Introduction
Studies conducted in our lab have indicated that
thalidomide cytotoxicity in the KG-1a human acute myelogenous
leukemia (AML) cell line was enhanced by combining it with arsenic
trioxide (As2O3). The current investigation
was conducted in order to evaluate the effect of thalidomide either
alone or in combination with As2O3 and
interleukin-2 (IL-2) on the release of tumor necrosis factor-α
(TNF-α) and vascular endothelial growth factor (VEGF) from this
cell line in an attempt to clarify its possible cytotoxic
mechanism(s).
TNF-α is a potent pro-inflammatory cytokine produced
primarily by monocytes and macrophages. Excessive or prolonged
production of TNF-α played a role in inflammatory processes as well
as in the pathogenesis of other human diseases (1). TNF-α appears to be particularly
important in abnormal apoptosis of hematopoietic progenitors
(2).
Abnormally high levels of TNF-α were associated with
the pathology and symptoms of a wide variety of diseases, including
erythema nodosum leprosum (ENL), acquired immune deficiency
syndrome (AIDS), cancer, graft vs. host disease (GVHD),
tuberculosis and malaria. Fever, weight loss and debility
associated with these diseases may be due to the macrophage
production of cytokines, one of which is TNF-α (3,4).
In vitro, thalidomide causes selective
inhibition of TNF-α production by human monocytes triggered by
lipopolysaccharides (LPS) or mycobacterial agonists (5). These effects occur at concentrations
comparable with serum levels achieved in vivo in dosing
regimens currently used, i.e., at doses of up to 400 mg/day. The
level of TNF-α inhibition in vitro is about 40% at the
clinically achievable concentration of 1 μg/ml (5).
Tumor cells with the angiogenic phenotype may
overexpress one or more angiogenic proteins, mobilize an angiogenic
protein from the extracellular matrix, recruit host cells that
produce their own angiogenic proteins and/or down-regulate negative
angiogenic regulators. The most common angiogenic proteins found in
tumors are basic fibroblast growth factor (bFGF) and VEGF.
Angiogenesis is a biological process whereby endothelial cells
divide and migrate to form new blood vessels. Angiogenesis is a key
step in various disease states, including tumor growth, invasion
and metastasis (6).
Thalidomide provides its anti-angiogenesis
inhibition by blocking bFGF and VEGF (7). Thalidomide inhibits angiogenesis in
corneal pellets containing the angiogenic factors bFGF and VEGF, in
a non-inflammatory model of angiogenesis, suggesting that
thalidomide directly affects endothelial cells (7,8). Kruse
et al (1998) concluded that thalidomide is a potent
angiogenesis inhibitor in vivo (7).
Materials and methods
Human KG-1a acute myeloid leukemia
cells
The KG-1a cells which are an early phenotype of
human AML (American Type Culture Collection, Manassas, VA, USA)
were grown in complete growth medium [Iscove’s Modified Dulbeco’s
Medium (American Type Culture Collection) supplemented with 20%
fetal bovine serum (Sigma-Aldrich, UK) and 1%
penicillin-streptomycin (Gibco Invitrogen Corporation, Carlsbad,
CA, USA) at 37°C in a humidified 5% CO2 incubator.
Treatment of human KG-1a acute myeloid
leukemia cells
The KG-1a cells were cultured for 48 h in 12-well
tissue culture plates, each containing complete growth medium at a
concentration of 2×106 cells/ml. Each well also
contained a total volume of 2 ml. Thalidomide (Tocris Bioscience,
Ellisville, MO, USA) was added at concentrations of 5 mg/l whether
used alone or in combination with other chemotherapeutic agents.
IL-2 (Proleukin®, Aldesleuken for injection) (Chiron
Therapeutics, Emeryville, CA, USA) was added at a concentration of
200 IU/ml whether used alone or in combination with thalidomide and
As2O3. As2O3
(Sigma-Aldrich, Inc., St. Louis, MO, USA) was added at two
concentrations of 2 and 4 μM with and without 100 μM of ascorbic
acid in the first flow cytometry study, and at 4 μM in the
remaining studies, either alone or combined with thalidomide and
IL-2. A control culture containing neither thalidomide nor IL-2 nor
As2O3 was set up in conditions otherwise
identical. The control and treated cultures were set in duplicate
and incubated for 48 h at 37°C in a humidified 5% CO2
incubator. The incubation time was selected to allow adequate time
for apoptosis and necrosis to occur in the KG-1a human myeloid
leukemia cells (9).
Analysis of biologically active tumor
necrosis factor
Treated and untreated KG-1a human leukemia cell line
culture supernates were assayed for biologically active TNF using
the Quantikine® Human TNF-α/TNFSF1A Immunoassay (R&D
Systems®, Minneapolis, MN, USA), which is used for the
quantitative determination of human TNF-α concentrations in cell
culture supernates. Analysis was carried out as described in the
Quantikine kit manual. Briefly, reagents and working standards were
prepared per kit instructions. Assay diluent RD1F (50 μl) was then
added to each well. Subsequently, 200 μl of the standards and
samples including the controls were added to each well. The TNF-α
microplate was incubated for 2 h at room temperature, after which
each well was aspirated and washed with the wash buffer four times.
TNF-α conjugate (200 μl) was then added to each well and the wells
were incubated for another hour. After that the wells were
aspirated and washed again four times with the washing buffer. This
was followed by the addition of 200 μl of substrate solution to
each well, and the microplate was incubated for 20 min away from
light. Stop solution (50 μl) was then added to each well. The
optical density of each well was determined within 30 min, using a
microplate reader (Power Wave X 340, Bio-Tek®
Instruments, Inc., Winooski, VT, USA) set to 450 nm (λ correction
at 570 nm) (10).
Measurements of the vascular endothelial
growth factor
Treated and untreated KG-1a human leukemia cell line
culture supernates were assayed to measure the concentration of
VEGF using the Quantikine Human VEGF Immunoassay (R&D Systems),
which is used for the quantitative determination of human VEGF
concentrations in cell culture supernates. Analysis was carried out
as described in the Quantikine kit manual. This assay employs the
quantitative sandwich enzyme immunoassay technique. Briefly,
reagents and working standards were prepared per kit instructions.
Assay diluent RD1W (50 μl) was added to each well. Subsequently,
200 μl of the standards and samples, including the controls, were
added to each well. The VEGF microplate was incubated for 2 h at
room temperature, after which each well was aspirated and washed
with the wash buffer three times. VEGF conjugate (200 μl) was then
added to each well and wells were incubated for 2 h. Following this
incubation, wells were aspirated and washed again three times with
the washing buffer. Substrate solution (200 μl) was added to each
well, and the microplate was incubated for 20 min away from light.
Stop solution (50 μl) was then added to each well. The optical
density of each well was determined within 30 min, using a
microplate reader (Power Wave X 340, Bio-Tek Instruments, Inc.) set
to 450 nm (λ correction at 570 nm) (11).
Statistical analysis
Results were subjected to one-way ANOVA. Statistical
significant differences between means was set at p<0.05.
Results
Human tumor necrosis factor-α immunoassay
(ELISA)
The objective of ELISA, which employs the
quantitative sandwich enzyme immunoassay technique, is to evaluate
the effect of thalidomide, IL-2, and As2O3 or
their combination on the level of TNF-α in the cell culture
supernate of the KG-1a human leukemia cell line. This evaluation
was conducted in an attempt to elucidate the possible mechanism of
thalidomide cytotoxicity as well as the enhancement mechanism of
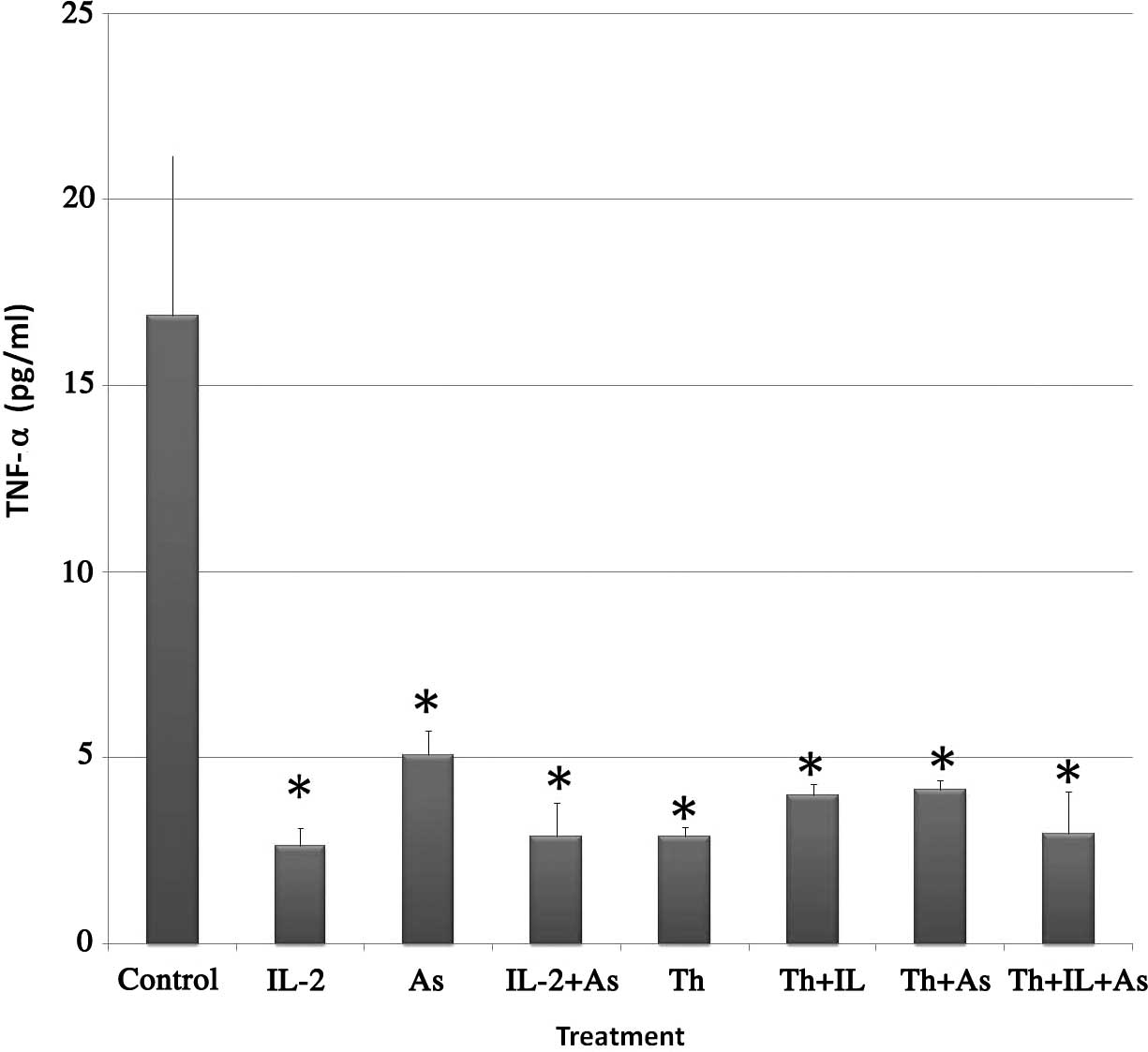
As2O3 on this cytotoxicity. Fig. 1 shows the results obtained. The
following levels of TNF-α were found in the supernatant of the
various treatment groups: i) Control, 16.89±4.43 pg/ml; ii) IL-2,
2.64±0.48 pg/ml; iii) As2O3, 5.07±0.69 pg/ml;
iv) IL-2 and As2O3, 2.89±0.91 pg/ml; v)
thalidomide, 2.89±0.25 pg/ml; vi) thalidomide and IL-2, 3.98±0.30
pg/ml; vii) thalidomide and As2O3, 4.15±0.25
pg/ml and viii) thalidomide, IL-2 and As2O3,
2.96±1.13 pg/ml.
From the results, it is evident that TNF-α levels
significantly decreased in KG-1a cells incubated with the
chemotherapeutic agents alone or in combination.
Human vascular endothelial growth factor
immunoassay (ELISA)
ELISA was used for the quantitative determination of
human VEGF concentration in the cell culture supernate of the KG-1a
human leukemia cell line following the application of the different
chemotherapeutic agents. This determination was performed to
evaluate the anti-angiogenic effect of thalidomide when used alone
or in combination with the remaining chemotherapeutic agents.
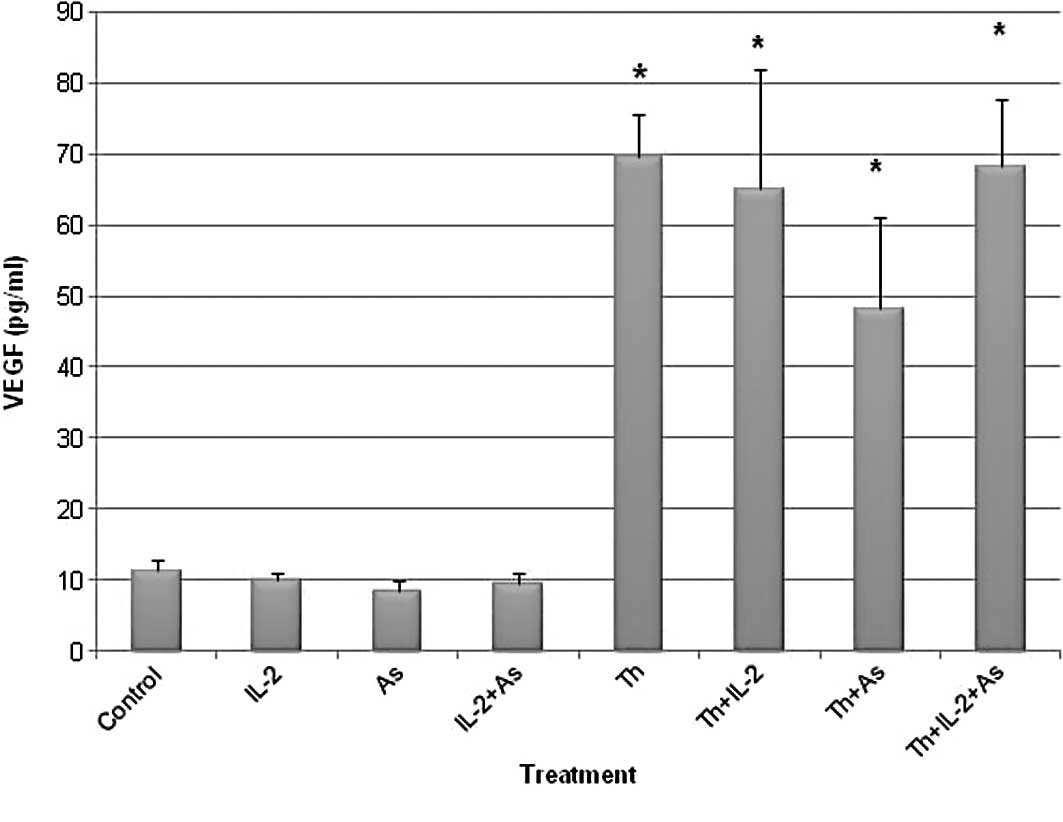
Fig. 2 shows the results obtained.
The VEGF levels observed in the supernatant of the various
treatment groups included: i) Control, 11.42±1.43 pg/ml; ii) IL-2,
9.99±1.15 pg/ml; iii) As2O3, 8.51±1.48 pg/ml;
iv) IL-2 and As2O3, 9.35±1.78 pg/ml; v)
thalidomide, 69.61±5.99 pg/ml; vi) thalidomide and IL-2,
65.07±16.83 pg/ml; vii) thalidomide and
As2O3, 48.13±12.82 pg/ml and viii)
thalidomide and IL-2 and As2O3, 68.19±9.57
pg/ml.
The results showed that thalidomide treatment
increased the VEGF levels in contrast to IL-2 and
As2O3 which did not affect the VEGF
levels.
Discussion
This study was conducted as a follow-up to our
previous study whose purpose was to evaluate the efficacy of
thalidomide in the management of AML and to test the possibility of
enhancing its cytotoxicity by combining it with other
chemotherapeutic agents such as IL-2 and
As2O3. The variant subline KG-1a of the human
acute myelogenous leukemia cell line KG-1 was used as a test model
in both the previous and current studies.
The present study hypothesized that the enhancement
of cytotoxicity induced by thalidomide is due to the effects on
TNF-α and VEGF. This hypothesis stems from the observation that
angiogenesis increases tumor growth via perfusion as well as the
paracrine production of growth factors by endothelial cells or
their release by macrophage and other host cells (12). The most common angiogenic proteins
noted in tumors are VEGF and bFGF. VEGF is a potent mitogen for
vascular endothelial cells that has been associated with
angiogenesis, growth, dissemination, metastasis, and poor outcome
in solid tumors (13). Fiedler
et al (14) found VEGF
transcription in 23 of 33 (69%) patients with AML. Leukemic cell
cultures from 24 of these patients produced significantly high VEGF
levels (14). In addition, TNF-α is
a potent pro-inflammatory cytokine produced primarily by monocytes
and macrophages. Excessive or prolonged production of TNF-α played
a role in inflammatory processes as well as in the pathogenesis of
other human diseases (1). TNF-α
appears to be particularly important in abnormal apoptosis of
hematopoietic progenitors (2).
Consequently, we attempted to clarify the role of TNF-α and VEGF in
cytotoxicity induced by thalidomide, As2O3
and IL-2.
The results obtained in the current study indicate
that thalidomide pretreatment decreased the levels of TNF-α in the
supernates from 16.9 pg/ml in the control to 2.9 pg/ml. This effect
was also detected when cells were pretreated with either IL-2 or
As2O3 (2.64 and 5.07 pg/ml, respectively).
When thalidomide was used concurrently with
As2O3 and IL-2 the levels of TNF-α were also
significantly lowered as compared to the control (4.15 and 3.98
pg/ml, respectively).
The results are consistent with other studies which
showed that thalidomide inhibits TNF-α (15–19).
Thalidomide inhibits the synthesis of TNF-α in vitro and
in vivo. Abnormally high levels of TNF-α have been
associated with the pathology and symptoms of a wide variety of
diseases, including ENL, AIDS, cancer, GVHD, tuberculosis and
malaria. Fever, weight loss, and debility associated with these
diseases may be due to the macrophage production of cytokines, one
of which is TNF-α (3,4). Animal models used to investigate the
activity of TNF-α have shown that infusion of purified recombinant
TNF-α reproduces the characteristics of septic shock syndrome,
including severe hypotension, lactic acidosis, third-space fluid
loss, and tissue injury (4).
Endothelial cell damage, disturbance of lipid metabolism and
interleukin-1 (IL-1) release also occur. Furthermore, chronic
sublethal injection of TNF-α produces cachexia in rats (4).
In vitro, thalidomide causes selective
inhibition of TNF-α production by human monocytes triggered by LPS
or mycobacterial agonists (5).
These effects occur at concentrations comparable with serum levels
achieved in vivo in dosing regimens currently used, i.e., at
doses of up to 400 mg/day. The level of TNF-α inhibition in
vitro is about 40% at the clinically achievable concentration
of 1 μg/ml (5). Thalidomide does
not directly influence the production of other cytokines (IL-1β,
IL-6 and GM-CSF) that may be important in conditions where
suppression of TNF-α is desirable, but immunity must otherwise
remain intact (16).
Thalidomide results in decreased TNF-α production by
accelerating the degradation of mRNA encoding the protein (16). This mechanism is different from the
mechanism of action proposed for pentoxifylline or corticosteroids,
which suppress LPS-induced TNF-α RNA transcription and translation,
respectively. Notably, the results are consistent with other
studies which showed that thalidomide inhibits TNF-α (15–19).
The ability of thalidomide to reduce elevated levels of TNF-α makes
the drug of potential therapeutic importance in any disease state
where high TNF-α levels cause primary problems or secondary
complications.
However, results obtained from the VEGF immunoassay
in the current study indicate that thalidomide pretreatment
increases VEGF levels in the supernates from 11.42 pg/ml in the
control to 69.61 pg/ml. This effect was also detected when cells
were pretreated with thalidomide combined with IL-2,
As2O3 or both (65.07, 48.13 and 68.19 pg/ml,
respectively). Moreover, when cells were pretreated with either
IL-2 or As2O3 VEGF levels were not different
from the control (9.99 and 8.51 pg/ml, respectively compared to
11.42 pg/ml for the control). These results confirm that
thalidomide has an important role in angiogenesis. Angiogenesis is
a biological process in which endothelial cells divide and migrate
to form new blood vessels. Anti-angiogenesis involves therapy
against biochemical targets on the neovasculature, inhibiting
proliferation and blood vessel formation. Angiogenesis is therefore
significant in various disease states, including tumor growth,
invasion and metastasis (6).
Thalidomide provides anti-angiogenesis inhibition by
blocking bFGF and VEGF (7).
Thalidomide inhibits angiogenesis in corneal pellets that contain
angiogenic factors, bFGF and VEGF, in a non-inflammatory model of
angiogenesis. Thus, thalidomide may directly affect endothelial
cells (7,8). Kruse et al concluded that
thalidomide is a potent angiogenesis inhibitor in vivo.
The increase of VEGF in our study was due to the
release of growth-promoting substances, including VEGF and bFGF, by
the surviving KG-1a leukemic cells. This increase of VEGF levels
likely reflects an autologous protective mechanism or mutation by
the surviving malignant leukemic cells to increase their resistance
to subsequent chemotherapy. Another comparable study that supports
our explanation regarding the increase of VEGF was conducted by
Brieger et al (20). These
authors reported on the time- and dose-dependent release of VEGF
and bFGF from squamous cell carcinoma (SCC) cells in cell culture
after irradiation. These authors also demonstrated that these
factors are possible protectors from the irradiation-induced cell
death of cancer cells. The hypothesis of autologous protection of
tumor cells from radiation-induced cell death by secreted factors
is supported by several studies. Shintani et al (21) reported increased VEGF-levels in oral
SCC in the case of non-responders to radiotherapy. Similar data
were noted by Koukourakis et al (22) in head and neck SCC. Geng et
al (23) showed that inhibition
of VEGF-signaling results in the reversal of tumor resistance to
radiotherapy. Taken together, high VEGF levels and consecutive
activation of the VEGF-receptor and downstream signal transduction
pathways may result in a direct cytoprotection of tumor cells or in
the protection of vessels and increased neoangiogenesis. Therefore,
ionizing radiation promotes tumor cell survival, as well as desired
effects such as the induction of apoptosis and growth arrest
(20).
In our study, the effects of thalidomide were
comparable to radiotherapy since the former showed considerable
cytotoxic effect as evidenced by flow cytometry studies.
Thalidomide also exhibited autologous protection of KG-1a leukemia
cells by increasing secretion of VEGF from the cells that
survived.
In conclusion, this study showed that thalidomide,
As2O3 and IL-2 decreased the level of tumor
necrosis-α in the cell supernatant in a treated KG-1a human acute
myelogenous leukemia cell line. On the other hand, it was shown
that thalidomide increased the release of vascular endothelial
growth factor in the cell supernatant. This increase suggests an
autologous protective mechanism or mutation by the surviving
malignant leukemic cells to increase their resistance to subsequent
chemotherapy.
Acknowledgements
This study was supported in part by NIH grant
RR03020.
References
|
1
|
McGeehan GM and Uhl J: TNF-α in human
diseases. Curr Pharm Des. 2:662–667. 1996.
|
|
2
|
Snoeck HW, Weekx S, Moulijn A, et al:
Tumor necrosis factor alpha is a potent synergistic factor for the
proliferation of primitive human hematopoietic progenitor cells and
induces resistance to transforming growth factor beta but not to
interferon gamma. J Exp Med. 183:705–710. 1996. View Article : Google Scholar
|
|
3
|
Girardin E, Grau GE, Dayer JM,
Roux-Lombard P and Lambert PH: Tumor necrosis factor and
interleukin-1 in the serum of children with severe infectious
purpura. N Eng J Med. 319:397–400. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tracey KJ, Wei H, Manogue KR, et al:
Cachectin/tumor necrosis factor induces cachexia, anemia and
inflammation. J Exp Med. 167:1211–1227. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sampaio EP, Sarno EN, Galilly R, Cohn ZA
and Kaplan G: Thalidomide selectively inhibits tumor necrosis
factor alpha production by stimulated human monocytes. J Exp Med.
173:699–703. 1991. View Article : Google Scholar
|
|
6
|
Minchinton AL, Fryer KH, Wendt KR, Clow KA
and Hayes MM: The effect of thalidomide on experimental tumors and
metastases. Anticancer Drugs. 7:339–343. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kruse FF, Joussen AM, Rohrschneider K,
Becker MD and Volcker HE: Thalidomide inhibits corneal angiogenesis
induced by vascular endothelial growth factor. Graefes Arch Clin
Exp Ophthalmol. 236:461–466. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
D’Amato RJ, Loughnan MS, Flynn E and
Folkman J: Thalidomide is an inhibitor of angiogenesis. Proc Natl
Acad Sci USA. 91:4082–4085. 1994.
|
|
9
|
Lu C and Hassan HT: Human stem cell
factor-antibody (anti-SCF) enhances chemotherapy cytotoxicity in
human CD34+ resistant myeloid leukemia cells. Leuk Res. 30:296–302.
2006.PubMed/NCBI
|
|
10
|
Renauld AE and Spengler RN: Tumor necrosis
factor expressed by primary hippocampal neurons and SH-SY5Y cells
is regulated by α2-adrenergic receptor activation. J Neurosci Res.
67:264–274. 2001.PubMed/NCBI
|
|
11
|
Ogasawara T, Narita C and Kawauchi K:
Production of vascular endothelial growth factor in T-cell
prolymphocytic leukemia. Leuk Res. 31:403–406. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Folkman J: Angiogenesis: mechanistic
insights, neovascular diseases in cancer, rheumatoid and other
disease. Nature Med. 1:27–31. 1995. View Article : Google Scholar
|
|
13
|
Aguayo A, Estey E, Kantarjian H, et al:
Cellular vascular endothelial growth factor is a predictor of
outcome in patients with acute myeloid leukemia. Blood.
94:3717–3721. 1999.PubMed/NCBI
|
|
14
|
Fiedler W, Graeven U, Ergun S, et al:
Vascular endothelial growth factor, a possible paracrine growth
factor in human acute myeloid leukemia. Blood. 89:1870–1875.
1997.PubMed/NCBI
|
|
15
|
Koch HP: Thalidomide and congeners as
anti-inflammatory agents. Prog Med Chem. 22:165–242. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Moreira AL, Sampaio EP, Zmuidzinas A,
Frindt P, Smith KA and Kaplan G: Thalidomide exerts its inhibitory
action on tumor necrosis factor alpha by enhancing mRNA
degradation. J Exp Med. 177:1675–1680. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sampaio EP, Kaplan G, Miranda A, Nery JA,
Miguel CP, Viana SM and Sarno EN: The influence of thalidomide on
the clinical and immunologic manifestation of erythema nodosum
leprosum. J Infect Dis. 168:408–414. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shannon EJ, Morales MJ and Sandoval F:
Immunomodulatory assays to study structure-activity relationships
of thalidomide. Immunopharmacology. 35:203–212. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tavares JL, Wangoo A, Dilworth P, Marshall
B, Kotecha S and Shaw RJ: Thalidomide reduces tumor necrosis factor
alpha production by human alveolar macrophages. Respir Med.
91:31–39. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brieger J, Schroeder P, Gosepath J and
Mann WJ: Vascular endothelial growth factor and basic fibroblast
growth factor are released by squamous cell carcinoma cells after
irradiation and increase resistance to subsequent irradiation. Intl
J Mol Med. 16:159–164. 2005.
|
|
21
|
Shintani S, Kiyota A, Mihara M, Nakahara
Y, Terakado N, Ueyama Y and Matsumura T: Association of
preoperative radiation effect with tumor angiogenesis and vascular
endothelial growth factor in oral squamous cell carcinoma. Jpn J
Cancer. 91:1051–1057. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Koukourakis MI, Giatromanolaki A, Sirvidis
E, Simopoulos K, Pissakas G, Gatter KC and Harris AL: Squamous cell
head and neck cancer: evidence of angiogenic regeneration during
radiotherapy. Anticancer Res. 2:4301–4309. 2001.PubMed/NCBI
|
|
23
|
Geng L, Donnelly E, McMahon G, Lin PC,
Sierra-River E, Oshinka H and Hallahan DE: Inhibition of vascular
endothelial growth factor receptor signaling leads to reversal of
tumor resistance to radiotherapy. Cancer Res. 61:2413–2419.
2001.PubMed/NCBI
|