Introduction
Gastrointestinal cancer ranks among the most common
cancer types worldwide. According to 2020 statistics from the World
Health Organization, gastrointestinal cancer accounts for ~26% of
all cancer cases globally and ~35% of all cancer-related deaths
(1). Despite notable advancements
in research on gastrointestinal malignancies in recent years, the
survival rate remains at only 25–30%, as most patients are
diagnosed at advanced or terminal stages of the disease (2,3). At
present, the primary clinical interventions for gastrointestinal
cancer include surgical resection, chemoradiotherapy and targeted
therapies. However, due to the high recurrence and metastatic
potential of this cancer type, the clinical prognosis for patients
remains unfavorable (4). The
exploration of novel diagnostic and therapeutic strategies for
gastrointestinal cancer has become a central focus of clinical
research. The clinical success of antitumor drugs targeting
histone-modifying enzymes, such as histone deacetylase inhibitors,
has rendered histone modification an increasingly prominent area of
interest in cancer research (5).
SET and MYND domain-containing protein 2 (SMYD2), a
lysine methyltransferase, has gained increasing attention due to
its dual ability to methylate both histone and non-histone
proteins, influencing critical processes such as transcription
regulation, cell cycle progression and apoptosis (6–8). The
activity of SMYD2 is essential for organismal development and
several cellular functions, with mutations of this gene frequently
associated with cardiovascular diseases and cancers (9–12). At
present, SMYD2 is actively being investigated as a potential target
for inhibitors that regulate gene transcription or signal
transduction pathways (13,14). The upregulation of SMYD2 is
associated with several cancer types, including hepatocellular
carcinoma (HCC), colorectal cancer (CRC) and gastric cancer (GC),
where it promotes tumorigenesis through both direct protein
modification and epigenetic modulation of oncogenic pathways
(15). Dysregulation of epigenetic
control is widely accepted as a cause of tumor malignancy;
therefore, the inhibition of epigenetic enzymes is a highly pursued
therapeutic strategy (16,17). SMYD2 inhibitors have shown promising
progress in cellular and animal studies across several cancer types
(18,19). Despite SMYD2 being regarded as an
oncogene, research on specific SMYD2 inhibitors remains limited
(20). However, current inhibitors,
such as AZ505 and BAY-598, have shown promise in preclinical
studies, albeit with challenges related to bioavailability and
pharmacokinetics (21).
In the last 10 years, SMYD2 has gained considerable
attention in gastrointestinal cancer research, resulting in a
growing number of related studies and publications (22). However, to the best of our
knowledge, there is currently no comprehensive review specifically
addressing the mechanisms of tumorigenesis and treatment strategies
associated with gastrointestinal malignancies. Consequently, the
present review primarily aims to emphasize the role of SMYD2 as a
key lysine methyltransferase and a promising therapeutic target in
different types of gastrointestinal cancer. The molecular structure
of SMYD2, its biological functions and the mechanisms through which
it contributes to the onset and progression of gastrointestinal
malignancies, as well as the current therapeutic strategies
targeting SMYD2, are thoroughly assessed. Furthermore, the
potential of SMYD2-targeted therapies for gastrointestinal cancer
and their possible impact on treatment outcomes are evaluated.
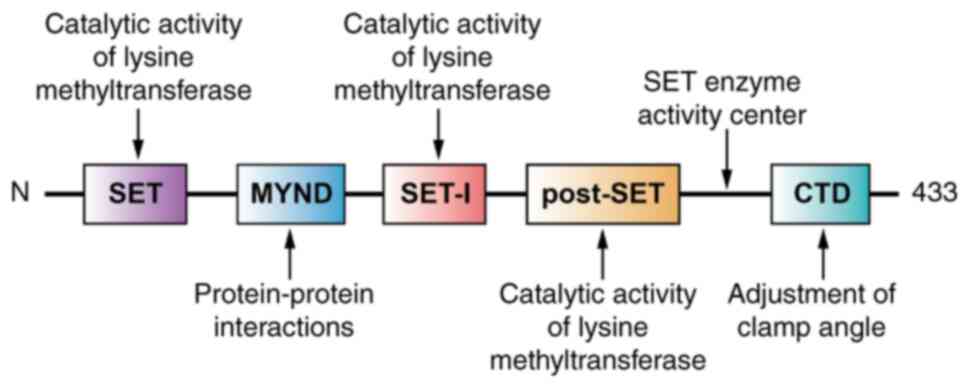
Structure of SMYD2
The SET and MYND domain-containing protein family,
which includes five members (SMYD1-5), is a class of lysine
methyltransferases. Among these, SMYD1, SMYD2 and SMYD3 exhibit the
highest homology and structural similarity, with their domains
organized sequentially as SET, MYND, SET-I, post-SET and C-terminal
domain (CTD) (23–25). The SMYD2 gene, located in the 1q32.3
region of the human genome, consists of 12 exons and encodes a
protein of 433 amino acids. SMYD2 was first identified by Brown
et al (6) in 2006.
Similar to other SMYD family members, SMYD2 consists
of two major structural components: The N-terminal domain and CTD.
The N-terminal domain (residues 1–271) includes four subdomains,
N-SET, MYND, SET-I and post-SET, whilst the CTD includes residues
272–433 (26,27). The N-terminal domain contains a
mixed structure of α-helices (α1-α6), β-strands (β1-β12) and
extended loops, whilst the CTD forms a twisted seven α-helical
bundle (α8-α14) (27).
Additionally, SMYD2 undergoes alternative splicing,
generating at least two known isoforms. The primary isoform,
SMYD2-1, consists of 433 amino acids and is considered the
canonical sequence. The alternative isoform, SMYD2-2, differs from
the canonical sequence, containing only 272 amino acids due to the
deletion of the 273–433 aa segment, resulting in a shorter protein
(28). However, detailed
descriptions of SMYD2-1 and SMYD2-2 remain limited in the
scientific literature. Further research is necessary to improve the
understanding of specific SMYD2 isoforms, including SMYD2-1 and
SMYD2-2, by investigating alternative splicing events and their
functional implications. This may involve transcriptome analysis
and protein characterization studies to elucidate the roles of
these isoforms in several physiological and pathological
contexts.
The SET, SET-I and post-SET domains are essential
for the catalytic activity of lysine methyltransferases (29), whilst the MYND domain, with a zinc
finger structure, primarily mediates protein-protein interactions
(30). Morphologically, the CTD is
arranged in antiparallel helices that include a tetratricopeptide
repeat (TPR)-like domain (31,32).
Together with the SET domain, the CTD contributes to the formation
of a two-lobed structure (26). The
angle between these lobes creates a ‘U’-shaped cavity that binds
substrates, with the bottom of this cavity serving as the active
site for SET enzyme activity (27).
The movement of this structure allows it to reach a specific state,
which influences the specificity of SMYD2 for its substrates
(27,33,34).
A previous study reported that under activation
stress, S-adenosylmethionine binding induced increased elasticity
in the CTD, causing SMYD2 to adopt an open conformation that
exposed the substrate-binding site (Fig. 1) (35). However, the full mechanisms
underlying the function of the CTD remain unclear, and its
potential as a drug target site has yet to be explored. Moreover, a
recent study identified a novel mixed allosteric site in SMYD2,
which exhibited mixed binding characteristics capable of
interacting with peptides, proteins, ethylene glycol polymers and
small molecules. This allosteric site could serve as a target for
future drug design research (36).
Biological function of SMYD2
In total, >50 proteins with SET domains are
encoded by the human genome and only a small fraction have been
reported to methylate histones, with the SMYD protein family being
among them (6). The first member of
this family, SMYD1, was identified and named by Gottlieb et
al (37) during the
investigation of genes expressed early in mouse heart development.
Since then, research on the SMYD protein family has grown. SMYD2 is
considered an essential protein for the development of skeletal
muscle and cardiac muscle cells in zebrafish (38); however, experiments with mouse
models suggest it does not serve a role in cardiac muscle
development (39). A previous study
reported that SMYD2 exhibits elevated expression levels in the
heart, brain, liver, kidneys, thymus and ovarian tissues. By
comparison, its expression in the gastrointestinal tract is
relatively low, except in the liver, where it is comparatively
higher, with its presence detected in both the cytoplasm and
nucleus of cells (6). SMYD2 is
present in the maternal genome of Xenopus laevis embryos and
persists until stage 40, with expression localized to the
dorsomedial lip, particularly in the face region. Loss-of-function
analysis using antisense morpholino oligonucleotide (MO) revealed
that Xenopus laevis embryos injected with SMYD1MO and
SMYD2MO exhibit abnormal somite, abnormal mandibular tissue and
severe malformations (40),
suggesting that SMYD2 may be involved in the development of
multiple systems, including the cardiovascular, nervous,
gastrointestinal, musculoskeletal, urinary and reproductive
systems.
Experimental evidence has also reported that SMYD2
specifically methylates the lysine 4 residue of histone H3 (H3K4),
lysine 36 residue of histone H3 (H3K36) and lysine 20 residue of
histone H4 (H4K20), and these methylation events may alter gene
transcription, thereby influencing a range of cellular activities
(41). Histone H3 contains the
majority of lysine residues targeted by known histone
methyltransferases, making it a key pathway member for the
regulation of epigenetic mechanisms (6). SMYD2 specifically demethylates H3K36,
generating H3K36Me2, which negatively regulates gene expression and
thereby inhibits cell proliferation (6). Additionally, in the presence of the
chaperone protein, heat shock protein 90α (HSP90α), SMYD2
specifically methylates H3K4, leading to gene activation;
conversely, in the absence of HSP90α, SMYD2 reverts to its specific
methylation of H3K36, resulting in gene repression (31). The SMYD2-mediated methylation of
H3K4 is typically associated with gene activation; however, the
precise mechanism by which this modification influences cell
proliferation has yet to be fully elucidated (41). H4K20 is an unrecognized target of
SMYD2, despite a previous study reporting that histone H4 is a more
effective substrate for SMYD2 than histone H3 (42). In a large-scale proteomic study of
SMYD2-mediated lysine monomethylation, it was reported that
H4K20me1 was downregulated in 4/7 cell lines following short
hairpin (sh)RNA-mediated knockdown and the use of SMYD2 inhibitors,
indicating a more widespread role of SMYD2 in H4K20 monomethylation
(43). Moreover, a previous study
reported that SMYD2 promoted monomethylation of H4K20 in the human
immunodeficiency virus (HIV)-1 promoter region, inhibiting HIV-1
transcription, highlighting its potential in HIV therapeutic
strategies (42).
SMYD2 also has a crucial role in the methylation of
non-histone proteins within cells (44–46).
For example, a previous study reported that SMYD2 methylated p53
and phosphatase and tensin homolog deleted on chromosome ten
(PTEN), thereby suppressing their tumor-inhibiting functions
(18,47). The retinoblastoma tumor suppressor
(RB) can be methylated by SMYD2 at lysine 860, a highly conserved
and novel site of modification, thereby influencing cell growth,
differentiation and the DNA damage response (48). Furthermore, SMYD2 can methylate the
lysine 810 of RB (49). The
methylation of RB increases the level of phosphorylation of Ser
807/811 and releases and activates the E2F transcription factor,
thereby promoting the cell cycle progression of cancer cells
(49). Additionally, SMYD2
suppresses the activity of p53 by monomethylating lysine 370, a
function that is dependent on the SMYD2 SET domain as the absence
of the conserved SET domain sequence abolishes this methylation
(7,31). However, the molecular chaperone
HSP90 appears to be unaffected by the SET domain, as the TPR-like
domain of SMYD2 interacts with HSP90 to form a chaperone complex,
thereby influencing cellular proliferation (50). The dual capacity of SMYD2 to
methylate both histone and non-histone proteins impacts key
processes such as transcriptional regulation, cell cycle
progression and apoptosis (7). The
aforementioned findings and further information on histone and
non-histone proteins are presented in Table I. These proteins are closely
associated with tumor development, underscoring the inextricable
link between SMYD2 and cancer.
 | Table I.Function of SET and MYND
domain-containing protein 2 histone and non-histone substrates in
cancer. |
Table I.
Function of SET and MYND
domain-containing protein 2 histone and non-histone substrates in
cancer.
| Substrate type | Substrate | Biological
function | (Refs.) |
|---|
| Histone | H3K36 | Inhibits
proliferation | (31) |
|
| H3K4 | Gene
activation | (41) |
|
| H4K20 | DNA repair | (43) |
| Non-histone | RB | Regulates the cell
cycle | (49) |
|
| p53 | Promotes
proliferation; inhibits apoptosis | (7) |
|
| PTEN | Promotes
proliferation and migration | (47) |
|
| Myc | Promotes
proliferation | (74) |
|
| EZH2 | Promotes
proliferation; inhibits senescence | (66) |
|
| Erα | Promotes
proliferation | (44) |
|
| HSP90 | Promotes
proliferation | (50) |
|
| β-catenin | Promotes
proliferation, migration and invasion | (59) |
|
| STAT3 | Promotes
proliferation | (45) |
|
| MAPKAPK3 | Promotes
proliferation | (12) |
|
| PARP1 | Promotes
proliferation | (46) |
SMYD2 and CRC
CRC ranks third globally in terms of both incidence
and mortality. The pathogenesis of CRC is multifactorial, with
genetic mutations, lifestyle choices and environmental factors
contributing to tumorigenesis. These factors include dietary
habits, smoking, alcohol consumption, lack of physical activity and
a family history of CRC (51).
Previous studies have revealed the association between SMYD2 and
CRC. Specifically, multiple investigations have demonstrated
elevated levels of SMYD2 mRNA and protein in CRC tissues, which are
associated with worse prognoses (52–54).
An in vitro study reported that SMYD2
promoted the proliferation and invasion of colon cancer cells,
whilst inhibiting apoptosis. Additionally, SMYD2 was reported to
enhance Erb-B2 receptor tyrosine kinase 2 (ERBB2) phosphorylation
and upregulate fucosyltransferase 4 (FUT4) expression in colon
cancer cells (55). Further in
vivo experiments reported that SMYD2 knockout markedly slowed
tumor growth in mice, indicated by the smaller tumor volumes
observed in the SMYD2-deficient group. These tumors exhibited
reduced expression levels of phosphorylated ERBB2, FUT4 and the
proliferation marker, Ki67. Mechanistically, the study demonstrated
that SMYD2 mediates colon cancer cell proliferation, invasion and
apoptosis through the ERBB2/FUT4 signaling pathway (55). Ren et al (56) reported that SMYD2 upregulation in
colorectal adenocarcinoma (COAD) tissues is often associated with
poor prognosis in patients undergoing oxaliplatin (L-OHP)
chemotherapy. In vitro and in vivo experiments
revealed that downregulation of SMYD2 sensitized COAD cells to
L-OHP exposure. Moreover, when SMYD2 expression was downregulated,
P-glycoprotein (P-gp) levels decreased, whereas its overexpression
led to P-gp upregulation, indicating that P-gp serves a role in the
SMYD2-mediated resistance to L-OHP (56). P-gp, a membrane-bound transporter,
serves as a marker for an unfavorable prognosis in patients with
COAD (5,56). Additional research on the role of
SMYD2 in L-OHP resistance revealed that its upregulation increased
MEK/ERK/activator protein 1 (AP-1) pathway activity, leading to
alterations in drug metabolism-associated receptors and signaling
cascades, ultimately influencing the responsiveness COAD cells to
L-OHP. Consequently, SMYD2 is suggested to facilitate P-gp
upregulation via the MEK/ERK/AP-1 pathway, thereby contributing to
L-OHP resistance in COAD cells (56). Additionally, SMYD2 has been reported
to inhibit the TNF-induced apoptosis and necrosis of colorectal
tumor cells by downregulating the receptor-interacting protein
kinase 1 phosphorylation signaling pathway, and inhibition of SMYD2
suppresses the growth of colorectal tumors (53).
Bioinformatics analysis has predicted that SMYD2
aberrantly modifies H3K4me3 and acetylated H3K27 (H3K27ac) on long
intergenic non-protein coding RNA 1605 (LINC01605) in CRC.
Subsequent experiments have reported that SMYD2 promotes LINC01605
expression through H3K27ac and H3K4me3 modifications, thereby
influencing the development and progression of CRC (57). Both in vitro and in
vivo experiments demonstrated that knocking down SMYD2 notably
suppressed CRC cell proliferation, migration and invasion as well
as tumor growth in nude mice, whilst overexpression of SMYD2
produced the opposite effects, indicating its role in promoting CRC
progression (58). Further in
vitro experiments reported that SMYD2 activated Mex-3 RNA
binding family member A (MEX3A) transcription by enhancing H3K36me2
modification in the region of its promoter. Silencing MEX3A
expression suppressed CRC cell proliferation and growth, as well as
reduced tumor growth in vivo. Rescue experiments
demonstrated that MEX3A silencing restored caudal type homeobox 2
(CDX2) expression, thereby blocking the oncogenic effects of SMYD2.
Conversely, silencing CDX2 expression rescued the malignant
behavior of CRC cells inhibited by MEX3A silencing. These findings
indicate that SMYD2 epigenetically activates MEX3A transcription
through H3K36me2 modification, leading to CDX2 downregulation and
the promotion of CRC development (58).
Furthermore, in a previous study, clinical data
indicated that high SMYD2 expression was positively associated with
tumor diameter in CRC and is a risk factor for tumor metastasis.
Further in vivo and in vitro experiments demonstrated
that SMYD2 promoted CRC metastasis by recruiting DNA
methyltransferase 1 in CRC cells to suppress adenomatous polyposis
coli 2 expression, thereby facilitating epithelial-mesenchymal
transition and activating the Wnt/β-catenin signaling pathway
(52,59). Another study reported that highly
expressed SMYD2 mediated the monomethylation of K422 of
heterogeneous nuclear ribonucleoprotein K, which in turn promoted
CRC angiogenesis by binding to and stabilizing epidermal growth
factor-like domain 7 mRNA. In xenograft experiments, combining a
SMYD2 inhibitor (BAY-598) with a VEGFR2 inhibitor (apatinib)
inhibited CRC angiogenesis (Fig.
2B). This indicates that targeting SMYD2 is a safe and
effective therapeutic strategy that can synergistically enhance the
anti-angiogenic effect of apatinib, demonstrating its potential for
CRC treatment (54).
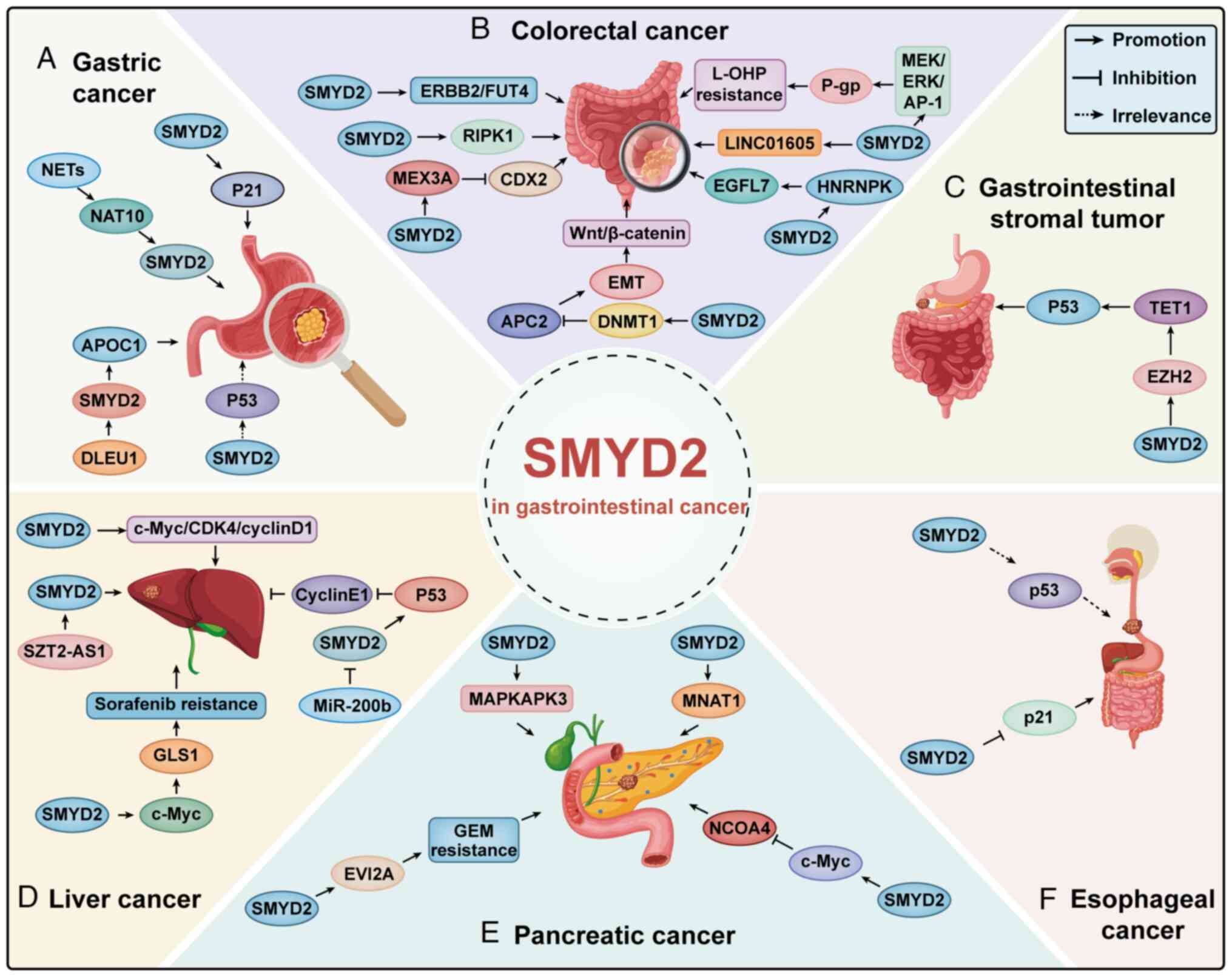 | Figure 2.SMYD2 promotes or inhibits the
progression of gastrointestinal cancer by targeting related
molecules in association with different proteins. (A) Gastric, (B)
colorectal, (C) gastrointestinal stromal tumor; (D) liver, (E)
pancreatic and (F) esophageal cancers. SMYD2, SET and MYND
domain-containing protein 2; NETs, neutrophil extracellular traps;
NAT10, N-acetyltransferase 10; DLEU1, deleted in lymphocytic
leukemia 1; APOC1, apolipoprotein C1; ERBB2, Erb-B2 receptor
tyrosine kinase 2; FUT4, fucosyltransferase 4; L-OHP, oxaliplatin;
P-gp, P-glycoprotein; RIPK1, receptor-interacting protein kinase 1;
LINC01605, long intergenic non-protein coding RNA 1605; MEX3A,
Mex-3 RNA binding family member A; CDX2, caudal type homeobox 2;
HNRNPK, heterogeneous nuclear ribonucleoprotein K; EGFL7, epidermal
growth factor-like domain 7; DNMT1, DNA methyltransferase 1; APC2,
adenomatosis polyposis coli 2; EMT, epithelial-mesenchymal
transition; EZH2, enhancer of zeste homolog 2; TET1, ten-eleven
translocation methylcytosine dioxygenase 1; CDK4, cyclin-dependent
kinase 4; GLS1, glutaminase 1; MAPKAPK3, mitogen-activated protein
kinase-activated protein kinase 3; MNAT1, MNAT1 component of CDK
activating kinase; NCOA4, nuclear receptor coactivator 4; EVI2A,
ecotropic viral integration site 2 A; GEM, gemcitabine. |
SMYD2 and GC
GC is one of the most common cancer types worldwide,
with the majority of patients diagnosed at the advanced stages of
disease and facing poor prognoses. The clinical staging of GC is
crucial in determining its prognosis. The 5-year survival rate for
early-stage gastric cancer can exceed 90%, whereas for
advanced-stage cases, it falls below 30% (60). The pathogenesis of GC is complex and
influenced by genetic, environmental and lifestyle factors.
Early-stage GC is primarily treated with surgery, whilst advanced
cases require chemotherapy, targeted therapy and immunotherapy.
Despite advancements in diagnosis and treatment, GC remains a major
cause of cancer-related mortality, highlighting the need for
further research and improved therapeutic strategies (61).
An analysis of public databases revealed that SMYD2
mRNA expression is notably upregulated in GC, with protein levels
consistent with the mRNA levels. Furthermore, SMYD2 expression is
negatively associated with both overall survival (OS) and
progression-free survival. Additionally, SMYD2 is closely
associated with the infiltration of six immune cell types:
Uncharacterized cells, CD8+ T cells, CD4+ T
cells, macrophages, endothelial cells and B cells. SMYD2 is also
markedly positively associated with tumor mutational burden and
microsatellite instability in GC (62). An additional analysis of publicly
available datasets identified a strong association between SMYD2
mRNA levels and both the occurrence of stomach adenocarcinoma and
the mutation status of P53. Moreover, SMYD2 was reported to be
negatively associated with CD8+ T cells and dendritic
cells (63).
Experimental data demonstrated that both SMYD2 mRNA
and protein are highly expressed in GC cell lines, and this
expression is independent of the TP53 mutation status and
expression levels. Furthermore, downregulation of SMYD2 was
reported to inhibit the proliferation, migration and invasion of GC
cells (10). In vitro
experiments revealed that this inhibitory effect was independent of
the P53 mutation and expression status, with SMYD2 gene
downregulation directly or indirectly inducing p21 expression,
leading to G0/G1 cell cycle phase arrest and the subsequent
suppression of GC cell proliferation (10). Clinical data analysis subsequently
reported that SMYD2 protein expression is associated with tumor
size, lymphatic infiltration, lymph node metastasis rate, invasion
depth and recurrence rate. Prognostic evaluation demonstrated that
patients with elevated SMYD2 expression exhibited significantly
worse outcomes than those with lower levels, suggesting that SMYD2
immunoreactivity may serve as an independent prognostic marker for
predicting OS in patients with GC (10).
Moreover, a clinical study revealed that the
secretion levels of neutrophil extracellular traps (NETs) were
notably elevated in 30 patients with GC compared with the healthy
control group, with these levels increasing as cancer progressed.
Both in vitro and in vivo experiments further
demonstrated that NETs promoted the proliferation, migration and
invasion of GC cells (22).
Mechanistically, NETs upregulate N-acetyltransferase 10
(NAT10), which enhances the mRNA and protein expression levels of
SMYD2 by increasing the half-life. Additionally, NAT10 promotes the
acetylation of SMYD2, further increasing SMYD2 stability and
promoting the proliferation, migration and invasion of GC cells
(22). Using a mouse model, it was
demonstrated that NETs facilitate the progression of GC and its
metastasis to the liver by increasing NAT10 expression (22). These findings suggest that NETs may
facilitate GC metastasis through the NAT10-mediated acetylation of
SMYD2.
Research by Xu et al (64) revealed that SMYD2 promotes the
expression of apolipoprotein C1 (APOC1) by regulating the
trimethylation of H3K4 at the APOC1 promoter. APOC1 has been
identified as a key factor in promoting glycolysis and driving the
proliferation of GC cells (64).
Additional research into the regulatory mechanisms of SMYD2 in GC
revealed that deleted in lymphocytic leukemia 1 (DLEU1) facilitates
SMYD2 recruitment to enhance APOC1 expression. Silencing DLEU1
expression disrupted the interaction of SMYD2 with the APOC1
promoter, leading to decreased H3K4me3 enrichment at this site.
These results highlight the pivotal role of the DLEU1-SMYD2-APOC1
axis in driving glycolysis and cell proliferation in GC (64) (Fig.
2A). This suggests that targeting this axis may represent a
novel therapeutic strategy for GC.
SMYD2 and gastrointestinal stromal tumor
(GIST)
GIST is the most prevalent mesenchymal tumor of the
gastrointestinal tract. GIST can vary in behavior from benign to
highly aggressive, depending on factors such as tumor size, mitotic
rate and location. Surgical resection is the primary treatment for
localized tumors, whilst targeted therapy has markedly improved the
outcomes of patients with advanced or metastatic disease. Combining
surgery with tyrosine kinase inhibitor therapy has increased the
5-year survival rate for localized GIST to 92%, while it is 50% for
metastatic cases (65).
A previous study reported that exposure to LLY-507
and AZ-505 in GIST-T1 cells induced the SMYD2-mediated methylation
of enhancer of zeste homolog 2 (EZH2) at K307, which subsequently
increased EZH2 stability by facilitating its degradation via the
proteasome pathway. Following treatment with LLY-507, there was a
notable reduction in demethylation at K307 and the abundance of
EZH2, alongside decreased methylation at K310 of p53. These
findings indicate that SMYD2 promotes the stability of EZH2 via
methylation at K307 (66). Previous
research in triple-negative breast cancer also indicated that EZH2
epigenetically regulates ten-eleven translocation methylcytosine
dioxygenase 1 (TET1) by mediating H3K27me3, leading to the
repression of TET1 expression. This repression inhibited the
activation of the p53 tumor suppressor signaling pathway, in which
TET1 serves a crucial role (67).
Expanding on this, the researchers further reported that EZH2
promoted GIST cell proliferation whilst suppressing senescence, an
effect that was achieved by elevating H3K27me3 at the TET1
promoter, thereby inhibiting TET1 expression and disrupting
downstream p53 signaling pathways. Furthermore, western blotting
revealed an increase in TET1 and p53 expression in GIST-T1 cells
treated with sh-EZH2, an effect that was reversed upon sh-TET1
treatment (66). Thus, suppressing
EZH2 expression elevated the TET1 and p53 levels, leading to cell
cycle arrest and enhanced senescence in GIST-T1 cells, whereas TET1
knockdown counteracted the effects of EZH2 inhibition (66). To further assess the associations
between SMYD2, EZH2 and TET1 expression in GIST,
immunohistochemistry was employed to analyze their expression
levels in tumor samples from patients classified as low,
intermediate or high risk. The results revealed a positive
association between SMYD2 and EZH2 expression, whilst TET1
expression was negatively associated with EZH2 expression (66). These findings further validated the
experimental results but from a clinical perspective. In summary,
SMYD2 promotes tumor progression in GIST, whilst it inhibits
cellular senescence and apoptosis through modulation of the
EZH2/TET1 signaling axis (Fig. 2C).
Moreover, inhibitors such as LLY-507 and AZ-505 can downregulate
SMYD2 activity, providing new insights for the potential clinical
treatment of GIST.
SMYD2 and liver cancer
HCC is an aggressive liver cancer characterized by
high rates of metastasis and recurrence, leading to a poor
prognosis. Over the past two decades, the incidence of liver cancer
has risen by 53.7%, while the mortality rate has increased by
48.0%. Despite rapid advancements in comprehensive treatment,
primarily centered on surgical resection, the overall 5-year
survival rate remains below 20% (68). HCC frequently arises in individuals
with chronic liver conditions, including hepatitis B or C
infections, alcohol-induced liver injury or non-alcoholic fatty
liver disease. Current treatment options include surgical
resection, liver transplantation, locoregional therapies and
systemic treatments such as targeted therapy and immunotherapy
(68).
Previous public database analysis indicated that
SMYD2 mRNA levels are positively associated with the incidence rate
and clinical staging of patients with HCC, whilst they are
negatively associated with OS. Further analysis of immune cell
infiltration and immune check sites revealed a positive association
between SMYD2 and CD4+ T cells, as well as macrophages
(63). Several studies have
reported that SMYD2 mRNA and protein levels are markedly elevated
in HCC tissues compared with adjacent non-cancerous tissues
(69–71). In one study, clinical samples and
data from patients with HCC were collected and univariate analysis
was performed, which demonstrated that elevated SMYD2 levels were
notably associated with a decreased OS. Multivariate analysis
revealed that SMYD2 could serve as an independent prognostic
marker, indicating a poor prognosis in patients with HCC (69). Functional experiments performed
using HCC cell lines demonstrated that silencing SMYD2 expression
led to a reduction in cell proliferation and triggered a G0/G1
phase cell cycle arrest, suggesting that SMYD2 promotes HCC cell
growth by regulating proliferation and cell cycle progression.
Moreover, cyclin D1 expression was markedly reduced upon SMYD2
downregulation, indicating that SMYD2 may influence HCC cell
proliferation through the regulation of cyclin D1 (69). However, the precise mechanisms
underlying the SMYD2 regulation of cyclin D1 remain unclear and
require further investigation.
A study by Xu et al (71) demonstrated that SMYD2 expression was
positively associated with tumor number, tumor size and patient
age. This study also reported that SMYD2 knockdown in HCC cell
lines induced G0/G1 phase cell cycle arrest, consistent with
previous findings. Additionally, SMYD2 knockdown inhibited the
expression of c-Myc, cyclin-dependent kinase (CDK)4 and cyclin D1
(71). Furthermore, the study
identified an association between SMYD2 expression and pathways
involved in glutamine metabolism. SMYD2 has been reported to
facilitate c-Myc methylation, leading to its stabilization and
subsequent upregulation of glutaminase 1 (GLS1), an essential
enzyme in glutamine metabolism (71). GLS1 mediates the conversion of
glutamine into glutamate, which enters the tricarboxylic acid cycle
to generate adenosine triphosphate. This metabolic pathway has been
reported to be enhanced by SMYD2 in HCC cells (71). Given the role of glutamine metabolic
reprogramming in sorafenib resistance, the study further
demonstrated that HCC cells with SMYD2 deletion were more sensitive
to sorafenib treatment. These findings suggest that SMYD2 promotes
HCC cell growth and enhances chemotherapy resistance to sorafenib
(71).
Furthermore, research on the relationship between
microRNA (miR)-200b and SMYD2 in HCC demonstrated that miR-200b
binds to the 3′-untranslated region of SMYD2, thereby suppressing
its transcription. Bioinformatics analysis predicted this
interaction, which was subsequently validated through a
dual-luciferase reporter assay (70). In vitro experiments reported
that overexpression of miR-200b reduced SMYD2 protein levels,
suppressed cell proliferation, increased p53 expression and
decreased cyclin E1 levels in HCC cells. These findings suggest
that miR-200b may limit SMYD2 expression, restore p53 levels and
induce cell cycle arrest by downregulating cyclin E1 expression
(70). Moreover, a recent study
reported that hypoxia-induced SZT2 antisense RNA 1 (SZT2-AS1)
regulated the levels of H3K4me3 and H3K36me3 in HCC cells. Rescue
experiments were performed to assess whether SMYD2-mediated
SZT2-AS1 regulates histone methylation, and western blotting
revealed that SZT2-AS1-knockdown decreased the levels of H3K4me3
and H3K36me3. These reductions were then rescued by the
overexpression of SMYD2 (72). This
finding suggests that SZT2-AS1 regulates H3K4me3 and H3K36me3
levels by recruiting SMYD2 in HCC cells under hypoxia, thereby
promoting HCC growth, metastasis and angiogenesis (Fig. 2D). In summary, SMYD2 is highly
expressed in HCC, where it exerts oncogenic effects and is
associated with adverse clinical characteristics and a poor
prognosis. Thus, SMYD2 protein levels may act as an independent
prognostic indicator of unfavorable outcomes in patients with HCC,
making it a potential target for therapeutic intervention.
SMYD2 and pancreatic cancer (PC)
PC is often diagnosed at the advanced stages of
disease due to its subtle early symptoms, leading to a poor
prognosis. Risk factors for PC include chronic pancreatitis,
smoking, obesity, diabetes and genetic predisposition. Treatment
strategies for PC involve surgical resection for eligible patients,
along with chemotherapy, radiation therapy and targeted therapy
(73).
SMYD2 is highly expressed in both PC tissues and
cell lines. SMYD2 mediates the methylation of c-Myc, promoting its
ubiquitination and subsequent degradation, which in turn reduces
c-Myc expression in PC (74).
Previous research has identified nuclear receptor coactivator 4
(NCOA4) as a specific receptor for ferritin, serving a crucial role
in ferritinophagy. This process facilitates iron release from
ferritin, contributing to iron homeostasis and the regulation of
ferroptosis (75). The regulation
of ferroptosis is achieved through c-Myc, which influences NCOA4
levels (76). Based on these
findings, the c-Myc/NCOA4 axis was further assessed and it was
reported that c-Myc inhibited NCOA4 expression by binding to NCOA4
mRNA, whilst a reduction in c-Myc led to an increase in NCOA4
expression (74). Furthermore, in
mouse experiments, knockdown of SMYD2 expression in PC tumor
tissues was associated with a notable reduction in tumor volume.
SMYD2 knockdown in PC tumor tissues also led to decreased c-Myc
levels, increased NCOA4 levels and heightened levels of
ferritinophagy and ferroptosis (74). Therefore, targeting SMYD2 may
promote ferritinophagy-dependent ferroptosis through the
c-Myc/NCOA4 axis, potentially inhibiting PC development.
Adenosquamous carcinoma of the pancreas (ASCP) is a
relatively rare histological subtype that includes both pancreatic
adenocarcinoma (PAAD) and pancreatic squamous cell carcinoma. In a
study by Lenkiewicz et al (77), whole-genome copy number variation
analysis of patient-derived xenografts (PDXs) from 3 patients with
ASCP and 3 patients with pancreatic ductal adenocarcinoma (PDAC)
revealed that SMYD2 displays distinct chromatin activity in ASCP
PDX samples, specifically targeting the active chromatin marker,
H3K4me1. Compared with PDAC, the specificity of SMYD2 for H3K4me1
in ASCP may serve as a distinguishing feature and a potential
biomarker for differentiating ASCP from PDAC (77).
PAAD accounts for ~85% of PCs and has a poor
prognosis, with nearly 80% of patients experiencing postoperative
recurrence and a 5-year survival rate of just 6% (78). An analysis of The Cancer Genome
Atlas database indicated that SMYD2 levels are markedly elevated in
PAAD tissues compared with adjacent normal tissues, with its
expression closely associated with patient prognosis. Further
research revealed that SMYD2 upregulates the MNAT1 component of CDK
activating kinase (MNAT1) expression in PAAD cells by modifying the
H3K36me2 marker in the region of the MNAT1 promoter. Silencing
MNAT1 expression inhibits the aggressive characteristics of PAAD
cells. Furthermore, rescue experiments demonstrated that
overexpression of MNAT1 restored the activity of PAAD cells
suppressed by sh-SMYD2 (79). MNAT1
functions as a substrate specificity determining factor, as well as
an assembly factor of cyclin-dependent kinase-activating kinase
(CAK) to promote CAK stability and activation, which further leads
to the phosphorylation and activation of CDKs to ensure cell cycle
progression. It has been reported that the phosphorylation of PI3K
and AKT in PAAD cells can be inhibited by sh-SMYD2 but restored by
overexpression of MNAT1, indicating that the SMYD2-MNAT1 axis is at
least partially involved in the activation of PI3K/AKT, thereby
promoting PAAD cell proliferation, migration and invasion (79). However, the exact mechanistic link
between MNAT1 and the PI3K/AKT pathway remains unclear,
necessitating further investigation. Another study reported that
SMYD2 increases ecotropic viral integration site 2A (EVI2A)
expression in PAAD cells via H3K36me2 modification. Elevated EVI2A
expression induced M2-like macrophage polarization, which inhibited
T-cell effector activation and promoted gemcitabine (GEM)
resistance and immune evasion in PAAD cells (80). These findings highlight the critical
role of SMYD2 in regulating PAAD invasiveness through mechanisms
involving GEM resistance and immune evasion.
Clinically, PDAC tends to originate from the
pancreatic ducts. A previous study reported that SMYD2 promoted
PDAC cell growth by methylating K355 of mitogen-activated protein
kinase-activated protein kinase 3 (MAPKAPK3) (12). Notably, SMYD2 expression is
undetectable in normal pancreatic tissue, but it is elevated in
pancreatic intraepithelial neoplasia (PanIN) tissues and PDAC
specimens from both mice and humans. In K-Ras mutant mouse models,
increased SMYD2 expression was associated with cancer development
and its deletion reduced acinar-to-ductal metaplasia and inhibited
PanIN development (12). Further
research indicated that suppressing SMYD2-driven MAPKAPK3
methylation using the small-molecule inhibitor, BAY598, slowed the
proliferation of PDAC cells harboring K-Ras/TP53 mutations, whereas
those with mutations in K-Ras/TP53/SMYD2 had a minimal response
(12). Additionally, the
combination of GEM and BAY598 markedly inhibited the growth of
K-Ras/TP53 mutant PDAC cells, whilst GEM alone inhibited the growth
of K-Ras/TP53/SMYD2 mutant PDAC cells (12). These findings suggest that combining
SMYD2 inhibitors with GEM could enhance therapeutic efficacy in
patients with PDAC (41) (Fig. 2E), highlighting the potential value
of targeting the SMYD2-MAPKAPK3 axis in PDAC treatment.
SMYD2 and esophageal cancer (EC)
EC is the eleventh most common cancer worldwide and
the seventh leading cause of cancer-related deaths, accounting for
2.6% of all new cancer cases and 4.6% of cancer deaths. Despite
significant advancements in the diagnosis and treatment of EC in
recent years, the 5-year survival rate remains only around 20%.
Esophageal squamous cell carcinoma (ESCC) accounts for ~90% of all
EC cases and is characterized by a high recurrence rate and poor
long-term prognosis (81). Due to
its asymptomatic early stages of disease, most ESCC cases are
diagnosed at an advanced stage, limiting treatment options. Current
therapeutic approaches include surgery, chemotherapy, radiotherapy
and emerging targeted and immunotherapy strategies (81).
Public database analysis revealed that SMYD2 mRNA
expression in EC is notably associated with incidence, clinical
stage, TP53 mutation status, OS and recurrence-free survival.
Furthermore, correlation analysis of immune cell infiltration and
immune checkpoints indicated a negative correlation between SMYD2
and CD4+ T cells (63).
Further experiments demonstrated that SMYD2 is highly expressed in
ESCC cell lines and tissues, with its protein levels markedly
associated with clinical features such as sex, venous invasion,
pathological tumor stage (depth of tumor invasion) in the TNM
classification and recurrence status (82). Multivariate analysis reported that
patients with ESCC harboring high SMYD2 expression had a worse OS
compared with those harboring low SMYD2 expression, and SMYD2
positivity was independently associated with a worse prognosis
(82). Knockdown of SMYD2
expression using specific small interfering RNAs suppressed the
proliferation of ESCC cell lines overexpressing SMYD2, largely
independent of TP53 mutation status (82). Moreover, SMYD2 downregulation
primarily arrested cells in the G0-G1 phase, and p21 protein was
detected in the KYSE790 cell line, in a previous study (82). This suggests that the oncogenic role
of SMYD2 in ESCC may involve the regulation of cell cycle proteins
such as p21, independent of p53 (Fig.
2F). However, the aforementioned study did not explore the
precise mechanism through which SMYD2 influences cell proliferation
via p21, indicating the need for further investigation. Overall,
studies on SMYD2 in ESCC remain limited and its precise mechanisms
of action require further investigation.
Future perspectives
Epigenetic modifications, such as DNA methylation
and histone modifications, have a crucial role in cancer
progression. The SMYD protein family of lysine methyltransferases
is integral to epigenetic regulation, with SMYD2 being a key
member. SMYD2 catalyzes the methylation of both histone and
non-histone proteins, influencing several cellular processes.
Structural studies of SMYD2 have provided valuable insights into
its functional domains: The SET domain drives its catalytic
activity, whilst the MYND domain facilitates protein-protein
interactions. SMYD2 methylates H3K4, H3K36 and H4K20, exerting
distinct effects on gene expression. The oncogenic properties of
SMYD2 primarily arise from its ability to methylate tumor
suppressor proteins, including p53, RB and PTEN, thereby inhibiting
their function and promoting tumor development. Numerous studies
have reported that SMYD2 is upregulated in several gastrointestinal
cancer types, including colorectal, gastric and liver cancer, with
specific effects depending on the cancer type (Table II).
| Table II.Function of SET and MYND
domain-containing protein 2 in gastrointestinal cancer. |
Table II.
Function of SET and MYND
domain-containing protein 2 in gastrointestinal cancer.
| Cancer type | Expression | Role | Targets | Biological
function | (Refs.) |
|---|
| Colorectal | Up | Oncogene | ERBB2/FUT4 | Promotes
proliferation and invasion; inhibits apoptosis | (55) |
|
|
| / | MEK/ERK/AP-1;
P-gp | Enhances the L-OHP
resistance of CRC cells | (56) |
|
| Up | Oncogene | RIPK1 | Inhibits apoptosis
and necrosis | (53) |
|
| Up | Oncogene | LINC01605 | Promotes
proliferation, migration and invasion | (57) |
|
| Up | Oncogene | MEX3A; CDX2 | Promotes
proliferation, migration and invasion | (58) |
|
| Up | Oncogene | DNMT1; APC2;
Wnt/β-catenin | Promotes
proliferation, migration and invasion | (52) |
|
| Up | Oncogene | HNRNPK; EGFL7 | Promotes
angiogenesis | (54) |
| Gastric | Up | Oncogene | p21 | Promotes
proliferation, migration and invasion | (10) |
|
| Up | Oncogene | APOC1 | Promotes glycolysis
and proliferation | (64) |
| Gastrointestinal
stromal tumor | Up | Oncogene | EZH2; TET1;
p53 | Promotes
proliferation; inhibits senescence | (66) |
| Liver | Up | Oncogene |
c-Myc/CDK4/cyclinD1 | Promotes
proliferation | (71) |
|
|
| / | c-Myc; GLS1 | Enhances the
sorafenib resistance of HCC cells | (71) |
|
| Up | Oncogene | p53; cyclinE1 | Promotes
proliferation | (70) |
| Pancreatic | Up | Oncogene | c-Myc; NCOA4 | Promotes
proliferation; inhibits ferroptosis | (75) |
|
| Up | Oncogene | MNAT1 | Promotes
proliferation, migration and invasion | (79) |
|
|
| / | EVI2A | Enhances the GEM
resistance of PAAD cells | (80) |
|
| Up | Oncogene | MAPKAPK3 | Promotes
proliferation; enhances the GEM resistance of PDAC cells | (12) |
| Esophageal | Up | Oncogene | p21 | Promotes
proliferation | (82) |
The present review assessed the multifaceted role of
SMYD2 in the progression of gastrointestinal cancer, its
contribution to drug resistance and emerging therapeutic strategies
aimed at inhibiting its activity. Several small molecule inhibitors
targeting the catalytic activity of SMYD2, such as AZ505 and
BAY-598, have demonstrated potent antitumor effects in preclinical
models, particularly when combined with other anticancer agents
such as GEM and the VEGFR2 inhibitor, apatinib. However, challenges
persist in improving the pharmacokinetics and cellular permeability
of these inhibitors to enhance their efficacy. Developing more
selective SMYD2 inhibitors is crucial to minimize off-target
effects and improve therapeutic outcomes. Additionally, identifying
biomarkers to predict SMYD2 inhibitor sensitivity is essential for
patient stratification and personalized treatment approaches.
Future research should focus on optimizing SMYD2 inhibitors and
exploring combination therapies targeting SMYD2 alongside other
oncogenic pathways. Despite the progress made with SMYD2 inhibitors
in the treatment of a number of diseases, especially cancer, no
safety or efficacy evaluations of SMYD2 inhibitors have been
performed in clinical trials to date, to the best of our knowledge.
Ongoing research into the molecular mechanisms of SMYD2, alongside
the advancement of innovative inhibitors, will be crucial for
translating these discoveries into effective cancer treatments.
Discussion and conclusions
SMYD2 has gained recognition as a crucial regulator
of several cellular functions, including the cell cycle and
differentiation, and has been implicated in several pathological
conditions, particularly cancer and cardiovascular diseases. SMYD2
is highly expressed in tissues such as the heart, brain, liver,
kidneys, thymus and ovaries, suggesting its involvement in the
development of the cardiovascular, nervous, gastrointestinal,
musculoskeletal, urinary and reproductive systems. Mechanistically,
SMYD2 acts as a methyltransferase, specifically methylating H3K4,
H3K3 and H4K20, as well as non-histone proteins, thereby regulating
key signaling pathways in tumor cells and influencing the
transcription and expression of downstream target genes.
Clinically, SMYD2 has been identified as an independent biomarker
for the diagnosis and prognosis of gastrointestinal malignancies.
Studies have reported that the aberrant regulation of SMYD2 is
associated with chemotherapy resistance and poor clinical outcomes
in certain patients with cancer, whilst its loss can sensitize
patients to chemotherapy and immunotherapy. This suggests that
SMYD2 antagonists may serve as potential adjuvants in cancer
treatment. SMYD2 is upregulated in several human tumors, and
increasing attention is being paid to understanding its molecular
mechanisms in tumorigenesis and to assessing its viability as a
therapeutic target for restricting tumor cell growth.
Despite the limited scope of research, pioneering
studies have demonstrated that SMYD2-mediated methylation of
histone and non-histone proteins serves a notable role in the
development and progression of gastrointestinal cancer types.
Ongoing research continues to explore the role of SMYD2 in these
cancer types, although challenges remain. These challenges include
the need for a more comprehensive understanding of the biological
functions of SMYD2, the development of small-molecule inhibitors
and probes, as well as clarification on the specific mechanisms by
which SMYD2 influences tumor development and treatment.
Furthermore, a critical challenge in SMYD2-targeted therapy is the
development of highly selective and potent inhibitors, as poor
bioavailability continues to limit their clinical efficacy. In
summary, SMYD2 serves as a crucial factor in the onset and
advancement of gastrointestinal malignancies, influencing both
treatment strategies and patient prognosis, making it a promising
therapeutic target.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
KX and YC contributed to the conceptualization and
design of this study and took responsibility for the integrity of
the work as a whole, from inception to published article. KX
collected the literature and wrote the article. YZ was involved in
drafting the manuscript. YX involved in revising it critically for
content. All authors read and approved the final manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Arnold M, Abnet CC, Neale RE, Vignat J,
Giovannucci EL, McGlynn KA and Bray F: Global burden of 5 major
types of gastrointestinal cancer. Gastroenterology.
159:335–349.e15. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang FF, Luo YH, Wang H and Zhao L:
Metastasis-associated long noncoding RNAs in gastrointestinal
cancer: Implications for novel biomarkers and therapeutic targets.
World J Gastroenterol. 22:8735–8749. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grady WM: Epigenetic alterations in the
gastrointestinal tract: Current and emerging use for biomarkers of
cancer. Adv Cancer Res. 151:425–468. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang DK, Zuo Q, He QY and Li B: Targeted
immunotherapies in gastrointestinal cancer: From molecular
mechanisms to implications. Front Immunol. 12:7059992021.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cao Z, Shu Y, Wang J, Wang C, Feng T, Yang
L, Shao J and Zou L: Super enhancers: Pathogenic roles and
potential therapeutic targets for acute myeloid leukemia (AML).
Genes Dis. 9:1466–1477. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brown MA, Sims RJ III, Gottlieb PD and
Tucker PW: Identification and characterization of Smyd2: A split
SET/MYND domain-containing histone H3 lysine 36-specific
methyltransferase that interacts with the Sin3 histone deacetylase
complex. Mol Cancer. 5:262006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang J, Perez-Burgos L, Placek BJ,
Sengupta R, Richter M, Dorsey JA, Kubicek S, Opravil S, Jenuwein T
and Berger SL: Repression of p53 activity by Smyd2-mediated
methylation. Nature. 444:629–632. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zeng Y, Qiu R, Yang Y, Gao T, Zheng Y,
Huang W, Gao J, Zhang K, Liu R, Wang S, et al: Regulation of EZH2
by SMYD2-mediated lysine methylation is implicated in
tumorigenesis. Cell Rep. 29:1482–1498.e4. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sajjad A, Novoyatleva T, Vergarajauregui
S, Troidl C, Schermuly RT, Tucker HO and Engel FB: Lysine
methyltransferase Smyd2 suppresses p53-dependent cardiomyocyte
apoptosis. Biochim Biophys Acta. 1843:2556–2562. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Komatsu S, Ichikawa D, Hirajima S, Nagata
H, Nishimura Y, Kawaguchi T, Miyamae M, Okajima W, Ohashi T,
Konishi H, et al: Overexpression of SMYD2 contributes to malignant
outcome in gastric cancer. Br J Cancer. 112:357–364. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hamamoto R, Toyokawa G, Nakakido M, Ueda K
and Nakamura Y: SMYD2-dependent HSP90 methylation promotes cancer
cell proliferation by regulating the chaperone complex formation.
Cancer Lett. 351:126–133. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Reynoird N, Mazur PK, Stellfeld T, Flores
NM, Lofgren SM, Carlson SM, Brambilla E, Hainaut P, Kaznowska EB,
Arrowsmith CH, et al: Coordination of stress signals by the lysine
methyltransferase SMYD2 promotes pancreatic cancer. Genes Dev.
30:772–785. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Eggert E, Hillig RC, Koehr S, Stöckigt D,
Weiske J, Barak N, Mowat J, Brumby T, Christ CD, Ter Laak A, et al:
Discovery and characterization of a highly potent and selective
aminopyrazoline-based in vivo probe (BAY-598) for the protein
lysine methyltransferase SMYD2. J Med Chem. 59:4578–4600. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hamamoto R, Saloura V and Nakamura Y:
Critical roles of non-histone protein lysine methylation in human
tumorigenesis. Nat Rev Cancer. 15:110–124. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zheng Q, Zhang W and Rao GW: Protein
lysine methyltransferase SMYD2: A promising small molecule target
for cancer therapy. J Med Chem. 65:10119–10132. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Copeland RA: Protein methyltransferase
inhibitors as precision cancer therapeutics: A decade of discovery.
Philos Trans R Soc Lond B Biol Sci. 373:201700802018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Copeland RA: Epigenetic medicinal
chemistry. ACS Med Chem Lett. 7:124–127. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tang M, Chen G, Tu B, Hu Z, Huang Y,
DuFort CC, Wan X, Mao Z, Liu Y, Zhu WG and Lu W: SMYD2
inhibition-mediated hypomethylation of Ku70 contributes to impaired
nonhomologous end joining repair and antitumor immunity. Sci Adv.
9:eade66242023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Meng J, Yang W, Li C and Li F: Synergistic
anticancer effects of SMYD2 inhibitor BAY-598 and doxorubicin in
non-small cell lung cancer. Heliyon. 10:e320152024. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Razmi M, Yazdanpanah A, Etemad-Moghadam S,
Alaeddini M, Angelini S and Eini L: Clinical prognostic value of
the SMYD2/3 as new epigenetic biomarkers in solid cancer patients:
A systematic review and meta-analysis. Expert Rev Mol Diagn. 1–15.
2022.(Epub ahead of print). PubMed/NCBI
|
|
21
|
Rubio-Tomás T: Novel insights into SMYD2
and SMYD3 inhibitors: From potential anti-tumoural therapy to a
variety of new applications. Mol Biol Rep. 48:7499–7508. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu D, Yang X and Wang X: Neutrophil
extracellular traps promote gastric cancer cell metastasis via the
NAT10-mediated N4-acetylcytidine modification of SMYD2. Cell
Signal. 116:1110142024. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Carr SR, Akerley W, Hashibe M and
Cannon-Albright LA: Evidence for a genetical contribution to
non-smoking-related lung cancer. Thorax. 70:1033–1039. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Spellmon N, Holcomb J, Trescott L,
Sirinupong N and Yang Z: Structure and function of SET and MYND
domain-containing proteins. Int J Mol Sci. 16:1406–1428. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Leinhart K and Brown M: SET/MYND lysine
methyltransferases regulate gene transcription and protein
activity. Genes (Basel). 2:210–218. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ferguson AD, Larsen NA, Howard T, Pollard
H, Green I, Grande C, Cheung T, Garcia-Arenas R, Cowen S, Wu J, et
al: Structural basis of substrate methylation and inhibition of
SMYD2. Structure. 19:1262–1273. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu S, Zhong C, Zhang T and Ding J:
Structure of human lysine methyltransferase Smyd2 reveals insights
into the substrate divergence in Smyd proteins. J Mol Cell Biol.
3:293–300. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
‘SMYD2-SET and MYND domain-containing
protein 2’. UniProt; Geneva: 2024, https://www.uniprot.org/uniprot/Q9NRG4
|
|
29
|
Herz HM, Garruss A and Shilatifard A: SET
for life: Biochemical activities and biological functions of SET
domain-containing proteins. Trends Biochem Sci. 38:621–639. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu J, Cheung T, Grande C, Ferguson AD, Zhu
X, Theriault K, Code E, Birr C, Keen N and Chen H: Biochemical
characterization of human SET and MYND domain-containing protein 2
methyltransferase. Biochemistry. 50:6488–6497. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Abu-Farha M, Lambert JP, Al-Madhoun AS,
Elisma F, Skerjanc IS and Figeys D: The tale of two domains:
Proteomics and genomics analysis of SMYD2, a new histone
methyltransferase. Mol Cell Proteomics. 7:560–572. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Spellmon N, Sun X, Sirinupong N, Edwards
B, Li C and Yang Z: Molecular dynamics simulation reveals
correlated inter-lobe motion in protein lysine methyltransferase
SMYD2. PLoS One. 10:e01457582015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu S, Wu J, Sun B, Zhong C and Ding J:
Structural and biochemical studies of human lysine
methyltransferase Smyd3 reveal the important functional roles of
its post-SET and TPR domains and the regulation of its activity by
DNA binding. Nucleic Acids Res. 39:4438–4449. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sirinupong N, Brunzelle J, Ye J, Pirzada
A, Nico L and Yang Z: Crystal structure of cardiac-specific histone
methyltransferase SmyD1 reveals unusual active site architecture. J
Biol Chem. 285:40635–40644. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chandramouli B and Chillemi G:
Conformational dynamics of lysine methyltransferase Smyd2. insights
into the different substrate crevice characteristics of Smyd2 and
Smyd3. J Chem Inf Model. 56:2467–2475. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang Y, Alshammari E, Sobota J, Spellmon
N, Perry E, Cao T, Mugunamalwaththa T, Smith S, Brunzelle J, Wu G,
et al: Structure of the SMYD2-PARP1 complex reveals both productive
and allosteric modes of peptide binding. bioRxiv [Preprint].
2024.12.03.626679. 2024.
|
|
37
|
Gottlieb PD, Pierce SA, Sims RJ, Yamagishi
H, Weihe EK, Harriss JV, Maika SD, Kuziel WA, King HL, Olson EN, et
al: Bop encodes a muscle-restricted protein containing MYND and SET
domains and is essential for cardiac differentiation and
morphogenesis. Nat Genet. 31:25–32. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sesé B, Barrero MJ, Fabregat MC, Sander V
and Izpisua Belmonte JC: SMYD2 is induced during cell
differentiation and participates in early development. Int J Dev
Biol. 57:357–364. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Diehl F, Brown MA, van Amerongen MJ,
Novoyatleva T, Wietelmann A, Harriss J, Ferrazzi F, Böttger T,
Harvey RP, Tucker PW and Engel FB: Cardiac deletion of Smyd2 is
dispensable for mouse heart development. PLoS One. 5:e97482010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kawamura S, Yoshigai E, Kuhara S and
Tashiro K: smyd1 and smyd2 are expressed in muscle tissue in
Xenopus laevis. Cytotechnology. 57:161–168. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Alshammari E, Zhang YX and Yang Z:
Mechanistic and functional extrapolation of SET and MYND
domain-containing protein 2 to pancreatic cancer. World J
Gastroenterol. 28:3753–3766. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Boehm D, Jeng M, Camus G, Gramatica A,
Schwarzer R, Johnson JR, Hull PA, Montan M, Sakane N, Pagans S, et
al: SMYD2-mediated histone methylation contributes to HIV-1
latency. Cell Host Microbe. 21:569–579.e6. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Olsen JB, Cao XJ, Han B, Chen LH, Horvath
A, Richardson TI, Campbell RM, Garcia BA and Nguyen H: Quantitative
profiling of the activity of protein lysine methyltransferase SMYD2
using SILAC-based proteomics. Mol Cell Proteomics. 15:892–905.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Weirich S, Schuhmacher MK, Kudithipudi S,
Lungu C, Ferguson AD and Jeltsch A: Analysis of the substrate
specificity of the SMYD2 protein lysine methyltransferase and
discovery of novel non-histone substrates. Chembiochem. 21:256–264.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li LX, Fan LX, Zhou JX, Grantham JJ,
Calvet JP, Sage J and Li X: Lysine methyltransferase SMYD2 promotes
cyst growth in autosomal dominant polycystic kidney disease. J Clin
Invest. 127:2751–2764. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Piao L, Kang D, Suzuki T, Masuda A, Dohmae
N, Nakamura Y and Hamamoto R: The histone methyltransferase SMYD2
methylates PARP1 and promotes poly(ADP-ribosyl)ation activity in
cancer cells. Neoplasia. 16:257–264. 264.e22014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Nakakido M, Deng Z, Suzuki T, Dohmae N,
Nakamura Y and Hamamoto R: Dysregulation of AKT pathway by
SMYD2-mediated lysine methylation on PTEN. Neoplasia. 17:367–373.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Saddic LA, West LE, Aslanian A, Yates JR
III, Rubin SM, Gozani O and Sage J: Methylation of the
retinoblastoma tumor suppressor by SMYD2. J Biol Chem.
285:37733–37740. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cho HS, Hayami S, Toyokawa G, Maejima K,
Yamane Y, Suzuki T, Dohmae N, Kogure M, Kang D, Neal DE, et al: RB1
methylation by SMYD2 enhances cell cycle progression through an
increase of RB1 phosphorylation. Neoplasia. 14:476–486. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Obermann WMJ: A motif in HSP90 and P23
that links molecular chaperones to efficient estrogen receptor α
methylation by the lysine methyltransferase SMYD2. J Biol Chem.
293:16479–16487. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ji K, Jia H, Liu Z, Yu G, Wen R, Zhang T,
Peng Z, Man W, Tian Y, Wang C, et al: New insight in immunotherapy
and combine therapy in colorectal cancer. Front Cell Dev Biol.
12:14536302025. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Meng F, Liu X, Lin C, Xu L, Liu J, Zhang
P, Zhang X, Song J, Yan Y, Ren Z and Zhang Y: SMYD2 suppresses APC2
expression to activate the Wnt/β-catenin pathway and promotes
epithelial-mesenchymal transition in colorectal cancer. Am J Cancer
Res. 10:997–1011. 2020.PubMed/NCBI
|
|
53
|
Yu YQ, Thonn V, Patankar JV, Thoma OM,
Waldner M, Zielinska M, Bao LL, Gonzalez-Acera M, Wallmüller S,
Engel FB, et al: SMYD2 targets RIPK1 and restricts TNF-induced
apoptosis and necroptosis to support colon tumor growth. Cell Death
Dis. 13:522022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhang Y, Zhou L, Xu Y, Zhou J, Jiang T,
Wang J, Li C, Sun X, Song H and Song J: Targeting SMYD2 inhibits
angiogenesis and increases the efficiency of apatinib by
suppressing EGFL7 in colorectal cancer. Angiogenesis. 26:1–18.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lai Y and Yang Y: SMYD2 facilitates cancer
cell malignancy and xenograft tumor development through
ERBB2-mediated FUT4 expression in colon cancer. Mol Cell Biochem.
477:2149–2159. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ren H, Wang Z, Chen Y, Liu Y, Zhang S,
Zhang T and Li Y: SMYD2-OE promotes oxaliplatin resistance in colon
cancer through MDR1/P-glycoprotein via MEK/ERK/AP1 pathway. Onco
Targets Ther. 12:2585–2594. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yue M, Liu T, Yan G, Luo X and Wang L:
LINC01605, regulated by the EP300-SMYD2 complex, potentiates the
binding between METTL3 and SPTBN2 in colorectal cancer. Cancer Cell
Int. 21:5042021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Pan L, Fan Y and Zhou L: SMYD2
epigenetically activates MEX3A and suppresses CDX2 in colorectal
cancer cells to augment cancer growth. Clin Exp Pharmacol Physiol.
49:959–969. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Deng X, Hamamoto R, Vougiouklakis T, Wang
R, Yoshioka Y, Suzuki T, Dohmae N, Matsuo Y, Park JH and Nakamura
Y: Critical roles of SMYD2-mediated β-catenin methylation for
nuclear translocation and activation of Wnt signaling. Oncotarget.
8:55837–55847. 2027. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ma X, Xu W, Qi L, Zhang Q, Sun X and Zhang
S: Clinical outcome of non-curative endoscopic submucosal
dissection for early gastric cancer. J Gastrointest Oncol.
15:566–576. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Liu Z, Xu H, You W, Pan K and Li W:
Helicobacter pylori eradication for primary prevention of gastric
cancer: Progresses and challenges. J Natl Cancer Cent. 4:299–310.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Liu D, Liu M, Wang W, Li X, Shi E, Zhang
C, Wang Y, Zhang Y, Wang L and Wang X: SMYD family members serve as
potential prognostic markers and correlate with immune infiltrates
in gastric cancer. J Oncol. 2023:60328642023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Liu D, Wang X, Shi E, Wang L, Nie M, Li L,
Jiang Q, Kong P, Shi S, Wang C, et al: Comprehensive analysis of
the value of SMYD family members in the prognosis and immune
infiltration of malignant digestive system tumors. Front Genet.
12:6999102021. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Xu H, Ba Z, Liu C and Yu X: Long noncoding
RNA DLEU1 promotes proliferation and glycolysis of gastric cancer
cells via APOC1 upregulation by recruiting SMYD2 to induce
trimethylation of H3K4 modification. Transl Oncol. 36:1017312023.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
He C, Wang Z, Yu J, Mao S and Xiang X:
Current drug resistance mechanisms and treatment options in
gastrointestinal stromal tumors: Summary and update. Curr Treat
Options Oncol. 25:1390–1405. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ji Y, Xu X, Long C, Wang J, Ding L, Zheng
Z, Wu H, Yang L, Tao L and Gao F: SMYD2 aggravates gastrointestinal
stromal tumor via upregulation of EZH2 and downregulation of TET1.
Cell Death Discov. 8:2742022. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Yu Y, Qi J, Xiong J, Jiang L, Cui D, He J,
Chen P, Li L, Wu C, Ma T, et al: Epigenetic co-deregulation of
EZH2/TET1 is a senescence-countering, actionable vulnerability in
triple-negative breast cancer. Theranostics. 9:761–777. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Hwang SY, Danpanichkul P, Agopian V, Mehta
N, Parikh ND, Abou-Alfa GK, Singal AG and Yang JD: Hepatocellular
carcinoma: Updates on epidemiology, surveillance, diagnosis and
treatment. Clin Mol Hepatol. 31 (Suppl):S228–S254. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zuo SR, Zuo XC, He Y, Fang WJ, Wang CJ,
Zou H, Chen P, Huang LF, Huang LH, Xiang H and Liu SK: Positive
expression of SMYD2 is associated with poor prognosis in patients
with primary hepatocellular carcinoma. J Cancer. 9:321–330. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Fang W, Song L, Li Z, Meng P, Zuo S and
Liu S: Effect of miRNA-200b on the proliferation of liver cancer
cells via targeting SMYD2/p53 signaling pathway. Zhong Nan Da Xue
Xue Bao Yi Xue Ban. 47:1303–1314. 2022.(In English, Chinese).
PubMed/NCBI
|
|
71
|
Xu K, Ding J, Zhou L, Li D, Luo J, Wang W,
Shang M, Lin B, Zhou L and Zheng S: SMYD2 promotes hepatocellular
carcinoma progression by reprogramming glutamine metabolism via
c-Myc/GLS1 axis. Cells. 12:252022. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Liu R, Guo Y, Wang L, Yin G, Tuo H, Zhu Y,
Yang W, Liu Q and Wang Y: A novel hypoxia-induced lncRNA, SZT2-AS1,
boosts HCC progression by mediating HIF heterodimerization and
histone trimethylation under a hypoxic microenvironment. Cell Death
Differ. Nov 22–2024.(Epub ahead of print).
|
|
73
|
Jiang Z, Zheng X, Li M and Liu M:
Improving the prognosis of pancreatic cancer: Insights from
epidemiology, genomic alterations, and therapeutic challenges.
Front Med. 17:1135–1169. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Tan J, Liao S, Yuan B, Liu X, Yu W, Zhan
H, Jiang Y and Liu Y: Targeting SMYD2 promotes ferroptosis and
impacts the progression of pancreatic cancer through the
c-Myc/NCOA4 axis-mediated ferritinophagy. Biochim Biophys Acta Gen
Subj. 1868:1306832024. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Mancias JD, Wang X, Gygi SP, Harper JW and
Kimmelman AC: Quantitative proteomics identifies NCOA4 as the cargo
receptor mediating ferritinophagy. Nature. 509:105–109. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Jin Y, Qiu J, Lu X and Li G: C-MYC
inhibited ferroptosis and promoted immune evasion in ovarian cancer
cells through NCOA4 mediated ferritin autophagy. Cells.
11:41272022. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Lenkiewicz E, Malasi S, Hogenson TL,
Flores LF, Barham W, Phillips WJ, Roesler AS, Chambers KR,
Rajbhandari N, Hayashi A, et al: Genomic and epigenomic landscaping
defines new therapeutic targets for adenosquamous carcinoma of the
pancreas. Cancer Res. 80:4324–4334. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Zhao Q, Ye Y, Zhang Q, Wu Y, Wang G, Gui Z
and Zhang M: PANoptosis-related long non-coding RNA signature to
predict the prognosis and immune landscapes of pancreatic
adenocarcinoma. Biochem Biophys Rep. 37:1016002023.PubMed/NCBI
|
|
79
|
Xu Z, Liu Y, Pan Z and Qin L: Epigenetic
upregulation of MNAT1 by SMYD2 is linked to PI3K/AKT activation and
tumorigenesis of pancreatic adenocarcinoma. Histol Histopathol.
39:263–277. 2024.PubMed/NCBI
|
|
80
|
Jin L, Qian D, Tang X, Huang Y, Zou J and
Wu Z: SMYD2 imparts gemcitabine resistance to pancreatic
adenocarcinoma cells by upregulating EVI2A. Mol Biotechnol.
66:2920–2933. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Jiang W, Zhang B, Xu J, Xue L and Wang L:
Current status and perspectives of esophageal cancer: A
comprehensive review. Cancer Commun (Lond). 45:281–331. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Komatsu S, Imoto I, Tsuda H, Kozaki KI,
Muramatsu T, Shimada Y, Aiko S, Yoshizumi Y, Ichikawa D, Otsuji E
and Inazawa J: Overexpression of SMYD2 relates to tumor cell
proliferation and malignant outcome of esophageal squamous cell
carcinoma. Carcinogenesis. 30:1139–1146. 2009. View Article : Google Scholar : PubMed/NCBI
|