Article
Cholesterol synthesis inhibitor RO 48-8071 suppresses transcriptional activity of human estrogen and
androgen receptor
- Authors:
- Benford Mafuvadze
- Yayun Liang
- Salman M. Hyder
-
View Affiliations / Copyright
Affiliations:
Department of Biomedical Sciences and Dalton Cardiovascular Research Center, University of Missouri, Columbia, MO 65211, USA
-
Pages:
1727-1733
|
Published online on:
July 15, 2014
https://doi.org/10.3892/or.2014.3332
- Expand metrics +
Metrics:
Total
Views: 0
(Spandidos Publications: | PMC Statistics:
)
Metrics:
Total PDF Downloads: 0
(Spandidos Publications: | PMC Statistics:
)
This article is mentioned in:
Abstract
Breast cancer cells express enzymes that convert cholesterol, the synthetic precursor of steroid hormones, into estrogens and androgens, which then drive breast cancer cell proliferation. In the present study, we sought to determine whether oxidosqualene cyclase (OSC), an enzyme in the cholesterol biosynthetic pathway, may be targeted to suppress progression of breast cancer cells. In previous studies, we showed that the OSC inhibitor RO 48-8071 (RO) may be a ligand which could potentially be used to control the progression of estrogen receptor-α (ERα)-positive breast cancer cells. Herein, we showed, by real-time PCR analysis of mRNA from human breast cancer biopsies, no significant differences in OSC expression at various stages of disease, or between tumor and normal mammary cells. Since the growth of hormone-responsive tumors is ERα-dependent, we conducted experiments to determine whether RO affects ERα. Using mammalian cells engineered to express human ERα or ERβ protein, together with an ER-responsive luciferase promoter, we found that RO dose-dependently inhibited 17β-estradiol (E2)-induced ERα responsive luciferase activity (IC50 value, ~10 µM), under conditions that were non-toxic to the cells. RO was less effective against ERβ-induced luciferase activity. Androgen receptor (AR) mediated transcriptional activity was also reduced by RO. Notably, while ERα activity was reduced by atorvastatin, the HMG-CoA reductase inhibitor did not influence AR activity, showing that RO possesses broader antitumor properties. Treatment of human BT-474 breast cancer cells with RO reduced levels of estrogen-induced PR protein, confirming that RO blocks ERα activity in tumor cells. Our findings demonstrate that an important means by which RO suppresses hormone-dependent growth of breast cancer cells is through its ability to arrest the biological activity of ERα. This warrants further investigation of RO as a potential therapeutic agent for use against hormone-dependent breast cancers.
View Figures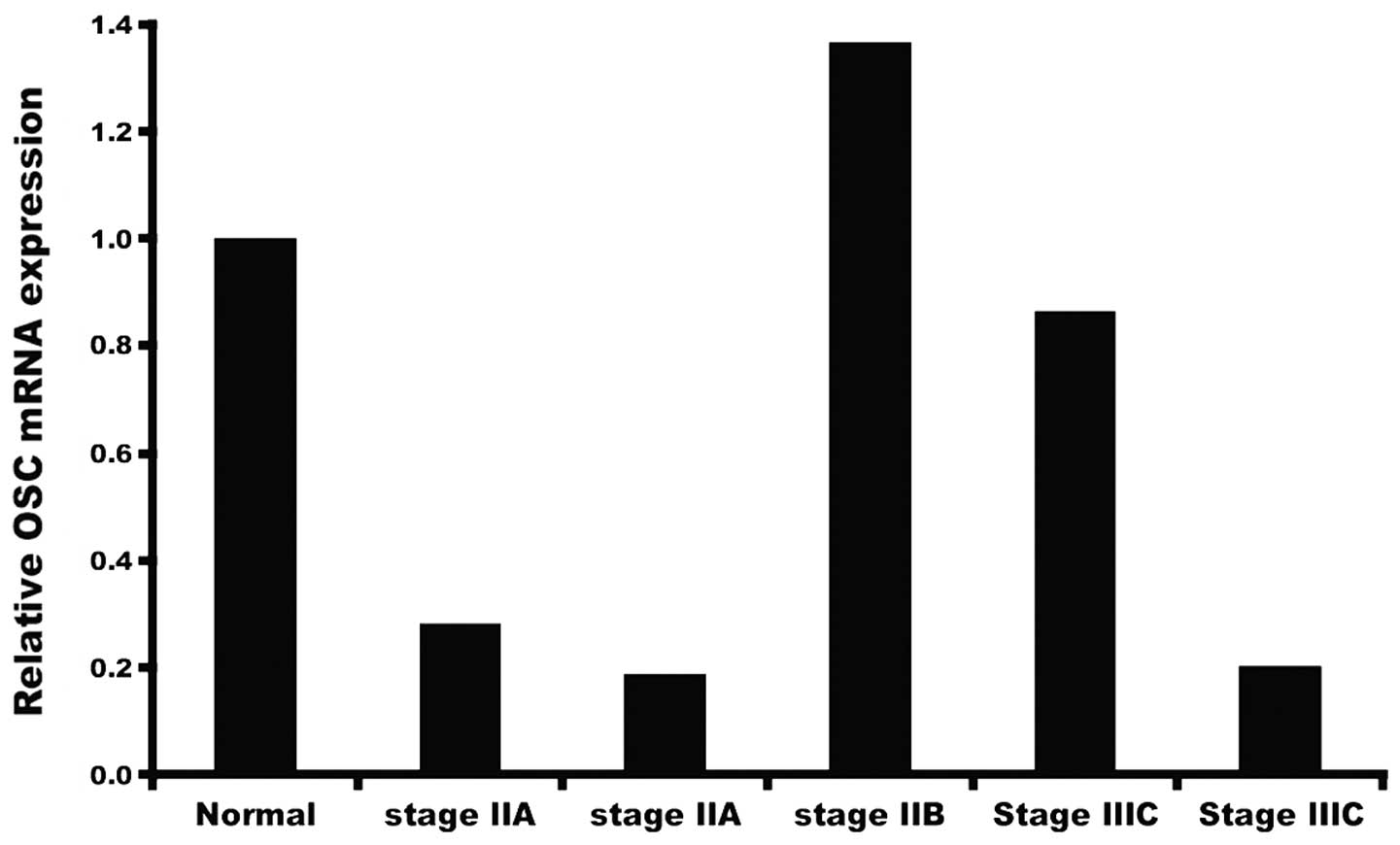 |
Figure 1
|
 |
Figure 2
|
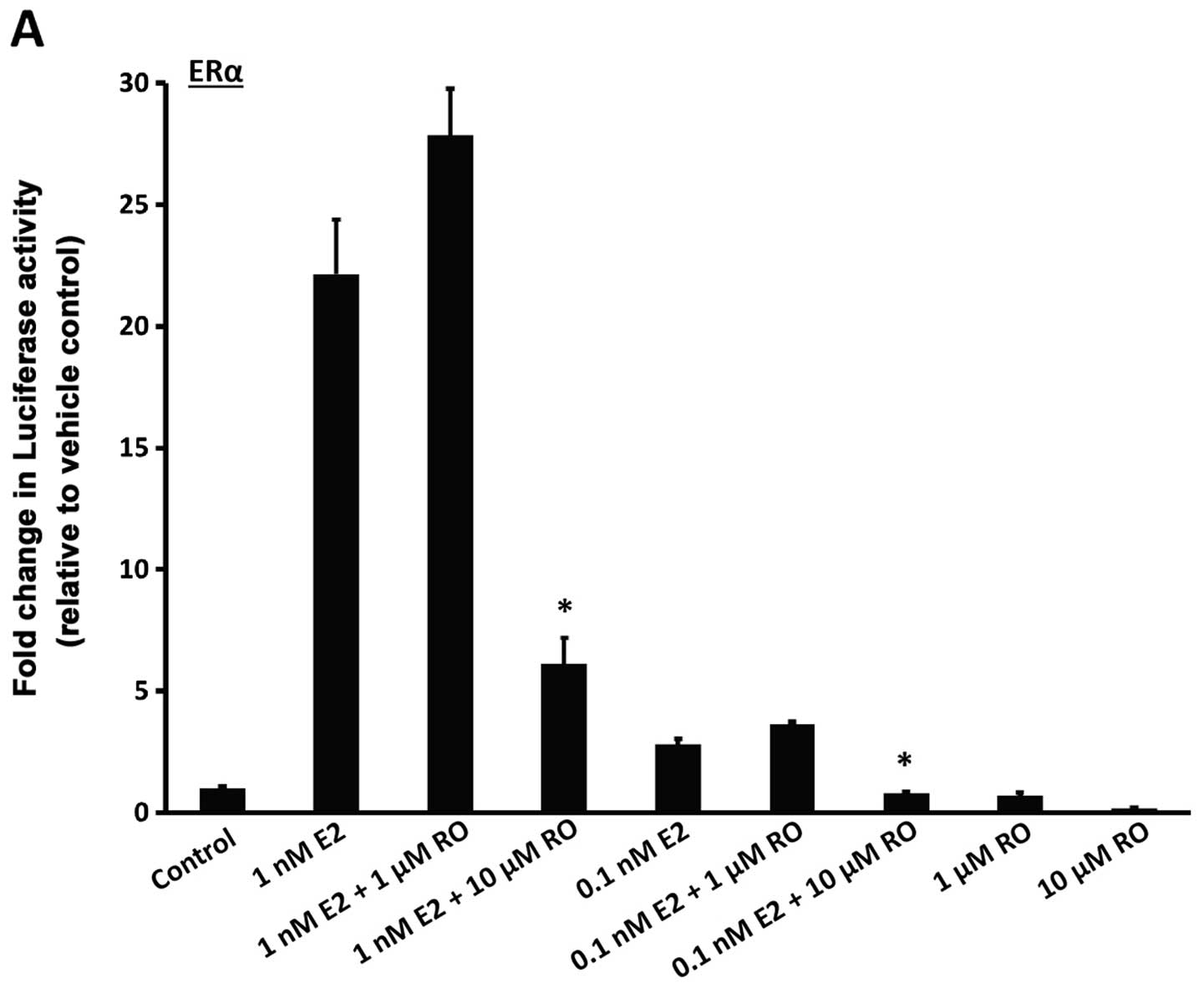 |
Figure 3
|
 |
Figure 4
|
 |
Figure 5
|
 |
Figure 6
|
View References
|
1
|
Sivente-Poirot S and Poirot M: Cholesterol
metabolism and cancer: the good, the bad and the ugly. Curr Opin
Pharmacol. 12:673–676. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Danilo C and Frank PG: Cholesterol and
breast cancer development. Curr Opin Pharmacol. 12:677–682. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wu Q, Ishikawa T, Sirianni R, Tang H, et
al: 27-Hydroxycholesterol promotes cell-autonomous, ER-positive
breast cancer. Cell Rep. 5:637–645. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Campagnoli C, Pasanisi P, Castellano I,
Abbà C, Brucato T and Berrino F: Postmenopausal breast cancer,
androgens, and aromatase inhibitors. Breast Cancer Res Treat.
139:1–11. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Umetani M, Domoto H, Gormley AK, Yuhanna
IS, et al: 27-Hydroxycholesterol is an endogenous SERM that
inhibits the cardiovascular effects of estrogen. Nat Med.
13:1185–1192. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
DuSell CD, Umetani M, Shaul PW,
Mangelsdorf DJ and McDonnell DP: 27-Hydroxycholesterol is an
endogenous selective estrogen receptor modulator. Mol Endocrinol.
22:65–77. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Michikawa M and Yanagisawa K: Inhibition
of cholesterol production but not of nonsterol isoprenoid products
induces neuronal cell death. J Neurochem. 72:2278–2285. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Grinter SZ, Liang Y, Huang SY, Hyder SM
and Zou X: An inverse docking approach for identifying new
potential anticancer targets. J Mol Graph Model. 29:795–799. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Geisler J, Suzuki T, Helle H, Miki Y, et
al: Breast cancer aromatase expression evaluated by the novel
antibody 677: correlations to intra-tumor estrogen levels and
hormone receptor status. J Steroid Biochem Mol Biol. 118:237–241.
2010. View Article : Google Scholar
|
|
10
|
Shah PD, Gucalp A and Traina TA: The role
of the androgen receptor in triple-negative breast cancer. Womens
Health. 9:351–360. 2013.PubMed/NCBI
|
|
11
|
Saceda M, Grunt TW, Colomer R, Lippman ME,
Lupu R and Martin MB: Regulation of estrogen receptor concentration
and activity by an erbB/HER ligand in breast carcinoma cell lines.
Endocrinology. 137:4322–4330. 1996.PubMed/NCBI
|
|
12
|
Klinge CM: Estrogen receptor interaction
with estrogen response elements. Nucleic Acids Res. 29:2905–2919.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen L, Monti S, Juszczynski P, Ouyang J,
et al: SYK inhibition modulates distinct PI3K/AKT-dependent
survival pathways and cholesterol biosynthesis in diffuse large B
cell lymphomas. Cancer Cell. 23:826–838. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fernández C, Martín M, Gómez-Coronado D
and Lasunción MA: Effects of distal cholesterol biosynthesis
inhibitors on cell proliferation and cell cycle progression. J
Lipid Res. 46:920–929. 2005.PubMed/NCBI
|
|
15
|
Bachelot T, McCool R, Duffy S, Glanville
J, et al: Comparative efficacy of everolimus plus exemestane versus
fulvestrant for hormone-receptor-positive advanced breast cancer
following progression/recurrence after endocrine therapy: a network
meta-analysis. Breast Cancer Res Treat. 143:125–133. 2014.
View Article : Google Scholar
|
|
16
|
Jensen EV and Jordan VC: The estrogen
receptor: a model for molecular medicine. Clin Cancer Res.
9:1980–1989. 2003.
|














