Introduction
Hepatocellular carcinoma (HCC) ranks third in
cancer-related mortality due to late diagnosis and poor treatment
options. To date, the therapies of liver cancer include surgery,
chemical therapy and target therapy, but there is no complete
treatment (1). An alternative
therapy for liver cancer is urgently required.
Autophagy was reported for the first time by Ashford
and Porter 50 years ago (2) and it
has recently gained considerable attention. Autophagy is a
lysosome-mediated protein and organelle degradation process that is
characterized by the formation of double-membrane vesicles,
referred to as autophagosomes (3,4).
Autophagy is involved in several pathophysiological processes and
contributes to numerous diseases, particularly to cancer (5,6).
However, the function of autophagy in cancer has yet to be fully
clarified, as it acts both as a tumor suppressor and as a tumor
promoter (7).
Reduced autophagy is associated with a malignant
phenotype and poor prognosis of HCC and activation of autophagy
contributes to the growth inhibition and cell death in human liver
cancer cells (8,9). Increasing evidence shows that
autophagy functions as a survival mechanism in liver cancer cells
against drug-induced apoptosis. Autophagy inhibition enhances
apoptosis induced by ginsenoside Rk1 (10), 3-bromopyruvate (11), BO-1051 (12), etoposide (13) in HCC cell lines. Targeting the
autophagy pathway is a promising therapeutic strategy to improve
chemotherapy efficiency. Thus, autophagy may play a dual and
apparently contradictory role in HCC and the exhibited function may
likely depend on the genetic composition of the cell and
environmental cues the cell is exposed to.
A number of studies have recently focused on the
regulation mechanism of autophagy. For example, TGF-β signaling
pathway (14) and IFN-γ (8) activate autophagy in HCC cells. STMN1
(Stathmin 1) (15), RAB GTPase 5A
(RAB5A) (16), autophagy-related
protein 4D (ATG4D) and mTOR (17)
have been reported to play an important role in autophagosome
formation. micro-RNAs (miRNAs), which play crucial roles in HCC
development and therapy, have been linked to autophagy. miR-375
inhibits autophagy by targeting autophagy-associated gene 7 (ATG7)
and impairs the viability of HCC cells under hypoxic conditions
both in vivo and in vitro(18). Downregulated miR-199a-5p enhanced
autophagy activation by targeting ATG7 in HCC cells (19). It is reported that miR-101 inhibits
basal, etoposide- and rapamycin-induced autophagy which can
sensitize breast cancer cells to 4-hydroxytamoxifen
(4-OHT)-mediated cell death. The targets of miR-101 in this process
are STMN1, RAB5A and ATG4D (15).
miR-101 sensitized HCC cell lines to chemotherapeutic drug-induced
apoptosis by targeting Mcl-1 (20).
However, the regulation mechanism of miR-101 and its function on
autophagy in HCC remains unclear.
In this study, we report that miR-101 inhibits
autophagy and enhances apoptosis induced by cisplatin in HCC cells.
The targets of miR-101 are STMN1, RAB5A, ATG4D and mTOR. This study
revealed that miR-101 which, inhibits autophagy, might be developed
as a potential novel therapy for HCC.
Materials and methods
Cell cultures
Human HCC cell line HepG2 was purchased from
Shanghai Cell Bank (Shanghai, China) and propagated in our
laboratory by culturing in Dulbecco’s modified Eagle’s medium
(DMEM) (Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum
(FBS) (Sigma, St. Louis, MO, USA), at 37°C with 5% CO2,
supplemented with 1% penicillin/streptomycin. Drug treatment
included cisplatin (0.25 mg/ml, Sigma) and bafilomycin (400 nM,
Sigma) for the indicated times.
miRNA transfection
miRNA transfection was performed using Lipofectamine
2000 (Invitrogen). Total RNA and protein were extracted at 24 h
post-transfection and were used for quantitative real-time PCR
(qRT-PCR) and western blot analysis. miR-101-mimic, inhibitor and
negative control groups were designed and synthesized by GenePharma
(Shanghai, China).
Reverse transcription quantitative
real-time polymerase chain reaction (RT-qPCR) for miRNA and mRNA
quantitation
Total RNA of cells and tissues was isolated using
TRIzol reagent (Invitrogen). For miRNA quantitation, cDNA was
synthesized with specific miRNA reverse transcriptase primers
(Applied Biosystems) using the TaqMan MicroRNA Reverse
Transcription kit (Applied Biosystems, Life Technologies Corp., CA,
USA). For mRNA quantitation, cDNA was synthesized using the
PrimeScript RT reagent kit (Takara, Dalian, China). Quantitative
RT-PCR was performed using an ABI 7500 (Applied Biosystems) with
FastStart Universal SYBR Green Master (Rox) (Roche, USA) for mRNA
quantitation and with TaqMan® MicroRNA Assay kit
(Applied Biosystems). The relative expression of miRNA and mRNA
were calculated as the inverse log of the ΔΔCT (21) and normalized to U6 and β-actin.
Probes for miRNA qPCR were purchased from Applied Biosystems,
primers for mRNA qPCR were synthesized by Invitrogen (Shanghai,
China); the sequences were: STMN1 sense: 5′-TCTGTCCCAATCTTACCA-3′,
antisense: 5′-GAGGCATCCAAACAAAGC-3′; RAB5A sense:
5′-GCTGGTCAAGAACGATAC-3′, antisense: 5′-CTTGCTTGCCTCTGAAGT-3′;
ATG4D sense: 5′-GCTGCCTGACCTCGGACTGT-3′, antisense:
5′-TCTGCCCAAGCTCCACCAG-3′; mTOR sense: 5′-CGCTGTCATCCCTTTATCG-3′,
antisense: 5′-ATGCTCAAACACCTCCACC-3′; β-actin sense:
TCACCCACACTGTGCCCATCTACGA, antisense:
CAGCGGAACCGCTCATTGCCAATGG.
Cell apoptosis analysis
Cell apoptosis was assessed by flow cytometry
(Becton-Dickinson, San Jose, CA, USA). For cell apoptosis, cells
were treated with cisplatin at a final concentration of 10 μM for
48 h. Then, cells were collected, washed, suspended in 100 μl 1X
binding buffer, stained with 5 μl fluorescein isothiocyanate
(FITC)-Annexin V and 1 μl PI at room temperature for 15 min in the
dark. The stained cells were immediately analyzed by flow
cytometry.
Luciferase reporter assay
Luciferase reporter constructs were made by ligating
60-bp-long synthetic oligonucleotides (Invitrogen, Shanghai, China)
containing putative miRNA binding sites from the 3′-UTR or their
mutant versions of STMN1, RAB5A, ATG4D and mTOR in
XbaI-FseI sites of the pGL3-control vector (Promega).
Cloning was verified by sequencing. HepG2 cells were plated at
1.5×105 cells/well in 24-well plates 24 h prior to
transfection. Each independent luciferase reporter plasmid (200 ng)
plus 80 ng pRL-TK (Promega) was transfected in combination with 60
pmol of miR-101-mimic, inhibitor and controls using Lipofectamine
2000 (Invitrogen). Luciferase activity was measured 48 h after
transfection by using the Dual-Luciferase Reporter assay system
(Promega). Firefly luciferase activity was normalized to Renilla
luciferase activity for each transfected well.
Western blotting
Cells were lysed using RIPA buffer with 1% PMSF on
ice. The concentration of total protein was determined using a BCA
kit (Keygen, Nanjing, China). Equal amounts of protein (30 μg) were
resolved with 10% SDS-PAGE and transferred to polyvinylidene
difluoride (PVDF) membranes (Millipore, Bedford, MA, USA) using a
mini trans-blot apparatus (Bio-Rad Laboratories, Hercules, CA,
USA). Membranes were probed with primary antibodies for 12 h at 4°C
and then incubated with secondary antibodies for 2 h at room
temperature. The primary antibodies used were:
microtubule-associated protein light chain 3 (LC3) and RAB5A rabbit
polyclonal antibodies (Novus Biologicals, Littleton, CO, USA),
STMN1 and mTOR antibodies (Cell Signaling Technology, Danvers, MA,
USA). ATG4D antibody (Abgent, San Diego, CA, USA). GAPDH and
tubulin (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were used
as an internal control. The secondary antibody was purchased from
Beyotime (Santa Cruz Biotechnology). Electrochemiluminescence was
performed with a ChemiImager 5500 imaging system (Alpha Innotech
Co., San Leandro, CA, USA).
Transmission electron microscopy
Representative images were taken of cells
transfected for 72 h as indicated and treated for 2 h with 200 nM
rapamycin prior to fixation. For ultrastructural examination, liver
samples ~1 mm3 were fixed with 2% OsO4 and
embedded in Araldite. Ultrathin sections were stained with uranyl
acetate and lead citrate and inspected using an electron microscope
(JEM-1010; Jeol, Tokyo, Japan).
Statistical analysis
All experiments were repeated in triplicate. All
values were the means ± standard deviation (SD). Statistical
significance was determined with a Student’s t-test using SPSS
15.0. P-values <0.05 were considered to indicate statistically
significant differences.
Results
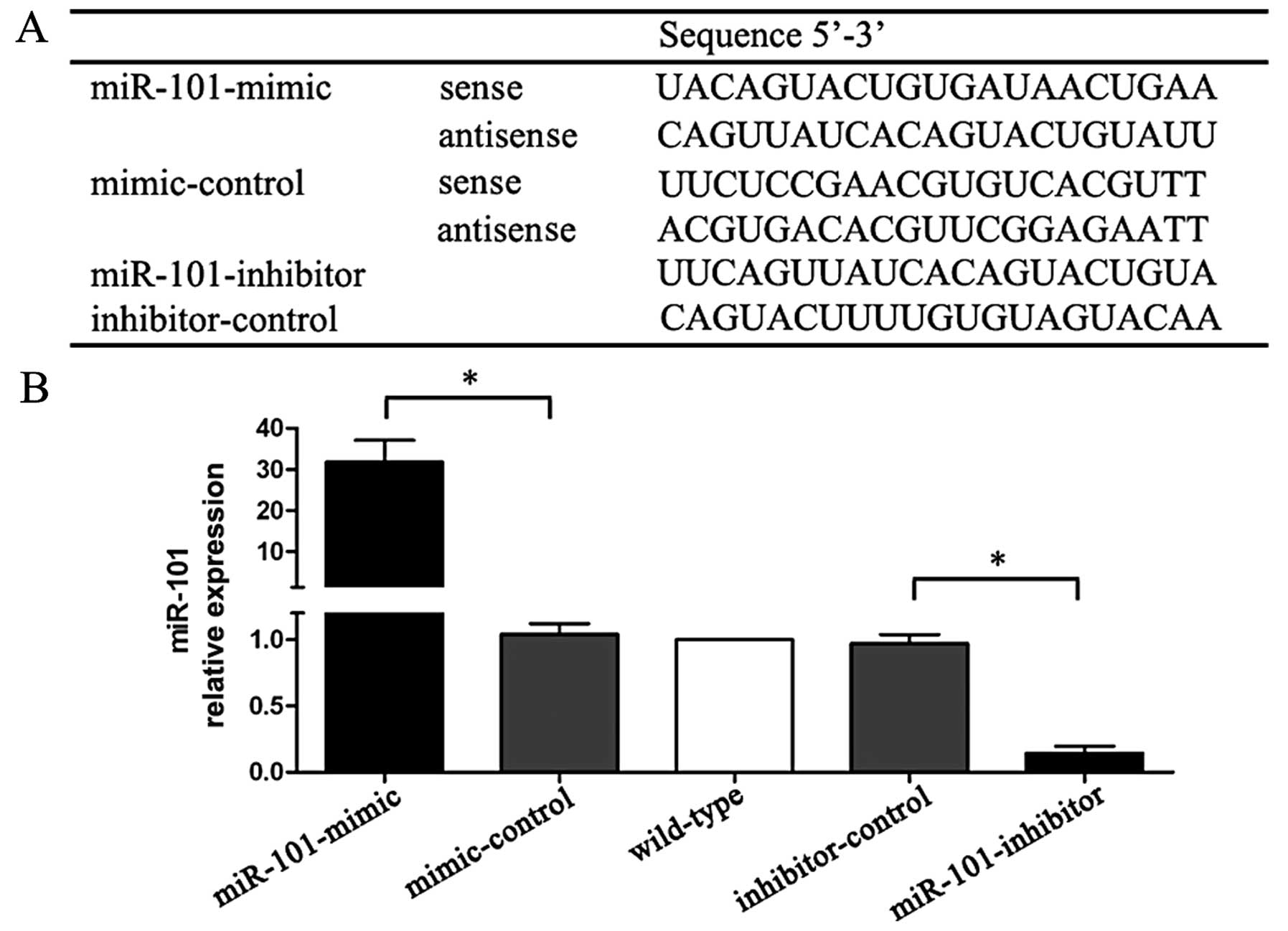
Verification of miR-101 transfection
The sequences of miR-mimic, miR-inhibitor and
controls are listed in Fig. 1A. The
expression levels of miR-101 were confirmed by qRT-PCR.
miR-mimic-treated cells showed a 31-fold higher miR-101 expression
than mimic-control treated cells, whereas miR-inhibitor cells had
an 85% lower expression when compared with inhibitor-control
treated cells (Fig. 1B).
miR-101 targets STMN1, RAB5A, ATG4D and
mTOR
To explore the regulation mechanism of miR-101, we
predicted four target genes (STMN1, RAB5A, ATG4D and mTOR) which
were matched with miR-101 on the website www.targetscan.org. Then, plasmids containing matching
or mutant sequences of each gene were constructed. These sequences
are listed in Fig. 2A. To establish
a direct molecular link between miR-101 and target genes,
luciferase reporter assay was performed. The data showed that
compared to the control group, miR-101 significantly reduced the
activity of the STMN1, RAB5A, ATG4D and mTOR 3′-UTR luciferase
plasmids, whereas plasmids containing mutant sequences were not
significantly affected (P<0.05; Fig.
2B). To examine the effect of miR-101 on endogenous mRNAs of
STMN1, RAB5A, ATG4D and mTOR, qPCR was carried out to detect mRNA
changes in miR-101-mimic transfected cells. The results showed that
compared with the control group, miR-101-mimic significantly
reduced STMN1, RAB5A, ATG4D and mTOR mRNA levels, whereas
miR-101-inhibitor elevated the mRNA levels of these four genes
(P<0.05; Fig. 2C). Finally, we
tested the effect of miR-101 on the protein of STMN1, RAB5A, ATG4D
and mTOR. As shown in Fig. 2D,
miR-101 downregulated STMN1, RAB5A, ATG4D and mTOR protein.
Therefore, miR-101 targets STMN1, RAB5A, ATG4D and mTOR,
downregulating them both at the mRNA and at the protein level.
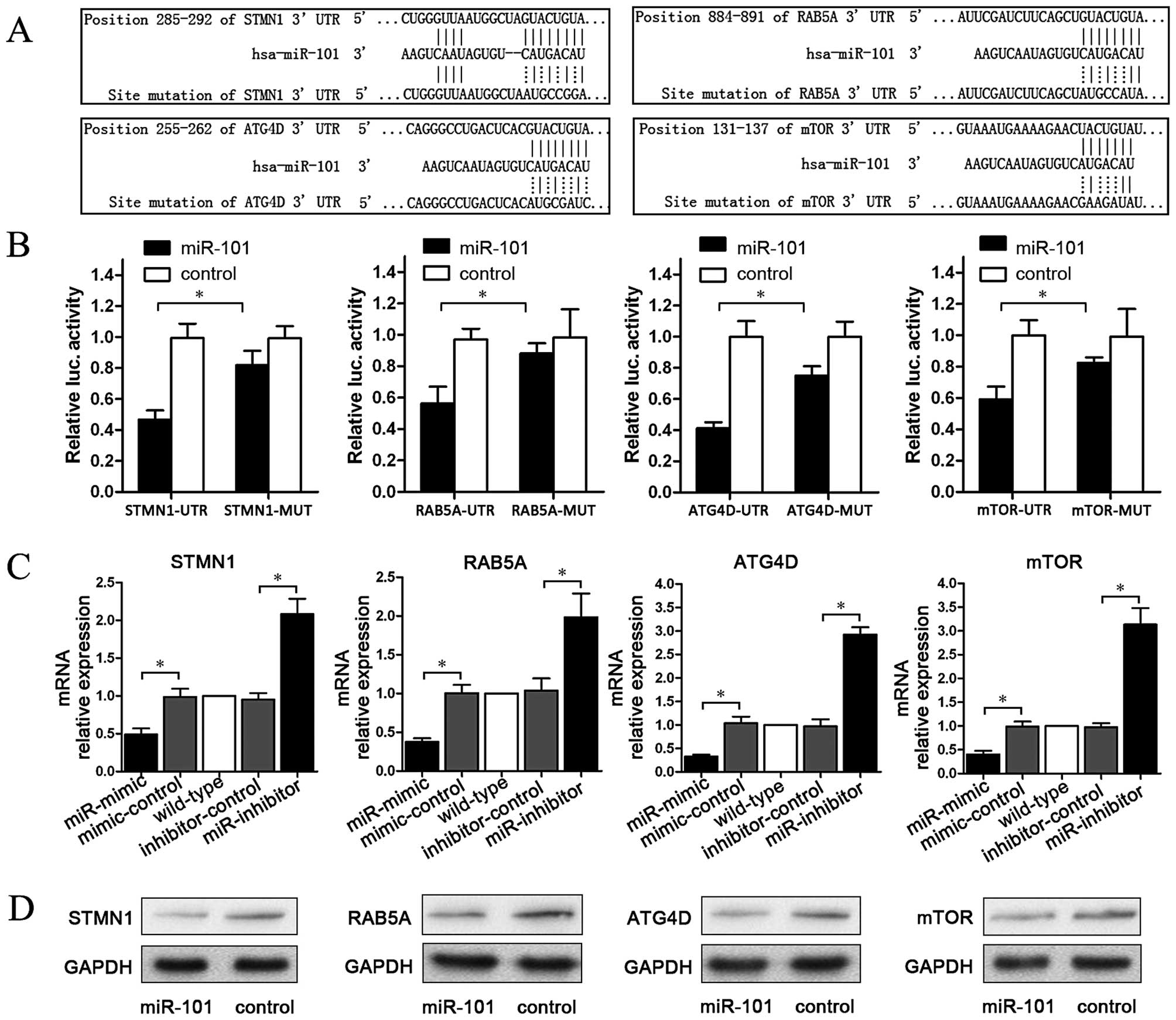 | Figure 2miR-101 targets STMN1, RAB5A, ATG4D
and mTOR. (A) Four target gene (STMN1, RAB5A, ATG4D and mTOR)
plasmids containing matching or mutant sequences of each gene were
constructed. (B) miR-101 significantly reduced the activity of the
STMN1, RAB5A, ATG4D and mTOR 3′-UTR luciferase plasmids, while
plasmids containing mutant sequences were not significantly
affected (*P<0.05). (C) miR-101-mimic significantly
reduced STMN1, RAB5A, ATG4D and mTOR mRNA levels, whereas
miR-101-inhibitor elevated the mRNA levels of these four genes
(*P<0.05). (D) Western blotting showed that miR-101
downregulated the expression levels of STMN1, RAB5A, ATG4D and mTOR
protein. |
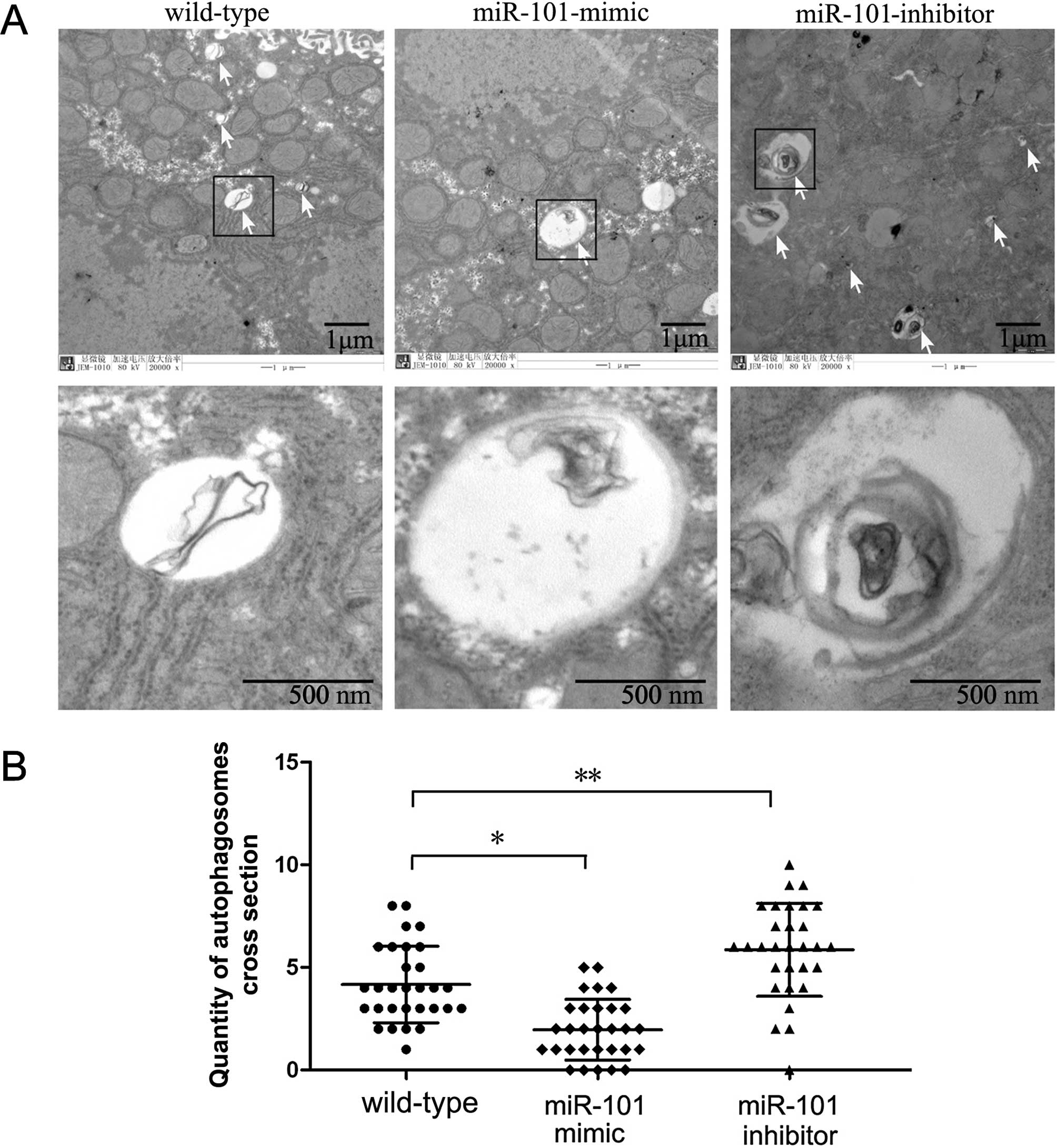
miR-101 suppresses autophagy
Since the formation of special double-membraned
structures containing undigested cytoplasmic contents
(autophagosomes) is the most important characteristic of autophagy,
demonstrating these structures by electron microscopy is considered
the gold standard for documenting autophagy. Hence, we examined the
ultrastructural changes in HepG2 cells undergoing
miR-101-mimic/inhibitor treatment. Prior to fixation, cells were
transfected with miR-101-mimic or miR-101-inhibitor for 72 h.
Non-transfected cells were used as a control. Quantification of
autophagosomes per cellular cross-section revealed that
autophagosomes were rarely detected in the non-transfected cell
group. However, the number of autophagosomes was significant
reduced in the miR-101-mimic group (Fig. 3). LC3 is also widely used to monitor
autophagy. The density of LC3-II band divided by the density of
tubulin band, which represented the expression level of LC3-II. The
ratio of LC3-II level/tubulin level was used as an indicator of
autophagic level. In our experiment, HepG2 cells were transfected
with miR-101-mimic or miR-101-inhibitor for 72 h. Cells were
treated with bafilomycin (400 nM, Sigma), which significantly
inhibits autophagy, for 2 h and used as an autophagy inhibitor.
Non-transfected cells were used as a control. Western blotting
results showed that, compared with the non-transfected group,
miR-101-mimic significantly reduced the ratio of LC3-II
level/tubulin levels, suggesting the process of autophagy was
inhibited. Meanwhile, miR-101-inhibitor elevated the ratio of
LC3-II level/tubulin significantly (Fig. 4). Therefore, miR-101 plays the
function of inhibition of autophagy.
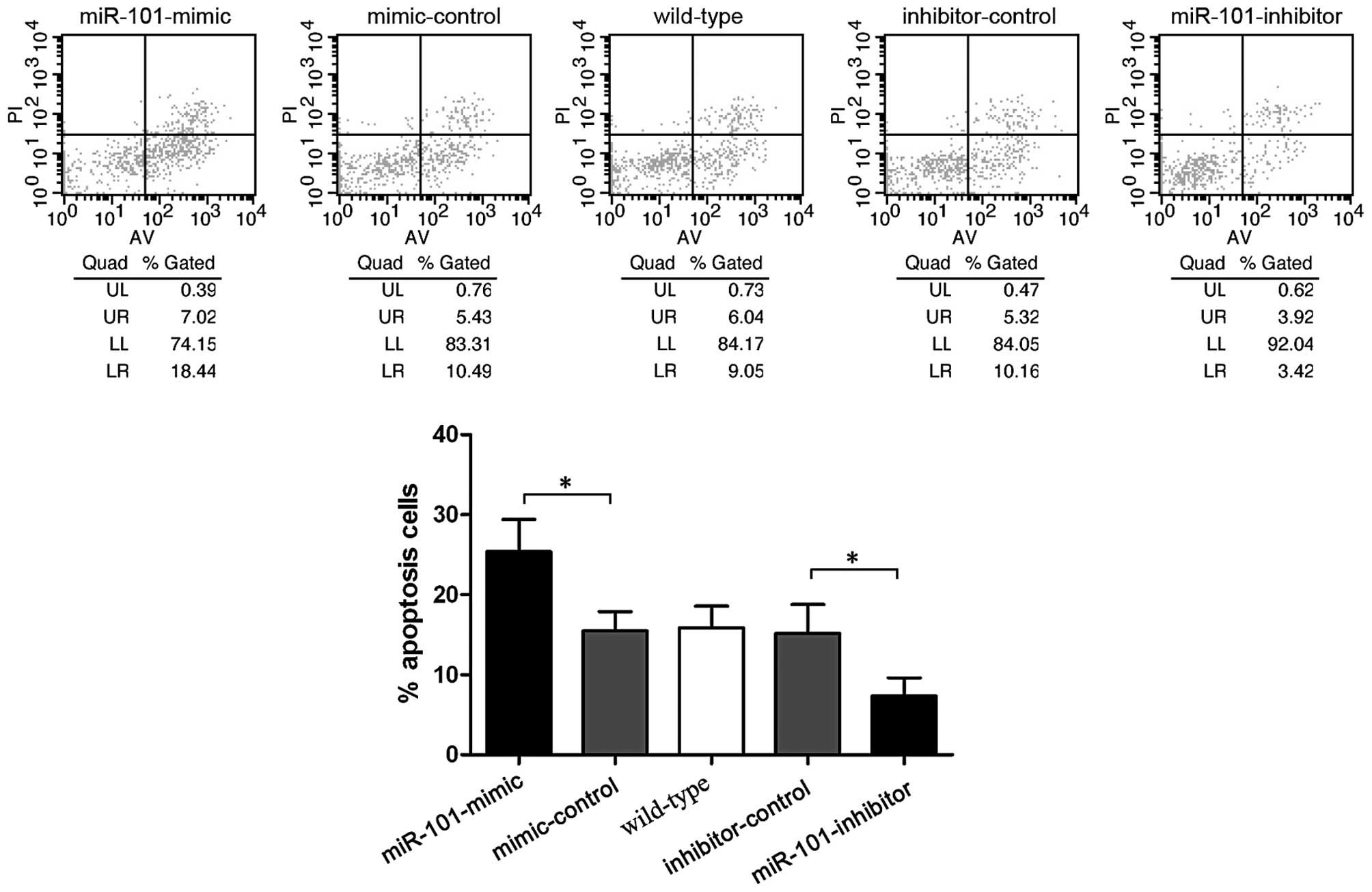
miR-101 enhances sensitization to
cisplatin
In order to investigate the role of miR-101 in
chemotherapy, cisplatin induced-apoptosis was assessed by flow
cytometry. Following treatment with 10 μM cisplatin for 48 h,
miR-101-mimic transfected cells showed a higher apoptosis ratio
(25.37±4.05%), whereas miR-101-inhibitor exhibited a lower
apoptosis ratio (7.35±2.25%; P<0.05) (Fig. 5). This indicated that miR-101
increases cisplatin sensitivity of HepG2 cells.
Discussion
In this study, we demonstrated that miR-101 enhances
apoptosis induced by cisplatin in HCC cells. In addition, our data
showed that the targets of miR-101 are STMN1, RAB5A, ATG4D and
mTOR. These results suggest that miR-101 plays an important role in
the cisplatin-induced apoptosis in HCC cells by inhibiting
autophagy.
It is reported that miR-101 is low expressed in
different types of cancer (20,
22–24). Emerging evidence suggests that
miR-101 induces apoptosis, suppresses tumorigenicity and inhibits
migration and invasion of gastric cancer cells (25). miR-101 sensitizes HCC cells to
apoptosis and impairs the ability of cancer cells to form colonies
both in vitro and in vivo(20). Moreover, myeloid cell leukemia
sequence 1 (Mcl-1) was characterized as a direct target of miR-101.
In addition, it was demonstrated that the TPA-induced ERK signaling
pathway in HepG2 cells upregulates expression of miR-101 (23). In this study, we investigated the
role of miR-101 in cisplatin-induced apoptosis of HCC cells. We
found that miR-101 enhanced apoptosis induced by cisplatin in HCC
cells, indicating the potential application of miR-101 in HCC
therapy.
Increasing evidence shows that autophagy functions
as a survival mechanism in liver cancer cells against drug-induced
apoptosis. A recent study showed that miRNA-101 is a potent
inhibitor of autophagy in breast cancer cells. In addition, STMN1,
RAB5A and ATG4D were identified as direct targets (26). In our study, miR-101 inhibited
autophagy in HCC cells. Aside from STMN1, RAB5A and ATG4D, we found
that mTOR was targeted directly by miR-101 as well. It is well
known that suppression of mTOR leads to activation of autophagy. It
seems controversial that miR-101 suppresses mTOR and inhibits
autophagy. It is also well documented that several targets exist
for a miRNA. Thus, the role of a miRNA is the result of changes in
all of these target genes. It may be that, although we found four
targets for miR-101 in autophagy, the exhibited role of miR-101 was
mainly based on STMN1, RAB5A and ATG4D protein level change.
In conclusion, we showed that miR-101 plays a key
role in enhancing apoptosis induced by cisplatin in HCC cells. The
possible mechanism of this effect may be through inhibition of
autophagy via targets including RAB5A, STMN1 and ATG4D. We propose
that gene therapy targeting miR-101/autophagy should be
investigated further as a potential alternative therapeutic
strategy for HCC.
Acknowledgements
This study was supported by grants from the
Department of Public Health of Jiangsu Province (no. RC2007056) and
the National Natural Science Foundation of China (no.
81170415).
References
|
1
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ashford TP and Porter KR: Cytoplasmic
components in hepatic cell lysosomes. J Cell Biol. 12:198–202.
1962. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mathew R, Karantza-Wadsworth V and White
E: Role of autophagy in cancer. Nat Rev Cancer. 7:961–967. 2007.
View Article : Google Scholar
|
|
6
|
Yang ZJ, Chee CE, Huang S and Sinicrope
FA: The role of autophagy in cancer: therapeutic implications. Mol
Cancer Ther. 10:1533–1541. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Eskelinen EL: The dual role of autophagy
in cancer. Curr Opin Pharmacol. 11:294–300. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li P, Du Q, Cao Z, et al: Interferon-gamma
induces autophagy with growth inhibition and cell death in human
hepatocellular carcinoma (HCC) cells through interferon-regulatory
factor-1 (IRF-1). Cancer Lett. 314:213–222. 2012. View Article : Google Scholar
|
|
9
|
Ding ZB, Shi YH, Zhou J, et al:
Association of autophagy defect with a malignant phenotype and poor
prognosis of hepatocellular carcinoma. Cancer Res. 68:9167–9175.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ko H, Kim YJ, Park JS, Park JH and Yang
HO: Autophagy inhibition enhances apoptosis induced by ginsenoside
Rk1 in hepatocellular carcinoma cells. Biosci Biotechnol Biochem.
73:2183–2189. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ganapathy-Kanniappan S, Geschwind JF,
Kunjithapatham R, et al: 3-Bromopyruvate induces endoplasmic
reticulum stress, overcomes autophagy and causes apoptosis in human
HCC cell lines. Anticancer Res. 30:923–935. 2010.PubMed/NCBI
|
|
12
|
Chen LH, Loong CC, Su TL, et al: Autophagy
inhibition enhances apoptosis triggered by BO-1051, an N-mustard
derivative and involves the ATM signaling pathway. Biochem
Pharmacol. 81:594–605. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xie BS, Zhao HC, Yao SK, et al: Autophagy
inhibition enhances etoposide-induced cell death in human hepatoma
G2 cells. Int J Mol Med. 27:599–606. 2011.PubMed/NCBI
|
|
14
|
Kiyono K, Suzuki HI, Matsuyama H, et al:
Autophagy is activated by TGF-beta and potentiates
TGF-beta-mediated growth inhibition in human hepatocellular
carcinoma cells. Cancer Res. 69:8844–8852. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Frankel LB, Wen J, Lees M, et al:
microRNA-101 is a potent inhibitor of autophagy. EMBO J.
30:4628–4641. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ravikumar B, Imarisio S, Sarkar S, O’Kane
CJ and Rubinsztein DC: Rab5 modulates aggregation and toxicity of
mutant huntingtin through macroautophagy in cell and fly models of
Huntington disease. J Cell Sci. 121:1649–1660. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jung CH, Ro SH, Cao J, Otto NM and Kim DH:
mTOR regulation of autophagy. FEBS Lett. 584:1287–1295. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chang Y, Yan W, He X, et al: miR-375
inhibits autophagy and reduces viability of hepatocellular
carcinoma cells under hypoxic conditions. Gastroenterology.
143:177–187.e8. 2012. View Article : Google Scholar
|
|
19
|
Xu N, Zhang J, Shen C, et al:
Cisplatin-induced downregulation of miR-199a-5p increases drug
resistance by activating autophagy in HCC cell. Biochem Biophys Res
Commun. 423:826–831. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Su H, Yang JR, Xu T, et al: MicroRNA-101,
down-regulated in hepatocellular carcinoma, promotes apoptosis and
suppresses tumorigenicity. Cancer Res. 69:1135–1142. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
|
|
22
|
Varambally S, Cao Q, Mani RS, et al:
Genomic loss of microRNA-101 leads to overexpression of histone
methyltransferase EZH2 in cancer. Science. 322:1695–1699. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chiang CW, Huang Y, Leong KW, et al:
PKCalpha mediated induction of miR-101 in human hepatoma HepG2
cells. J Biomed Sci. 17:352010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Buechner J, Tomte E, Haug BH, et al:
Tumour-suppressor microRNAs let-7 and mir-101 target the
proto-oncogene MYCN and inhibit cell proliferation in
MYCN-amplified neuroblastoma. Br J Cancer. 105:296–303. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang HJ, Ruan HJ, He XJ, et al:
MicroRNA-101 is down-regulated in gastric cancer and involved in
cell migration and invasion. Eur J Cancer. 46:2295–2303. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gui T and Shen K: miRNA-101: A potential
target for tumor therapy. Cancer Epidemiol. 36:537–540. 2012.
View Article : Google Scholar : PubMed/NCBI
|