Introduction
Tamoxifen (Tam), a selective estrogen receptor (ER)
modulator, is the most frequently used anti-hormonal drug for the
treatment of both early and advanced ER-positive breast cancer in
pre- and post-menopausal women. Studies concerning adjuvant therapy
reveal that Tam significantly decreases recurrence and mortality in
patients with ER-positive breast cancer and could make a great
contribution to the reduction in the odds of breast cancer
incidence in healthy women at an increased risk (1–3). De
novo and acquired resistant to Tam remains a problems, and
several reasons have been proposed to be responsible for the
development of Tam resistance. The reduction or loss of ER
signaling, the function of recently discovered G protein-coupled
estrogen receptor 1 (GPER) also known as G protein-coupled receptor
30 (GPR30), quantitative or qualitative change in ER-related
co-factors, overexpression of epidermal growth factor receptor
(EGFR) can contribute to the development of endocrine-resistance in
breast cancer (4–7). However, these studies have failed to
focus on the dose-effect of Tam on the proliferation of
Tam-resistant (TAM-R) cells.
To date, Tam has no effect on the proliferation of
TAM-R cells derived from MCF-7 breast cancer cells by means of
continuous Tam exposure (8).
However, it has been proven that a relatively low concentration of
Tam has a stimulated effect on the proliferation of TAM-R cells
in vitro(9,10), and can promote tumor growth in
MCF-7/HER2-18 xenografts (11), in
Tam-resistant animal models (12).
More importantly, it has been confirmed that a high concentration
of Tam (≥10 μM) is able to inhibit TAM-R cell proliferation
(10). Recently, Ward et al
reported that exposure to 10 μM Tam was sufficient to induce G1
arrest in both MCF-7 and TAM-R cells, which demonstrated the
preserved sensitivity of TAM-R cells to Tam at a relatively high
concentration (13). This evidence
indicates that a relatively low concentration and a high
concentration of Tam have specific effects on the proliferation of
TAM-R cells.
In the present study, we investigated the
differential effects of different concentrations of Tam on the
proliferation of TAM-R cells, an issue that has not received
sufficient attention in the past. We proposed that Tam treatment at
a low concentration may not have beneficial effects on
Tam-resistant breast cancer cells, instead it may promote cell
growth. In addition, Tam treatment at a relatively high
concentration has the ability to inhibit TAM-R cell proliferation
in the experimental condition. Furthermore, we determined that
different signaling pathways may account for this dose-effect of
Tam in TAM-R cells.
Materials and methods
Cell culture and treatment
MCF-7 cells obtained from the American Type Culture
Collection (ATCC, Manassas, VA, USA) were routinely cultured in
RPMI-1640 media containing 5% fetal calf serum (FCS). Tam-resistant
(TAM-R) cells were derived from MCF-7 cells by continuous culture
in phenol-red-free media containing 5% steroid-depleted FCS,
10−7 M Tam and 0.1% ethanol. The media for control cells
contained 0.1% ethanol. For all experiments, the cells were seeded
in 6-well plates and cultured in phenol-red-free medium
supplemented with 5% steroid-depleted FCS.
Isolation of primary tumor cells from
pleural effusion
After obtaining informed consent, primary breast
cancer cells were collected from pleural effusion of a patient
diagnosed with breast carcinoma with liver and lung metastases
[HER2(+++), ER(+), PR(+)]. Cells in the pleural fluid were diluted
with DMEM, centrifuged, and the pellet was washed twice with PBS.
Breast cancer cells were isolated to apply density gradient
centrifugation using Percoll fluid. The freshly isolated primary
breast cancer cells were resuspended in RPMI-1640 media with 5%
fetal calf serum, 50 μg/ml penicillin and 100 μg/ml
streptomycin.
RNA interference (RNAi)
The following small-interfering RNA oligonucleotides
were used for knockdown of the stimulatory G protein α subunit
(Gαs): sense, 5′-GGCGCAGCGUGAGGC CAACTT-3′ and antisense,
5′-GUUGGCCUCACGCUGCG CCTT-3′. A random siRNA (5′-GUGUCUCCCAGUCCUG
CGCCTT-3′) was used as the control. The RNAi oligonucleotides were
transfected into cells using Lipofectamine 2000™ (Invitrogen,
Carlsbad, CA, USA) and all experiments were performed 48 h
later.
Growth curves
MCF-7 and TAM-R cells were treated with various
concentrations (0 nM, 10 nM, 100 nM, 1 μM, 5 μM, 10 μM, 15 μM and
20 μM) of Tam or 17β-estradiol (E2) for 5 days, and the number of
cells were counted using a cell counting chamber according to the
manufacturer's instructions as previously described (10). The cell number was expressed as a
percentage (%) of the control.
Immunoblotting
Total proteins were extracted using total protein
mammalian cell lysis buffer, separated using 10% SDS-PAGE gels and
transferred to nitrocellulose filter membranes. The membranes were
blocked with 5% skimmed milk in Tris-buffered saline containing
0.1% Tween-20 and incubated with primary antibodies overnight at
4°C, followed by peroxidase-conjugated secondary antibodies for 2 h
at room temperature. The bands were detected using enhanced
chemiluminescence. The rabbit anti-Gαs polyclonal antibody,
anti-β-actin antibody and secondary antibodies were purchased from
Abcam (Cambridge, UK), and the total and phosphorylated ERK1/2 and
AKT antibodies, ER and EGFR antibodies were purchased from Bioworld
Technology Inc. (Visalia, CA, USA).
Statistical analysis
Each experiment was repeated at least three times,
and the results are expressed as means ± SD. Statistical analysis
was performed using the Student's t-test; P<0.05 was considered
to indicate a statistically significant result.
Results
Generation of TAM-R cells and
dose-response of Tam
MCF-7 cells were continuously cultured in medium
containing 10 nM Tam dissolved in 0.1% ethanol for 6 months to
generate a Tam-resistant (TAM-R) cell line. MCF-7 cells
continuously cultured with 0.1% ethanol were used as controls.
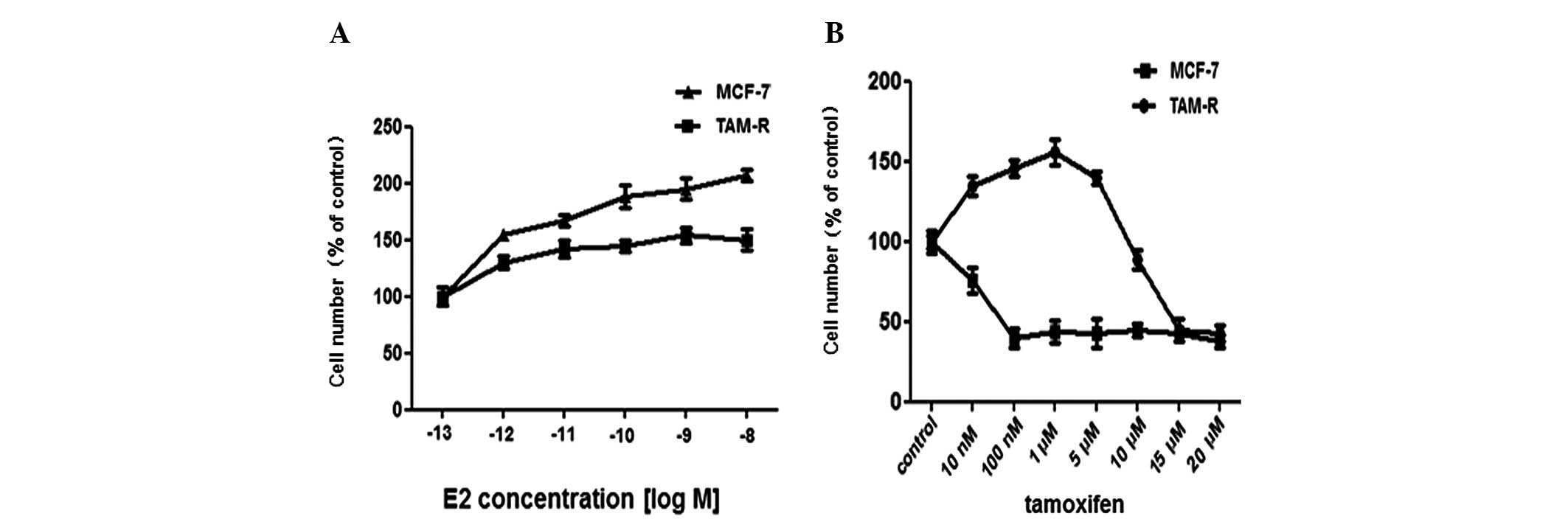
Since breast tumors are dependent on E2 for growth and Tam acts as
an anti-estradiol drug to inhibit cell growth in vivo, the
growth effect of Tam and E2 was tested. As expected, there was a
difference in the proliferation rate between MCF-7 and TAM-R cells
following treatment with 10 nM E2. MCF-7 cells had a higher
proliferation rate as measured by direct cell counting compared
with the TAM-R cells (Fig. 1A).
Exposure to 1 μM Tam was sufficient to inhibit the proliferation of
MCF-7 cells, thus many previous studies selected 1 μM as the final
concentration of Tam for their experiments. Compared with MCF-7
cells, the growth of TAM-R cells was inhibited completely by Tam at
a high concentration >10 μM, while low doses (10 nM-5 μM) of Tam
stimulated the growth of TAM-R cells; 1 μM Tam promoted TAM-R cell
growth most significantly (Fig.
1B). Overall, we identified the dose-response of Tam in TAM-R
cells in our experiment.
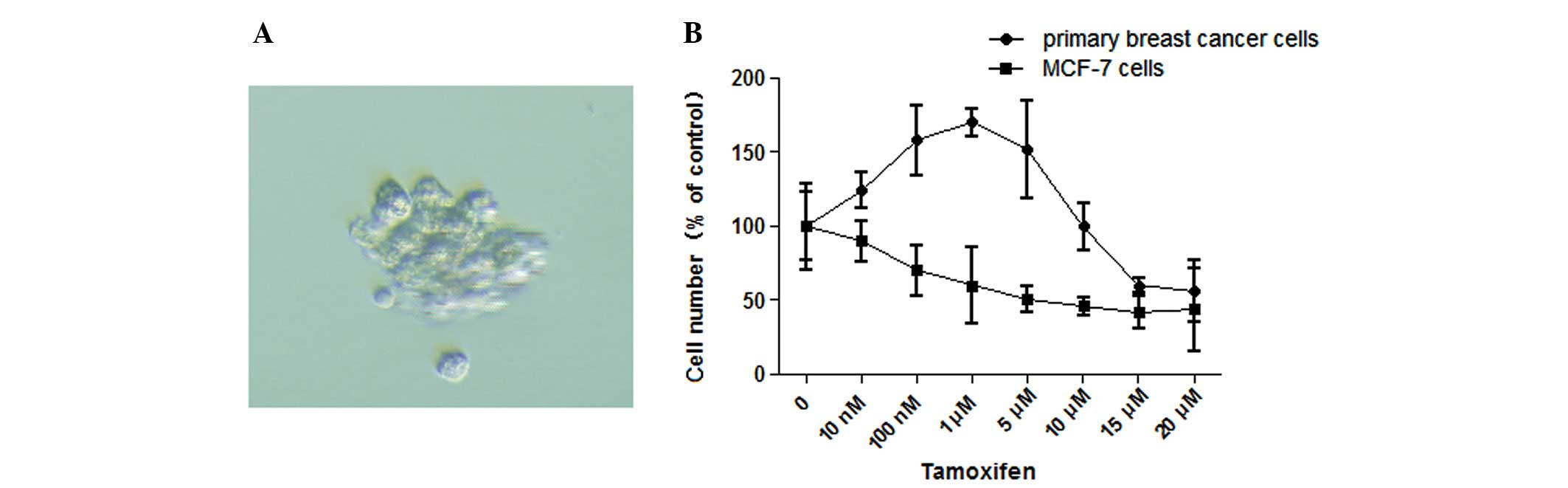
Isolation of Tam-resistant primary breast
cancer cells and the dose-effect of Tam
To further confirm the results shown in the above
experiment, we isolated the primary breast cancer cell line shown
in Fig. 2A from the pleural
effusion of a patient following long-term use of Tam and diagnosed
with breast carcinoma with liver and lung metastases [HER2 (+++),
ER (+), PR (+)]. The patient was diagnosed as Tam-resistant in the
clinic (14). The acquired
resistance to Tam was measured by dose-dependent growth assays.
When primary breast cancer and MCF-7 cells were treated with Tam at
increasing concentrations from 10 nM to 20 μM, the survival ratios
showed marked differences between the two cell lines. For example,
at 10 nM Tam, the cell growth was stimulated in the primary breast
cancer cells, and this trend continued until the Tam concentration
reached 10 μM where the growth of primary breast cancer cells was
blocked; however, at 1 μM Tam, the proliferation of MCF-7 cells was
markedly inhibited (Fig. 2B).
Moreover, the proliferation responses of the primary breast cancer
cells to Tam were similar to the TAM-R cells. Taken together, we
conclude that there is a dose-dependent relationship between Tam
and the growth responses of TAM-R cells.
Effect of a low-dose of Tam on the ERK1/2
and AKT signaling pathways in TAM-R cells
As the activation of the ERK1/2 and AKT kinase
signaling pathways is thought to be an important regulating factor
(8,11,15,16),
we analyzed the ability of E2 and Tam to induce ERK1/2 and AKT
phosphorylation in TAM-R and control MCF-7 cells. ERK1/2
phosphorylation increased in the TAM-R cells treated with 10 nM E2
within 5–10 min and disappeared rapidly, whereas ERK1/2
phosphorylation was observed after 10 min and continued until 30
min in MCF-7 cells following 10 nM E2 treatment (Fig. 3A), which implied that E2 had a more
lasting effect on MCF-7 cells than on TAM-R cells. Treatment with 1
μM Tam resulted in a significant attenuation of ERK1/2
phosphorylation in MCF-7 cells, whereas 1 μM Tam stimulated ERK1/2
phosphorylation within 5–20 min in TAM-R cells, in a similar manner
to the effect of E2 on ERK1/2 phosphorylation in MCF-7 cells. This
indicated that 1 μM Tam may act as E2, to stimulate ERK1/2
phosphorylation in TAM-R cells (Fig.
3B). However, after treated with E2 or Tam, the phosphorylation
level of AKT remained constant in the TAM-R cells as well as
control MCF-7 cells (Fig. 3). These
results demonstrated that low-dose Tam may act as an agonist, such
as E2, to stimulate ERK1/2 phosphorylation and induce TAM-R cell
proliferation, whereas AKT activity may be unrelated with the
growth or survival of TAM-R cells in the presence of low-dose
Tam.
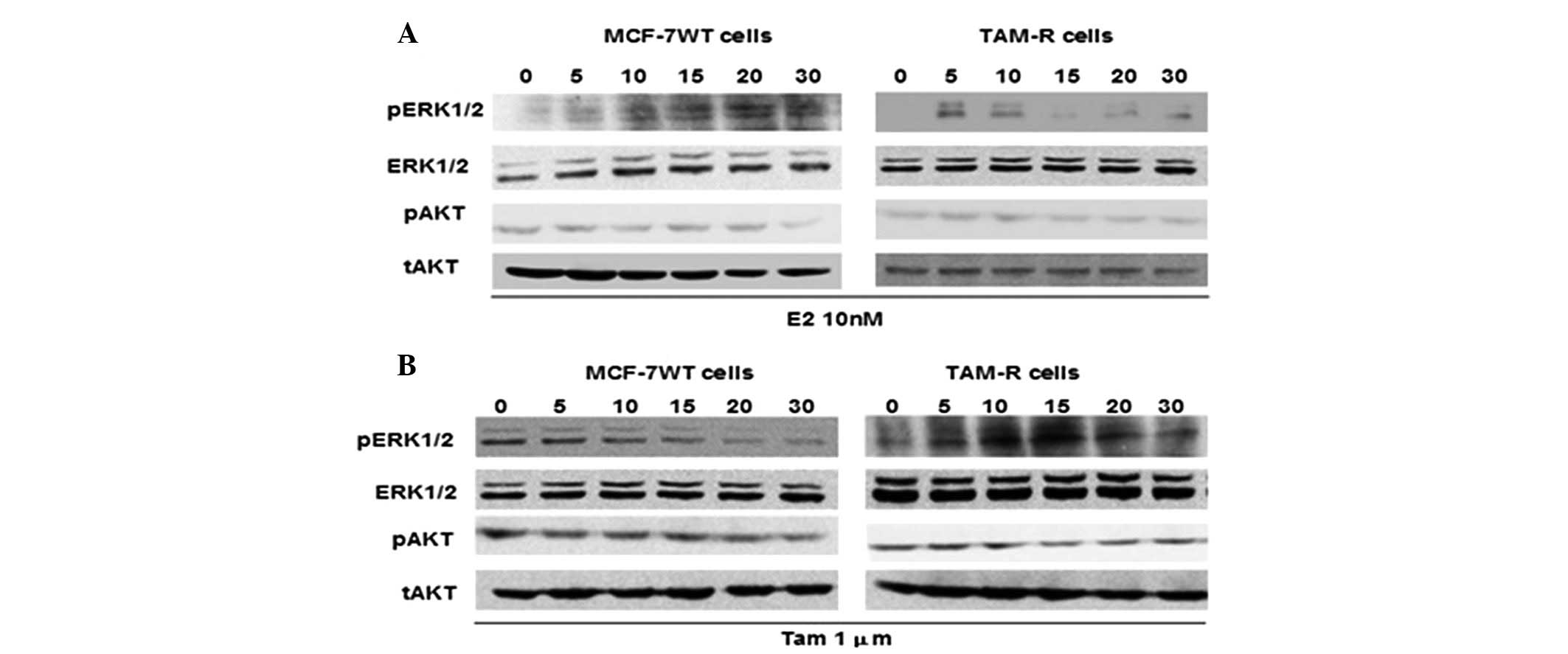 | Figure 3Effect of low-dose tamoxifen on the
ERK1/2 and AKT signaling pathways in TAM-R cells. (A) MCF-7 and
TAM-R cells were treated with 10 nM 17β-estradiol (E2) for 0, 5,
10, 15, 20 or 30 min. (B) TAM-R and MCF-7 cells were treated with 1
μM tamoxifen for 0, 5, 10, 15, 20 or 30 min, and after the
preparation of cell lysates, western blot analysis was performed.
The expression of phosphorylated (p) or total (t) ERK1/2 and AKT
proteins was examined by western blot analysis using specific
antibodies. |
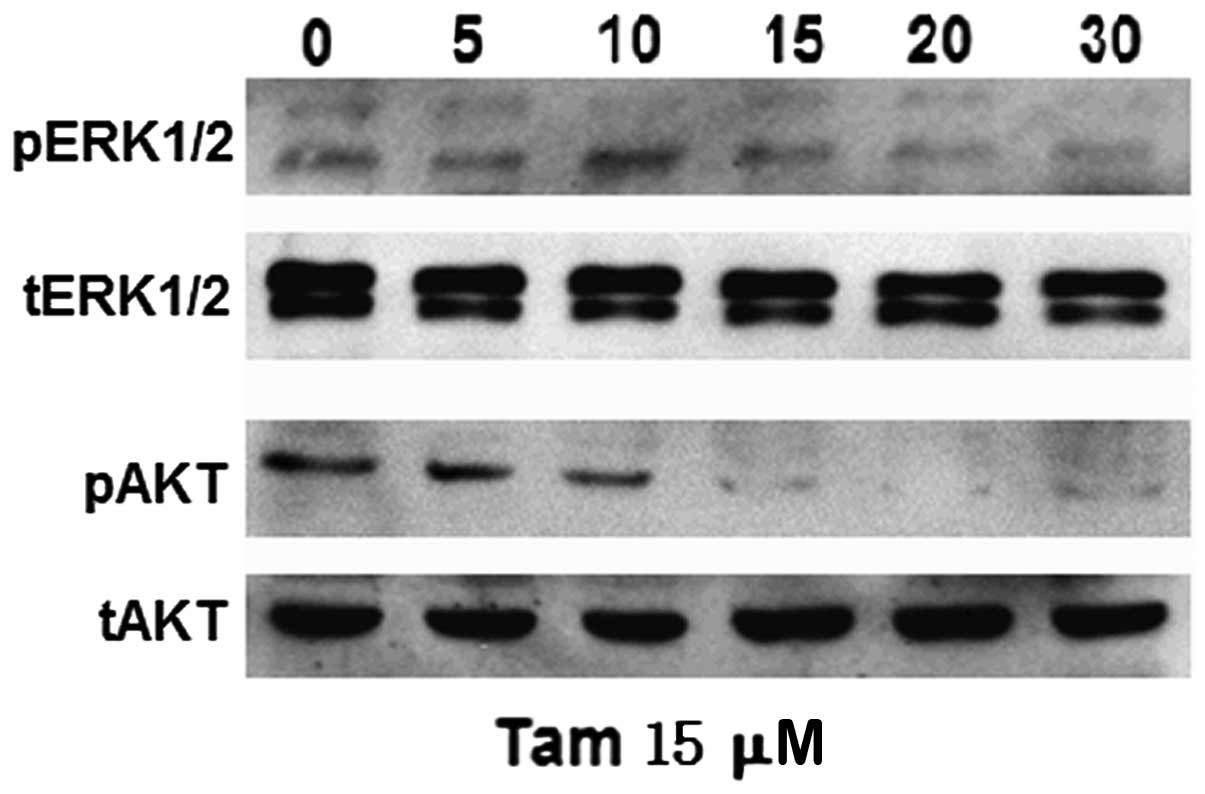
High-dose Tam downregulates ERK1/2 and
AKT activity in TAM-R cells
In order to investigate whether high-dose and
low-dose Tam have differential effects on ERK1/2 and AKT signaling
pathways in TAM-R cells, we analyzed the phosphorylation of ERK1/2
and AKT induced following treatment with 15 μM Tam. The activation
of ERK1/2 was markedly and sustainably suppressed under this
condition, which was different compared to treatment with 1 μM Tam
in TAM-R cells (Fig. 3B). In
addition, the phosphorylation of AKT was inhibited in TAM-R cells
following treatment with 15 μM Tam for 10 min (Fig. 4); however, after exposure to 1 μM
Tam, the levels of p-AKT in the TAM-R cells barely changed
(Fig. 3B). These results suggested
that downregulation of p-ERK1/2 and p-AKT was caused by high-dose
Tam to inhibit TAM-R cell proliferation. Taken together, a
dose-response effect of Tam was noted on TAM-R cell proliferation
and different doses of Tam had differential effects on ERK1/2 and
AKT signaling pathways in TAM-R cells; thus we hypothesized that
different doses of Tam stimulate or inhibit TAM-R cell growth via
different signaling pathways.
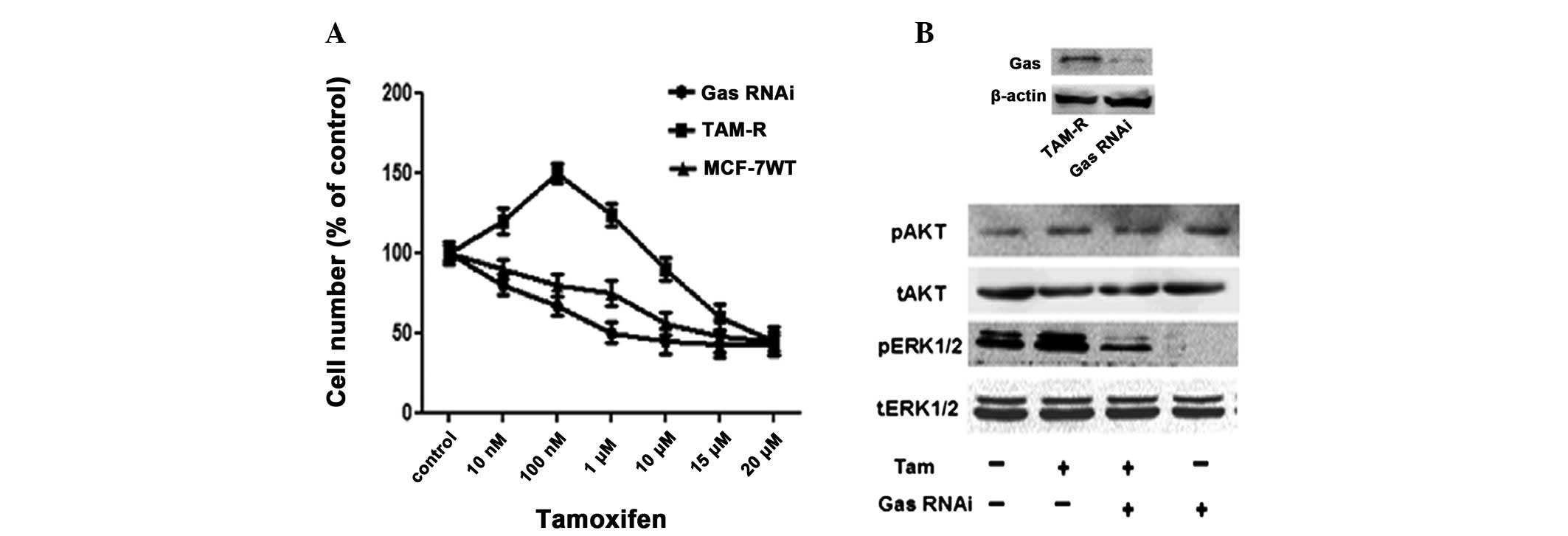
Gαs knockdown alters Tam-mediated
proliferation responses and ERK1/2 phosphorylation in TAM-R
cells
It has been confirmed that the MEK/ERK pathway is
involved in Tam resistance (15),
and Gαs stimulates MEK/ERK via the AC/cAMP/PKA pathway (17,18).
Therefore, we investigated whether Gαs is involved in the
activation of ERK induced by Tam at a low concentration and is
related with Tam resistance in TAM-R cells. We used siRNA to
silence the expression of Gαs in TAM-R cells. Knockdown of Gαs in
TAM-R cells significantly enhanced the anti-proliferative activity
of Tam (Fig. 5A). To investigate
whether Gαs is required for Tam-induced ERK1/2 and AKT
phosphorylation in TAM-R cells, Gαs RNAi oligonucleotides were used
to knock down Gαs in TAM-R cells. TAM-R cells were transfected with
Gαs RNAi oligonucleotides for 48 h, and the cells were treated with
1 μM Tam for 15 min. As shown in Fig.
5B, silencing of Gαs expression inhibited the ability of Tam to
induce ERK1/2 phosphorylation but not AKT phosphorylation in TAM-R
cells, when compared to the control cells. These results indicate
that the ability of Tam to stimulate ERK1/2 phosphorylation in
TAM-R cells was mediated by Gαs.
Discussion
In the present study, we confirmed that different
concentrations of Tam have opposite effects via different signaling
pathways on the proliferation of TAM-R cells. Our results indicate
that relatively low doses of Tam promote the proliferation of TAM-R
cells by activation of ERK1/2. On the contrary, relatively high
doses of Tam inhibit the proliferation of TAM-R cells by
downregulating the phosphorylation of ERK1/2 and AKT. To our
knowledge, this is the first study demonstrating the different
mechanisms involved in the differential effects of high and low
concentrations of Tam on TAM-R cells. These unexpected findings
enrich the limited data concerning the effects of different doses
of Tam on TAM-R cells, and provide a novel and helpful perspective
to identify the molecular targets of high-dose Tam compared with a
relatively low-dose Tam in TAM-R cells which was used to
investigate the mechanism of Tam resistance in a previous study
(19). Additionally, our
observations not only identify the harmful effects of a low dose of
Tam for the therapy of Tam-resistant patients but also provide a
convenient and workable approach to overcome resistance to Tam.
Although much research has been focused on the
mechanisms of Tam resistance in breast cancer (20–23),
this issue still remains to be elucidated. In order to further
elucidate this issue, and identify the role of Tam in TAM-R cells,
we first tested the stability of TAM-R cells that were obtained
after six months of screening (10,24).
In agreement with previous studies (9–10,25,26),
we found a Tam-stimulated growth effect in TAM-R cells after
treatment with low-dose Tam, which implied that Tam at a low
concentration may promote tumor growth in Tam-resistant patients.
Nevertheless, the specific mechanism was only partially
characterized; a possible reason for this phenomenon may be that
Tam at a low concentration plays an estrogen-like role in this
process.
At standard dosages, Tam has minimal or no effect on
Tam-resistant breast cancer cells (27). At higher concentrations of Tam
beyond the dose that promotes tumor growth most effectively, we
found that the growth-promoting effect of Tam decreased
accordingly, with gradually increasing concentrations of Tam.
However, when the concentration of Tam reached a certain high
value, Tam exerted an inhibitory effect on the proliferation of
TAM-R cells. Leung et al reported that high-dose Tam did not
function to block TAM-R cell proliferation (28), while our data challenged the
traditional view on the role of Tam. The results from our
experiments suggest that Tam at a high concentration still has an
inhibitory effect on the proliferation of TAM-R cells. In order to
provide further evidence for our unexpected results, we collected
the pleural effusion of a patient with advanced breast cancer
recurrence and metastasis, who underwent long-term use of Tam and
had been diagnosed as Tam resistant in the clinic (14). We treated the primary breast cancer
cells isolated from the pleural effusion of the Tam-resistant
patient with different concentrations of Tam. As expected, similar
effects of Tam at different concentrations on the proliferation of
primary TAM-R cells were observed (Fig.
2B). Taken together, Tam may still exhibit complicated yet
significant effects on tumor growth after Tam resistance is
abrogated in breast tumors.
Studies have shown that high ERK1/2 or Akt activity
in breast carcinoma is associated with a poor patho-phenotype, as
well as hormone and Tam resistance (8,11,15,16).
Moreover, Tam at a low concentration is suggested to have an
estrogen-like role in TAM-R cells (10). We assayed the effects of E2 and Tam
on the phosphorylation of ERK1/2 and AKT in TAM-R and control
cells. We found that Tam at 1 μM, similar to estrogen, induced
phosphorylation of ERK1/2 in Tam-R cells; however, it behaved as an
antagonist in the parental cells. However, to our surprise, the
phosphorylation level of AKT remained constant in TAM-R cells as
well as control cells after being induced with E2 and 1 μM Tam. Our
data suggest that the activation of AKT is not involved in the
function of a low concentration of Tam as an agonist in the
proliferation of TAM-R cells. This is not consistent with previous
studies that found the phosphorylation of AKT as an important
element in the proliferation of TAM-R cells (8,29).
Gαs stimulates MEK/ERK via the AC/cAMP/PKA pathway
(17,18). Therefore, we investigated whether
Gαs is involved in Tam resistance in TAM-R cells. We found that Tam
failed to activate the phosphorylation of ERK1/2, and that Tam at a
low concentration inhibited the proliferation of TAM-R cells
significantly after knockdown of Gαs expression. Thus, Gαs is
involved in the Tam resistance in TAM-R cells and should be a
prerequisite for Tam at a low concentration regarding its
estrogen-like effect on TAM-R cells. In addition, research has now
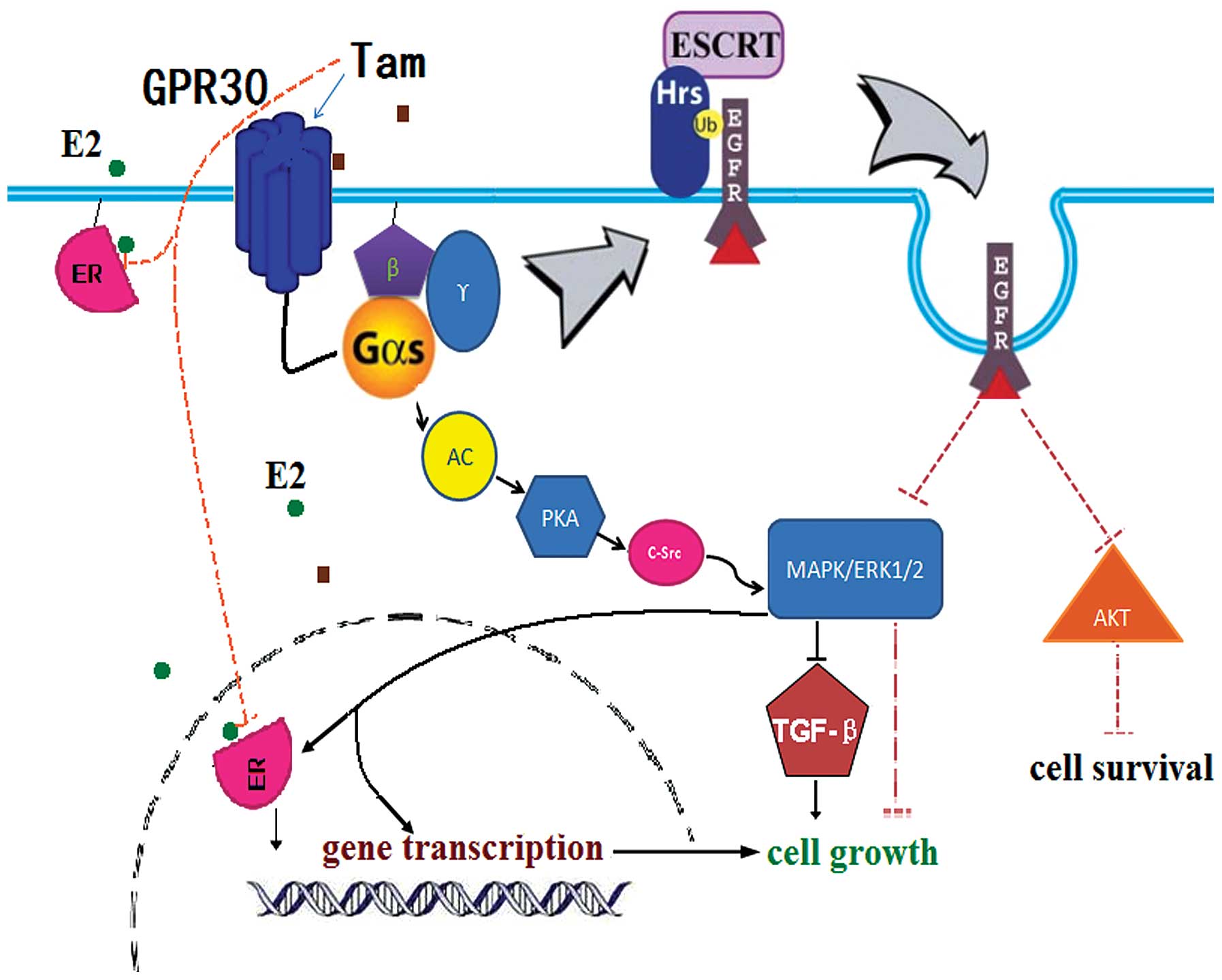
demonstrated that there exists a cross-communication between GPR30
and the development of breast cancer resistance in response to Tam
(30–32). Tam not only has high binding
affinities to GPR30, but also mimicks the actions of E2 to
stimulate cell growth (33). Tam
binding to GPR30 induced MAPK/ERK1/2 phosphorylation via G protein
and suppressed TGF-β signaling to promote cell proliferation
(34). Tam at a low concentration
was found to promote the proliferation of TAM-R cells through the
above-mentioned pathway.
In addition, many studies have confirmed that
overexpression of EGFR or the aberrant activation of the EGFR
signaling pathways are responsible for Tam-resistance (35,36).
Activated Gαs promotes the degradation of EGFR (37,38),
which reduces the expression of EGFR and blocks the EGFR signaling
pathways indirectly, and then inhibits cell growth. Thus,
consistently overactive Gαs inhibits the proliferation of TAM-R
cells by promoting the degradation of EGFR. Combined with our
results, we provide an explanation for the estrogen-like activity
of low-dose Tam in TAM-R cells and why high-dose Tam inhibits TAM-R
cell proliferation (Fig. 6). In
TAM-R cells, the sensitivity of ER to Tam decreases, and the
expression of GPR30 is upregulated (39). Therefore, Tam at a low concentration
can promote the proliferation of TAM-R cells by further
upregulating the expression and activation of GPR30. However, in
this case, Gαs is consistently overactive when GPR30 binds to
high-dose Tam, and then continuous activation of Gαs promotes the
degradation of EGFR, whose expression decreases quickly and
obviously. The degradation of EGFR results in the arrest of the
activation of ERK1/2 and AKT directly, which has an inhibitory
effect on the proliferation of TAM-R cells. When the inhibitory
effect of overactive Gαs which induces EGFR degradation overrides
the stimulating effect on proliferation via GPR30, the growth of
TAM-R cells is blocked. This hypothesis will provide answers to
whether the estrogen-like effect of low-dose Tam or Tam at a high
concentration inhibits the proliferation of TAM-R cells via
suppression of the activation of ERK1/2 and AKT.
The controversy concerning a standardized guideline
for Tam dosage has not been reached, and the mechanism of Tam
resistance is still partially characterized. Although we have not
yet acquired a large-scale epidemiological survey of the outcome of
different doses of Tam, there is no doubt that our data will help
to promote the appropriate clinical application of Tam and
elucidate the mechanism of Tam resistance. We identified the
dose-effects of different doses of Tam in the therapy of breast
cancer especially TAM-R breast cancer. We also provide a new
insight into the study of the mechanism of Tam resistance. However,
the mechanisms responsible for the dose-response of Tam and the
reason for differences in the survival rates of patients treated
with different dosages of Tam need further elucidation.
Many studies have shown that the compliance to Tam
dosage has a close relationship with the survival rate of breast
cancer patients (40). According to
our results, we hypothesized that patients with an irregular
medication schedule have a poor survival rate as the drug
concentration in blood fails to maintain a relatively high level,
and a low dose of Tam may play an estrogen-like role in promoting
tumor growth after patients acquire breast cancer with Tam
resistance. Therefore, to ensure a safe dose range, a relatively
high level of Tam is better for the control of disease and delays
the progression to Tam-resistant breast cancer. In order to confirm
our data and hypothesis, and more importantly to provide a powerful
conclusion to the standardized use of Tam, a large-scale
epidemiological survey should be implemented to identify the
differences in the survival rates and disease progression in
different Tam concentration groups. If possible, the optimal dosage
of Tam should be identified by further investigation as soon as
possible to delay the occurrence of the Tam resistance, even to
overcome the Tam resistance.
In summary, we describe a novel phenomenon that
different doses of Tam have exactly opposite effects on TAM-R cells
and identified a previously unappreciated role of high-dose Tam
which has significant anti-proliferative effect on TAM-R cells.
More importantly, we found that the phosphorylation of ERK1/2 and
AKT may play an important role in this process. Further research is
warranted to identify the molecular targets of high doses of Tam,
and the practical implications that may be derived from these data.
First, clinical workers should strictly avoid the stimulating
effects of low-dose Tam for Tam-resistant patients in clinical
application, as low-dose Tam does not block tumor growth but
instead may accelerate disease progression. Second, the special
anti-proliferative effect of high-dose Tam on TAM-R cells should be
investigated and considered as a breakthrough point in the
mechanistic study of Tam resistance. More studies must be carried
out to further confirm the disadvantages of the application of
low-dose Tam for Tam-resistant patients and assess the feasibility
of high-dose Tam therapy for Tam-resistant patients.
Acknowledgements
This work was financially supported by National
Natural Science Foundation of China (81072117).
Abbreviations:
|
Tam
|
tamoxifen
|
|
ER
|
estrogen receptor
|
|
PR
|
progesterone receptor
|
|
TAM-R
|
tamoxifen-resistant
|
|
GPER
|
G protein-coupled estrogen receptor
1
|
|
GPR30
|
G protein-coupled receptor 30
|
|
EGFR
|
epidermal growth factor receptor
|
|
E2
|
17β-estradiol
|
|
RNAi
|
RNA interference
|
|
Gαs
|
stimulatory G protein α subunit
|
|
FCS
|
fetal calf serum
|
References
|
1
|
Tamoxifen for early breast cancer: an
overview of the randomised trials. Early Breast Cancer Trialists'
Collaborative Group. Lancet. 351:1451–1467. 1998. View Article : Google Scholar
|
|
2
|
Powles TJ, Ashley S, Tidy A, Smith IE and
Dowsett M: Twenty-year follow-up of the Royal Marsden randomized,
double-blinded tamoxifen breast cancer prevention trial. J Natl
Cancer Inst. 99:283–290. 2007.PubMed/NCBI
|
|
3
|
Cuzick J, Powles T, Veronesi U, et al:
Overview of the main outcomes in breast-cancer prevention trials.
Lancet. 361:296–300. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kurebayashi J: Resistance to endocrine
therapy in breast cancer. Cancer Chemother Pharmacol. 56(Suppl 1):
39–46. 2005. View Article : Google Scholar
|
|
5
|
Campbell RA, Bhat-Nakshatri P, Patel NM,
Constantinidou D, Ali S and Nakshatri H: Phosphatidylinositol
3-kinase/AKT-mediated activation of estrogen receptor alpha: a new
model for anti-estrogen resistance. J Biol Chem. 276:9817–9824.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gee JM, Robertson JF, Ellis IO and
Nicholson RI: Phosphorylation of ERK1/2 mitogen-activated protein
kinase is associated with poor response to anti-hormonal therapy
and decreased patient survival in clinical breast cancer. Int J
Cancer. 95:247–254. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ignatov A, Ignatov T, Weissenborn C, et
al: G-protein-coupled estrogen receptor GPR30 and tamoxifen
resistance in breast cancer. Breast Cancer Res Treat. 128:457–466.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Miller TW, Balko JM and Arteaga CL:
Phosphatidylinositol 3-kinase and antiestrogen resistance in breast
cancer. J Clin Oncol. 29:4452–4461. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brunner N, Frandsen TL, Holst-Hansen C, et
al: MCF7/LCC2: a 4-hydroxytamoxifen resistant human breast cancer
variant that retains sensitivity to the steroidal antiestrogen ICI
182,780. Cancer Res. 53:3229–3232. 1993.
|
|
10
|
Ignatov A, Ignatov T, Roessner A, Costa SD
and Kalinski T: Role of GPR30 in the mechanisms of tamoxifen
resistance in breast cancer MCF-7 cells. Breast Cancer Res Treat.
123:87–96. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shou J, Massarweh S, Osborne CK, et al:
Mechanisms of tamoxifen resistance: increased estrogen
receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J
Natl Cancer Inst. 96:926–935. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Benz CC, Scott GK, Sarup JC, et al:
Estrogen-dependent, tamoxifen-resistant tumorigenic growth of MCF-7
cells transfected with HER2/neu. Breast Cancer Res Treat. 24:85–95.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ward A, Balwierz A, Zhang JD, et al:
Re-expression of microRNA-375 reverses both tamoxifen resistance
and accompanying EMT-like properties in breast cancer. Oncogene.
Apr 16–2012.(Epub ahead of print).
|
|
14
|
Hurtado A, Holmes KA, Geistlinger TR, et
al: Regulation of ERBB2 by oestrogen receptor-PAX2 determines
response to tamoxifen. Nature. 456:663–666. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Britton DJ, Hutcheson IR, Knowlden JM, et
al: Bidirectional cross talk between ERalpha and EGFR signalling
pathways regulates tamoxifen-resistant growth. Breast Cancer Res
Treat. 96:131–146. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Clark AS, West K, Streicher S and Dennis
PA: Constitutive and inducible Akt activity promotes resistance to
chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol
Cancer Ther. 1:707–717. 2002.PubMed/NCBI
|
|
17
|
Gerits N, Kostenko S, Shiryaev A,
Johannessen M and Moens U: Relations between the mitogen-activated
protein kinase and the cAMP-dependent protein kinase pathways:
comradeship and hostility. Cell Signal. 20:1592–1607. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu AM, Lo RK, Wong CS, Morris C, Wise H
and Wong YH: Activation of STAT3 by G alpha(s) distinctively
requires protein kinase A, JNK, and phosphatidylinositol 3-kinase.
J Biol Chem. 281:35812–35825. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Selever J, Gu G, Lewis MT, et al:
Dicer-mediated upregulation of BCRP confers tamoxifen resistance in
human breast cancer cells. Clin Cancer Res. 17:6510–6521. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schafer JM, Bentrem DJ, Takei H, Gajdos C,
Badve S and Jordan VC: A mechanism of drug resistance to tamoxifen
in breast cancer. J Steroid Biochem Mol Biol. 83:75–83. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wiebe VJ, Osborne CK, Fuqua SA and
DeGregorio MW: Tamoxifen resistance in breast cancer. Crit Rev
Oncol Hematol. 14:173–188. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Clarke R, Liu MC, Bouker KB, et al:
Antiestrogen resistance in breast cancer and the role of estrogen
receptor signaling. Oncogene. 22:7316–7339. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schwartz JL, Shajahan AN and Clarke R: The
role of interferon regulatory factor-1 (IRF1) in overcoming
antiestrogen resistance in the treatment of breast cancer. Int J
Breast Cancer. 2011:9121022011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou C, Zhong Q, Rhodes LV, et al:
Proteomic analysis of acquired tamoxifen resistance in MCF-7 cells
reveals expression signatures associated with enhanced migration.
Breast Cancer Res. 14:R452012. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Canney PA, Griffiths T, Latief TN and
Priestman TJ: Clinical significance of tamoxifen withdrawal
response. Lancet. 1:361987. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Howell A, Dodwell DJ, Anderson H and
Redford J: Response after withdrawal of tamoxifen and progestogens
in advanced breast cancer. Ann Oncol. 3:611–617. 1992.PubMed/NCBI
|
|
27
|
Banerjee S, Kambhampati S, Haque I and
Banerjee SK: Pomegranate sensitizes tamoxifen action in ER-alpha
positive breast cancer cells. J Cell Commun Signal. 5:317–324.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Leung E, Kannan N, Krissansen GW, Findlay
MP and Baguley BC: MCF-7 breast cancer cells selected for tamoxifen
resistance acquire new phenotypes differing in DNA content,
phospho-HER2 and PAX2 expression, and rapamycin sensitivity. Cancer
Biol Ther. 9:717–724. 2010. View Article : Google Scholar
|
|
29
|
deGraffenried LA, Friedrichs WE, Russell
DH, et al: Inhibition of mTOR activity restores tamoxifen response
in breast cancer cells with aberrant Akt Activity. Clin Cancer Res.
10:8059–8067. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vivacqua A, Bonofiglio D, Recchia AG, et
al: The G protein-coupled receptor GPR30 mediates the proliferative
effects induced by 17beta-estradiol and hydroxytamoxifen in
endometrial cancer cells. Mol Endocrinol. 20:631–646. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Carmeci C, Thompson DA, Ring HZ, Francke U
and Weigel RJ: Identification of a gene (GPR30) with homology to
the G-protein-coupled receptor superfamily associated with estrogen
receptor expression in breast cancer. Genomics. 45:607–617. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Filardo EJ, Graeber CT, Quinn JA, et al:
Distribution of GPR30, a seven membrane-spanning estrogen receptor,
in primary breast cancer and its association with clinicopathologic
determinants of tumor progression. Clin Cancer Res. 12:6359–6366.
2006. View Article : Google Scholar
|
|
33
|
Li Y, Birnbaumer L and Teng CT: Regulation
of ERRalpha gene expression by estrogen receptor agonists and
antagonists in SKBR3 breast cancer cells: differential molecular
mechanisms mediated by g protein-coupled receptor GPR30/GPER-1. Mol
Endocrinol. 24:969–980. 2010. View Article : Google Scholar
|
|
34
|
Kleuser B, Malek D, Gust R, Pertz HH and
Potteck H: 17-Beta-estradiol inhibits transforming growth
factor-beta signaling and function in breast cancer cells via
activation of extracellular signal-regulated kinase through the G
protein-coupled receptor 30. Mol Pharmacol. 74:1533–1543. 2008.
View Article : Google Scholar
|
|
35
|
Massarweh S, Osborne CK, Creighton CJ, et
al: Tamoxifen resistance in breast tumors is driven by growth
factor receptor signaling with repression of classic estrogen
receptor genomic function. Cancer Res. 68:826–833. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hutcheson IR, Knowlden JM, Madden TA, et
al: Oestrogen receptor-mediated modulation of the EGFR/MAPK pathway
in tamoxifen-resistant MCF-7 cells. Breast Cancer Res Treat.
81:81–93. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zheng B, Lavoie C, Tang TD, et al:
Regulation of epidermal growth factor receptor degradation by
heterotrimeric Galphas protein. Mol Biol Cell. 15:5538–5550. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Stryjek-Kaminska D, Piiper A and Zeuzem S:
Epidermal growth factor regulates adenylate cyclase activity via Gs
and Gi1–2 proteins in pancreatic acinar membranes. Biochem J.
316:87–91. 1996.PubMed/NCBI
|
|
39
|
Maggiolini M and Picard D: The unfolding
stories of GPR30, a new membrane-bound estrogen receptor. J
Endocrinol. 204:105–114. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huiart L, Dell'Aniello S and Suissa S: Use
of tamoxifen and aromatase inhibitors in a large population-based
cohort of women with breast cancer. Br J Cancer. 104:1558–1563.
2011. View Article : Google Scholar : PubMed/NCBI
|