Introduction
Gastric cancer (GC) is the second leading cause of
cancer-related mortality globally (1). There are ~750,000 new cases diagnosed
annually around the world and 5-year overall survival rates are
<25% (2). In spite of the
progress in understanding the pathophysiological mechanisms and
treatment for GC in recent years, the overall survival time of GC
patients has not changed significantly. Thus, a deeper
understanding of the molecular and genetic networks that control
the initiation and progression of GC is imperative.
Long non-coding RNAs (lncRNAs) are a class of
newfound non-coding RNAs, <200 nucleotides in length (3). LncRNAs, such as H19, HOTAIR and MEG3,
have been suggested to have a functional role in tumorigenesis and
tumor progression (4). Highly
upregulated in liver cancer (HULC) is ~1.6 k nucleotides long
containing two exons but not translated (5,6). It
has been shown that HULC might function as a miRNA sponge for
miRNA372 and could thereby regulate gene expression at a
post-transcriptional level (7).
Moreover, Matouk et al(8)
reported for the first time that HULC is not HCC-specific and is
high in liver nodule of colon cancer origin but not in primary
colorectal carcinoma samples and corresponding normal counterparts.
However, investigations of HULC in GC are scarce. The expression
pattern of HULC in human GC tissues and cell lines, its biological
roles, and potential mechanisms in GC progression still need to be
addressed.
Epithelial-to-mesenchymal transition (EMT), an
essential cell-biological program during embryonic development,
contributes to cancer invasion and metastasis (9,10).
Autophagy is generally thought to be a double-edged sword in the
regulation of tumor progression and some studies showed that
autophagy activation is upregulated in cancer cells and contributes
to tumor cell survival (11,12).
Furthermore, it has been shown that EMT involves the autophagy
activation of several important pathways that help tumors survive
and evolve into highly invasive and metastatic variants (13). In the present study, we examined the
role of HULC in GC and the potential mechanisms involved by a
retrospective analysis of 58 GC patients, and by carrying out in
vitro experiments to clarify the contribution of HULC to
various aspects of the malignant phenotype of human GC and its
effect on autophagy and EMT.
Materials and methods
Tissue specimens
Fifty-eight specimens of GC tissues and adjacent
non-cancer tissues were surgically obtained between January 2012
and May 2013 at the First Affiliated Hospital of Nanjing Medical
University (median age, 64; range, 45–84). Written informed consent
was obtained from all patients prior to sample collection. The
matched normal gastric tissue samples were obtained from tissues
that were 5 cm from the edge of the tumor and there were no obvious
tumor cells, as evaluated by a pathologist. Tissue specimens were
immediately frozen in liquid nitrogen after surgery and stored at
−80°C until the extraction of total RNA. TNM disease stage was
classified according to the American Joint Committee on Cancer
(AJCC), 7th Edition. None of the patients recruited in the present
study received any preoperative treatments.
Cell lines and cell culture
Human gastric epithelial mucosa cell line GES-1, GC
cell lines SGC7901, MKN28, MKN45, AGS and BGC823 were maintained in
our laboratory. The cells were cultured in RPMI-1640 containing 10%
fetal bovine serum (FBS) (Gibco, Grand Island, NY, USA), 100 U/ml
of penicillin sodium at 37°C in a humidified environment containing
5% CO2.
Quantitative real-time (RT)-PCR
Total RNA from cells and tissues was extracted using
TRIzol reagent (Invitrogen, Carlsbad, CA, USA). cDNA was
synthesized using the PrimeScript RT kit (Takara, Dalian, China).
Quantitative RT-PCR was performed with FastStart Universal
SYBR-Green Master (Rox) (Roche Diagnostics, Indianapolis, IN, USA)
with an ABI 7500 (Applied Biosystems, Life Technologies
Corporation, Carlsbad, CA, USA). The qPCR cycling was performed as
follows: initial denaturation at 95°C for 10 min followed by 40
cycles of denaturation at 95°C for 10 sec, annealing for 60 sec at
55°C (HULC), 50°C (E-cadherin) or 60°C (vimentin) and finally a
melting curve profile was set at 60°C (30 sec). Primers for qRT-PCR
were synthesized by Invitrogen (Shanghai, China) and the sequences
were: HULC sense, 5′-ACTCTGAAGTAAAGGCCGGA-3′ and antisense,
5′-TGCCAGGAAACTTCTTGCTTG-3′; E-cadherin sense,
5′-GTGTCATCCAACGGAATGC-3′ and antisense, 5′-TGG
CGGCATTGTAGGTGTTC-3′; vimentin sense, 5′-ATGAC CGCTTCGCCAACTAC-3′
and antisense, 5′-CGGGCTTTG TCGTTGGTTAG-3′. β-actin was used as an
internal control, and the following primer sequences were used to
amplify β-actin: forward, 5′-CTACAATGAGCTGCGTGTGG-3′ and reverse,
5′-AAGGAAGGCTGGAAGAGTGC-3′. PCR amplifications were performed in
three duplicates for each sample.
Lentivirus packaging and stable
transfection cell line generation
To further investigate the function of HULC, HULC
expression was modified by gene knockdown and overexpression via
lentivirus vector. We modified the commercial LV-HULC vector and
LV-HULC-30 vector lentiviral constructs (Shanghai Genepharma Co.,
Ltd., Shanghai, China) to overexpress or knock down HULC in GC
cells. For knockdown, LV-HULC-30 (target sequence: 5′-GCCTTTACA
AGGGAATGAAGA-3′), with ≥75% knockdown efficiency, was used for
further studies. All lentiviral vectors expressed GFP and the
efficiency of infection was measured under a fluorescent microscope
based on GFP expression.
Western blot analysis
Cells were collected and lysed with RIPA lysis
buffer (Beyotime Institute of Biotechnology, Shanghai, China).
Equal amount of protein (30 μg) was loaded and separated on
SDS-PAGE, and then transferred to polyvinylidene fluoride membranes
(Millipore, Bedford, MA, USA). The membranes were blocked with 5%
non-fat milk in Tris-buffered saline solution containing 0.05%
Tween-20 and then incubated with antibodies specific for
E-cadherin, vimentin, LC3-I and LC3-II (1:5,000; Cell Signaling
Technology). Following incubation with goat anti-rabbit IgG
(1:1,000; Cell Signaling Technology) at 37°C for 2 h, bound
proteins were visualized using ECL (Pierce) and detected using a
Bio-Imaging System. Protein levels were normalized to GAPDH
(1:10,000; Cell Signaling Technology) and changes were
determined.
Cell proliferation assay
Cell proliferation was evaluated using the Cell
Counting Kit-8 (CCK-8; Beyotime Institute of Biotechnology) and
following the manufacturer’s instructions. Briefly, cells were
plated in 96-well plates in medium containing 10% FBS at
~5×103 cells/well. Then 10 μl CCK-8 solution was added
to each well and incubated for 1 h. The absorbance at 450 nm was
measured using a microplate reader. Results are representative of
three individual experiments in triplicate.
Wound healing assay
Cells (5×105) were seeded in 6-well
plates and cultured in complete medium. After 24 h, when the cells
were grown to 90–100% confluency, a single wound was created in the
center of the well by removing the attached cells with a sterile
200 μl pipette tip. After 24 h of culturing, the cells which
migrated into the wounded area were visualized and photographed
under an inverted microscope. Each experiment was performed at
least three times independently.
Transwell migration and invasion
assay
Transwell invasion assay was performed using
Boyden’s chambers. Cells were planted in the upper chamber
consisting of 8-mm membrane filter inserts coated with Matrigel (BD
Biosciences). The chemoattractant in the lower chamber was
supplemented with medium containing 10% FBS. Cells on the upper
surface were removed by a wet cotton swab after 24 h, and those
attached on the lower side of the membrane were fixed and stained
with crystal violet before counting under a microscope in five
randomly selected fields. Migration assays were performed the same
way as the invasion assays, using Transwell compartment, with the
exception that Matrigel was not included. At least three chambers
from three different experiments were analyzed.
Flow cytometric analysis of apoptosis and
autophagy
Cells (7,000,000) treated with LV-HULC or LV-NC-2 or
5 mM 3-methyladenine (3-MA; Sigma) were seeded into 6-cm tissue
culture dishes for 24 h. For detection of apoptosis, adherent cells
were both collected and resuspended in cold PBS for analysis.
Apoptosis was detected using the Alexa Fluor® 647/7-AAD
apoptosis kit (BioLegend, San Diego, CA, USA) according to the
manufacturer’s instructions. Data were assessed by flow cytometry
(Becton-Dickinson, San Jose, CA, USA).
Statistical analysis
Statistical Program for Social Sciences (SPSS) 20.0
software (IBM, SPSS, Inc., Chicago, IL, USA) was used for the
statistical analysis. Data are expressed as mean ± standard
deviation (SD) from at least three separate experiments.
Statistical analyses were performed with the Student’s t-test. ROC
curve was established to evaluate the diagnostic value for
differentiating between GC tissues and normal tissues. Differences
were considered to be statistically significant at P<0.05.
Results
HULC is upregulated in GC tissues and
cell lines
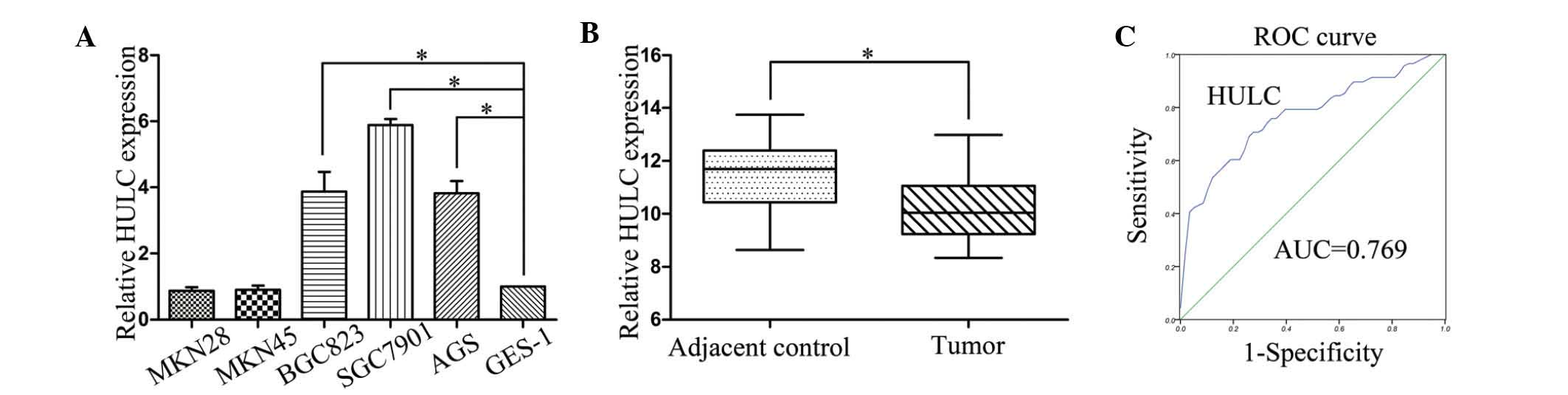
To assess the role of HULC in GC progression, we
first examined HULC expression levels in the GC cell lines and the
human gastric epithelial mucosa cell line GES-1 using qRT-PCR. As
presented in Fig. 1A, the
expression of HULC was increased in three GC cell lines (SGC7901,
BGC823 and AGS) relative to the expression in the human gastric
epithelial mucosa cell line GES-1, but there was no significant
difference for MKN28 and MKN45. Next, qRT-PCR assays were further
developed to quantify HULC in 58 pairs of GC tissues and
pair-matched adjacent normal tissues. HULC levels were markedly
upregulated in cancerous tissues compared with noncancerous tissues
(Fig. 1B). Then, we verified that
expression of HULC was significantly correlated with lymph node
metastasis, distant metastasis and TNM stages. However, we did not
find any association between HULC expression levels and other
clinicopathological features including age, gender,
differentiation, tumor size, tumor location and CEA values
(Table I). Finally, we examined
whether HULC could be used as a marker of GC. We used corresponding
adjacent non-tumorous tissues as a control to produce a ROC curve.
The cut-off value was 10.88. The area under the ROC curve (AUC) was
0.769 (P<0.001). The sensitivity and specificity was 0.707 and
0.724. The Youden index was 0.431 (Fig.
1C).
 | Table IThe relationship between HULC
expression levels (ΔCT) and clinicopathological factors
of 58 GC patients. |
Table I
The relationship between HULC
expression levels (ΔCT) and clinicopathological factors
of 58 GC patients.
| Characteristics | No. of patients
(%) | Mean ± SD | P-value |
|---|
| Age (years) |
| ≥60 | 32 (55.17) | 10.11±1.08 | 0.87 |
| <60 | 26 (44.83) | 10.16±1.83 | |
| Gender |
| Male | 44 (75.86) | 10.26±1.16 | 0.12 |
| Female | 14 (24.14) | 9.73±0.86 | |
| Diameter (cm) |
| ≥5 (large) | 35 (60.34) | 10.09±1.01 | 0.7 |
| <5 (small) | 23 (39.66) | 10.21±1.27 | |
| Location |
| Cardia or
body | 22 (37.93) | 10.10±1.12 | 0.83 |
| Antrum | 36 (62.07) | 10.16±1.13 | |
|
Differentiation |
| Poor or not | 34 (58.62) | 9.98±1.07 | 0.20 |
| Well or
moderate | 24 (41.38) | 10.36±1.16 | |
| Lymphatic
metastasis |
| Present | 41 (70.69) | 9.83±1.09 | 0.001 |
| Absent | 17 (29.31) | 10.87±0.82 | |
| Distal
metastasis |
| Present | 6 (10.34) | 9.06±0.46 | 0.01 |
| Absent | 52 (89.66) | 10.26±1.10 | |
| AJCC clinical
stage |
| I + II | 20 (34.48) | 10.73±0.91 | 0.003 |
| III + IV | 38 (65.52) | 9.83±1.10 | |
| Serum CEA
value |
| ≥5 μg/l | 38 (65.52) | 10.13±1.14 | 0.95 |
| <5 μg/l | 20 (34.48) | 10.15±1.09 | |
Abnormally expressed HULC promotes the
proliferation of GC cells
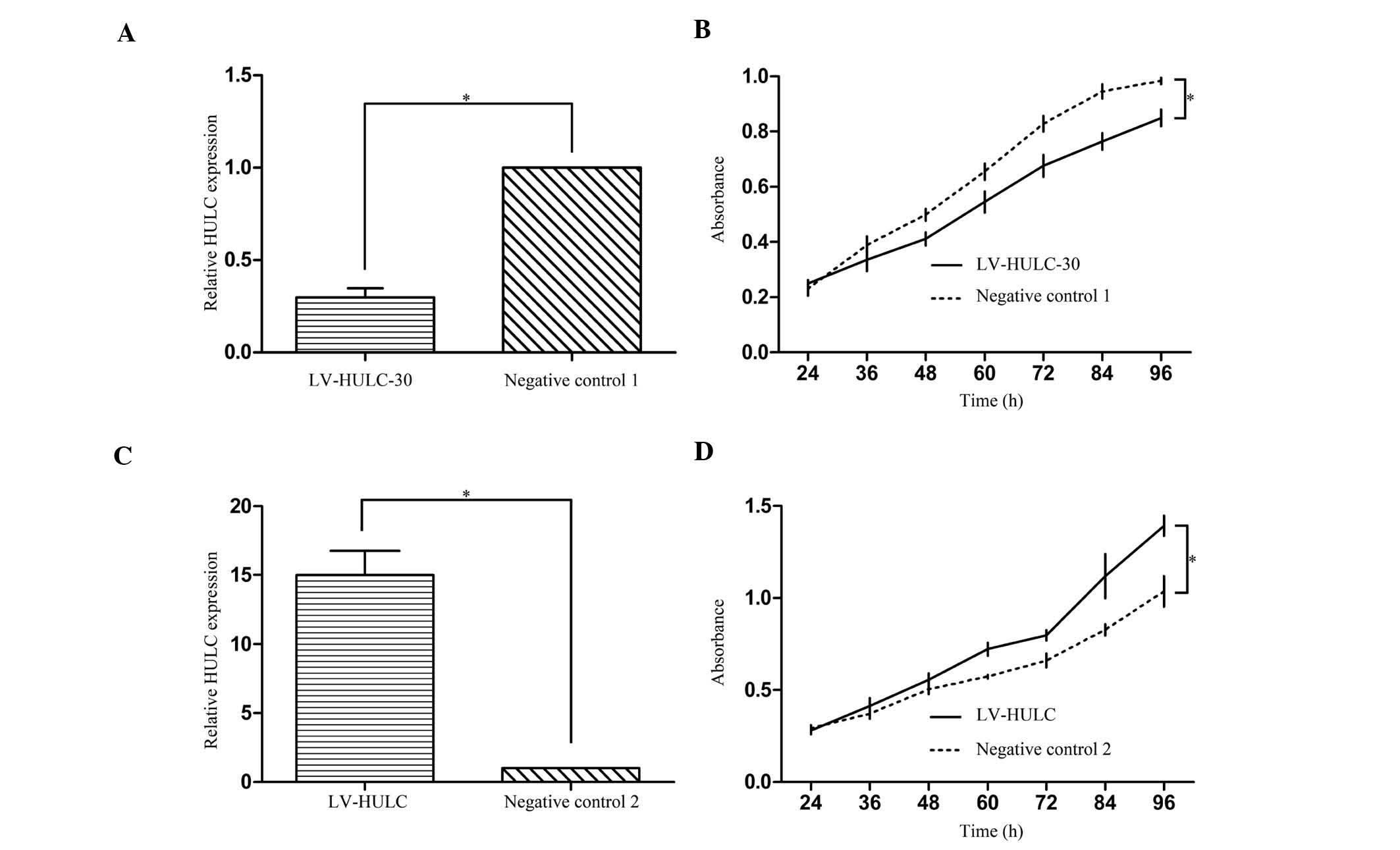
To characterize the functional importance of HULC in
GC tumorigenesis, we infected SGC7901 cells with either HULC
overexpression vector (LV-HULC) or lentivirus-mediated HULC
silencing vector (LV-HULC-30) to increase or knock down the HULC
expression, respectively. qRT-PCR was performed to examine the mRNA
levels of HULC in the derived cells (Fig. 2A and C). CCK-8 assays indicated that
enhanced expression of HULC promoted cell proliferation in SGC7901
cells. The inhibition of HULC, on the other hand, inhibited cell
proliferation (Fig. 2B and D).
These results suggest that HULC plays an important role in
regulating cell proliferation.
Knockdown of HULC suppresses cell
invasion and reverses EMT in GC
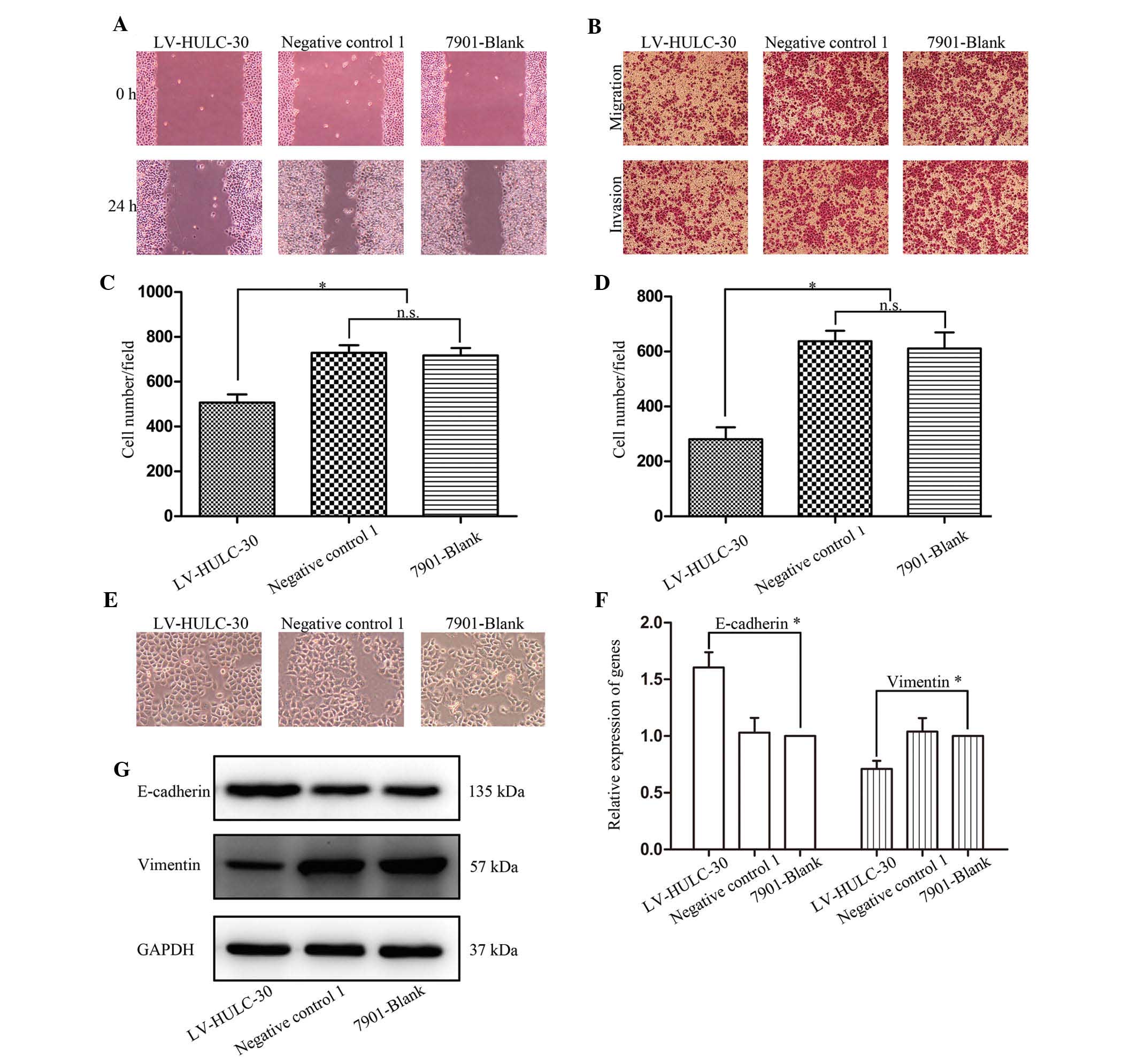
Studies indicated that knockdown of HULC inhibited
the proliferation of SGC7901. We subsequently investigated the
migration and invasion of SGC7901 cells following LV-HULC-30
transfection. The effect of HULC on the migration of SGC7901 cells
determined by wound healing assay demonstrated that knockdown of
HULC significantly inhibited the migration of SGC7901 cells
compared with the SGC7901 only or negative control 1
vector-transfected cells (Fig. 3A).
In analogical results observed in the Transwell assay, in
comparison with original SGC7901 and negative control cells, the
LV-HULC-30 cells showed decreased migration and invasion ability
(Fig. 3B–D).
To further define the role of HULC in the
progression of cell metastasis in GC cells, we then transfected the
LV-HULC-30 vector and assessed the expression of EMT markers at
mRNA and protein levels. As shown in Fig. 3E, silencing of HULC in SGC7901 cells
induced round spheroids with no or few protrusions. Depletion of
HULC expression upregulated E-cadherin and downregulated vimentin
expressions at mRNA and protein levels, respectively (Fig. 3F and G). Our in vitro study
confirmed that HULC positively regulates GC cell migration and
invasion and deletion of HULC reverses EMT, indicating that HULC
could act as a possible regulator of EMT.
Upregulated HULC inhibits cell apoptosis
by activating autophagy
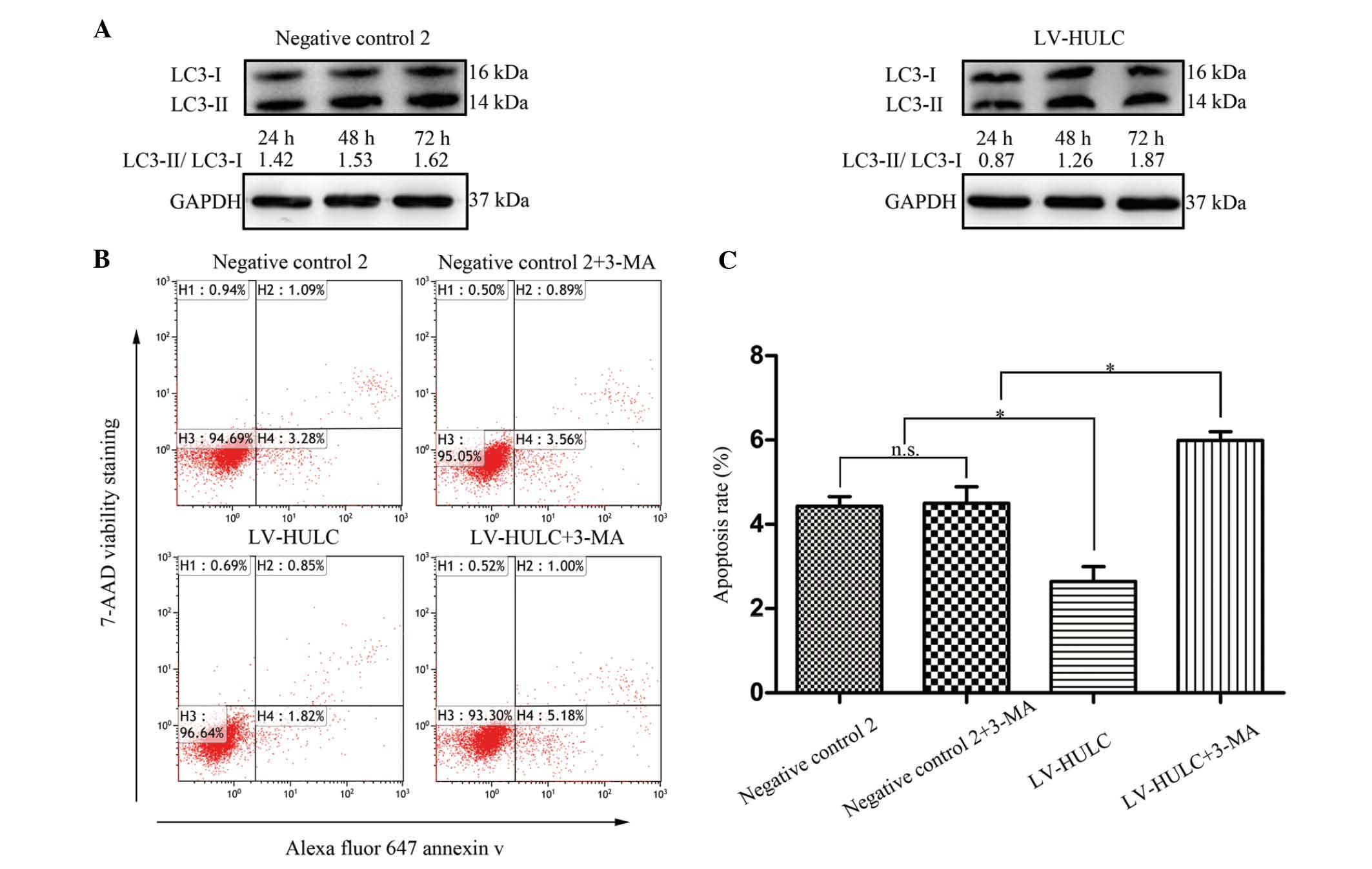
To explore the mechanisms of HULC in regulating
tumorigenesis, we then investigated whether HULC regulates cell
apoptosis-related signals, focusing on autophagy. For that purpose,
we initially determined a change in the expression level of the
microtubule-associated protein 1 light chain 3 (LC3)-II, a marker
for the presence of autophagosomes. As shown in Fig. 4A, HULC overexpression resulted in an
increase in the ratio of LC3-II/LC3-I. These data suggest that
upregulated HULC contributes to autophagy activation in SGC7901.
Subsequently, to further study the role of HULC in the regulation
of cell apoptosis, SGC7901 cells were treated with LV-HULC or 3-MA
(an inhibitor of autophagic sequestration blocker). As depicted in
Fig. 4B and C, enhanced expression
of HULC inhibited SGC7901 cell apoptosis, whereas autophagy
inhibition increased cell apoptosis in SGC7901 cells treated with
LV-HULC.
Discussion
Emerging evidence suggests that lncRNAs play a
crucial role in the modulation of tumor behavior through various
complex mechanisms such as modulating gene transcription and
epigenetic signaling; however, limited data are available on the
expression and function of lncRNAs in GC (14–17).
Hence, in the present study, we tested the expression of a novel
lncRNA HULC in GC tissues and cell lines. We also identified the
function of HULC in GC cells by applying gain- and loss-of-function
approaches. Our results demonstrated that HULC is upregulated in GC
tissues in comparison with the adjacent normal gastric tissues. Of
note, its level was significantly associated with lymph node
metastasis, distant metastasis and TNM stages. Alteration of HULC
expression provided further functional evidence, supporting the
stimulatory effect of HULC on GC via enhancing aggressive
biological behavior of cancer cells. There were certain limitations
in the present study; all the patients enrolled were from a single
institution and the number of samples was not sufficient to make
subgroup analysis. For example, only six patients were at M1
status. In future studies, we will expand the samples for further
investigation and seek to elucidate the association between HULC
expression and overall survival of GC patients since the present
study was limited to patients who underwent complete resection and
the follow-up period after surgery was short.
Metastasis is a complex and multistep process, which
has to be divided into 2 phases; namely, physical translocation of
a tumor cell from the primary tumor to a distant tissue to seed and
colonization of disseminated tumor cell in the tissue (18). A crucial mechanism by which
carcinoma cells enhance their invasive capacity is the dissolution
of intercellular adhesions and the acquisition of a more motile
mesenchymal phenotype as part of an epithelial-to-mesenchymal
transition (EMT) (19). During EMT,
epithelial markers that are downregulated during this process
include E-cadherin, ZO-1 and MUC1 (20,21).
Molecules that are upregulated in this process include the
transcription factors Snail, Slug, Twist as well as N-cadherin and
vimentin (22,23). To determine whether these tumor
characteristics initiated by HULC are associated with the EMT, we
focused on hallmarks of the EMT phenotype. Notably, in the present
study, we showed for the first time that HULC is a positive
regulator of EMT. This conclusion was based on the observation that
silencing HULC induced a repertoire of biochemical (increased
E-cadherin and decreased vimentin) and morphological (growth
pattern, decreased formation of lamellipodia) changes that reverse
EMT. Furthermore, we confirmed that downregulation of HULC
expression decreased the invasion and migration ability of SGC7901
cells. These results indicated that alterative expression of HULC,
at least in part, had an impact on the process of EMT by regulating
the expression of E-cadherin and vimentin in GC cells, suggesting
that HULC plays a causative role in epithelial characteristics
weakening. However, the concrete mechanisms of how HULC regulates
E-cadherin and vimentin expression in SGC7901 cells remain to be
clarified in our future studies
Programmed cell death (PCD), including two classical
forms, apoptosis and autophagy, is a crucial mechanism for
maintaining cell homeostasis of multicellular organisms. Autophagy,
termed type II PCD, is generally activated by conditions of
nutrient deprivation, but it has also been associated with a number
of physiological processes including development, differentiation,
infection and cancer (24–27). However, in cancer development,
autophagy has been shown to have a dual role. In some cases,
autophagy is a tumor-suppressive mechanism, but in others,
autophagy promotes tumorigenesis (28–30).
Therefore, one question that has emerged from our studies is
whether autophagy displays a cytotoxic or cytoprotective role.
Autophagosome marker light chain 3 (LC3) was originally identified
as a subunit of microtubule-associated proteins 1A and 1B and was
subsequently found to be similar to the yeast protein
Apg8/Aut7/Cvt5 critical for autophagy (31,32).
The conversion of LC3 to the lower migrating form, LC3-II, has been
used as an indicator of autophagy (33). Studies are beginning to elucidate
the association between autophagy and lncRNA. Ying et
al(34) demonstrated that
downregulated MEG3 promotes cell proliferation by activating
autophagy in bladder cancer. In the present study, we provided a
line of evidence that HULC may be involved in a molecular switch
mechanism between apoptosis and autophagy in SGC7901 cells.
However, despite data from the present study, several issues remain
to be clarified, such as the relationship between autophagy and
apoptosis, especially in cancer cells, the role of PI3K-Akt-mTOR
signaling pathway in autophagy, and the parameters that determine
whether autophagy is pro-survival or pro-death, tumor promoting or
tumor suppressive. Future studies are required to better understand
the function of this novel gene and its role in GC tumorigenesis
in vivo.
In the present study, for the first time, we found
that high expression levels of HULC, a cancer-related lncRNA,
correlated clinically with GC progression. Additionally, HULC
contributed to the malignant phenotype of GC cells through its
regulation of diverse cellular processes, including proliferation,
apoptosis, migration and invasion. Therefore, the above findings
not only suggest a useful candidate molecular marker for GC and an
indicator for advanced-stage GC, but they also provide new insights
into the role of lncRNA in cancer biology.
Acknowledgements
The present study was supported by the Department of
Health of the Jiangsu Province Fund.
Abbreviations:
|
lncRNA
|
long non-coding RNA
|
|
GC
|
gastric cancer
|
|
HULC
|
highly upregulated in liver cancer
|
|
AJCC
|
American Joint Committee on Cancer
|
|
EMT
|
epithelial-to-mesenchymal
transition
|
|
3-MA
|
3-methyladenine
|
|
mRNA
|
messenger RNA
|
References
|
1
|
Konishi H, Ichikawa D, Komatsu S, et al:
Detection of gastric cancer-associated microRNAs on microRNA
microarray comparing pre- and post-operative plasma. Br J Cancer.
106:740–747. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cheng Y, Jin Z, Agarwal R, et al: LARP7 is
a potential tumor suppressor gene in gastric cancer. Lab Invest.
92:1013–1019. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Costa FF: Non-coding RNAs: new players in
eukaryotic biology. Gene. 357:83–94. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Batista PJ and Chang HY: Long noncoding
RNAs: cellular address codes in development and disease. Cell.
152:1298–1307. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Panzitt K, Tschernatsch MM, Guelly C, et
al: Characterization of HULC, a novel gene with striking
up-regulation in hepatocellular carcinoma, as noncoding RNA.
Gastroenterology. 132:330–342. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu D, Yang F, Yuan JH, et al: Long
noncoding RNAs associated with liver regeneration 1 accelerates
hepatocyte proliferation during liver regeneration by activating
Wnt/β-catenin signaling. Hepatology. 58:739–751. 2013.PubMed/NCBI
|
|
7
|
Wang J, Liu X, Wu H, et al: CREB
up-regulates long non-coding RNA, HULC expression through
interaction with microRNA-372 in liver cancer. Nucleic Acids Res.
38:5366–5383. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Matouk IJ, Abbasi I, Hochberg A, Galun E,
Dweik H and Akkawi M: Highly upregulated in liver cancer noncoding
RNA is overexpressed in hepatic colorectal metastasis. Eur J
Gastroenterol Hepatol. 21:688–692. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li J, Yang B, Zhou Q, et al: Autophagy
promotes hepatocellular carcinoma cell invasion through activation
of epithelial-mesenchymal transition. Carcinogenesis. 34:1343–1351.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ledford H: Cancer theory faces doubts.
Nature. 472:2732011. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Enomoto M, Tsuchida A, Miyazawa K, et al:
Vitamin K2-induced cell growth inhibition via autophagy formation
in cholangiocellular carcinoma cell lines. Int J Mol Med.
20:801–808. 2007.PubMed/NCBI
|
|
12
|
Lv Q, Hua F and Hu ZW: DEDD, a novel tumor
repressor, reverses epithelial-mesenchymal transition by activating
selective autophagy. Autophagy. 8:1675–1676. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Akalay I, Janji B, Hasmim M, et al:
Epithelial-to-mesenchymal transition and autophagy induction in
breast carcinoma promote escape from T-cell-mediated lysis. Cancer
Res. 73:2418–2427. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nie Y, Liu X, Qu S, Song E, Zou H and Gong
C: Long non-coding RNA HOTAIR is an independent prognostic marker
for nasopharyngeal carcinoma progression and survival. Cancer Sci.
104:458–464. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang F, Bi J, Xue X, et al: Up-regulated
long non-coding RNA H19 contributes to proliferation of gastric
cancer cells. FEBS J. 279:3159–3165. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang H, Zhong Y, Xie H, et al: Induction
of the liver cancer-down-regulated long noncoding RNA uc002mbe.2
mediates trichostatin-induced apoptosis of liver cancer cells.
Biochem Pharmacol. 85:1761–1769. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wahlestedt C: Targeting long non-coding
RNA to therapeutically upregulate gene expression. Nat Rev Drug
Discov. 12:433–446. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu Q, Huang J, Zhou N, et al: LncRNA
loc285194 is a p53-regulated tumor suppressor. Nucleic Acids Res.
41:4976–4987. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lv Q, Wang W, Xue J, et al: DEDD interacts
with PI3KC3 to activate autophagy and attenuate
epithelial-mesenchymal transition in human breast cancer. Cancer
Res. 72:3238–3250. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shin NR, Jeong EH, Choi CI, et al:
Overexpression of Snail is associated with lymph node metastasis
and poor prognosis in patients with gastric cancer. BMC Cancer.
12:5212012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lin X, Shang X, Manorek G and Howell SB:
Regulation of the epithelial-mesenchymal transition by claudin-3
and claudin-4. PLoS One. 8:e674962013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Davidson B, Trope CG and Reich R:
Epithelial-mesenchymal transition in ovarian carcinoma. Front
Oncol. 2:332012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zeisberg M and Neilson EG: Biomarkers for
epithelial-mesenchymal transitions. J Clin Invest. 119:1429–1437.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Levine B and Yuan J: Autophagy in cell
death: an innocent convict? J Clin Invest. 115:2679–2688. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
He H, Zang LH, Feng YS, et al: Physalin A
induces apoptotic cell death and protective autophagy in HT1080
human fibrosarcoma cells. J Nat Prod. 76:880–888. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ye L, Zhao X, Lu J, Qian G, Zheng JC and
Ge S: Knockdown of TIGAR by RNA interference induces apoptosis and
autophagy in HepG2 hepatocellular carcinoma cells. Biochem Biophys
Res Commun. 437:300–306. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Oh SY, Choi SJ, Kim KH, Cho EY, Kim JH and
Roh CR: Autophagy-related proteins, LC3 and Beclin-1, in placentas
from pregnancies complicated by preeclampsia. Reprod Sci.
15:912–920. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mann SS and Hammarback JA: Molecular
characterization of light chain 3. A microtubule binding subunit of
MAP1A and MAP1B. J Biol Chem. 269:11492–11497. 1994.PubMed/NCBI
|
|
32
|
Lang T, Schaeffeler E, Bernreuther D,
Bredschneider M, Wolf DH and Thumm M: Aut2p and Aut7p, two novel
microtubule-associated proteins are essential for delivery of
autophagic vesicles to the vacuole. EMBO J. 17:3597–3607. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kabeya Y, Mizushima N, Yamamoto A,
Oshitani-Okamoto S, Ohsumi Y and Yoshimori T: LC3, GABARAP and
GATE16 localize to autophagosomal membrane depending on form-II
formation. J Cell Sci. 117:2805–2812. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ying L, Huang Y, Chen H, et al:
Downregulated MEG3 activates autophagy and increases cell
proliferation in bladder cancer. Mol Biosyst. 9:407–411. 2013.
View Article : Google Scholar : PubMed/NCBI
|