Introduction
Peripheral neuropathy is among the most common and
frequently encountered neurological conditions. Notably,
multiregional epidemiological studies have reported that the
prevalence of peripheral neuropathy increases with age, ranging
from 1–3% in the general population and increasing to 7% in
individuals >50 years of age (1). Although peripheral neuropathy is
highly prevalent, it can be triggered by a wide range of causes
(2). Clinically, neurologists often
manage diverse presentations of peripheral neuropathy, including
diabetic peripheral neuropathy indicated by foot numbness in
diabetes mellitus, chemotherapy-induced peripheral neuropathy in
patients with cancer, or Guillain-Barré syndrome marked by abrupt
limb weakness and respiratory difficulty (3). Notably, thorough screening of the
underlying causes is crucial in the accurate diagnosis and
effective treatment of peripheral neuropathy; however, determining
the cause can be challenging and the outcome is sometimes
unexpected (4). The present study
describes the complex diagnosis of a patient with plasma cell
myeloma, who initially presented with peripheral neuropathy upon
admission to Hainan Hospital of Chinese PLA General Hospital
(Sanya, China).
Case report
A 48-year-old woman was admitted to Hainan Hospital
of Chinese PLA General Hospital in September 2020 with a 2-month
history of lower limb numbness and gait instability. The patient
first noticed numbness in both feet in July 2020, which
progressively worsened and spread further. By early September 2020,
the numbness extended to the knees, further compromising their gait
stability. The patient had been in good health prior to this
episode. Upon admission, neurological examination showed clear
consciousness, fluent speech and normal cognitive function. Cranial
nerve assessment revealed no abnormalities. Muscle strength was
grade 5 in all four limbs (5), with
normal muscle tone. Pain and temperature sensations were intact in
the upper limbs, but diminished in a stocking-like distribution in
the lower limbs, accompanied by reduced vibration sense below the
ankles. Finger-to-nose testing was accurate, although heel-to-shin
testing showed instability. Deep tendon reflexes were normal in all
four limbs. The patient exhibited a broad-based gait, a positive
Romberg sign and bilateral pathological reflexes (6). Laboratory evaluations revealed a
reduced vitamin B12 level, whereas other tests, such as
blood chemistry, thyroid function and tumor markers, were within
normal limits (Table I). Nerve
conduction studies indicated peripheral nerve impairment in the
lower limbs. Sensory nerve conduction was absent and motor nerve
conduction velocity was markedly reduced in the lower limbs.
Imaging evaluations, such as brain, cervical, thoracic and lumbar
magnetic resonance imaging (MRI); lung and abdominal CT; and
thyroid and vascular ultrasound, showed no notable abnormalities.
Based on the clinical presentation and physical examination of the
patient, they were initially diagnosed with subacute combined
degeneration (SCD). Since the patient initially declined a lumbar
puncture, methylcobalamin (500 µg intramuscular injection once a
day), vitamin B1 (100 mg intramuscular injection once a
day), vitamin B6 (10 mg orally twice a day) and folic
acid (5 mg orally once a day), were administered.
 | Table I.Main laboratory parameters during
hospitalization. |
Table I.
Main laboratory parameters during
hospitalization.
| Primary lab indices
(reference values) | Initial hospital stay
results (September 2020) | Subsequent hospital
stay results (December 2020) |
|---|
| Red blood cells
(3.9–5.2×1012/l) |
4.34×1012/l |
4.95×1012/l |
| White blood cells
(3.5–10×109/l) |
4.09×109/l |
9.82×109/l |
| Platelets
(100–300×109/l) |
121×109/l |
514×109/l |
| Alanine
aminotransferase (0–40 U/l) | 5.3 U/l | 9.2 U/l |
| Serum albumin (35–50
g/l) | 33.4 g/l | 26.4 g/l |
| Creatinine (30–110
µmol/l) | 61 µmol/l | 49 µmol/l |
| Serum thyroxine
(66–181 nmol/l) | 75.56 nmol/l | 61.27 nmol/l |
| Vitamin
B12 (187–1,059 pg/ml) | 148.6 pg/ml | >2,000 pg/ml |
| Tumor markers |
|
|
| CEA (0–5
µg/l) | 0.281 µg/l | 0.407 µg/l |
| AFP (0–20
µg/l) | 6.22 µg/l | 2.8 µg/l |
| CA125
(0.1–35 U/ml) | 20.59 U/ml | 27.39 U/ml |
| Cerebrospinal
fluid |
|
|
| White
cell count (0–20×106/l) |
2×106/l |
1×106/l |
| Glucose
(2.8–4.48 mmol/l) | 3.79 mmol/l | 4.63 mmol/l |
| Chloride
(119–127 mmol/l) | 128.9 mmol/l | 119.7 mmol/l |
| Protein
(150–400 mg/l) | 1,146 mg/l | 1,044 mg/l |
Despite treatment, the symptoms worsened, leading to
severe weakness in all four limbs. By mid-September 2020, muscle
strength had declined to grade 4 in the distal upper limbs and
grade 3 in the distal lower limbs, accompanied by reduced deep
tendon reflexes in all four limbs. Given the ongoing deterioration,
the patient ultimately agreed to undergo a lumbar puncture, which
revealed albuminocytologic dissociation in the cerebrospinal fluid.
All other cerebrospinal fluid parameters, including biochemical
markers (glucose, chloride and protein levels, detected using
hexokinase, ion-selective electrode and immunoturbidimetry methods,
respectively), immunoglobulins (IgA, IgM and IgG, detected using
nephelometry) and smear tests (to detect bacteria, fungi,
parasites, tuberculosis and cancer cells), were within normal
values. Further investigations were carried out to refine the
diagnosis. Autoimmune peripheral neuropathy antibody panels (GM1,
GM2, GD1a, GD1b, etc., detected using western blotting), Langerhans
cell-related antibodies (neurofascin 155, neurofascin 186,
contactin-associated protein 1, human contactin 1, etc. detected
using a cytometric bead array) and genetic screening for hereditary
neuropathies (detected using high-throughput sequencing) were all
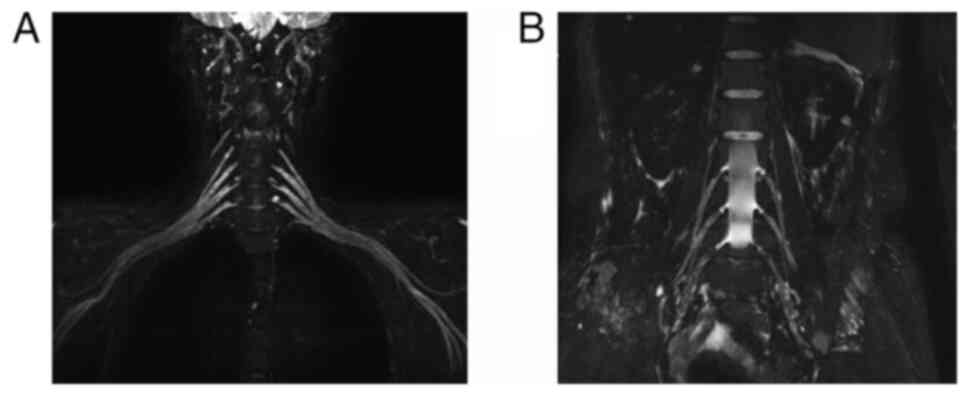
negative. MRI of the brachial and lumbosacral plexuses showed mild
nerve root thickening (Fig. 1).
Integrating the cerebrospinal fluid results and imaging findings, a
possible diagnosis of chronic inflammatory demyelinating
polyneuropathy (CIDP) was arrived at. The treatment regimen was
revised to include high-dose methylprednisolone pulse therapy
(initially 1 g/day with gradual tapering) in combination with
intravenous (IV) immunoglobulin at 0.4 g/kg for 5 days. Following
this therapy, the symptoms of the patient markedly improved. At
discharge, muscle strength had improved to grade 5− in
the distal upper limbs and grade 4+ in the distal lower
limbs. Although the area of numbness remained largely unchanged,
its severity had diminished.
Notably, the clinical symptoms of the patient flared
again in mid-to-late November 2020, culminating in a second
hospitalization in early December 2020. In contrast to their first
admission, the second hospital stay was marked by hyperhidrosis,
dry mouth, facial flushing, muscle atrophy and reduced muscle tone
on physical examination. Muscle strength was grade 4 in the
proximal limbs, grade 4 in the distal upper limbs and grade 2 in
the distal lower limbs. Deep tendon reflexes were absent across all
four limbs. Laboratory tests indicated thrombocytosis
(514×109/l, reference values: 100–300×109/l)
and a decreased serum thyroxine level (61.27 nmol/l, reference
values: 66–181 nmol/l) (Table I).
Cerebrospinal fluid analysis continued to show albuminocytologic
dissociation (CSF white cell count, 1×106/l; protein,
1,044 mg/l; Table I). Nerve
conduction studies demonstrated marked sensory and motor nerve
involvement in both upper and lower limbs, with the lower limbs
more severely affected. In light of the notable progression of
peripheral neuropathy following treatment and the involvement of
multiple organ systems (endocrine, hematological and cutaneous),
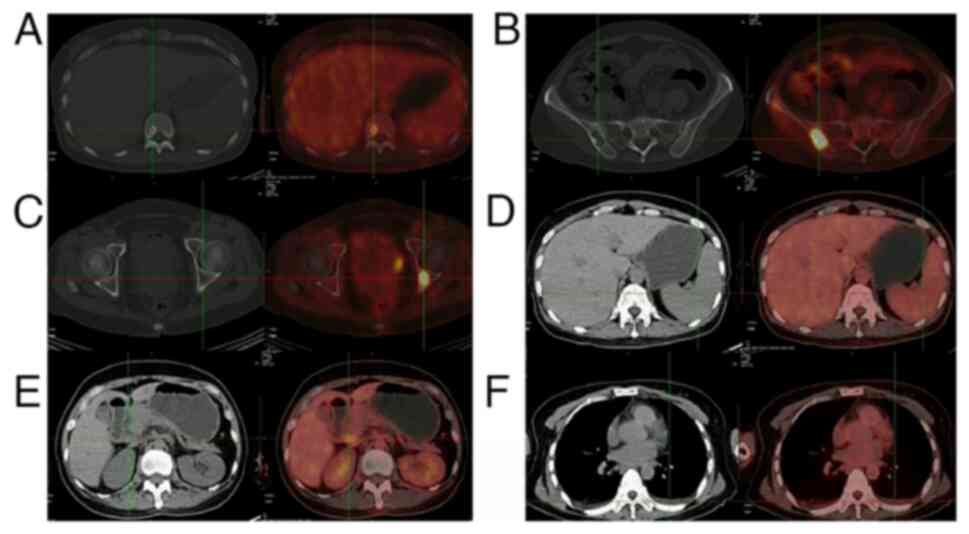
further specialized tests and evaluations were performed. A PET-CT
scan uncovered multiple hypermetabolic lesions accompanied by
sclerotic and osteolytic bone changes throughout the body,
hepatomegaly, splenomegaly, multiple enlarged hypermetabolic lymph
nodes in both supraclavicular regions, around the portal vein and
in the retroperitoneum, as well as bilateral pleural effusions
(Fig. 2). Serum protein
electrophoresis revealed an M-protein band, and serum
immunofixation electrophoresis indicated a monoclonal
immunoglobulin of the IgG-λ subtype (data not shown). Vascular
endothelial growth factor (VEGF) concentration was 998 pg/ml
(reference values: 0–300 pg/ml, detected by ELISA (cat. no.
KL-VEGF-Hu; Shanghai KangLang Biological Technology Co., Ltd.],
according to the manufacturer's protocol. Integrating the clinical
features with these findings led to a suspected diagnosis of
polyneuropathy, organomegaly, endocrine dysfunction, monoclonal
plasma cell proliferation and skin changes syndrome (POEMS
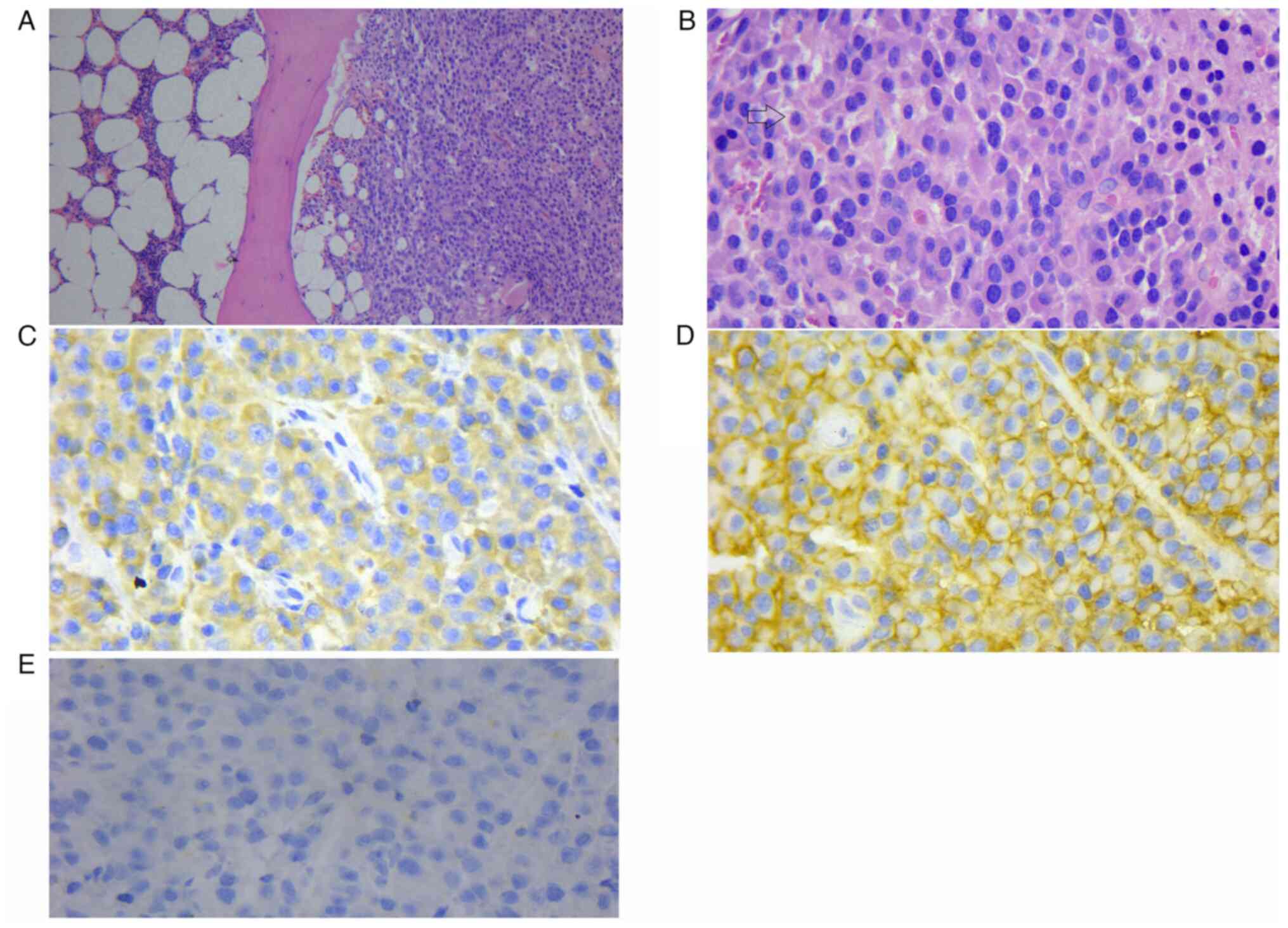
syndrome). A pathological biopsy, including H&E (Wuhan Jinhong
Biotech Development Co., Ltd.), lambda (cat. no. 760-2515; Roche
Tissue Diagnostics), CD38 (cat. no. ZM-0422; OriGene Technologies,
Inc.) and kappa (cat. no. 760-2514; Roche Tissue Diagnostics)
staining of the iliac lesion was performed (7). Kappa and lambda stains are
immunoglobulin light chains used primarily to diagnose
hematological diseases. The final pathological diagnosis of this
patient was plasma cell myeloma. (Fig.
3).
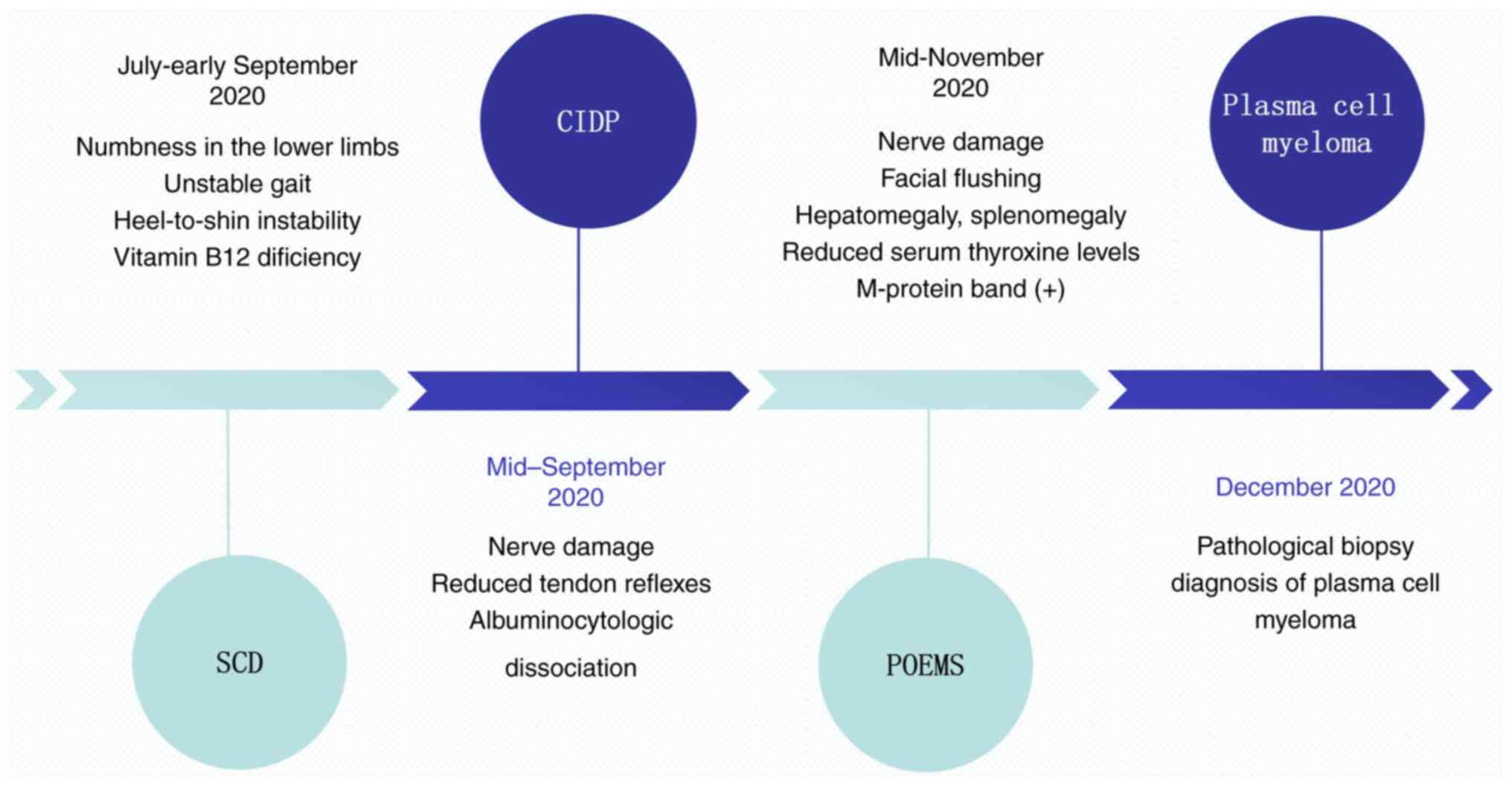
In the current case report, the patient initially
presented with peripheral neuropathy, and underwent a long and
complex diagnostic and therapeutic course, initially being
considered for SCD, CIDP and POEMS syndrome, before ultimately
receiving a definitive diagnosis of plasma cell myeloma (Fig. 4). The patient was then transferred
to the hematology department to receive combination therapy with
bortezomib (1.3 mg/m2 IV on days 1, 4, 8 and 11),
lenalidomide (25 mg/day orally on days 1–21) and dexamethasone (20
mg/day orally on days 1–4, 8–11 and 15–18), for a total of three
cycles, in conjunction with early rehabilitation intervention.
Since then, the patient was last followed up in June 2024 and has
been followed up for a cumulative total of 3.5 years, during which
early diagnosis and timely, comprehensive treatment has allowed
them to regain independence in daily activities. In addition, their
limb strength has substantially improved, enabling the patient to
run and hike on their own, with only mild residual sensory
deficits.
Discussion
Peripheral neuropathy has a notably high incidence
in the general population and ranks among the most frequently
encountered neurological conditions. Its underlying causes are
highly diverse, and may involve factors such as nutritional
deficiencies, chronic diseases, environmental toxins, prolonged
alcohol misuse, genetic predisposition and medication-related side
effects (8–11). In rare cases, peripheral neuropathy
may be the starting form of a tumor. Nonetheless, diagnosing and
treating peripheral neuropathy can be challenging, and in up to 25%
of cases, the underlying cause remains elusive (12).
During the initial phase of peripheral neuropathy,
patients frequently exhibit non-specific clinical symptoms and
ancillary test findings. To some degree, the clinical
manifestations and auxiliary evaluations may misdirect the
diagnostic process. In the present case, the patient initially
manifested with ataxia and severe sensory deficits in both lower
limbs. Laboratory results demonstrated vitamin B12
deficiency, while nerve conduction studies revealed peripheral
nerve impairment in the lower limbs. SCD is primarily characterized
by deficits in proprioception and vibratory sensation, often
accompanied by ataxia and gait disturbances, symptoms closely
mirroring the initial presentation of the current patient (13).
Notably, as the condition evolved, the current
patient acquired marked sensorimotor deficits in the limbs,
diminished tendon reflexes and albuminocytologic dissociation in
the cerebrospinal fluid. Accordingly, the diagnosis was modified to
CIDP. Classical CIDP symptoms consist of progressively worsening
sensory disturbances and limb weakness, potentially accompanied by
muscle atrophy, reduced muscle tone and diminished tendon reflexes
(14). In patients with CIDP,
cerebrospinal fluid analysis frequently demonstrates
albuminocytologic dissociation (15,16).
Electromyographic evaluation typically indicates multifocal
peripheral neuropathy in the upper and lower limbs, involving both
sensory and motor fibers, with evidence of demyelination often
coexisting with axonal damage. Notably, after treatment with
immunoglobulin and methylprednisolone, the symptoms of the patient
were markedly improved, reinforcing the provisional diagnosis of
CIDP and inadvertently leading to de-prioritization of additional
evaluations for alternative conditions.
POEMS syndrome is an uncommon paraneoplastic
disorder linked to monoclonal plasma cell proliferation. Its
incidence is low, and its initial presentation can be complex and
varied, often with limited clinical specificity and a wide range of
severity (17). Although the
precise etiology and pathogenesis of POEMS syndrome remain
incompletely clarified, research has suggested potential
associations with monoclonal plasma cell hyperplasia, translocation
at chromosome 14q32 and deletion of 13q14, Epstein-Barr virus and
human herpesvirus 8 infections, proinflammatory cytokines, and
VEGF. POEMS syndrome is mainly characterized by multisystem
involvement. In the present case, comprehensive follow-up and
additional evaluations demonstrated peripheral nerve involvement,
hepatomegaly, splenomegaly, lymph node enlargement, thyroid hormone
abnormalities, clonal plasma cell disease and cutaneous changes.
Other notable features included pleural effusion, thrombocytosis,
hyperhidrosis and reduced vitamin B levels, leading to the
conclusion that the patient fulfilled the criteria for POEMS
syndrome.
The diagnostic criteria for POEMS syndrome are as
follows: i) Mandatory criteria: multiple instances of peripheral
neuropathy and a monoclonal plasma cell proliferative disorder; ii)
major criteria: Sclerotic bone lesions, Castleman's disease and
elevated serum VEGF levels; iii) minor criteria: Organomegaly
(involving the spleen, liver or lymph nodes), increased
extravascular fluid volume, endocrine dysfunction, cutaneous
changes, papilledema and thrombocytosis; and iv) other symptoms:
Hyperhidrosis, weight loss, digital clubbing and pulmonary
hypertension. A definitive diagnosis of POEMS syndrome calls for
the presence of two mandatory criteria, at least one major
criterion, and at least one minor criterion (18). In the present case, the patient
fulfilled two mandatory criteria, one major criterion, four minor
criteria and two additional symptoms, thereby meeting the
diagnostic requirements for POEMS syndrome. In general, confirming
a POEMS syndrome diagnosis can take upwards of 6 months; however,
in this case, rigorous follow-up and prompt assessments narrowed
that timeframe to just 3 months.
Notably, the clinical features of CIDP and POEMS
syndrome are markedly similar, sharing a number of overlapping
characteristics that frequently result in misdiagnoses (19,20).
For example, both disorders manifest as multifocal peripheral
neuropathies featuring gradually worsening sensorimotor deficits,
with the lower limbs often more severely involved than the upper
limbs. Sensory impairments generally follow a glove-and-stocking
distribution, accompanied by pronounced muscle weakness, the
principal contributor to disability. Laboratory evaluations show
that patients with POEMS syndrome nearly always demonstrate
albuminocytologic dissociation in the cerebrospinal fluid, a
finding also noted in CIDP. Likewise, nerve conduction studies in
both conditions can reveal multifocal peripheral nerve involvement,
reduced conduction velocities in both motor and sensory fibers, and
concomitant axonal damage. Pathological biopsies in both disorders
commonly show axonal degeneration alongside segmental demyelination
(19,20). Despite these similarities, key
differences also exist between CIDP and POEMS syndrome. First, CIDP
primarily affects peripheral nerves and may occasionally involve
the cranial nerves, but generally does not produce the multisystem
manifestations commonly seen in POEMS syndrome. By contrast, POEMS
syndrome necessarily involves multiple organ systems in addition to
peripheral neuropathy, although cranial nerve involvement is
typically not observed. Furthermore, when compared with CIDP, nerve
biopsies in POEMS syndrome indicate more pronounced axonal damage
and more markedly reduced nerve conduction velocities (21).
M protein, which is essentially an immunoglobulin or
immunoglobulin fragment, is produced by the clonal malignant
proliferation of plasma cells or B lymphocytes. It is widely
regarded as a key cornerstone in diagnosing POEMS syndrome and
serves as one of the primary laboratory markers distinguishing
POEMS syndrome from CIDP (22).
Monoclonal gammopathy of undetermined significance (MGUS) refers to
the presence of M protein in individuals without evidence of
multiple myeloma, amyloidosis or other related diseases. M proteins
are monoclonal immunoglobulins secreted by a neoplastic clone of
plasma cells. In MGUS, these plasma cells are clonal but not
malignant, whereas in multiple myeloma, macroglobulinemia and
plasmacytoma, the clone has undergone malignant transformation
(23). MGUS is also one of the
important differential diagnoses of POEMS syndrome. MGUS is a
premalignant precursor of multiple myeloma. Notably, patients with
MGUS often present with peripheral neuropathy. MGUS can present
with multisystemic manifestations due to monoclonal gammopathy,
with the main clinical features manifesting as motor, sensory and
autonomic dysfunction due to peripheral nerve damage (19). It is important to note that the
combination of MGUS with peripheral neuropathy is predominantly
seen in individuals >50 years of age. Despite the presence of
protein M, MGUS is not associated with bone radiological or skin
changes.
PET-CT serves a pivotal role in diagnosing POEMS
syndrome (24). As a whole-body
imaging technique that integrates morphological and cellular
metabolic data, PET-CT provides substantial benefits for diagnosing
and managing multisystem disorders. This modality can identify bone
lesions in patients with POEMS syndrome early and comprehensively.
These lesions primarily affect the spine, pelvis and ribs,
exhibiting single or multiple nodular osteogenic or mixed
destructive changes, sometimes with osteolytic lesions bordered by
sclerotic margins. PET-CT also has considerable clinical utility
for visualizing lymph node lesions, thus facilitating early
identification of lymph nodes with abnormal metabolic activity
(25). Additionally, it is highly
valuable for guiding biopsy localization, given that sampling
hypermetabolic bone lesions and lymph nodes can aid in establishing
a diagnosis. In the present case, PET-CT offered critical insights
into the overall metabolic status of the patient and informed the
selection of an appropriate biopsy site.
In summary, the present study provides a review of a
rare case of plasma cell myeloma that initially mimicked SCD and
CIDP, subsequently evolving into POEMS syndrome. This case
highlights the complexity and multiple challenges in the diagnosis
and treatment of plasma cell myeloma, and suggests that
neurologists should be aware of the possibility of the tumor having
a non-specific peripheral neuropathy as its starting form.
Furthermore, it underscores the need to screen for M protein, to
conduct imaging to detect potential bone destruction, and, when
indicated, to perform PET-CT to evaluate metabolic activity in
patients with chronic peripheral neuropathies. For patients
presenting with POEMS syndrome and bone destruction, timely
pathological biopsy is essential to secure an early diagnosis and
prevent delays in optimal treatment.
Notably, there are several limitations in the
present case report. Although the presentation closely resembled
SCD, CIDP and POEMS syndrome at various stages, this does not imply
that all peripheral neuropathies manifest in such a manner; by
contrast, this particular course of disease is rare among
peripheral neuropathies.
In conclusion, diagnosing and treating peripheral
neuropathy is inherently challenging because the early symptoms of
numerous conditions associated with peripheral neuropathy are
misleading. Clinicians need to be alert to the possibility of
plasma cell myeloma with peripheral neuropathy as a form of disease
initiation.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SW, JL and FC designed the report. TS, NZ, YL and JL
investigated and followed up the patient. YL and TS supervised the
project. SW, NZ and YL wrote the original draft. SW, NZ and JL
reviewed, revised and edited the original draft. SW, NZ and JL
confirmed the authenticity of all the raw data. All authors have
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Written informed consent was obtained from the
patient for the publication of this case report and any
accompanying images.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
SCD
|
subacute combined degeneration
|
|
CIDP
|
chronic inflammatory demyelinating
polyneuropathy
|
|
MRI
|
magnetic resonance imaging
|
|
POEMS
|
syndrome, polyneuropathy,
organomegaly, endocrine dysfunction, monoclonal plasma cell
proliferation and skin changes syndrome
|
|
MGUS
|
monoclonal gammopathy of undetermined
significance
|
References
|
1
|
Hanewinckel R, van Oijen M, Ikram MA and
van Doorn PA: The epidemiology and risk factors of chronic
polyneuropathy. Eur J Epidemiol. 31:5–20. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Callaghan BC, Price RS, Chen KS and
Feldman EL: The importance of rare subtypes in diagnosis and
treatment of peripheral neuropathy: A review. JAMA Neurol.
72:1510–1518. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lehmann HC, Wunderlich G, Fink GR and
Sommer C: Diagnosis of peripheral neuropathy. Neurol Res Pract.
2:202020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
McDonald CM: Clinical approach to the
diagnostic evaluation of hereditary and acquired neuromuscular
diseases. Phys Med Rehabil Clin N Am. 23:495–563. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cuthbert SC and Goodheart GJ Jr: On the
reliability and validity of manual muscle testing: A literature
review. Chiropr Osteopat. 15:42017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lanska DJ and Goetz CG: Romberg's sign:
Development, adoption, and adaptation in the 19th century.
Neurology. 55:1201–1206. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Suvarna KS, Layton C and Bancroft JD:
Bancroft's Theory and Practice of Histological Techniques. 7th
Edition. Churchill Livingstone; 2012
|
|
8
|
Rubens O, Logina I, Kravale I, Eglîte M
and Donaghy M: Peripheral neuropathy in chronic occupational
inorganic lead exposure: A clinical and electrophysiological study.
J Neurol Neurosurg Psychiatry. 71:200–204. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Weimer LH: Medication-induced peripheral
neuropathy. Curr Neurol Neurosci Rep. 3:86–92. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ammendola A, Tata MR, Aurilio C, Ciccone
G, Gemini D, Ammendola E, Ugolini G and Argenzio F: Peripheral
neuropathy in chronic alcoholism: A retrospective cross-sectional
study in 76 subjects. Alcohol Alcohol. 36:271–275. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
López-Hernández N, García-Escrivá A,
Pampliega-Pérez A, Alvarez-Saúco M, Martín-Estefanía C and
Asensio-Asensio M: Peripheral and optical myeloneuropathy in a
folic acid deficient alcoholic patient. Rev Neurol. 37:726–729.
2003.(In Spanish). PubMed/NCBI
|
|
12
|
Dyck PJ, Oviatt KF and Lambert EH:
Intensive evaluation of referred unclassified neuropathies yields
improved diagnosis. Ann Neurol. 10:222–226. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hemmer B, Glocker FX, Schumacher M,
Deuschl G and Lücking CH: Subacute combined degeneration: Clinical,
electrophysiological, and magnetic resonance imaging findings. J
Neurol Neurosurg Psychiatry. 65:822–827. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stino AM, Naddaf E, Dyck PJ and Dyck PJB:
Chronic inflammatory demyelinating polyradiculoneuropathy
diagnostic pitfalls and treatment approach. Muscle Nerve.
63:157–169. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Illes Z and Blaabjerg M: Cerebrospinal
fluid findings in Guillain-Barré syndrome and chronic inflammatory
demyelinating polyneuropathies. Handb Clin Neurol. 146:125–138.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dalakas MC, Latov N and Kuitwaard K:
Intravenous immunoglobulin in chronic inflammatory demyelinating
polyradiculoneuropathy (CIDP): Mechanisms of action and clinical
and genetic considerations. Expert Rev Neurother. 22:953–962. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brown R and Ginsberg L: POEMS syndrome:
Clinical update. J Neurol. 266:268–277. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dispenzieri A: POEMS syndrome. Blood Rev.
21:285–299. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chaudhry HM, Mauermann ML and Rajkumar SV:
Monoclonal gammopathy-associated peripheral neuropathy: Diagnosis
and management. Mayo Clin Proc. 92:838–850. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Moshe-Lilie O, Ensrud E, Ragole T, Nizar
C, Dimitrova D and Karam C: CIDP mimics: A case series. BMC Neurol.
21:942021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mauermann ML, Sorenson EJ, Dispenzieri A,
Mandrekar J, Suarez GA and Dyck PJ and Dyck PJ: Uniform
demyelination and more severe axonal loss distinguish POEMS
syndrome from CIDP. J Neurol Neurosurg Psychiatry. 83:480–486.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang Q, Liu P, Ji LL, Wu S, Feng GD, Wang
X and Dong JH: Clinical and electrophysiological profiles in early
recognition of polyneuropathy, organomegaly, endocrinopathy,
M-protein, and skin changes syndrome. Chin Med J (Engl).
132:1666–1672. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kyle RA and Rajkumar SV: Monoclonal
gammopathy of undetermined significance. Neoplastic Diseases of the
Blood. Wiernik P, Goldman J, Dutcher J and Kyle R: 5th Edition.
Springer; New York, NY: 2013, View Article : Google Scholar
|
|
24
|
Pan Q, Li J, Li F, Zhou D and Zhu Z:
Characterizing POEMS syndrome with 18F-FDG PET/CT. J Nucl Med.
56:1334–1337. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shi XF, Hu SD, Wu LL, Chen XY, Wu JN, Yu
XQ, Li DY, Chen M, Liu YC, Zhu Y and Xi XD: Lymphadenopathy in
POEMS syndrome: A correlation between clinical features and imaging
findings. Int J Clin Exp Pathol. 13:21–25. 2020.PubMed/NCBI
|