Introduction
Colorectal cancer (CRC) is the third most common
cause of cancer-related mortalities worldwide. The conventional
therapies of CRC start with surgical resection followed by adjuvant
therapies, such as radiotherapy and chemotherapy (1,2).
Oxaliplatin (OX), one of the most used chemotherapeutic agents in
CRC treatment, is a platinum-based antineoplastic agent that
promotes apoptosis of tumor cells by blocking DNA replication and
transcription processes. Empirical evidence indicates that
resistance to OX commonly emerges during treatment, thereby
undermining the efficacy of therapeutic interventions (3,4). The
main causes for the progression of drug resistance depend on
regulations of molecular mechanisms such as apoptosis, DNA damage
repair, autophagy, drug uptake and epithelial-mesenchymal
transition (EMT) (5). EMT is a
biological process in which epithelial cells undergo a phenotypic
transformation into a mesenchymal state, and this holds critical
relevance in cancer progression, as neoplastic cells develop
characteristics linked to increased aggressiveness and drug
resistance, facilitating their dissemination (6). Previous studies have substantiated the
pivotal involvement of several established signaling pathways,
including TGF-β, PI3K/AKT, Ras/MAPK, Wnt/β-catenin, NF-κB and
Notch, in orchestrating EMT. Thus, it is imperative to direct
efforts toward identifying novel agents capable of reducing or
inhibiting EMT and its concomitant role in fostering resistance to
chemotherapy, including OX-based resistance (7–10).
Therefore, one strategy for overcoming the progression of drug
resistance is the targeting of EMT.
The insulin-like growth factor (IGF)-1 axis consists
of IGF-1, IGF-2, their receptors [mainly IGF-1 receptor (IGF-1R)]
and binding proteins. IGF-1 binds to its receptor, IGF-1R, a
receptor tyrosine kinase, which activates several downstream
signaling pathways, including the PI3K/AKT, Ras/MAPK, Wnt/β-catenin
and NF-κB pathways, all of which are crucial for cell survival,
proliferation and EMT (11–13).
The IGF-1 axis serves a pivotal role in promoting
EMT by activating key signaling pathways such as PI3K/AKT and
MAPK/ERK. The activation of AKT stabilizes transcription factors
such as Snail and Slug, which suppress epithelial markers and
induce mesenchymal traits, thus enhancing cell survival and
migration during EMT. Similarly, ERK activation contributes to EMT
by promoting mesenchymal marker expression and further repressing
E-cadherin, enhancing invasion and motility. The IGF-1 axis also
interacts with pathways such as TGF-β and Wnt/β-catenin, amplifying
EMT and driving more aggressive cancer behaviors (14,15).
Given its central role in metastasis and therapy resistance,
targeting the IGF-1 axis and related pathways offers potential for
therapeutic strategies in cancer treatment (16).
The activation of the IGF-1 axis occurs when IGFs
stimulate IGF-1R, which is overexpressed in several cancer types,
including CRC, promoting cell cycle progression and inhibiting
apoptosis (17–19). Several studies have demonstrated
elevated IGF-1R expression in colon adenocarcinoma tissues compared
with normal mucosa, linking this receptor to key cancer hallmarks,
such as proliferation and survival. In the study by Shiratsuchi
et al (20), 66% of patients
with CRC displayed positive IGF-1R expression, which was markedly
associated with tumor size and depth of invasion. Zhang et
al (21) reported that IGF-1R
gene expression was markedly higher in CRC samples compared with
the adjacent normal tissues, and their analysis revealed a strong
association between increased IGF-1R expression levels and key
clinicopathological features of CRC, including cancer stage and
metastasis. Furthermore, in murine models, overexpression of IGF-1R
in HCT116/IGF-1R cells led to highly invasive tumors and distant
metastases, such as parental cells (22). These findings indicate that IGF-1R
inhibitors hold potential as therapeutic strategies to inhibit
cancer progression, metastasis and EMT-related processes, as they
target pathways such as PI3K/AKT, Ras/MAPK and Wnt/β-catenin, all
of which are key factors in cancer development and drug
resistance.
Studies investigating the effects of these
inhibitors on colon cancer cells remain relatively limited,
although several research efforts have focused on targeting the IGF
signaling pathway using IGF-1R antagonists such as NVP-AEW541,
BMS-754807, OSI-906 (linsitinib), picropodophyllin (PPP), AMG 479
(ganitumab) and MK-0646 (dalotuzumab). These studies underscore the
potential therapeutic value of disrupting IGF-1R signaling in
cancer treatment, suggesting that these inhibitors may serve a
significant role in enhancing anticancer efficacy by inhibiting
tumor growth and overcoming resistance mechanisms (12,23–30).
For instance, NVP-AEW541, a small molecule IGF-1R inhibitor, was
tested in CRC cell lines, such as SW480 and HT-29. The inhibitor
effectively blocked IGF-1R signaling, reducing cancer cell
proliferation and inducing apoptosis. Additionally, it increased
sensitivity to chemotherapy agents, such as 5-fluorouracil, by
disrupting critical survival pathways, namely PI3K/AKT and MAPK
(23). Similarly, research by
Pennarun et al (24,25) reported that inhibiting IGF-1R
sensitized colon cancer cells to apoptosis mediated by death
receptor 5, enhancing apoptotic signaling and reducing cell
survival through inhibition of the PI3K/AKT pathway. Similarly,
BMS-754807, a dual IGF-1R/insulin receptor inhibitor, was reported
to induce cell cycle arrest and apoptosis in CRC cell lines, such
as HCT-116 (26,27). Its combination with cetuximab
enhanced anticancer effects, showing greater inhibition of cell
growth (26). OSI-906 has also
demonstrated strong inhibition of IGF-1R signaling, leading to
reduced tumor growth and increased apoptosis in both in
vitro and in vivo models (27). Furthermore, MK-0646 (dalotuzumab)
combined with OSI-906 has shown enhanced efficacy in CRC xenograft
models by reducing cell proliferation and tumor growth (28).
PPP, a selective inhibitor of IGF-1R, has shown
promising anticancer effects in several studies on CRC. In the
study by Lee et al (29),
PPP was tested on HCT-116 CRC cells, where it induced G1 phase cell
cycle arrest and triggered apoptosis through reactive oxygen
species generation. Additionally, PPP activated the p38 MAPK
signaling pathway, contributing to its anticancer effects.
Similarly, in the study by Sipos et al (30), PPP inhibited IGF-1R, leading to the
disruption of toll-like receptor 9 signaling and autophagy, which
reduced the survival of HT-29 CRC cells. Moreover, Feng et
al (12) reported that PPP,
whether used as a standalone treatment or in combination with other
therapies, blocked IGF-1R signaling, reducing cell viability and
inducing apoptosis in colon cancer cells. These findings underscore
the potential of PPP as a therapeutic agent for treating CRC by
targeting key survival and proliferation pathways.
There is limited literature on enhancing
chemotherapy responses through IGF-1R inhibition in
chemotherapy-resistant CRC cells. However, a study by Codony-Servat
et al (31) reported that
chemotherapy application increased IGF-1R expression, which was
notably elevated in OX-resistant cells. These findings suggest that
IGF-1R may serve a critical role in the development of chemotherapy
resistance and that targeting IGF-1R could be a potential strategy
for enhancing treatment efficacy in resistant CRC cells.
Due to the marked involvement of the IGF axis in
both drug resistance and EMT, and in EMT-associated drug
resistance, blocking IGF-1 signaling may reduce EMT-based
phenotypes and resensitize tumors to therapy. However, there is a
scarcity of studies investigating agents targeting IGF-1R to
enhance the efficacy of chemotherapy in chemotherapy-resistant
cancers (32–34). Therefore, the aim of the present
study was to develop a more effective treatment strategy for
OX-resistant CRC by combining PPP, an IGF-1R inhibitor, with OX.
The rationale for this approach was based on the notable role that
the IGF-1 axis serves in cancer progression, metastasis and
resistance to chemotherapy, particularly through its involvement in
EMT and EMT-associated drug resistance.
Materials and methods
Cell culture
Colorectal carcinoma HCT-116 cells (CCL-247;
American Type Culture Collection) and OX-resistant colorectal
carcinoma cells (named as HCT116-R), were cultured in DMEM
(Cegrogen Biotech GmbH) supplemented with 10% fetal bovine serum
(Cegrogen Biotech GmbH), 1% penicillin/streptomycin (Cegrogen
Biotech GmbH) and incubated at 5% CO2 and 37°C in a
humidified incubator. To generate OX-resistant HCT-116-R cells,
parental HCT-116 cells were exposed to gradually increasing
concentrations of OX (cat. no. O9512; Sigma-Aldrich; Merck KGaA)
(1–52 µM), over a 6-month period, established as described in our
recent study (10). During the
resistance development process, to maintain the balance between
proliferation and cell death, cells were cultured in culture media
supplemented with gradually increasing doses of OX (1–52 µM) at 5%
CO2 and 37°C in a humidified incubator. After each OX
treatment, the cells were maintained in drug-free culture media for
1–2 days to allow recovery and support the development of
resistance. The characterization of resistance was confirmed by the
higher IC50 value of the resistant cells compared with
that of the parental cells, along with an increased expression of
mesenchymal marker vimentin in the resistant cells relative to the
parental cells as assessed by immunofluorescence staining (10).
Cell viability assay
Resazurin, a cell-permeable blue dye, was utilized
as a redox indicator to assess cell viability and evaluate the
effects of treatment conditions on the HCT116 and HCT116-R cells
(10,35,36).
Viable cells with active metabolism reduce resazurin to red
fluorescent resorufin through the action of mitochondrial
dehydrogenase enzymes (35). To
determine cell viability, resazurin salt (cat. no. 14322; Cayman
Chemical Company) diluted in phosphate buffered saline (PBS)
(Cegrogen Biotech GmbH) was added to the culture medium of cells
treated with OX and/or PPP (cat. no. S7668; Selleck Chemicals) (1
µM) for 48 h at 37°C. The cells were then incubated at 37°C for 4
h. Fluorescence changes in the medium were measured using a
microplate reader (Ex/Em 570/580 nm; Varioskan™ LUX Multimode
Microplate Reader; Thermo Fisher Scientific, Inc.). The viability
of the control group was set to 100%, and the viability of treated
groups was calculated relative to the control.
Determination of OX and PPP
concentrations
OX was prepared by dissolving it in saline to a
concentration of 5 mg/ml and stored at 4°C. PPP was dissolved in
dimethyl sulfoxide (DMSO) (cat. no. D4540; Sigma-Aldrich; Merck
KGaA) to a final concentration of 82 mg/ml, and stored at
−80°C.
In the context of the present study, the combination
of PPP and OX involved the determination of the non-toxic maximal
concentration of PPP and IC50 of OX in both parental and
OX-resistant CRC cells. For IC50 determination, HCT116
cells were treated with varying concentrations of OX at 3, 6, 12,
24, 48, 96 and 192 µM at 37°C (33–35).
Meanwhile HCT116-R cells were exposed to OX at 100, 200, 300, 400,
500, 600 and 700 µM for 48 h at 37°C (37–39).
To determine the non-toxic highest concentration for PPP, cells
were exposed to concentrations of 1, 2, 3, 4, 5, 6 and 7 µM PPP for
48 h at 37°C (40–42). To assess the effect of PPP on
enhancing OX sensitivity, OX and PPP were used as single agents and
in combination in both HCT116 and HCT116-R cells. The experimental
design involved treating both HCT116 parental and HCT116-R
OX-resistant CRC cells with OX and/or PPP at varying
concentrations. For the HCT116 parental cells, the treatment groups
included a Control (untreated), 53 µM OX alone, 1 µM PP alone, and
a combination of 53 µM OX + 1 µM PPP. In the HCT116-R OX-resistant
cells, the groups consisted of a Control (untreated), 53 µM OX
alone, a combination of 53 µM OX + 1 µM PPP, 1 µM PPP alone, 324 µM
OX alone, and a combination of 324 µM OX + 1 µM PPP.
To evaluate the potential of enhancing chemotherapy
efficacy through the use of lower doses of OX in the presence of
PPP, combination studies were performed in OX-resistant HCT116-R
cells, using both the OX IC50 value determined for
resistant cells (324 µM), and the OX IC50 value
determined for parental HCT116 cells (53 µM) in combination with
PPP (1 µM). The aim of this strategy was to provide insights into
the ability of PPP to increase sensitivity to OX, potentially
enabling the use of lower chemotherapy doses and thereby reducing
treatment-related side effects.
Wound healing analysis
A wound-healing model was used to assess the
migratory capacities of HCT116 and HCT116-R cells. Once the seeded
cells reached 80–90% confluence in 24-well plates, a scratch was
generated in the center of the well using a pipette tip. After
washing with PBS, HCT116 and HCT116-R cells were treated with one
of the following: 1 µM PPP, OX (53 or 324 µM), or a combination of
OX (53 or 324 µM) + PPP (1 µM). Subsequently, the migration of
cells on either side of the scratch into the open space was
monitored under an inverted light microscope (Axio Vert.A1; Zeiss
GmbH) at 0 and 48 h (12). Cells
were serum-starved throughout the assay. Wound closure was
quantified using Image J Software version 1.53 (National Institutes
of Health). The wound area at each time point was measured by
manually outlining the scratch boundaries using the freehand
selection tool in ImageJ (version 1.54h; National Institutes of
Health), and the open wound area was quantified in mm after setting
the appropriate scale using the 50 µm scale bar present in the
original wound healing images. The percentage of wound closure was
calculated using the following formula: % Wound closure=[Wound
opening (t0) - Wound opening (tx)*]/Wound opening (t0) ×100, where
* refers to 48 h.
Immunofluorescence staining for
epithelial-mesenchymal markers
Immunofluorescence staining was used to assess the
EMT specific E-cadherin (epithelial) and vimentin (mesenchymal)
markers on HCT116 and HCT116-R cells. The seeded cells treated with
1 µM PPP, OX (53 or 324 µM), or a combination of OX (53 or 324 µM)
+ PPP (1 µM) for 48 h at 37°C. After fixation with 4%
paraformaldehyde (Thermo Fisher Scientific, Inc.) at room
temperature for 20 min, the cells were permeabilized with 0.1%
Triton X100 (Thermo Fisher Scientific, Inc.) solution in 1X PBS for
5 min at room temperature. To prevent non-specific antibody
binding, the cells were blocked with 1% bovine serum albumin (BSA)
(cat. no. D0050-4090; Cegrogen Biotech) in 1X PBS for 30 min at
room temperature. Permeabilized cells were incubated overnight at
4°C with primary antibodies against E-cadherin (cat. no. 14472S;
1:250; Cell Signaling Technology, Inc.) and vimentin (cat. no.
NB300-223; 1:250; Novus Biologicals, LLC; Bio-Techne) in the
staining buffer (1% BSA diluted in 1X PBS). The cells were then
incubated at room temperature for 1 h with secondary antibodies,
including Alexa Fluor™-488-goat anti-mouse (cat. no. ab150171;
1:1,000; Abcam) and Alexa Fluor-647 goat anti-chicken (cat. no.
4408S; 1:1,000; Cell Signaling Technology, Inc.) in PBS.
Subsequently, the cells were washed with 1X PBS and incubated for
10 min at room temperature with nucleus stain DAPI (1:1,000; Thermo
Fisher Scientific, Inc.). The stained cells were observed using a
confocal microscope (Zeiss, LSM 800 Confocal, USA), and
fluorescence intensities were analyzed with the accompanying
microscope software [Zen 2.1 (Blue Edition); Zeiss AG].
Statistical analysis
All experiments were performed with ≥3 biological
and technical replicates. The collected data was subjected to
comprehensive analysis and visualization using GraphPad Prism
Version 10.3.1 (Dotmatics). The data are presented as mean ±
standard deviation. Statistical significance between two
experimental groups was determined using the nonparametric
Mann-Whitney U test, whilst multiple comparisons were analyzed
using the Mann-Whitney U test followed by Bonferroni's correction.
The use of this nonparametric test was justified by assessing data
normality using the Shapiro-Wilk test, which confirmed that the
data did not follow a normal distribution. P<0.05 was considered
to indicate a statistically significant difference.
Results
Established concentrations for OX, PPP
and combined therapy
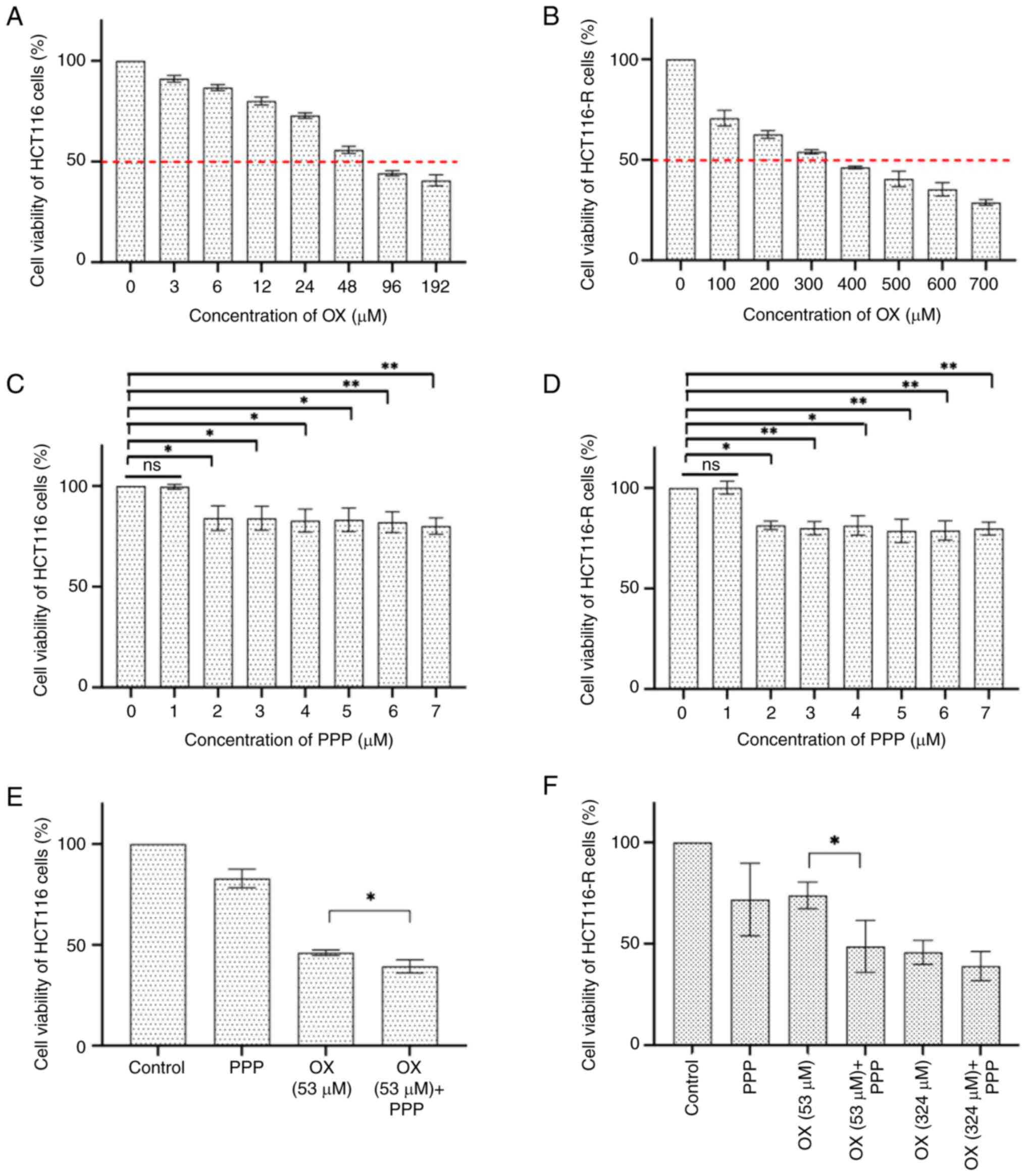
The collected data revealed that the IC50
values of OX for HCT116 (Fig. 1A)
and HCT116-R (Fig. 1B) cells were
53 and 324 µM, respectively. Furthermore, to evaluate the potential
alteration of OX efficacy without interference from PPP on cell
viability, the highest non-toxic highest concentration of PPP was
determined for both HCT116 (Fig.
1C) and HCT116-R cells (Fig.
1D) by applying PPP at a concentration range of 1–7 µM. PPP
treatment resulted in a dose-dependent reduction in cell viability
in both HCT116 and HCT116-R cells. In HCT116 cells, no significant
difference was observed between the control and 1 µM PPP groups
(P>0.999). However, a significant decrease in cell viability was
demonstrated at 2 µM (P=0.0373), 3 µM (P=0.0385), 4 µM (P=0.0137),
5 µM (P=0.0200), 6 µM (P=0.0072) and 7 µM (P=0.0018), compared with
the control. Similarly, in HCT116-R cells, 1 µM PPP did not induce
a significant change (P>0.999), whilst a significant reduction
in cell viability was demonstrated at 2 µM (P=0.049), 3 µM
(P=0.0060), 4 µM (P=0.0127), 5 µM (P=0.0012), 6 µM (P=0.0012) and 7
µM (P=0.0027). These results suggest a dose-dependent effect of PPP
on both HCT116 and HCT116-R cells, with cytotoxicity observed at
concentrations of ≥2 µM when each PPP concentration group was
compared separately with the control group. Therefore, the highest
non-toxic concentration for PPP was determined to be 1 µM, which
was used in combination with OX in subsequent experiments.
PPP potentiates OX sensitivity in
resistant CRC cells
To assess the effect of PPP (1 µM) on enhancing OX
sensitivity, the determined OX IC50 value (53 µM) in
HCT116 parental cells was used in combination therapy.
Additionally, two different combination strategies were applied in
OX-resistant HCT116-R cells. First, the OX IC50 dose
(324 µM) for HCT116-R cells was combined with 1 µM PPP in resistant
cells. Second, to assess the effect of 1 µM PPP at lower OX
concentrations, the combination of 53 µM OX + 1 µM PPP was also
evaluated. The aim of this approach was to provide insight into the
potential of using lower doses of OX in the presence of PPP to
enhance chemotherapy efficacy, and the findings suggest a treatment
strategy that could improve the effectiveness of low-dose
chemotherapy whilst reducing side effects.
The combined treatment of 53 µM OX + 1 µM PPP in
HCT116 cells was associated with a reduction in cell viability
compared with 53 µM OX treatment alone (P=0.0286; Fig. 1E). Moreover, treatment of HCT116-R
cells with 53 µM OX demonstrated a marked reduction in cell
viability compared with the group treated with 53 µM OX alone
(0.65-fold decrease; P=0.029; Fig.
1F). In HCT116 cells, the combined treatment of 324 µM OX + 1
µM PPP also showed a similar trend of reduced viability compared
with 324 µM OX alone, indicating the potential sensitizing effect
of 1 µM PPP on OX treatment, although statistical significance was
not reached (P>0.05; Fig.
1F).
PPP attenuates migration in resistant
CRC cells
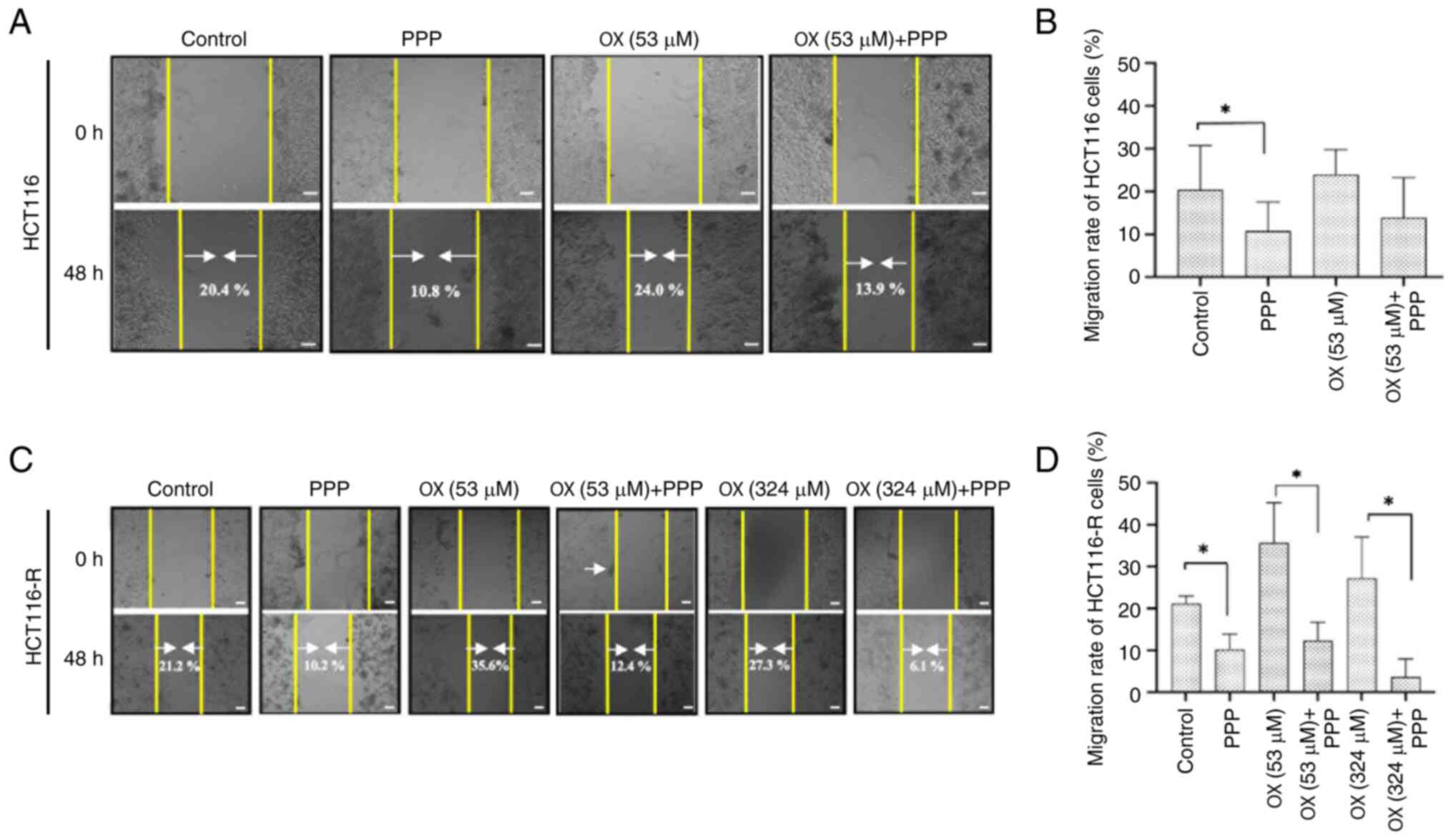
Wound healing assays were performed to assess the
migration of HCT116 and HCT116-R cells. In HCT116 cells, wound
closure (migration rate) was 20.4±12.57% in the control group,
23.98±7.11% in the OX (53 µM) group, 10.80±8.30% in the PPP (1 µM)
group and 13.89±11.42% in the 53 µM OX + 1 µM PPP group (Fig. 2A and B). The PPP (1 µM) group showed
a significant reduction in wound closure compared with that in the
control group (P=0.020), demonstrating that PPP (1 µM) alone could
limit cell migration. In the 53 µM OX + 1 µM PPP group, wound
closure was decreased further compared with OX alone, indicating
that the combination more effectively impaired migration; however,
this reduction was not statistically significant (P=0.200).
In HCT116-R cells, wound closure in the control, PPP
(1 µM), OX (53 µM), 53 µM OX + 1 µM PPP, OX (324 µM) and 324 µM OX
+ 1 µM PPP groups were determined to be 21.17±2.21, 10.22±4.49,
35.64±11.74, 12.35±5.37, 27.27±12 and 6.05±2.69%, respectively
(Fig. 2C and D). PPP (1 µM) alone
significantly reduced migration in comparison with the control
group, indicating its effectiveness in limiting migration in
resistant cells (P=0.028). The combination treatment, 53 µM OX + 1
µM PPP, further significantly decreased the migration rate in
comparison with OX (53 µM) alone (0.34-fold decrease; P=0.029).
Moreover, the combination of 324 µM + 1 µM PPP resulted in a
significantly lower wound closure percentage compared with OX (324
µM) alone, highlighting the greater degree of migration reduction
achieved using the combination therapy (0.22-fold decrease;
P=0.028). Overall, the combination of OX (53 µM or 324 µM) and PPP
(1 µM) reduced the migration rate of HCT116-R cells compared with
OX (52 µM or 324 µM) alone (P<0.05), indicating that PPP
effectively impairs the migration of OX-resistant cells, even at
different concentrations of OX, making it a promising adjunct in
overcoming the migration of OX-resistant CRC cells.
PPP preserves the epithelial phenotype
against the mesenchymal phenotype in resistant CRC cells
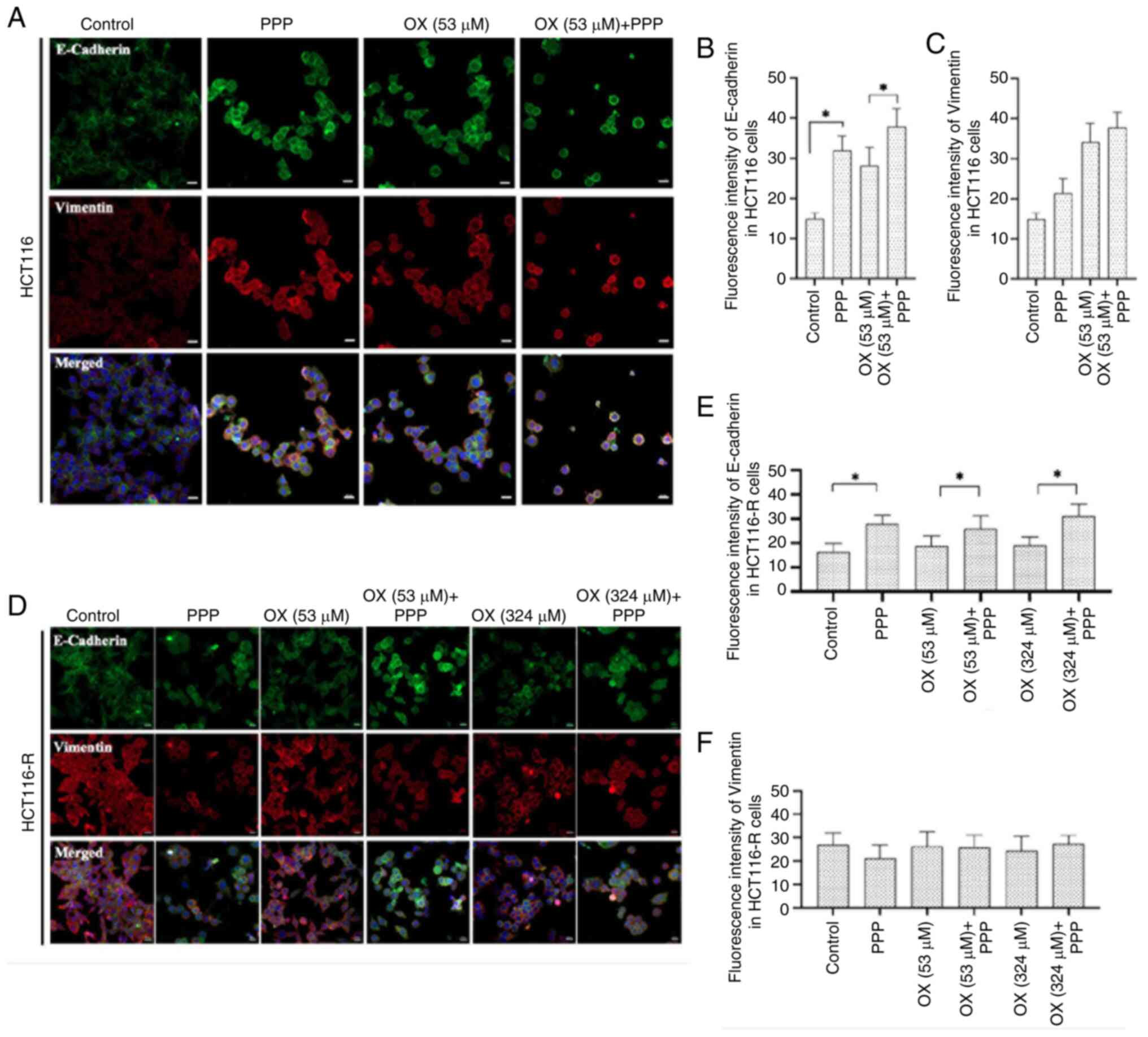
Immunofluorescence staining was performed on HCT116
and HCT116-R cells to evaluate the expression levels of epithelial
biomarker E-cadherin and the mesenchymal biomarker vimentin
following single and combination treatments.
The fluorescence intensity of E-cadherin in HCT116
cells was significantly increased in the combination of 53 µM OX +
1 µM PPP group compared with that in the OX (53 µM) group alone
(P<0.001), suggesting a synergistic effect of the combination
treatment in promoting epithelial characteristics. The PPP (1 µM)
group also demonstrated significantly increased fluorescence
intensity for E-cadherin expression compared with the control
(P<0.001) (Fig. 3A and B).
Furthermore, the combination of 53 µM OX + 1 µM PPP revealed a
slight but statistically insignificant increase in the fluorescence
intensity of vimentin levels compared with OX (53 µM) treatment
alone (P>0.05). Treatment with PPP (1 µM) also slightly
increased vimentin levels compared with the control, but the change
was not statistically significant (P>0.05) (Fig. 3C).
In HCT116-R cells, the 53 µM OX + 1 µM PPP group
demonstrated a statistically significant increase in E-cadherin
levels compared with the OX (53 µM) group alone (1.37-fold
increase; P=0.0104). This indicates the ability of PPP to enhance
epithelial marker expression in resistant cells even with low doses
of OX (Fig. 3D and E). The 324 µM
OX + 1 µM PPP group also demonstrated a significant increase in
E-cadherin expression compared with the OX (324 µM) group alone
(1.63-fold increase; P=0.001). This result suggests that PPP (1 µM)
can enhance the ability of OX to induce epithelial features in
resistant cells at a higher dose of OX. The PPP (1 µM) group also
revealed significantly increased fluorescence intensity for
E-cadherin expression compared with the control group (P<0.001).
However, the fluorescence intensity of vimentin in HCT116-R cells
demonstrated no significant differences between the following
groups: Control and PPP; OX (53 µM) and 53 µM OX + 1 µM PPP; and OX
(324 µM) and 324 µM OX + 1 µM PPP (P>0.05). The expression
levels of vimentin remained consistent across all treatment groups,
suggesting that PPP does not affect its expression in the presence
or absence of OX in HCT116-R cells (Fig. 3F).
The aforementioned findings suggest that PPP induces
an enhancement in epithelial characteristics in both HCT116 and
HCT116-R cells and supports the potential use of PPP (1 µM) to
improve the efficacy of OX in CRC treatment.
Discussion
Despite advancements in novel treatments, OX-based
chemotherapy combinations continue to serve a key role in CRC
treatment. The major issue in the OX treatment of CRC is acquired
resistance. The IGF-1R signaling axis serves a critical role in the
proliferation and growth of tumor cells in an EMT-based metastatic
phenotype. Different studies have reported that IGF-1 production
and IGF-1R expression can inhibit cell death and cause drug
resistance by upregulating survival protein in cancers. As EMT
serves a crucial role in acquired resistance, there is an
increasing trend towards agents that increase treatment sensitivity
by targeting EMT inhibition (16,43–45).
In line with this, the present study aimed to propose a novel
combination strategy using an IGF-1R inhibitor with the aspect of
proliferation, drug resistance and EMT-supportive IGF-1R signaling,
to increase the chemotherapeutic efficacy of OX-resistant CRC.
Based on the results, the present study aimed to propose a more
effective treatment strategy by using PPP in combination with OX to
reduce the proliferative and EMT-based aggressive phenotype of
OX-resistant CRC cells. We hypothesized that, by targeting IGF-1R,
PPP may inhibit the proliferation and EMT-driven aggressiveness of
OX-resistant CRC cells, thus re-sensitizing the tumors to
chemotherapy and enhancing treatment efficacy. This dual approach
seeks to overcome chemotherapy resistance, which is a major
obstacle in CRC treatment, by attacking both survival and drug
resistance pathways.
IGF-1R proteins have emerged as important targets
for cancer therapy. Previous studies have been performed with
IGF-1R-targeted agents such as NVP-AEW541, OSI-906 (linsitinib),
PPP, AMG 479 (ganitumab) and MK-0646 (dalotuzumab) in several human
cancers, showing that these compounds can inhibit tumor cell growth
and induce apoptosis (12,23–30).
However, the substantial role of the IGF axis in both drug
resistance and the EMT has led to a notable lack of studies
exploring agents that target the IGF-1R to augment the
effectiveness of chemotherapy in chemotherapy-resistant cancers
(32–34,46).
Therefore, the present study specifically focused on assessing the
impact of PPP in combination with OX to enhance drug sensitivity
and attenuate the drug resistance-promoting EMT phenotype in
OX-resistant CRC cells.
Initially, the aim of the present study was to
determine the highest non-toxic dose of PPP to prevent any
PPP-induced cytotoxicity when utilized in combination with OX,
forming part of the combination therapeutic strategy. Feng et
al (12) investigated the
anticancer effect of PPP in colon carcinoma cell lines and reported
a net reduction in viable cells when treated with PPP at
concentrations ≥1 µM. Another study by Wang et al (41) investigated the therapeutic response
of PPP in colorectal carcinoma, and reported that PPP markedly
suppressed the growth of CRC cells at a concentration of 1 µM. The
analysis in the present study demonstrated that 2 µM PPP affected
cell viability, therefore 1 µM PPP was considered as the highest
possible concentration that had no cytotoxic effect on CRC cell
lines. Furthermore, the present study used the maximum non-toxic
concentration of PPP in combination with OX to enhance OX
sensitivity and overcome OX-resistance. In the present study,
cytotoxic effects were observed at concentrations >1 µM which is
consistent with previous studies that reported a reduction in cell
viability at concentrations ≥1 µM (12,14).
These findings suggested that PPP begins to exhibit cytotoxic
effects at concentrations ≥1 µM.
Although OX is a standard treatment option in CRC,
it leads to toxicity (especially peripheral neurotoxicity), a
serious dose-limiting problem in the clinic. Due to the OX-related
side effects and toxicities, the treatment may need to be
interrupted before remission can be achieved (47). Therefore, the dosage should be
decreased for OX to prevent other toxicities in healthy tissue, but
this decrease will affect other parameters, the most crucial being
the treatment capacity. Therefore, PPP could be used as an adjuvant
agent at its nontoxic concentration to enhance the treatment effect
of OX and provide high efficacy in low dosages by avoiding
OX-related healthy tissue toxicities in patients with CRC.
To evaluate the impact of PPP on enhancing
sensitivity to OX in CRC, the present study used the
IC50 value of OX (53 µM) in HCT116 parental cells for
combination therapy. In OX-resistant HCT116-R cells, two different
combination strategies were tested. First, the IC50 dose
of OX for resistant cells (324 µM) was combined with PPP (1 µM) to
assess the potential of overcoming resistance. Second, to assess
the effect of PPP (1 µM) at lower OX concentrations, OX was tested
at the lower parental IC50 value (53 µM) in combination
with PPP (1 µM). This latter approach aimed to explore whether PPP
(1 µM) could enhance OX efficacy at lower drug concentrations,
potentially improving treatment outcomes while minimizing side
effects. The results of this strategy offer a promising avenue for
reducing chemotherapy dosages, minimizing toxicity, whilst
maintaining or enhancing therapeutic efficacy in resistant cancer
cells.
The results of the present study demonstrate that
PPP significantly enhances the cytotoxic effects of OX in both
HCT116 and OX-resistant HCT116-R CRC cells. In non-resistant HCT116
cells, the combination of 53 µM OX + 1 µM PPP led to a
statistically significant decrease in cell viability compared with
OX alone. Similarly, in resistant HCT116-R cells, the same
combination further reduced cell viability, indicating that PPP
improves sensitivity to OX. Whilst the higher concentration of OX
(324 µM) combined with PPP (1 µM) showed a trend toward greater
cytotoxicity in resistant cells, this difference was not
statistically significant. Overall, PPP appeared to enhance the
effectiveness of OX, particularly at lower doses.
The results of the current study, namely, that PPP
enhances the cytotoxic effects of OX, are consistent with prior
research highlighting the therapeutic potential of PPP. PPP has
been widely recognized for its role as a selective IGF-1R
inhibitor, effectively disrupting cancer cell survival pathways,
such as the PI3K/AKT and MAPK/ERK pathways, which are often
upregulated in chemotherapy-resistant cancer cells. Feng et
al (12) demonstrated that PPP
effectively inhibits IGF-1R signaling, leading to reduced viability
and increased apoptosis in colon cancer cells, both as a standalone
treatment and in combination with other therapies. Similar to the
findings of the present study, Lee et al (29) reported that PPP enhances the
anticancer effects of chemotherapeutic agents by inducing apoptosis
and inhibiting cell proliferation in CRC cells, further supporting
the synergistic effect observed in the combination of OX and PPP.
Moreover, the finding that PPP increases OX sensitivity at both
lower (53 µM) and higher (324 µM) concentrations aligns with study
by Sipos et al (30) showing
that PPP can overcome drug resistance mechanisms, particularly in
cells exhibiting an EMT phenotype, which is closely associated with
chemotherapy resistance. The previous study demonstrated that PPP
disrupts EMT signaling and autophagy, reducing the survival of
chemo-resistant CRC cells, further supporting the potential role of
PPP in combination therapies targeting drug-resistant cancers.
To the best of our knowledge, there are no studies
specifically addressing the combination of PPP with chemotherapy in
CRC, but a limited number of studies focus on combination therapies
involving PPP in other types of cancer. Tarnowski et al
(40) evaluated the effects of PPP
in combination with actinomycin-D and cisplatin in rhabdomyosarcoma
cells and demonstrated that PPP increased the sensitivity to
chemotherapy. Moreover, in another study by Duan et al
(48), PPP was reported to enhance
the cytotoxic effects of doxorubicin in osteosarcoma cell lines
that had developed resistance to doxorubicin. In addition,
downregulation of IGF-1R expression in drug-resistant cell lines by
small interfering RNA led to the restoration of sensitivity to
doxorubicin (48). Therefore, PPP
holds promise as a potent adjunct to OX in treating chemo-resistant
CRC, potentially improving patient outcomes by sensitizing tumors
to lower doses of chemotherapy, reducing both drug toxicity and
resistance (27,49).
The wound healing assays in the present study
demonstrated that PPP effectively reduces the migration of both
HCT116 and OX-resistant HCT116-R CRC cells. The findings,
demonstrating that PPP was associated with a significant reduction
in the migration of both types of CRC cells, align with earlier
research that highlights the ability of PPP to impair cancer cell
migration and metastasis. For example, Feng et al (12) reported that PPP reduced the
migration of HCT116 cells in a dose-dependent manner. This is
consistent with the results of the present study, where PPP reduced
migration in combination with OX, especially in resistant cells.
Additionally, whilst the combination of PPP and OX (53 µM) led to a
reduction in migration in HCT116 cells compared with OX alone, this
decrease did not reach statistical significance. By contrast, the
combination of PPP with either low-dose OX (53 µM) or high-dose OX
(324 µM) led to a statistically significant reduction in migration
in HCT116-R cells, compared with OX alone (P<0.05). This
suggests that PPP not only enhances the cytotoxic effects of OX,
but also significantly reduces the migratory potential of
OX-resistant CRC cells, making it a promising adjunct for
overcoming their migratory capacity. The observed differences in
the effects of the combinatorial use of PPP with OX on HCT116 and
HCT116-R cells may be attributed to two main factors. Firstly, the
wound healing assay used in the present study may lack the
sensitivity to detect subtle changes in migration, particularly in
non-resistant HCT116 cells, where the migration rate might not be
significantly altered by PPP and OX under these specific
experimental conditions. Secondly, biological mechanisms likely
serve a role in these differences. Non-resistant HCT116 cells may
lack EMT-related changes, which are commonly associated with
increased migration and invasiveness (50). By contrast, HCT116-R cells, which
are resistant to OX, exhibit significant differences in migration,
suggesting the involvement of EMT or other resistance-associated
mechanisms in driving migration. These findings highlight the
distinct biological and functional characteristics of resistant and
non-resistant cell populations and underscore the potential of PPP
in targeting migration in chemo-resistant cancer cells.
The paradoxical finding that OX alone increased the
migration of resistant cells, potentially through mechanisms
related to EMT, is supported by previous studies. EMT is often
linked to increased cell migration and metastasis, particularly in
chemo-resistant cancers, where it enhances the invasiveness of
tumor cells (50–52). Previous studies have demonstrated
that IGF-1R inhibition, as achieved through PPP, can suppress EMT
and reduce the metastatic potential of cancer cells (29). This reversal of OX-induced migration
by PPP in resistant cells further underscores its potential role as
an adjunct therapy to prevent metastasis-related phenotypes in
OX-resistant CRC. Moreover, similar findings from Sipos et
al (30) reinforce the
hypothesis that PPP can disrupt EMT processes by targeting IGF-1R
signaling, thereby reducing drug resistance and metastatic behavior
in cancer cells. By targeting these pathways, PPP not only enhances
the cytotoxicity of OX but also limits the metastatic potential,
making it a promising approach for overcoming the dual challenges
of drug resistance and metastasis in CRC treatment.
The immunofluorescence results of the present study,
which demonstrated that PPP preserved the epithelial phenotype in
both HCT116 and OX-resistant HCT116-R CRC cells by promoting
E-cadherin expression, align with prior research that highlights
the ability of PPP to influence EMT markers. In the present study,
PPP was demonstrated to significantly increase E-cadherin levels,
an important epithelial marker, without significantly affecting the
mesenchymal marker vimentin, particularly when combined with OX.
These findings are consistent with reports by Lee et al
(29) and Feng et al
(12), who reported that PPP not
only induces apoptosis, but also inhibits key EMT markers, thereby
promoting epithelial traits in cancer cells.
Previous studies have also reported that EMT is a
critical mechanism by which cancer cells acquire drug resistance
and increased metastatic potential (14). Sipos et al (30) reported that targeting the IGF-1R
pathway with PPP disrupted EMT signaling, reinforcing epithelial
characteristics such as E-cadherin expression, which corresponds
with the findings of the present study. The ability of PPP to
promote E-cadherin expression in both non-resistant and resistant
cell lines suggests that it could serve a vital role in combating
chemoresistance by inhibiting EMT, a key process involved in drug
resistance and metastasis. Additionally, Zhang et al
(21) reported that IGF-1R serves a
pivotal role in maintaining EMT traits, contributing to the
aggressive and resistant nature of CRC cells. The findings of the
present study, where PPP enhances E-cadherin expression without
significantly affecting vimentin, further support the notion that
targeting IGF-1R with PPP could potentially reverse EMT-associated
resistance and enhance the effectiveness of chemotherapy. In
congruence with these outcomes, Li et al (16,46)
aimed to enhance the drug sensitivity of epidermal growth factor
receptor-tyrosine kinase inhibitor-resistant non-small cell lung
cancer cells through the inhibition of insulin-such as growth
factor 1 receptor via PPP. The study reported a diminution in
mesenchymal markers including Snail, Slug, zinc finger E-box
binding homeobox 1 and Vimentin, and an elevation in the epithelial
marker E-cadherin following treatment with PPP.
The combination treatment of 53 µM OX + and 1 µM PPP
in parental HCT116 cells resulted in a slight, statistically
non-significant increase in vimentin fluorescence intensity
compared with OX treatment alone, whilst PPP (1 µM) alone also led
to a modest, non-significant increase relative to the control.
These findings may reflect inherent biological variability and the
limitations of a small sample size, potentially contributing to the
lack of statistical significance. Additionally, it is possible that
PPP may interact with alternative signaling pathways or
compensatory mechanisms, such as stress responses or overlapping
EMT pathways (such as, TGF-β/Smad and Wnt/β-catenin), which could
sustain or slightly enhance vimentin expression. Moreover, specific
genetic or epigenetic characteristics of the HCT116 cell line may
influence vimentin expression, making it less responsive to PPP. In
HCT116-R cells, vimentin fluorescence intensity did not demonstrate
significant changes across treatment groups, including PPP, OX and
their combinations. This lack of response may indicate the
possibility of intrinsic resistance in HCT116-R cells, potentially
involving a stabilized EMT phenotype or constitutive activation of
EMT pathways. Additionally, the doses of PPP and OX used may not
have been optimal for modulating vimentin expression, and cellular
heterogeneity or other regulatory mechanisms could contribute to
the observed outcomes. In summary, the preservation of epithelial
markers such as E-cadherin and the lack of significant changes in
mesenchymal traits such as vimentin upon PPP treatment,
particularly in combination with OX, suggest that PPP could be an
influential adjunct to chemotherapy by mitigating EMT-associated
drug resistance in CRC.
In concordance with analogous results from the
literature, the data from the present study indicate that PPP
markedly augments therapeutic efficacy in CRC by potentially
counteracting mechanisms of drug resistance. This underscores the
need for an innovative dual therapy strategy for the management of
OX-resistant CRC. The use of PPP in the treatment protocol is an
innovative strategy aimed at increasing the chemotherapy efficacy
of OX. This strategy aims to reverse the mesenchymal properties of
cancer cells by sensitizing them to OX. The proposed dual therapy
not only addresses the OX resistance in CRC but also offers a
promising way to improve the overall efficacy of chemotherapy in
this context.
However, the present study has several limitations
that need to be addressed in future research. First, the
experiments were performed solely on in vitro models using
two CRC cell lines, HCT116 and OX-resistant HCT116-R. This approach
may not fully capture the complexity and heterogeneity of tumors in
in vivo conditions. Moreover, there was no attempt to
account for the tumor microenvironment, which can notably influence
drug resistance and response (53,54).
Additionally, there is a need to further explore the long-term
effects of PPP on cell behavior and the molecular mechanisms
underlying its action, particularly regarding EMT and IGF-1R
signaling. Furthermore, vimentin may not be a primary marker
affected by PPP, which could instead predominantly target other
mesenchymal markers such as N-cadherin. Evaluating additional EMT
markers and increasing sample sizes are crucial to better
understand the broader effects of PPP on EMT regulation. Future
studies should include IGF-1R knockdown and overexpression
experiments to improve the understanding of the role of this
pathway in OX resistance. These experiments could provide deeper
insights into how IGF-1R signaling influences cancer cell survival,
migration and EMT. Additionally, future studies should include
in vivo models and a broader range of CRC subtypes to better
understand the efficacy of PPP in overcoming OX resistance.
Detailed mechanistic studies are also needed to provide a clearer
picture of how PPP modulates cancer cell survival, migration and
resistance pathways.
In summary, PPP has emerged as a promising
adjunctive agent to enhance the therapeutic efficacy of OX in CRC.
The present study highlights potential avenues for future research,
particularly in exploring novel treatment modalities. Specifically,
integrating EMT suppressive agents with conventional
chemotherapeutic approaches could further improve treatment
effectiveness. However, due to the complex and multifactorial
nature of CRC, numerous therapeutic targets remain to be explored
for optimizing therapeutic outcomes. Therefore, it is essential to
identify next-generation drug targets and refine current treatment
protocols to develop comprehensive therapeutic strategies aligned
that address the intricate pathogenesis of CRC.
Acknowledgements
The authors would like to thank Professor Hulya
Ellidokuz and Dr Mehmet Emin Arayici (Department of Biostatistics
and Medical Informatics, Faculty of Medicine, Dokuz Eylul
University, Izmir, Turkey) for their support in statistical
analysis, including data normalization assessment and selection of
appropriate statistical tests.
Funding
The present work was supported by the Dokuz Eylul University,
Scientific Research Projects Coordination Unit (grant no.
TYL-2022-2851). NK was supported by the TUBITAK in the field of
Biotechnological Pharmaceutical Technologies (scholarship no.
1649B022204091).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
NK, YB and GCK contributed to the conceptualization
and design of the study. NK, HK, TS and GCK were responsible for
data acquisition and analysis, while NK, HK, TS, YB and GCK
contributed to the interpretation of the data. All authors were
involved in drafting the manuscript and critically reviewing the
manuscript for intellectual content. All authors have reviewed and
approved the final version of the manuscript and agree to be
accountable for the accuracy and integrity of the work. NK, YB and
GCK confirm the authenticity of all raw data.
Ethics approval and consent to
participate
The present research, performed as a Master of
Science thesis, was approved by the Dokuz Eylül University
Non-Interventional Research Ethics Committee (14.07.2021; approval
no. 2021/21-13). Obtaining ethical approval is mandatory for all
thesis studies performed under the Dokuz Eylül University Institute
of Health Sciences. The present study did not involve the use of
human or animal materials, and all necessary institutional
permissions and ethical committee approvals were obtained.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
IGF
|
insulin-like growth factor
|
|
IGF-1R
|
IGF-1 receptor
|
|
PPP
|
picropodophyllin
|
|
OX
|
oxaliplatin
|
|
EMT
|
epithelial-mesenchymal transition
|
References
|
1
|
Hammond WA, Swaika A and Mody K:
Pharmacologic resistance in colorectal cancer: A review. Ther Adv
Med Oncol. 8:57–84. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Biller LH and Schrag D: Diagnosis and
treatment of metastatic colorectal cancer: A review. JAMA.
325:669–685. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li Y, Gan Y, Liu J, Li J, Zhou Z, Tian R,
Sun R, Liu J, Xiao Q, Li Y, et al: Downregulation of MEIS1 mediated
by ELFN1-AS1/EZH2/DNMT3a axis promotes tumorigenesis and
oxaliplatin resistance in colorectal cancer. Signal Transduct
Target Ther. 7:872022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jin Q, Feng J, Yan Y and Kuang Y:
Prognostic and immunological role of adaptor related protein
complex 3 subunit mu2 in colon cancer. Sci Rep. 14:4832024.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hashemi M, Esbati N, Rashidi M, Gholami S,
Raesi R, Bidoki SS, Goharrizi MASB, Motlagh YSM, Khorrami R,
Tavakolpournegari A, et al: Biological landscape and nanostructural
view in development and reversal of oxaliplatin resistance in
colorectal cancer. Transl Oncol. 40:1018462024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lu J, Kornmann M and Traub B: Role of
epithelial to mesenchymal transition in colorectal cancer. Int J
Mol Sci. 24:148152023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ho KH, Chen PH, Shih CM, Lee YT, Cheng CH,
Liu AJ, Lee CC and Chen KC: miR-4286 is involved in connections
between IGF-1 and TGF-β signaling for the mesenchymal transition
and invasion by glioblastomas. Cell Mol Neurobiol. 42:791–806.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sabbah M, Emami S, Redeuilh G, Julien S,
Prévost G, Zimber A, Ouelaa R, Bracke M, De Wever O and Gespach C:
Molecular signature and therapeutic perspective of the
epithelial-to-mesenchymal transitions in epithelial cancers. Drug
Resist Updat. 11:123–151. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ngo MT, Peng SW, Kuo YC, Lin CY, Wu MH,
Chuang CH, Kao CX, Jeng HY, Lin GW, Ling TY, et al: A
yes-associated protein (YAP) and insulin-like growth factor 1
receptor (IGF-1R) signaling loop is involved in sorafenib
resistance in hepatocellular carcinoma. Cancers (Basel).
13:38122021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kurter H, Basbinar Y, Ellidokuz H and
Calibasi-Kocal G: The Role of Cyanidin-3-O-glucoside in
modulating oxaliplatin resistance by reversing mesenchymal
phenotype in colorectal cancer. Nutrients. 15:47052023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hosseini SA, Zand H and Cheraghpour M: The
influence of curcumin on the downregulation of MYC, insulin and
IGF-1 receptors: A possible mechanism underlying the anti-growth
and anti-migration in chemoresistant colorectal cancer cells.
Medicina (Kaunas). 55:902019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Feng X, Aleem E, Lin Y, Axelson M, Larsson
O and Strömberg T: Multiple antitumor effects of picropodophyllin
in colon carcinoma cell lines: Clinical implications. Int J Oncol.
40:1251–1258. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pollak M: The insulin and insulin-like
growth factor receptor family in neoplasia: An update. Nat Rev
Cancer. 12:159–169. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nieto MA, Huang RY, Jackson RA and Thiery
JP: EMT: 2016. Cell. 166:21–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ianza A, Sirico M, Bernocchi O and
Generali D: Role of the IGF-1 axis in overcoming resistance in
breast cancer. Front Cell Dev Biol. 9:6414492021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li H, Batth IS, Qu X, Xu L, Song N, Wang R
and Liu Y: IGF-IR signaling in epithelial to mesenchymal transition
and targeting IGF-IR therapy: Overview and new insights. Mol
Cancer. 16:62017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Freier S, Weiss O, Eran M, Flyvbjerg A,
Dahan R, Nephesh I, Safra T, Shiloni E and Raz I: Expression of the
insulin-like growth factors and their receptors in adenocarcinoma
of the colon. Gut. 44:704–708. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nosho K, Yamamoto H, Taniguchi H, Adachi
Y, Yoshida Y, Arimura Y, Endo T, Hinoda Y and Imai K: Interplay of
insulin-like growth factor-II, insulin-like growth factor-I,
insulin-like growth factor-I receptor, COX-2, and matrix
metalloproteinase-7, play key roles in the early stage of
colorectal carcinogenesis. Clin Cancer Res. 10:7950–7957. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yamamoto N, Oshima T, Yoshihara K, Aoyama
T, Hayashi T, Yamada T, Sato T, Shiozawa M, Yoshikawa T, Morinaga
S, et al: Clinicopathological significance and impact on outcomes
of the gene expression levels of IGF−1, IGF−2 and
IGF-1R, IGFBP−3 in patients with colorectal cancer:
Overexpression of the IGFBP−3 gene is an effective predictor
of outcomes in patients with colorectal cancer. Oncol Lett.
13:3958–3966. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shiratsuchi I, Akagi Y, Kawahara A,
Kinugasa T, Romeo K, Yoshida T, Ryu Y, Gotanda Y, Kage M and
Shirouzu K: Expression of IGF-1 and IGF-1R and their relation to
clinicopathological factors in colorectal cancer. Anticancer Res.
31:2541–2545. 2011.PubMed/NCBI
|
|
21
|
Zhang Z, Zhang Y, Lao S, Qiu J, Pan Z and
Feng X: The clinicopathological and prognostic significances of
IGF-1R and Livin expression in patients with colorectal cancer. BMC
Cancer. 22:8552022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sekharam M, Zhao H, Sun M, Fang Q, Zhang
Q, Yuan Z, Dan HC, Boulware D, Cheng JQ and Coppola D: Insulin-like
growth factor 1 receptor enhances invasion and induces resistance
to apoptosis of colon cancer cells through the Akt/Bcl-x(L)
pathway. Cancer Res. 63:7708–7716. 2003.PubMed/NCBI
|
|
23
|
García-Echeverría C, Pearson MA, Marti A,
Meyer T, Mestan J, Zimmermann J, Gao J, Brueggen J, Capraro HG,
Cozens R, et al: In vivo antitumor activity of NVP-AEW541-A novel,
potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell.
5:231–239. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pennarun B, Kleibeuker JH, Oenema T,
Stegehuis JH, de Vries EG and de Jong S: Inhibition of
IGF-1R-dependent PI3K activation sensitizes colon cancer cells
specifically to DR5-mediated apoptosis but not to rhTRAIL. Anal
Cell Pathol (Amst). 33:229–244. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pennarun B, Kleibeuker JH, Oenema T,
Stegehuis JH, de Vries EG and de Jong S: Inhibition of
IGF-1R-dependent PI3K activation sensitizes colon cancer cells
specifically to DR5-mediated apoptosis but not to rhTRAIL. Cell
Oncol (Dordr). 34:245–259. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Carboni JM, Wittman M, Yang Z, Lee F,
Greer A, Hurlburt W, Hillerman S, Cao C, Cantor GH, Dell-John J, et
al: BMS-754807, a small molecule inhibitor of insulin-like growth
factor-1R/IR. Mol Cancer Ther. 8:3341–3349. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fuentes-Baile M, Ventero MP, Encinar JA,
García-Morales P, Poveda-Deltell M, Pérez-Valenciano E, Barberá VM,
Gallego-Plazas J, Rodríguez-Lescure Á, Martín-Nieto J and Saceda M:
Differential effects of IGF-1R small molecule tyrosine kinase
inhibitors BMS-754807 and OSI-906 on human cancer cell lines.
Cancers (Basel). 12:37172020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Leiphrakpam PD, Agarwal E, Mathiesen M,
Haferbier KL, Brattain MG and Chowdhury S: In vivo analysis of
insulin-like growth factor type 1 receptor humanized monoclonal
antibody MK-0646 and small molecule kinase inhibitor OSI-906 in
colorectal cancer. Oncol Rep. 31:87–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee SO, Kwak AW, Lee MH, Seo JH, Cho SS,
Yoon G, Chae JI, Joo SH and Shim JH: Picropodophyllotoxin induces
G1 cell cycle arrest and apoptosis in human colorectal cancer cells
via ROS generation and activation of p38 MAPK signaling pathway. J
Microbiol Biotechnol. 31:1615–1623. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sipos F, Bohusné Barta B, Simon Á, Nagy L,
Dankó T, Raffay RE, Petővári G, Zsiros V, Wichmann B, Sebestyén A
and Műzes G: Survival of HT29 cancer cells is affected by IGF1R
inhibition via modulation of Self-DNA-Triggered TLR9 signaling and
the autophagy response. Pathol Oncol Res. 28:16103222022.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Codony-Servat J, Cuatrecasas M, Asensio E,
Montironi C, Martínez-Cardús A, Marín-Aguilera M, Horndler C,
Martínez-Balibrea E, Rubini M, Jares P, et al: Nuclear IGF-1R
predicts chemotherapy and targeted therapy resistance in metastatic
colorectal cancer. Br J Cancer. 117:1777–1786. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sun R, Tanino R, Tong X, Haque EF, Amano
Y, Isobe T and Tsubata Y: Picropodophyllin inhibits the growth of
pemetrexed-resistant malignant pleural mesothelioma via microtubule
inhibition and IGF-1R-, caspase-independent pathways. Transl Lung
Cancer Res. 11:543–559. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Singh RK, Gaikwad SM, Jinager A, Chaudhury
S, Maheshwari A and Ray P: IGF-1R inhibition potentiates cytotoxic
effects of chemotherapeutic agents in early stages of
chemoresistant ovarian cancer cells. Cancer Lett. 354:254–262.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Du J, Shi HR, Ren F, Wang JL, Wu QH, Li X
and Zhang RT: Inhibition of the IGF signaling pathway reverses
cisplatin resistance in ovarian cancer cells. BMC Cancer.
17:8512017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Präbst K, Engelhardt H, Ringgeler S and
Hübner H: Basic colorimetric proliferation assays: MTT, WST, and
Resazurin. Methods Mol Biol. 1601:1–17. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ourhzif EM, Decombat C, Abrunhosa-Thomas
I, Delort L, Khouili M, Akssira M, Caldefie-Chezet F, Chalard P and
Troin Y: Synthesis and biological evaluation of new naphthoquinones
derivatives. Curr Org Synth. 17:224–229. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu T, Zhang X, Du L, Wang Y, Liu X, Tian
H, Wang L, Li P, Zhao Y, Duan W, et al: Exosome-transmitted
miR-128-3p increase chemosensitivity of oxaliplatin-resistant
colorectal cancer. Mol Cancer. 18:432019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Boot A, van Eendenburg J, Crobach S, Ruano
D, Speetjens F, Calame J, Oosting J, Morreau H and van Wezel T:
Characterization of novel low passage primary and metastatic
colorectal cancer cell lines. Oncotarget. 7:14499–14509. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
McCarthy B, Singh R and Levi-Polyachenko
N: Oxaliplatin-resistant colorectal cancer models for nanoparticle
hyperthermia. Int J Hyperthermia. 38:152–164. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tarnowski M, Tkacz M, Zgutka K, Bujak J,
Kopytko P and Pawlik A: Picropodophyllin (PPP) is a potent
rhabdomyosarcoma growth inhibitor both in vitro and in vivo. BMC
Cancer. 17:5322017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang Q, Wei F, Lv G, Li C, Liu T,
Hadjipanayis CG, Zhang G, Hao C and Bellail AC: The association of
TP53 mutations with the resistance of colorectal carcinoma to the
insulin-like growth factor-1 receptor inhibitor picropodophyllin.
BMC Cancer. 13:5212013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dong L, Du M and Lv Q: Picropodophyllin
inhibits type I endometrial cancer cell proliferation via
disruption of the PI3K/Akt pathway. Acta Biochim Biophys Sin
(Shanghai). 51:753–760. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kim SY, Toretsky JA, Scher D and Helman
LJ: The role of IGF-1R in pediatric malignancies. Oncologist.
14:83–91. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Oh SH, Jin Q, Kim ES, Khuri FR and Lee HY:
Insulin-like growth factor-I receptor signaling pathway induces
resistance to the apoptotic activities of SCH66336 (lonafarnib)
through Akt/mammalian target of rapamycin-mediated increases in
survivin expression. Clin Cancer Res. 14:1581–1589. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jones HE, Gee JM, Barrow D, Tonge D,
Holloway B and Nicholson RI: Inhibition of insulin receptor
isoform-A signalling restores sensitivity to gefitinib in
previously de novo resistant colon cancer cells. Br J Cancer.
95:172–180. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Li L, Gu X, Yue J, Zhao Q, Lv D, Chen H
and Xu L: Acquisition of EGFR TKI resistance and EMT phenotype is
linked with activation of IGF1R/NF-κB pathway in EGFR-mutant NSCLC.
Oncotarget. 8:92240–92253. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cheng F, Zhang R, Sun C, Ran Q, Zhang C,
Shen C, Yao Z, Wang M, Song L and Peng C: Oxaliplatin-induced
peripheral neurotoxicity in colorectal cancer patients: Mechanisms,
pharmacokinetics and strategies. Front Pharmacol. 14:12314012023.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Duan Z, Choy E, Harmon D, Yang C, Ryu K,
Schwab J, Mankin H and Hornicek FJ: Insulin-like growth factor-I
receptor tyrosine kinase inhibitor cyclolignan picropodophyllin
inhibits proliferation and induces apoptosis in multidrug resistant
osteosarcoma cell lines. Mol Cancer Ther. 8:2122–2130. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Arcaro A: Targeting the insulin-like
growth factor-1 receptor in human cancer. Front Pharmacol.
4:302013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yang AD, Fan F, Camp ER, van Buren G, Liu
W, Somcio R, Gray MJ, Cheng H, Hoff PM and Ellis LM: Chronic
oxaliplatin resistance induces epithelial-to-mesenchymal transition
in colorectal cancer cell lines. Clin Cancer Res. 12((14 Pt 1)):
4147–4153. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Skarkova V, Kralova V, Krbal L, Matouskova
P, Soukup J and Rudolf E: Oxaliplatin and irinotecan induce
heterogenous changes in the EMT markers of metastasizing colorectal
carcinoma cells. Exp Cell Res. 369:295–303. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Li X, Zhang ZS, Zhang XH, Yang SN, Liu D,
Diao CR, Wang H and Zheng FP: Cyanidin inhibits EMT induced by
oxaliplatin via targeting the PDK1-PI3K/Akt signaling pathway. Food
Funct. 10:592–601. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kurter H, Yesil J, Daskin E,
Calibasi-Kocal G, Ellidokuz H and Basbinar Y: Drug resistance
mechanisms on colorectal cancer. J Basic Clin Health Sci. 1:88–93.
2021. View Article : Google Scholar
|
|
54
|
Chen Y, Zheng X and Wu C: The role of the
tumor microenvironment and treatment strategies in colorectal
cancer. Front Immunol. 12:7926912021. View Article : Google Scholar : PubMed/NCBI
|