Introduction
Glioblastoma multiforme (GBM) is a rare tumor but
one of the most frequent and the most malignant among brain tumors.
Despite recent therapeutic advances the prognosis still remains
very poor, with most patients (>70%) dying within 1 year after
diagnosis. Five-year overall survival is ~2% (1). Currently available treatment options,
such as surgery, radiotherapy and chemotherapy, can only minimally
prolong life expectancy.
The GBM risk factors include e.g. chemicals,
pesticides and therapeutic ionizing radiation (2). In one study, children irradiated
during leukemia treatment had increased risk of brain tumors
including GBM (3,4). Passive smoking also increases risk of
GBM (5).
GBM tumors consist of poorly differentiated
oncogenic astrocytes, and are often heterogenic and highly invasive
(6). The loss of PTEN, EGF receptor
and changes in the expression of PDGF receptors, p16 and p53 are
found in GBM (7). GBM tumors have
been shown to express MET receptor and its ligand hepatocyte growth
factor (HGF) (8).
MET has been reported as one of the receptors
responsible for proliferation, inhibition of apoptosis and
dissemination of cancer cells. It is encoded by MET proto-oncogene
with tyrosine kinase activity (9).
MET activation by its ligand HGF leads to pleiotropic biological
effects on various target cells, including the induction of cell
proliferation, morphogenetic transformation, cell motility and
invasiveness under both normal and pathological conditions
(9–11). In transformed tissues, the
activation of MET by HGF triggers tumor growth, invasion and
metastasis (9,12).
Both HGF and MET are overexpressed in human GBM, and
it was shown that levels of expression are upregulated during
transition from low grade to malignant GBM and positively correlate
with GBM malignancy grade and vascularity (8). In vitro, HGF stimulates GBM and
endothelial cell migration and proliferation as well as endothelial
tube formation. The overexpression of HGF and/or MET promotes GBM
growth and angiogenesis in vivo (8).
The heat shock protein 90 (HSP90) plays a key role
in the process of protein folding, stabilization and degradation.
It is a molecular chaperone that regulates the maturation and
intracellular trafficking of several signaling proteins and
receptors, including Her2/Neu, mutant EGFR, steroid receptors, AKT,
RAF, SRC, mutant p53, Bcr-Abl and MET receptor (13,14).
In cancer cells, HSP90 is present entirely in multichaperone
complexes with high ATPase activity, being involved in the
processing of oncoproteins critical to cancer progression (15).
Geldanamycin, a naturally occurring benzoquinone
ansamycin antibiotic produced by yeast, and its analogues belong to
a new class of anticancer agents that inhibit the molecular
chaperone HSP90. Ansamycin antibiotics bind to the ATP pocket of
HSP90, altering the conformation of the multichaperone complex
(16). Consequently,
HSP90-dependent protein folding and maturation are arrested and
proteins are targeted for degradation. In cancer cells, HSP90
inhibitors (geldanamycin, radicicol, herbimycin A, and
17-allylamino-17-demethoxygeldanamycin (17-AAG)), promote the
degradation of HSP90-dependent oncoproteins, eventually leading to
cell cycle arrest and cell death (17). Interestingly, geldanamycin
derivatives accumulate and bind preferentially to HSP90 in tumor
cells compared with normal cells, promoting selective cancer cell
death. These properties make this class of agents suitable for
clinical development as anticancer therapeutic agents (18).
In this study we evaluated the use of GAs as a
potential treatment option for GBM patients. We investigated the
influence of GAs on GBM growth and HGF-dependent motility of tumor
cells.
Materials and methods
Cell lines and cell culture
Experiments were performed on three human
glioblastoma cell lines (LN18, LN229, T98G). Cells were kindly
provided by Dr K. Reiss from Temple University, (Philadelphia, PA,
USA). The cells were cultured in DMEM (LN18, LN229) and in MEM
(T98G) (PAA), supplemented with 100 IU/ml penicillin and 100 μg/ml
streptomycin (Polfa Tarchomin, Poland) in the presence of 10% FBS
(PAA) at 37˚C, 5% of CO2 and 95% humidity.
HSP90 inhibitors
Geldanamycin, 17-AAG
(17-allylamino-17-demethoxygeldanamycin), a GA analogue, both
insoluble in H2O and soluble in DMSO. 17DMAG
[17-(dimethylaminoethylamino)-17-demethoxygeldanamycin], a
water-soluble 17-AAG analogue; 17AEP-GA
[17-(2-(pyrrolidin-1-yl)ethyl)aminno-17-demethoxygeldanamycin], a
water-soluble GA analogue containing an alkylamino group instead of
the methoxy moiety at C17; 17DMAP-GA
[17-(dimethylaminopropylamino)-17-demethoxygeldanamycin], a
water-soluble GA analogue synthesized based on binding affinity to
HSP90. GA, 17AAG, 17DMAG, 17AEP-GA and 17DMAP-GA were purchased
from InvivoGen, Inc. (USA).
MTT assay
The influence of GAs (InvivoGen, Inc.) on
viability/proliferation was checked by CellTiter 96®
AQueous One Solution assay (Promega). Cells were seeded in growth
medium (containing 10% FBS) on 96-well plates at
1×103/well (LN18 and LN229) and at 2×103/well
(T98G) density in the presence of different concentration of GAs
(1, 10, 100, 1,000 nM). After 24, 48, 72 and 96 h, 20 μl of
CellTiter 96® Aqueous One Solution reagent was added to
each well and plates were incubated for 3 h. Subsequently, plates
were read at 490 nm using the ELx800 Universal Microplate Reader
(Bio-Tek) and analyzed with KC4 v3.0 with PowerReports
software.
Apoptosis
The influence of GAs on glioblastoma cells was
studied by staining with Annexin V according to the manufacturer’s
instructions. Briefly, after drug treatments for 24, 48 and 72 h,
cells were stained with propidium iodide (PI) and Annexin V-FITC in
100 μl of staining solution at room temperature for 15 min in the
dark. Samples were diluted with binding buffer and were analyzed
within 1 h. In some experiments cells were tested for the activity
of caspase-3 (BD Pharmingen). For this staining, cells were
permeabilized for 20 min, washed and subsequently stained with
anti-caspase-3-PE mAb for 30 min on ice (both Abs from BD
Pharmingen). Cytofluorometric analysis was performed by FACS Canto
(Becton-Dickinson) and analyzed with FASC Diva software
(Becton-Dickinson).
RNA extraction and reverse
transcription
Total RNA was extracted using RNeasy mini kit
(Qiagen), followed by DNAse treatment (Promega). The reverse
polymerase transcription was performed using M-MLV reverse
transcriptase (Invitrogen) according to the manufacturer’s
protocol.
Quantitative real-time PCR analysis
The detection of MET mRNA level was performed by
quantitative real-time PCR analysis on ABI PRISM® 7300
Sequence Detection System (Applied Biosystems, Inc.) using a
commercially available TaqMan PCR Master mix and primers for human
MET gene (Hs01565589_m1; Applied Biosystems, Inc.). The mRNA
expression level for all samples was normalized to the housekeeping
gene GAPDH (Hs99999905_m1; Applied Biosystems, Inc.).
Western blot analysis
Western blot analyses were carried out on extracts
prepared from GBM cells. The cells were lysed on ice with MPER
lysing buffer (Pierce) containing protease and phosphatase
inhibitors (Sigma). Extracted proteins were separated on a 12%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE
gel) and transferred to a PVDF membrane (Bio-Rad). The activation
of AKT and MAPK was detected using mouse and rabbit
phospho-specific antibodies (AKT, MAPK; Cell Signaling Technology,
Inc.) followed by HRP-conjugated goat anti-mouse IgG or goat
anti-rabbit IgG as secondary antibodies (Santa Cruz Biotechnology,
Inc.). The membranes were developed with an ECL reagent and exposed
to HyperFilm (Amersham Life Sciences). An equal loading in the
lanes was evaluated by probing with an anti-GAPDH antibody (Santa
Cruz Biotechnology, Inc.).
Chemotaxis studies
The direct migration of GBM cells towards HGF
gradient was studied using modified Boyden’s chamber with 8-μm pore
polycarbonate membrane inserts. The cells were seeded into the
upper chamber of a Transwell inserts (Costar Transwell). The lower
chamber was filled with medium containing 0.5% BSA and HGF (20
ng/ml). After 24 h, inserts were removed from transwells and cells
were fixed with methanol. Cells that transmigrated to the lower
side of the membrane were stained with Wright solution (Merck) and
counted under high power field (HPF) with an inverted
microscope.
Invasion assay
The ability of GBM cells to invade was examined by
BD BioCoat™ Matrigel invasion inserts (Becton-Dickinson) where
cells migrated through Matrigel layer towards HGF gradient. After
24 h, cells that invaded the Matrigel were counted on the
undersides of filters after fixation and staining with Wright
solution. As a control of invasion the same number of control
inserts (no GFR Matrigel coating) were used.
Flow cytometry
The presence of MET receptor on the surface of
target cells were demonstrated by staining with rabbit
anti-MET-FITC antibody (R&D Systems) and analyzed by FACS.
Briefly, 1×105 cells were suspended in 100 μl of
staining buffer (PBS, 2% FBS) and antibody was added. Next, the
cells were incubated in the dark for 30 min at 4˚C. The stained
cells were washed, collected using FACSCanto cytometer
(Becton-Dickinson, USA) and analyzed with FACS Diva software
(Becton-Dickinson, USA).
Statistical analysis
All results are mean ± SD. Statistical analysis was
performed using the two-tailed Student’s t-test. P-values <0.05
were considered as significant, unless otherwise indicated.
Results
Geldanamycin and its analogs inhibit
proliferation of glioblastoma cells
Several in vitro studies indicate that GAs
inhibit the proliferation of a wide range of cancer cells (18,19).
The effects of GAs on the growth rate of GBM cell lines (LN18,
LN229, T98G) were determined using the CellTiter 96®
Aqueous One Solution assay at 96 h. GAs were used in 1 and 10 nM
concentrations. As shown in Fig. 1,
GAs inhibited growth of all investigated cells already at the
concentration of 1 nM. However, the level of inhibition differs
between cell lines and geldanamycin analogs. We observed the
strongest inhibition for LN18 and LN229 cell lines where all
investigated analogs were able to inhibit cell growth (Fig. 1A and B). Moreover, we noted similar
level of inhibition after 96-h incubation for higher concentration
of GAs, 100 and 1,000 nM for these two cell lines (data not shown).
However, T98G seemed to be less sensitive to investigated analogs
at the concentration of 1 and 10 nM (Fig. 1C). Growth of T98G cell line was
inhibited by 50% in comparison to untreated control after
incubation with 100 and 1,000 nM of GAs (data not shown). The best
results for all investigated cell lines were obtained using
17-AEP-GA.
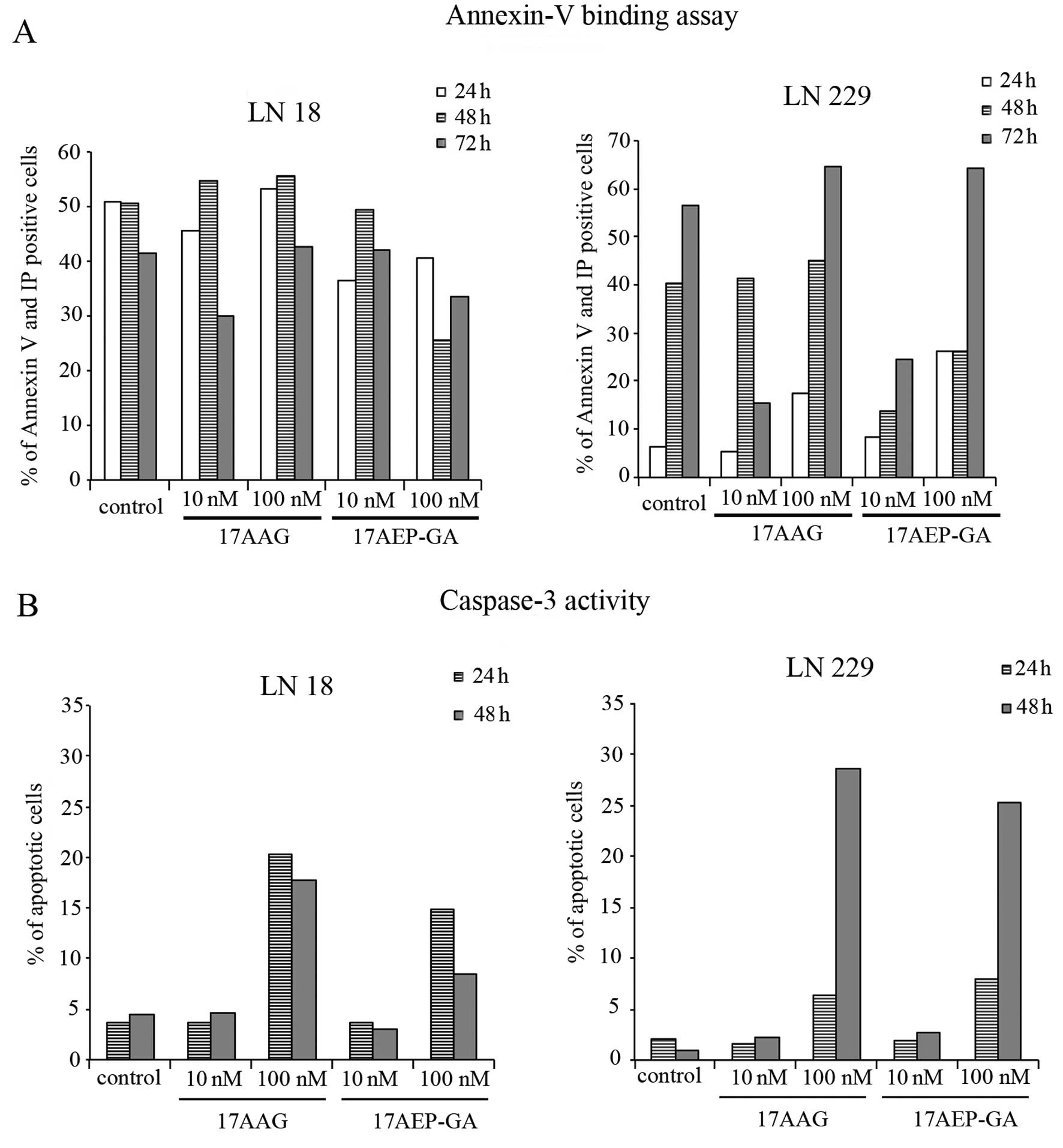
Induction of glioblastoma cell apoptosis
by HSP90 inhibitors
We became interested whether the inhibition of cell
growth is due to the induction of apoptosis. Therefore, in order to
answer this question we used Annexin V and activated caspase-3
staining to detect apoptotic cells after GAs treatment. After
incubation with 10 and 100 nM of 17-AAG and 17-AEP-GA cells became
Annexin V positive (Fig. 2A). These
data were confirmed with caspase-3 staining. We observed a high
number of cells positive for activated caspase-3 after 17-AAG and
17-AEP-GA treatment at the concentration of 100 nM (Fig. 2B).
Glioblastoma cell lines express active
MET receptor
The expression of MET receptor mRNA was estimated by
real-time PCR and we have observed the expression of MET gene for
all tested cell lines (Fig. 3A).
LN229 cell line, which is the most malignant one, had the highest
expression of mRNA for MET receptor. LN18 and T98G cell lines had a
similar level of expression. There were no significant differences
between tested cell lines. To determine whether MET receptor is
functional on investigated cell lines we stimulated them by HGF and
checked phosphorylation of MAPK 42/44 and AKT Ser 473 by western
blot analysis (Fig. 3B). For all
tested cell lines we observed strong phosphorylation of AKT and
MAPK after 5 min of stimulation by HGF. However, LN18 cell line did
not show any differences in AKT phosphorylation between control
cells and cells after stimulation. It might suggest constitutive
activation of AKT.
HSP90 inhibitors downregulate expression
of MET receptor
It has been shown that MET receptor is a client
protein for HSP90 (14,19). Thus, we checked if incubation of GBM
cells with GAs had any effect on the surface expression of MET
receptor. Cells were grown in the presence of GAs for 24 h and the
level of MET receptor expression was evaluated by cytofluorometric
analysis. The downregulation of MET receptor expression was
observed (Fig. 3C). The strongest
reduction was in LN229 cell line. These results confirm that the
expression of MET receptor is dependent on the presence of HSP90
protein.
Geldanamycin and its analogs inhibit
HGF-dependent chemotaxis and invasion of glioblastoma cells
Since GBM expresses high level of MET receptor and
its expression can be modulated by GAs, we tested the influence of
GAs on chemotactic response of GBM cells towards HGF. The GBM cells
were subjected to 10 and 100 nM concentrations of GAs and their
ability to migrate was evaluated. We observed decreased migration
of treated GBM cells towards HGF gradient (Fig. 4A). This effect was further enhanced
when cells were preincubated for 6 h with GAs (Fig. 4B).
We also evaluated invasive properties of GBM cells
after GAs treatment. LN229 cell line was treated with inhibitors in
the concentration of 100 nM. We observed a strong decrease in the
ability of GBM to transmigrate through extracellular matrix
proteins after HGF stimulation (Fig.
4C).
Discussion
Despite substantial research the GBM is still an
untreatable disease. Thus, finding new ways of GBM treatment is
necessary. In this study we tested GAs as new potential agents for
GBM therapy. Our results showed that GAs were able to block
proliferation of GBM cells and to induce apoptosis of tumor cells.
Importantly, GAs were also able to inhibit migration and invasion
of GBM cells toward HGF gradient.
In this study we used five different HSP90
inhibitors, geldanamycin and its analogs. Some of these inhibitors
e.g. 17-AAG is already in clinical trials (20) GA and 17AAG was also tested in GBM
cells (21,22).
All five inhibitors were able to inhibit
proliferation and induced apoptosis of GBM cells. As we expected,
based on our previous studies and published data (23), the inhibition of proliferation was
observed already at the concentration of 1 nM and achieving up to
50% for 10 nM, depending on a cell line (Fig. 1). The strongest inhibition was
observed for LN18 and LN229 cell lines. However, contrary to other
studies in which GAs at concentrations of ≥100 nM were able almost
completely to block growth of tumor cells (14,23),
we did not observe complete inhibition of GBM growth even at 1,000
nM concentration (data not shown). The efficiency of GA analogs in
comparison to geldanamycin revealed the strongest inhibition of
tumor cell proliferation in the case of 17-AEP-GA. This is in the
agreement with our previous studies on rhabdomyosarcoma cell lines
where we also observed the strongest inhibition of tumor cell
proliferation with 17-AEP-GA (23).
The study of apoptosis revealed that the most
effective HSP90 inhibitors that were able to induce apoptosis in
GBM cells are 17-AAG and 17-AEP-GA. The concentration of 100 nM
induced apoptosis in LN18 and LN229 cell lines. Annexin V staining
was confirmed by caspase-3 staining assay. These data show
different effectiveness of tested analogs in inducing apoptosis in
tumor cells (14,23). In these experiments, 17-AEP-GA was
again the most effective analog among those tested. Based on our
previous data and other publications we can postulate that the
induction of apoptosis could be due to reduction in the expression
of survival proteins including AKT that were shown to be the client
proteins for HSP90, the direct target of GA and analogs (13,23,24).
Interestingly, we also noticed some Annexin V-positive cells under
control conditions. The reason for the presence of positive cells
under control condition might be caused by a change in plasma
membrane structure by surface exposure of phosphatidylserine (PS),
while the membrane integrity remains unchallenged. Surface exposed
PS can be detected by its affinity for Annexin V, a phospholipid
binding protein (25).
MET receptor has been shown to be expressed on GBM
cells and to induce various cellular responses in these cells
(8). In this study, using real-time
RT-PCR and flow cyto-metry, we showed that GBM cells express the
MET receptor and that HGF stimulates the phosphorylation of MAPK
and AKT kinases in GBM cells, two kinases involved not only in
tumor growth but also in tumor cell migration (26,27).
Moreover, we have shown that GAs can downregulate the surface
expression on MET receptor.
Next, we confirmed that HGF can stimulate the
migration and invasion of GBM cells as shown previously (8,28). In
our previous studies on rhabdomyosarcoma cells we showed that HGF
induced migration of these cells can be blocked by HSP90 inhibitors
(19). Thus, in this study, we
tested whether HSP90 inhibitors, such as GAs, can block the ability
of GBM cells to migrate and invade. Strong inhibition of GBM
migration and invasion was noted after treatment of tumor cells
with 10 and 100 nM of 17-AAG and 17-AEP that was comparable to
inhibition observed with geldanamycin. This block of tumor cell
migration is due to inhibition of MET receptor expression (mRNA
expression and FACS analysis in this study). Similar results have
been obtained for different tumors (19,22,23,28).
GAs are able to inhibit the expression of MAPK and AKT kinases
which were shown previously to be involved in activating the
motility of tumor cells (26,27).
Thus, parallel blocking of MET receptor expression and activation
of kinases involved in cell migration by GAs could be responsible
for observed inhibition of GBM migration and invasion after HGF
stimulation.
GA and its analogs have been shown to block growth
of tumor cells and the expression of various oncoproteins including
tyrosine kinase receptors (17,19,23).
However, most of the studies on GBM cells were conducted using
geldanamycin and its analog 17-AAG. Due to its high toxicity,
geldanamycin cannot be considered a useful therapeutic (23,29)
and 17-AAG has also some drawbacks, e.g. it is insoluble in water.
In this study, we tested other geldanamycin analogs with some
characteristics preferable in clinical use such as water
solubility. Particularly, 17-AEP-GA seems to possess the best
clinical features. It is water-soluble and the efficacy of blocking
tumor growth and migration is in similar range to geldanamycin and
17-AAG. Importantly, our previous studies showed that the use of
17-AEP-GA gives less toxic side effects against normal cells
(23).
Based on our results presented here and our previous
studies (23), we postulate that
17-AEP-GA could be considered as a new potential therapeutic for
the treatment of glioblastoma multiforme.
Acknowledgements
This study was supported by research grant from the
Polish Ministry of Science and Higher Education (NN 401 010036, N N
401 054839, N N401 142339).
References
|
1
|
McLendon RE and Halperin EC: Is the
long-term survival of patients with intracranial glioblastoma
multiforme overstated? Cancer. 98:1745–1748. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Navas-Acién A, Pollán M, Gustavsson P,
Floderus B, Plato N and Dosemeci M: Interactive effect of chemical
substances and occupational electromagnetic field exposure on the
risk of gliomas and meningiomas in Swedish men. Cancer Epidemiol
Biomarkers Prev. 11:1678–1683. 2002.
|
|
3
|
Galloway TJ, Indelicato DJ, Amdur RJ,
Swanson EL, Smith AA and Marcus RB: Second tumors in pediatric
patients treated with radiotherapy to the central nervous system.
Am J Clin Oncol. 35:279–283. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Krishnan S, Wade R and Moorman AV:
Temporal changes in the incidence and pattern of central nervous
system relapses in children with acute lymphoblastic leukaemia
treated on four consecutive Medical Research Council trials,
1985–2001. Leukemia. 24:450–459. 2010.PubMed/NCBI
|
|
5
|
Cordier S, Monfort C, Filippini G, et al:
Parental exposure to polycyclic aromatic hydrocarbons and the risk
of childhood brain tumors: The SEARCH International Childhood Brain
Tumor Study. Am J Epidemiol. 159:1109–1116. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Molina JR, Hayashi Y, Stephens C and
Georgescu MM: Invasive glioblastoma cells acquire stemness and
increased Akt activation. Neoplasia. 12:453–463. 2010.PubMed/NCBI
|
|
7
|
Sauvageot CM, Weatherbee JL, Kesari S, et
al: Efficacy of the HSP90 inhibitor 17-AAG in human glioma cell
lines and tumorigenic glioma stem cells. Neuro Oncol. 11:109–121.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Abounader R and Laterra J: Scatter
factor/hepatocyte growth factor in brain tumor growth and
angiogenesis. Neuro Oncol. 7:436–451. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mazzone M and Comoglio PM: The Met
pathway: master switch and drug target in cancer progression. FASEB
J. 20:1611–1621. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lesko E and Majka M: The biological role
of HGF-MET axis in tumor growth and development of metastasis.
Front Biosci. 13:1271–1280. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jiang W, Hiscox S, Matsumoto K and
Nakamura T: Hepatocyte growth factor/scatter factor, its molecular,
cellular and clinical implications in cancer. Crit Rev Oncol
Hematol. 29:209–248. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Weidner KM, Sachs M and Birchmeier W: The
Met receptor tyrosine kinase transduces motility, proliferation,
and morphogenic signals of scatter factor/hepatocyte growth factor
in epithelial cells. J Cell Biol. 121:145–154. 1993. View Article : Google Scholar
|
|
13
|
Pratt WB and Toft DO: Regulation of
signaling protein function and trafficking by the hsp90/hsp70-based
chaperone machinery. Exp Biol Med (Maywood). 228:111–133.
2003.PubMed/NCBI
|
|
14
|
Maulik G, Kijima T, Ma PC, et al:
Modulation of the c-Met/hepatocyte growth factor pathway in small
cell lung cancer. Clin Cancer Res. 8:620–627. 2002.PubMed/NCBI
|
|
15
|
Workman P: Altered states: selectively
drugging the Hsp90 cancer chaperone. Trends Mol Med. 10:47–51.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Whitesell L, Mimnaugh EG, De Costa B,
Myers CE and Neckers LM: Inhibition of heat shock protein
HSP90-pp60v-src heteroprotein complex formation by benzoquinone
ansamycins: essential role for stress proteins in oncogenic
transformation. Proc Natl Acad Sci USA. 91:8324–8328. 1994.
View Article : Google Scholar
|
|
17
|
Goetz MP, Toft DO, Ames MM and Erlichman
C: The Hsp90 chaperone complex as a novel target for cancer
therapy. Ann Oncol. 14:1169–1176. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ochel HJ, Eichhorn K and Gademann G:
Geldanamycin: the prototype of a class of antitumor drugs targeting
the heat shock protein 90 family of molecular chaperones. Cell
Stress Chaperones. 6:105–112. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lesko E, Gozdzik J, Kijowski J, Jenner B,
Wiecha O and Majka M: HSP90 antagonist, geldanamycin, inhibits
proliferation, induces apoptosis and blocks migration of
rhabdomysoracoma cells in vitro and seeding into bone marrow in
vivo. Anticancer Drugs. 18:1173–1181. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ramanathan RK, Trump DL, Eiseman JL, et
al: Phase I pharmacokinetic-pharmacodynamic study of
17-(allylamino)-17-demethoxygeldanamycin (17AAG, NSC 330507), a
novel inhibitor of heat shock protein 90, in patients with
refractory advanced cancers. Clin Cancer Res. 11:3385–3391. 2005.
View Article : Google Scholar
|
|
21
|
Xie Q, Thompson R, Hardy K, et al: A
highly invasive human glioblastoma pre-clinical model for testing
therapeutics. J Transl Med. 6:772008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zagzag D, Nomura M, Friedlander DR, Blanco
CY, Gagner JP, Nomura N and Newcomb EW: Geldanamycin inhibits
migration of glioma cells in vitro: a potential role for
hypoxia-inducible factor (HIF-1alpha) in glioma cell invasion. J
Cell Physiol. 196:394–402. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lukasiewicz E, Miekus K, Kijowski J, et
al: High anti tumor activity against rhabdomyosarcoma cells and low
normal cells cytotoxicity of heat shock protein 90 inhibitors, with
special emphasis on 17-
(2-(pyrrolidin-1-yl)ethyl)-aminno-17-demethoxy-geldanamycin. J
Physiol Pharmacol. 60:161–166. 2009.
|
|
24
|
Solit DB, Basso AD, Olshen AB, Scher HI
and Rosen N: Inhibition of heat shock protein 90 function
down-regulates Akt kinase and sensitizes tumors to Taxol. Cancer
Res. 63:2139–2144. 2003.PubMed/NCBI
|
|
25
|
van Engeland M, Nieland LJ, Ramaekers FC,
Schutte B and Reutelingsperger CP: Annexin V-affinity assay: a
review on an apoptosis detection system based on phosphatidylserine
exposure. Cytometry. 31:1–9. 1998.PubMed/NCBI
|
|
26
|
Hardy KM, Yatskievych TA, Konieczka J,
Bobbs AS and Antin PB: FGF signalling through RAS/MAPK and PI3K
pathways regulates cell movement and gene expression in the chicken
primitive streak without affecting E-cadherin expression. BMC Dev
Biol. 11:202011. View Article : Google Scholar
|
|
27
|
Miekus K, Jarocha D, Trzyna E and Majka M:
Role of I-TAC-binding receptors CXCR3 and CXCR7 in proliferation,
activation of intracellular signaling pathways and migration of
various tumor cell lines. Folia Histochem Cytobiol. 48:104–111.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xie Q, Gao CF, Shinomiya N, et al:
Geldanamycins exquisitely inhibit HGF/SF-mediated tumor cell
invasion. Oncogene. 24:3697–3707. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Supko JG, Hickman RL, Grever MR and
Malspeis L: Preclinical pharmacologic evaluation of geldanamycin as
an antitumor agent. Cancer Chemother Pharmacol. 36:305–315. 1995.
View Article : Google Scholar : PubMed/NCBI
|