In the study, 39 CD-1 nude mice were divided into 13
groups (n=3/group), with each group representing a different time
point. Mice were anesthetized with inhalational isoflurane, with
induction using 3–4% isoflurane in oxygen and maintenance using
1–2% isoflurane in oxygen prior to dosing. Then, an intravenous
bolus of 50 mg/kg IDD-1040 (5 mg/ml solution, 10 ml/kg dose volume)
was administered via the tail vein. Blood samples were collected at
time points of 0 h (pre-dose), 5 min, and 1, 3, 6, 8, 10, 12, 24,
48, 72, 120 and 168 h post-dose from the tail vein. A sparse
sampling schedule was used in which 3 mice were tested at each time
point and two samples were collected from each mouse. The mice were
anesthetized by isoflurane inhalation prior to blood withdrawal.
The blood samples were transferred to a heparinized tube and
centrifuged at 7,500 × g at 4°C for 10 min. The resulting plasma
was separated and kept frozen at −80°C prior to analysis. The
plasma samples were analyzed for detection of the prodrug IDD-1040
and its PTX hydrolysis product. Pharmacokinetic parameters
(Tables I and II) were computed from the data using PK
Solutions-2 software, version 2 (Summit Research Services). The
clearance (Cl) and volume of distribution (Vd) for IDD-1040 were
calculated as follows: Cl=dose/area under the concentration-time
curve) and Vd=Cl/ke (elimination rate constant).
 | Table I.Pharmacokinetic parameters for
IDD-1040. |
Table I.
Pharmacokinetic parameters for
IDD-1040.
| Statistical
measure | C initial, ng/ml | T1/2 A
phase, h | AUC, ng.h/ml | T1/2 E
phase, h | T1/2
according to Vd and Cl, h |
|---|
| Mean | 25,880 | 0.484 | 29,589 | 8.64 | 8.63 |
| SEM | 2,492 | 0.111 | 2,256 | 0.74 | 0.73 |
| Table II.Pharmacokinetic parameters for
paclitaxel. |
Table II.
Pharmacokinetic parameters for
paclitaxel.
| Statistical
measure | Cmax at
3 h, ng/ml | T1/2 A
phase, h | AUC, ng.h/ml | T1/2 E
phase, h | T1/2
according to Vd and Cl |
|---|
| Average | 140 | 0.57 | 2001 | 16.69 | 16.70 |
| SEM | 3.4 | 0.15 | 102 | 2.29 | 2.28 |
Development of IDD-1040 formulations: A
formulation development experiment was conducted to address the
poor aqueous solubility of IDD-1040. Initially, the solubility of
IDD-1040 was assessed in various excipients suitable for parenteral
administration, including dimethylacetamide (DMA), propylene glycol
(PG), polyethylene glycol 400 (PEG 400), and ethanol. Subsequently,
different formulations were prepared using these excipients, along
with cosolvents, surfactants, and cyclodextrin. The excipients and
their precise quantities for each formulation type are detailed in
Tables III and IV.
| Table III.Composition of clear aqueous
formulations organized by IDD-1040 concentration. |
Table III.
Composition of clear aqueous
formulations organized by IDD-1040 concentration.
| A, 1 mg/g
IDD-1040 |
|---|
|
|---|
|
|
Formulation
no. |
|---|
|
|
|
|---|
| Components | F56 | F57 | F58 | F60 | F62 | F63 | F65 | F67 | F68 | F70 | F72 |
|---|
| DMA, % w/w | - | - | 1 | 1 | 1 | 2 | 2 | 2 | 4 | 4 | 4 |
| PG, % w/w | 40 | 50 | 29 | 39 | 49 | 28 | 38 | 48 | 26 | 36 | 46 |
| HPCD, % w/w | 27 | 22.5 | 31.5 | 27 | 22.5 | 31.5 | 27 | 22.5 | 31.5 | 27 | 22.5 |
|
| B, 1.8–3.2 mg/g
IDD-1040 |
|
|
| Formulation
no. |
|
|
|
|
Components | F44 | F90 | F91 | F92 | F93 | F94 | F40 | F17 | F36 |
|
| IDD-1040,
mg/kg | 1.8 | 2 | 2 | 2 | 2 | 2 | 2.8 | 3.2 | 3.2 |
| DMA, % w/w | 4 | 4 | 4 | - | 4 | - | 4 | 7 | 7 |
| Tween 80, %
w/w | - | - | - | - | - | - | - | 10 | - |
| PG, % w/w | 50 | 40 | 30 | 20 | 20 | 20 | 50 | 40 | 50 |
| HPCD, % w/w | 20.7 | 25.2 | 25.2 | 27 | 25.2 | 27 | 20 | - | 19.4 |
| Ethanol, % w/w | - | - | 10 | 20 | - | - | - | - | - |
| PEG 400, % w/w | - | - | - | - | 20 | 20 | - | - | - |
|
| C, 4–10 mg/g
IDD-1040 |
|
|
| Formulation
no. |
|
|
|
|
Components | F16 | F30 | F12 | F18 | F27 | F49 | F45 | F46 | F48 | F87 | F88 |
|
| IDD-1040,
mg/kg | 4 | 4.5 | 5 | 5 | 5 | 5 | 5.7 | 7 | 7 | 10 | 10 |
| DMA, % w/w | 7 | 10 | 7 | 7 | 7 | 5 | 10 | 10 | 10 | 10 | 10 |
| Tween 80, %
w/w | 10 | 10 | 10 | - | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| Cremophor EL, %
w/w | - | - | - | 10 | - | - | - | - | - | - | - |
| PG, % w/w | 40 | 40 | 40 | 40 | 50 | 40 | 40 | 40 | 30 | 30 | 15 |
| HPCD, % w/w | - | 16 | - | - | - | 20.3 | 18 | 18 | 22.5 | 22.5 | 22.5 |
| Ethanol, % w/w | - | - | - | - | - | - | - | - | - | 10 | 15 |
| Table IV.Homogeneous formulations of
IDD-1040. |
Table IV.
Homogeneous formulations of
IDD-1040.
|
| IDD-1040, mg/g |
|---|
|
|
|
|---|
| Variable | 5 | 6.5 | 8 | 9 | 10.5 | 12 |
|---|
| Formulation
no. | F32 | F80 | F81 | F82 | F83 | F95 | F96 |
| DMA 100 mg/g | 50 | 50 | 50 | 75 | 75 | 90 | 85 |
| PG 10 mg/g | - | 150 | 100 | 150 | 100 | 100 | 150 |
| Ethanol 20
mg/g | - | - | 100 | - | 100 | 100 | 100 |
| PEG 400 | 150 | - | - | - | - | - | - |
| Lipofundin | 800 | 800 | 750 | 775 | 725 | 710 | 665 |
Statistical analysis
One-way ANOVA followed by Tukey's HSD post-hoc test
was employed to analyze the results of in vivo
pharmacokinetic study using SPSS version 29 (IBM Corp.). P<0.05
was considered to indicate a statistically significant
difference.
Results and Discussion
Purity of IDD-1040
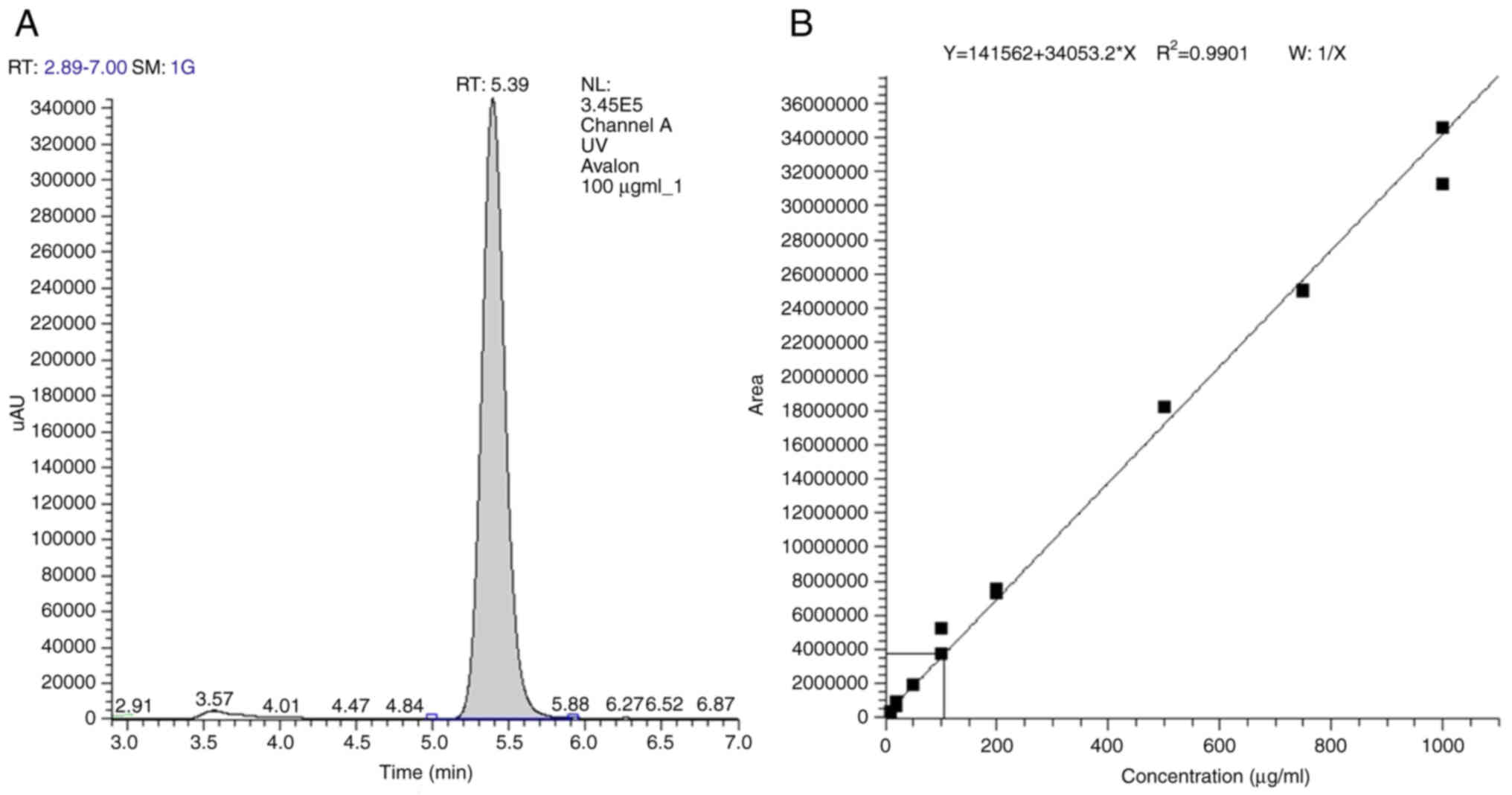
The purity of IDD-1040 used in the present study was
assessed using HPLC and LC-MS analyses. A single symmetric eluting
peak is evident in the chromatogram, with a retention time of 5.39
min (Fig. 2). The R2
value of 0.9901 indicates excellent linearity, confirming the
method's reliability for quantifying IDD-1040 within the tested
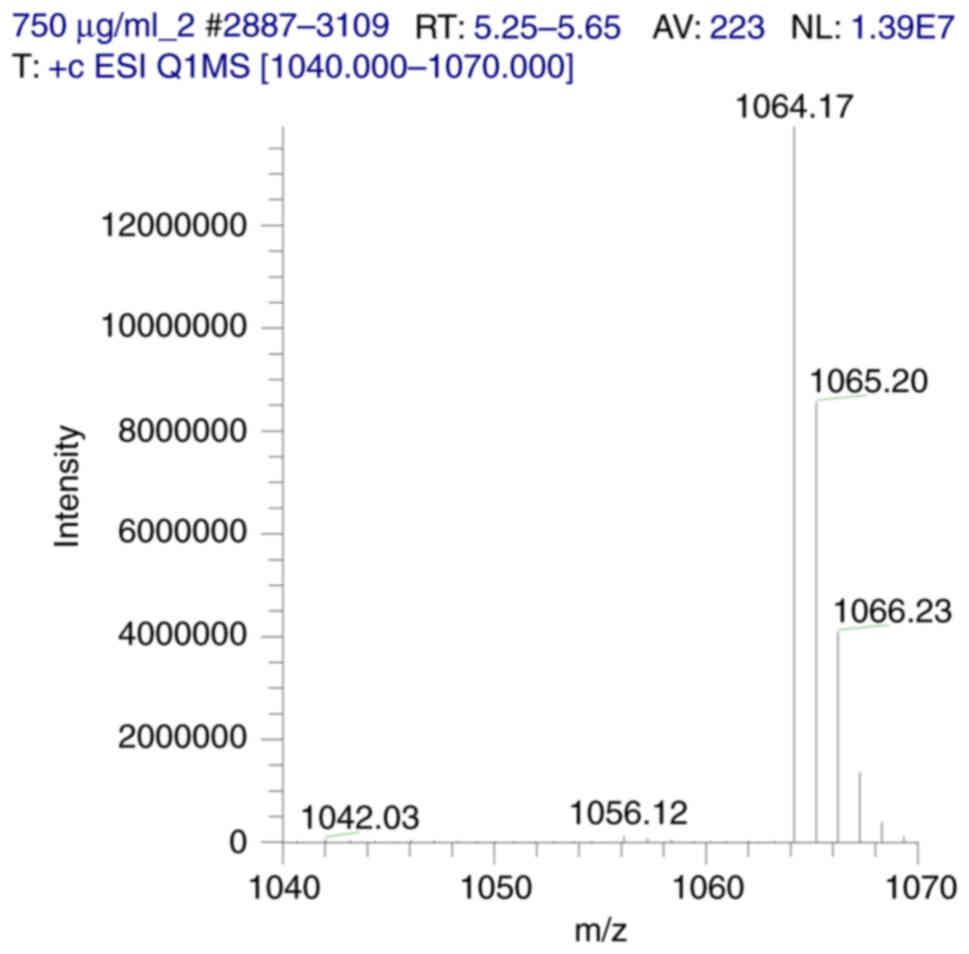
concentration range. ESI-MS analysis revealed the presence of the
sodiated stable adduct ion [M+Na]+ as the prominent
peak, at an m/z value of 1,064 Da. Additionally, another, weaker
peak, corresponding to the protonated IDD-1040 conjugate
[M+H]+, is present, with a m/z value of 1,042 Da
(Fig. 3).
Pharmacokinetic analysis of IDD-1040
and PTX concentrations in mice following the intravenous
administration of IDDD-1040
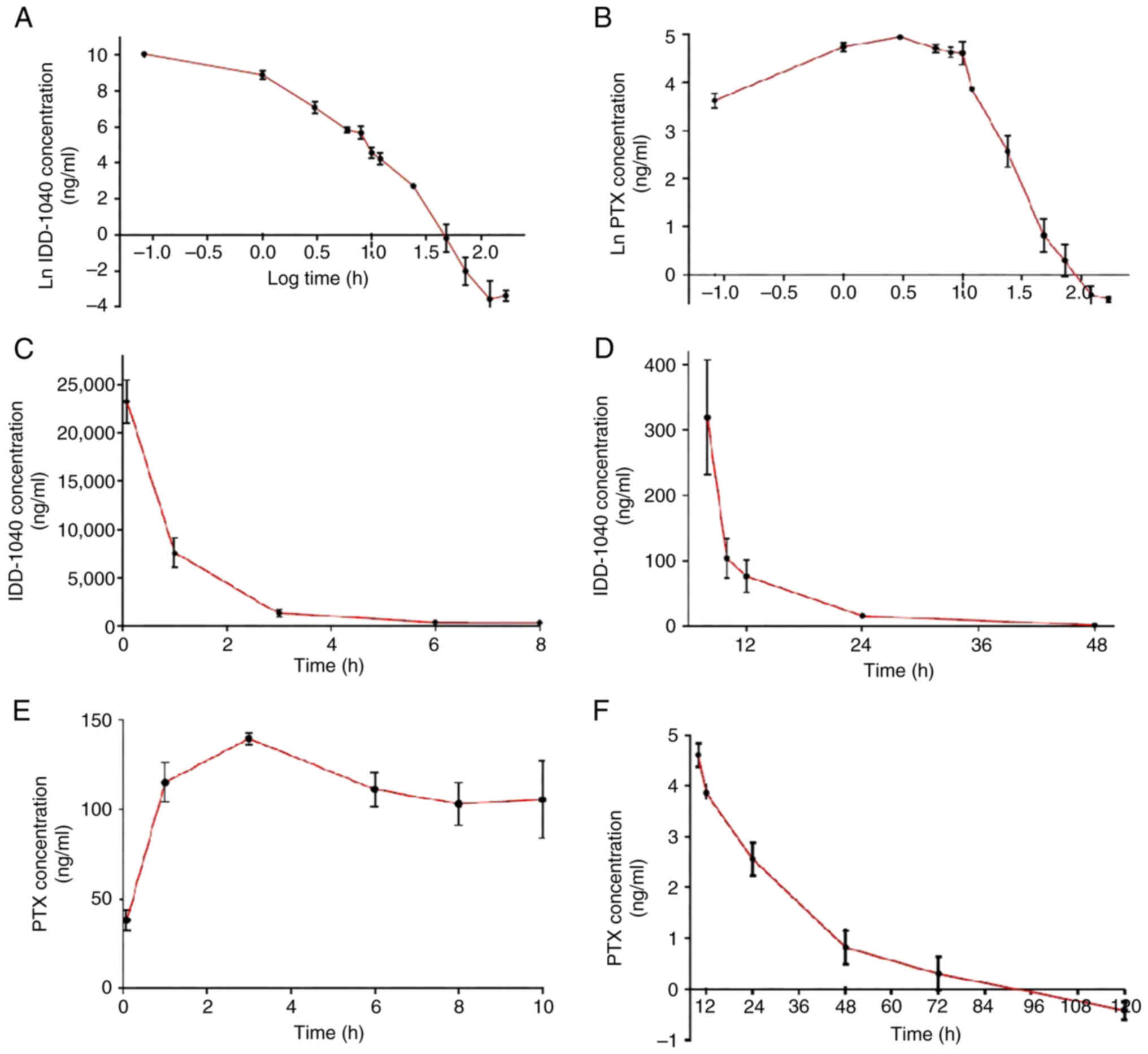
Female mice were intravenously dosed with a bolus of
50 mg/kg IDD-1040, and blood samples were collected at 13 time
points, ranging from 0 h to 7 days. Plasma from these blood samples
was analyzed to determine the concentration of the pro-drug
IDD-1040 and its hydrolysis product, PTX, at each time point. Given
the extended observation period, encompassing the metabolism and
elimination of IDD-1040, the concentrations spanned six orders of
magnitude. Consequently, a natural log-common log (ln-log)
conversion was applied to facilitate the visualization of all the
data collectively (Fig. 4A). Three
distinct phases can be observed. First, the initial 1 h following
administration is the distribution phase of the drug. Second,
between 1 and 10 h post-injection, the drug undergoes a phase
characterized by both metabolism and elimination. Third, the final
phase is the terminal elimination phase, extending to 5 days. A
parallel pattern is observed when the ln-log transformation of
concentration data for the hydrolysis product PTX is examined. Up
to 10 h, a primary phase marked by an increase in PTX concentration
in the blood is evident, followed by a terminal elimination phase
extending up to 5 days following the administration of IDD-1040
(Fig. 4B).
The line graphs in Fig.
4C and D show the initial and terminal phases of the
pharmacokinetics of IDD-1040. Both phases are shown as exponential
decay curves from which corresponding rate constants (k values) can
be determined. Parallel representations of the PTX profiles are
presented in Fig. 4E and F.
Pharmacokinetic parameters were computed from the data using PK
Solutions-2 software version 2 and are presented in Tables I and II.
As indicated by the area under the curve (AUC)
values in Table I and the
calculated using the corresponding pharmacokinetic equations, the
clearance for IDD-1040 is 1.689 l/h.kg, while the volume of
distribution is 1.93 l/kg. This distribution exceeds the volume of
systemic blood by >30-fold, which suggests extensive tissue
distribution beyond the bloodstream. Utilizing these parameters, a
half-life of 1.14 h was calculated for IDD-1040 in the central
elimination phase, which is notably shorter than the terminal
elimination half-life of 8.64 h.
Given that PTX is derived from the administration of
IDD-1040, it is not feasible to estimate its clearance and volume
of distribution using the data obtained (Table II). A notable observation arises
when the data for IDD-1040 and PTX are compared: The AUC of
IDD-1040 is >14-fold higher than that of PTX. This suggests that
the conversion of IDD-1040 into its metabolite occurs slowly, and
hints at the possibility that some of the biological activity
previously observed for IDD-040 may be attributed to the prodrug
itself.
While the data on the half-life of PTX in the
literature vary and are challenging to compare against (28,29),
it is generally noted that PTX exhibits a significantly shorter
half-life than IDD-1040. Consequently, it is evident that the
extended half-life of IDD-1040 observed in the current study was
associated with the continuous release of PTX by the prodrug
IDD-1040.
MoA assessment by in vitro tubulin
polymerization assay
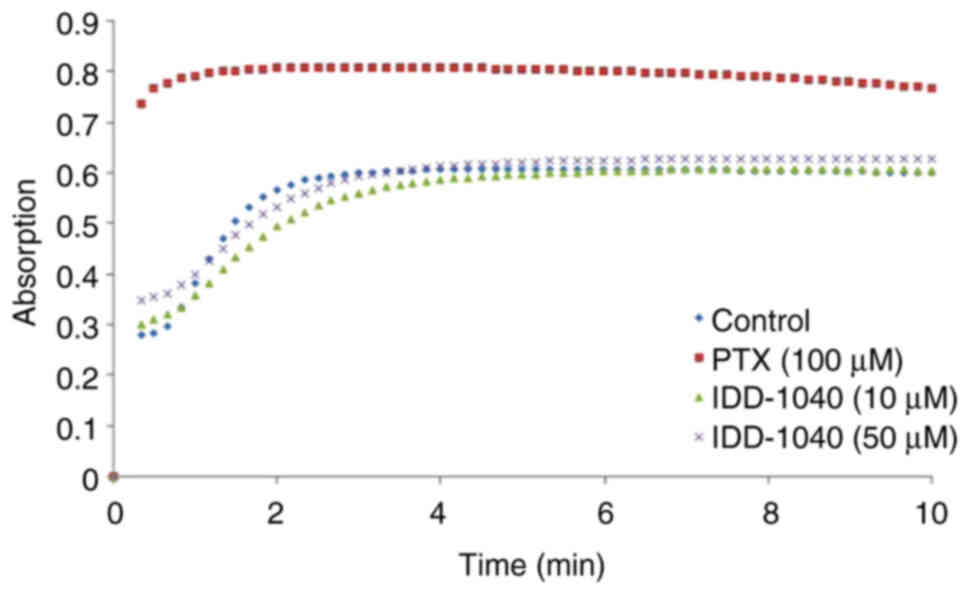
The results of the in vitro tubulin
polymerization assay used to evaluate the MoA of IDD-1040 are
presented in Fig. 5. The assay was
conducted using PTX as a positive control, along with IDD-1040 at
two different concentrations. The results demonstrated that PTX at
a concentration of 10 µM increased tubulin polymerization by ~41.8%
compared with that of the negative control. In comparison, IDD-1040
at a concentration of 50 µM increased tubulin polymerization by
only ~9.1% compared with that of the negative control. The
tubulin-binding characteristics exhibited by IDD-1040 appear to
differ from those of PTX, suggesting a different MoA. The results
of this assay provide valuable insights into the MoA of IDD-1040
and its interaction with tubulin, highlighting its potential as an
anticancer agent with distinctive properties.
Clinical formulation development
The development of an effective formulation for a
therapeutic agent with low aqueous solubility, such as IDD-1040, is
a critical aspect of drug delivery, as it can markedly improve the
pharmacokinetics and therapeutic efficacy of the drug. An aim of
the present study was to improve the formulation of IDD-1040 to
address the limitations associated with its current formulation,
which comprises a vehicle primarily composed of Cremophor EL and
dehydrated alcohol. As the aqueous solubility of PTX is very low,
Cremophor EL was included in the formulation to increase the
solubility of PTX. However, it is toxic and causes serious side
effects (17), such as
hypersensitivity, myelosupression, nephrotoxicity and
neurotoxicity.
As aforementioned, IDD-1040 has low aqueous
solubility and lacks ionizable functionalities, which renders pH
adjustment ineffective for increasing its solubility; therefore, it
is crucial to find another means of improving its delivery. Using
the current formulation, the maximal concentration that resulted in
a clear solution was 0.4 mg/ml; higher drug concentrations were
milky. In an attempt to achieve a suitable parenteral formulation
for IDD-1040, a formulation development experiment was conducted.
In the first stage, the solubility of IDD-1040 in excipients
suitable for parenteral administration was tested. IDD-1040 was
found to be soluble in dimethylacetamide (DMA; 100 mg/g), propylene
glycol (PG; 16 mg/g), polyethylene glycol 400 (PEG 400; 10 mg/g)
and ethanol (20 mg/g). This profile differs from that reported in
the literature for PTX, which is considered to be insoluble
(<0.1 mg/ml) in PG and PEG 400 (30). The solvents found to solubilize the
drug, together with cosolvents, surfactants and cyclodextrin, were
then used to prepare and test several different IDD-1040
formulations. These were diluted with either water or Lipofundin (a
commercial parenteral nutrition nanoemulsion). A total of 31
formulations diluted with water were found to be clear after
preparation (Table III). These
included 1–3 mg/g formulations using solvents and cyclodextrin.
Formulations with higher drug concentrations (4–10 mg/g) containing
the surfactant Tween™ 80 were also clear and exhibited a
25-fold increase in solubility compared with that of the reference
formulation. When the drug was solubilized in solvent mixtures
followed by dilution with Lipofundin instead of water, higher drug
concentrations could be achieved (12 mg/g), resulting in visually
homogeneous formulations (Table
IV) with a high drug content. These were obtained without the
use of surfactants and, overall, had fewer excipients compared with
the formulations diluted with water.
Based on these results, the following are the key
findings and their implications for the development of IDD-1040
formulations.
IDD-1040 solubility profile
The investigation began with an assessment of the
solubility of IDD-1040 in various excipients suitable for
parenteral administration. Notably, appreciable solubility was
observed in DMA, PG, PEG 400 and ethanol. This solubility profile
differs slightly from that of PTX, its parent compound, which is
generally considered to be insoluble in PG and PEG 400.
Understanding these solubility characteristics is crucial for
formulating an effective delivery system.
Formulation approaches
In the subsequent stages of the present study, to
develop a suitable parenteral formulation, various agents that had
the potential to enhance the solubility of IDD-1040, including
co-solvents, surfactants and cyclodextrin, were explored.
Co-solvents
It was observed that the formulations containing
IDD-1040 in combination with the solvents DMA, PG, PEG 400 and
ethanol resulted in clear solutions at drug concentrations of 1–3
mg/g. This suggests that co-solvent systems have the potential to
increase the solubility of IDD-1040, which presents a method for
improving the formulations.
Surfactants
The addition of Tween 80, a surfactant, resulted in
an increase in IDD-1040 solubility in certain formulations,
particularly at higher drug concentrations (4–10 mg/g). This
increase in solubility indicates that this as a promising strategy
for the development of formulations with a higher drug content and
enhanced therapeutic efficacy.
Cyclodextrin
Cyclodextrin-based formulations also warrant
consideration, as they yielded clear solutions at certain drug
concentrations. The use of cyclodextrin may offer an alternative
route for the improvement of IDD-1040 formulations.
Lipofundin
Notably, when the drug was solubilized in solvent
mixtures and diluted with Lipofundin, higher drug concentrations
(12 mg/g) could be achieved, resulting in visually homogeneous
formulations. This approach is particularly appealing, as it
achieves high levels of drug content without the need for
additional surfactants and can reduce the amount of excipient
required.
Clinical implications of alternative
IDD-1040 formulations
The findings for the formulations prepared in the
present study have important clinical implications. The low
solubility and toxic nature of Cremophor EL, which is included in
the current Taxol® formulation, have been associated
with serious side effects. The successful development of
alternative IDD-1040 formulations that have improved solubility and
do not contain Cremophore EL should mitigate these side effects and
increase patient safety. Furthermore, the ability to achieve higher
drug concentrations in formulations is crucial for optimizing the
therapeutic effects of IDD-1040. This should enable the reduction
of dosing volumes and improve convenience of administration for
patients.
Summary of findings
In the present comprehensive study, the compound
IDD-1040, formed through the conjugation of lipoic acid and PTX,
was investigated. The study has provided valuable insights into the
analytical, pharmacokinetic and pharmacological aspects of
IDD-1040, which aid in understanding the improved antitumor
efficacy it has previously demonstrated compared with PTX alone.
Pharmacokinetic analysis using PK Solutions-2 software revealed key
parameters that characterize the behavior of IDD-1040 in
vivo. When intravenously administered to mice, IDD-1040
exhibited a total clearance of 1.689 l/h.kg, a volume of
distribution of 1.93 l/kg, an average half-life of 1.14 h, and a
terminal half-life of 8.64 h. Particularly noteworthy is the
markedly (>14-fold) higher AUC of IDD-1040 compared with that of
PTX, indicating a slower conversion of IDD-1040 to its metabolites.
This suggests that a considerable proportion of the antitumor
activity of IDD-1040 can be attributed to the prodrug. The
substantial volume of distribution observed for IDD-1040 reflects
efficient absorption into tissues in the secondary compartment,
possibly contributing to its enhanced antitumor effects.
Furthermore, the results revealed a notably extended half-life of
PTX when delivered via IDD-1040, indicating sustained release from
the prodrug, which should offer a prolonged therapeutic effect and
contribute to its enhanced therapeutic potential. To gain further
insights into the MoA of IDD-1040, an in vitro tubulin
polymerization assay was conducted, in which IDD-1040 was compared
with PTX. This assay indicated that tubulin-binding by IDD-1040 is
much lower than that by PTX, suggesting a unique MoA for the
prodrug and highlighting its potential as an innovative anticancer
agent.
Despite the extremely lipophilic nature of IDD-1040,
its formulation with solvent mixtures and Lipofundin provided a
reasonably high drug concentration (12 mg/g) with few excipients
and no surfactant, indicating its potential for future use. Certain
formulations explored show promise in improving the solubility and
formulation of IDD-1040 and provide a foundation for further
research aimed at optimizing the drug delivery of IDD-1040 and
enhancing its therapeutic outcomes while minimizing toxicity
concerns. In future studies, it is essential to assess the
stability, pharmacokinetics and pharmacodynamics of these IDD-1040
formulations to determine their suitability for clinical use.
Additionally, the compatibility of these formulations with
different routes of administration requires investigation to
explore their versatility for addressing various clinical
scenarios.
IDD-1040 represents a marked advancement in cancer
therapy, offering improved pharmacokinetics, enhanced anticancer
efficacy and reduced side effects compared with conventional PTX
formulations. The extended circulation, efficient tissue
distribution, reduced metabolite formation and unique MoA of
IDD-1040, as revealed through the in vitro tubulin
polymerization assay, underscore the importance of further
investigations into the MoA and therapeutic potential of IDD-1040.
These findings strongly support the promise of IDD-1040 as a
candidate for cancer treatment. Given its potential to make a
substantial impact in the field of oncology, its advancement into
clinical trials is highly recommended. Future studies should focus
on optimizing the formulation, stability and clinical application
of IDD-1040 to harness its full therapeutic potential.
Acknowledgements
Not applicable.
Funding
The current study was partially supported by the Israel
Innovation Authority as an incubator project that was granted to
IDD Therapeutics, Ltd., between 2010 and 2014, and the Israel
Cancer Association (grant nos. 20191644 and 20180071,
respectively). The Al-Qasemi Research Foundation covered the costs
of language editing.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author. Samples of the compounds
may also be requested.
Authors' contributions
AR conceived the study, raised funds, designed the
study and edited the final version of the manuscript. MR, SAL, MF
and MZ designed parts of the study, interpreted the data, wrote up
the conclusions and drafted the first version of the manuscript. AR
and MF confirm the authenticity of all the raw data. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The Israel Board for Animal Experiments Committee
approved in vivo experiment and protocols (certificate no.
GB06/68708).
Patient consent for publication
Not applicable.
Competing interests
AR is the founder and chief executive officer of IDD
Therapeutics, Ltd., which is the owner of intellectual property on
IDD-1010. The other authors declare that they have no competing
interests.
References
|
1
|
Reyes-Farias M and Carrasco-Pozo C: The
anti-cancer effect of quercetin: Molecular implications in cancer
metabolism. Int J Mol Sci. 20:31772019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Alipour V, Rezapour A, Adel A, Pourtaleb
A, Bazrafshan M, Ghaem Mohammadi MS and Jahangiri R: Economical
evaluation of cancer types using intensity-modulated radiation
therapy compared to 3d conformal radiation therapy: A systematic
review. Iran J Public Health. 52:1355–1366. 2023.PubMed/NCBI
|
|
3
|
Siegel RL, Giaquinto AN and Jemal A:
Cancer statistics, 2024. CA Cancer J Clin. 74:12–49. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Manthalkar L, Ajazuddin and Bhattacharya
S: Evidence-based capacity of natural cytochrome enzyme inhibitors
to increase the effectivity of antineoplastic drugs. Discov Oncol.
13:1422022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tauro S, Dhokchawle B, Mohite P, Nahar D,
Nadar S and Coutinho E: Natural anticancer agents: Their
therapeutic potential, challenges,s and promising outcomes. Curr
Med Chem. May 2–2023.(Epub ahead of print). PubMed/NCBI
|
|
6
|
Ahmed Khalil A, Rauf A, Alhumaydhi FA,
Aljohani ASM, Javed MS, Khan MA, Khan IA, El-Esawi MA, Bawazeer S,
Bouyahya A, et al: Recent developments and anticancer therapeutics
of paclitaxel: An update. Curr Pharm Des. 28:3363–3373. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wani MC, Taylor HL, Wall ME, Coggon P and
McPhail AT: Plant antitumor agents. VI. The isolation and structure
of taxol, a novel antileukemic and antitumor agent from Taxus
brevifolia. J Am Chem Soc. 93:2325–2327. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Seidman AD: Single-agent use of Taxol
(paclitaxel) in breast cancer. Ann Oncol. 5 (Suppl 6):S17–S22.
1994.PubMed/NCBI
|
|
9
|
Weaver BA: How Taxol/paclitaxel kills
cancer cells. Mol Biol Cell. 25:2677–2681. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wilkinson SP: The kidney in cirrhosis.
Tijdschr Gastroenterol. 19:155–169. 1976.PubMed/NCBI
|
|
11
|
Ozols RF: Carboplatin and Taxol
(paclitaxel) in advanced ovarian carcinoma. Ann Oncol. 5 (Suppl
6):S39–S43. 1994.PubMed/NCBI
|
|
12
|
Einzig AI: Review of phase II trials of
Taxol (paclitaxel) in patients with advanced ovarian cancer. Ann
Oncol. 5 (Suppl 6):S29–S32. 1994.PubMed/NCBI
|
|
13
|
Johnson DH, Chang AY and Ettinger DS:
Taxol (paclitaxel) in the treatment of lung cancer: The eastern
cooperative oncology group experience. Ann Oncol. 5 (Suppl
6):S45–S50. 1994.PubMed/NCBI
|
|
14
|
Fanale D, Bronte G, Passiglia F, Calò V,
Castiglia M, Di Piazza F, Barraco N, Cangemi A, Catarella MT,
Insalaco L, et al: Stabilizing versus destabilizing the
microtubules: A double-edge sword for an effective cancer treatment
option? Anal Cell Pathol (Amst). 2015:6909162015.PubMed/NCBI
|
|
15
|
Mukhtar E, Adhami VM and Mukhtar H:
Targeting microtubules by natural agents for cancer therapy. Mol
Cancer Ther. 13:275–284. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Amaya C, Smith ER and Xu XX: Low intensity
ultrasound as an antidote to taxane/paclitaxel-induced
cytotoxicity. J Cancer. 13:2362–2373. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Walker FE: Paclitaxel (TAXOL): Side
effects and patient education issues. Semin Oncol Nurs. 9 (Suppl
2):S6–S10. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nagumo Y, Villareal MO, Isoda H and Usui
T: RSK4 confers paclitaxel resistance to ovarian cancer cells,
which is resensitized by its inhibitor BI-D1870. Biochem Biophys
Res Commun. 679:23–30. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Januškevičienė I and Petrikaitė V:
Interaction of phenotypic sublines isolated from triple-negative
breast cancer cell line MDA-MB-231 modulates their sensitivity to
paclitaxel and doxorubicin in 2D and 3D assays. Am J Cancer Res.
13:3368–3383. 2023.PubMed/NCBI
|
|
20
|
Li Y, Zeng Y, Mooney SM, Yin B, Mizokami
A, Namiki M and Getzenberg RH: Resistance to paclitaxel increases
the sensitivity to other microenvironmental stresses in prostate
cancer cells. J Cell Biochem. 112:2125–2137. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Patel A, Kalachand R, Busschots S, Doherty
B, Kapros E, Lawlor D, Hall N and Stordal BK: Taxane monotherapy
regimens for the treatment of recurrent epithelial ovarian cancer.
Cochrane Database Syst Rev. 7:CD0087662022.PubMed/NCBI
|
|
22
|
Wang L, Chen H, Wang F and Zhang X: The
development of peptide-drug conjugates (PDCs) strategies for
paclitaxel. Expert Opin Drug Deliv. 19:147–161. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wender PA, Galliher WC, Bhat NM, Pillow
TH, Bieber MM and Teng NNH: Taxol-oligoarginine conjugates overcome
drug resistance in-vitro in human ovarian carcinoma. Gynecol Oncol.
126:118–123. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nakamura J, Nakajima N, Matsumura K and
Hyon SH: Water-soluble taxol conjugates with dextran and targets
tumor cells by folic acid immobilization. Anticancer Res.
30:903–909. 2010.PubMed/NCBI
|
|
25
|
Falah M, Rayan M and Rayan A: A novel
paclitaxel conjugate with higher efficiency and lower toxicity: A
new drug candidate for cancer treatment. Int J Mol Sci.
20:49652019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shelanski ML, Gaskin F and Cantor CR:
Microtubule assembly in the absence of added nucleotides. Proc Natl
Acad Sci USA. 70:765–768. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee JC and Timasheff SN: In vitro
reconstitution of calf brain microtubules: Effects of solution
variables. Biochemistry. 16:1754–1764. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu X, Sun J, Chen X, Wang S, Scott H,
Zhang X and Zhang Q: Pharmacokinetics, tissue distribution and
anti-tumour efficacy of paclitaxel delivered by
polyvinylpyrrolidone solid dispersion. J Pharm Pharmacol.
64:775–782. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shin HC, Cho H, Lai TC, Kozak KR, Kolesar
JM and Kwon GS: Pharmacokinetic study of 3-in-1 poly(ethylene
glycol)-block-poly(D, L-lactic acid) micelles carrying paclitaxel,
17-allylamino-17-demethoxygeldanamycin, and rapamycin. J Control
Release. 163:93–99. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Andersson BS and Way C: Parenternal
Paclitaxel in a stable non-toxic formulation. Patent US5877205A.
Filed June 28 1996; issued March 2, 1999.
|